


I. Introduction
Actions of acetylcholine in the periphery are the result of activation of either the ionotropic nicotinic receptor or the metabotropic muscarinic receptor. In the mammalian central nervous system (CNS)c, both nicotinic and muscarinic receptor subtypes are present on neurons, although there is as yet very limited evidence for a physiological role for nicotinic receptors in synaptic function in the mammalian brain (Role and Berg, 1996). In the periphery, among other effects, muscarinic receptors mediate smooth muscle contraction, glandular secretion, and modulation of cardiac rate and force. In the CNS, there is evidence that muscarinic receptors are involved in motor control, temperature regulation, cardiovascular regulation, and memory. Interest in the classification of muscarinic receptors involved in functions at different locations has been heightened by the potential therapeutic application of selective agents in areas such as Alzheimer’s disease, Parkinson’s disease, asthma, analgesia, and disorders of intestinal motility and cardiac and urinary bladder function.
Historically, the first indications of the existence of muscarinic receptor subtypes were the cardioselective actions of gallamine (Riker and Wescoe, 1951) and the sympathetic ganglionic stimulant behavior of (4-hydroxy-2-butynyl)-1-trimethylammonium-m-chlorocarbanilate chloride (McN-A-343) (Roszkowski, 1961). Subsequently, Barlow et al. (1976) demonstrated significant differences in the pharmacological properties of ileal and atrial muscarinic receptors. The introduction of pirenzepine, a drug used in the treatment of peptic ulcer disease, had a major role in the appreciation of the existence of muscarinic receptor subtypes. Its selectivity in binding (Hammeret al., 1980) and functional studies (Brown et al., 1980; Hammer and Giachetti, 1982) provided an explanation of its in vivo selectivity. It seemed as if there were at least three subclasses of muscarinic receptors (Birdsall and Hulme, 1983).
Knowledge of the potential functions and roles of muscarinic receptors and their subtypes was advanced significantly by the cloning of five mammalian genes encoding muscarinic receptors. Their expression in cell lines has resulted in the generation of much information on potential coupling mechanisms, the production of selective antibodies, and the ability to localize sites of expression of messenger ribonucleic acids (mRNAs) encoding the receptors. Notwithstanding this progress, the definition of the receptor subtype(s) involved in a particular functional response still is accomplished best by the use of selective pharmacological tools. Although it is true that the presence of receptor gene-specific mRNA, or receptor-specific immunoreactivity can provide evidence supporting the pharmacological demonstration of a functional receptor subtype, it is equally true that firm pharmacological evidence for the involvement of a particular subtype stands alone: lack of supporting molecular data is not sufficient justification for rejecting the pharmacological evidence.
This review describes the naming and classification of muscarinic receptors in line with the guidelines of NC-IUPHAR. The main features of muscarinic receptor structure, pharmacology, and function that provide the basis for the classification are summarized. Because this review does not set out to be a comprehensive review of the literature, readers seeking more detail should refer to the many relevant reviews in the field (table 1).
Recent reviews on muscarinic receptors
II. Nomenclature
The previous nomenclature was recommended by the Fourth Symposium on Subtypes of Muscarinic Receptors and the NC-IUPHAR Subcommittee on Muscarinic Acetylcholine Receptors (Levine and Birdsall, 1989). This nomenclature used a lower case ‘m’ followed by its number to describe a subtype when the muscarinic receptor gene or gene product was known unambiguously (for example, by transfection into a nonexpressing cell line). On the other hand, when the properties of the receptor were defined by its pharmacology and the molecular species contributing to these properties was not known unambiguously the receptor was denoted by ‘M’ and a subscript number (this being the nomenclature used before the cloning of the receptor genes). The aim was that, with the discovery of antagonists of greater selectivity, the dual molecular and pharmacological descriptors of muscarinic receptor subtypes would merge into a single definition.
Based on existing knowledge, summarized in this review, it is now recommended that M1, M2, M3, M4, and M5 be used to describe both the pharmacological subtypes (as defined previously) and the molecular subtypes (defined previously as m1–m5, respectively).
A pharmacological characterization of endogenous M5 receptors in whole-tissue functional studies is still lacking. However, under the revised guidelines of the NC-IUPHAR Committee on Receptor Nomenclature and Drug Classification (Vanhoutte et al., 1998), it is viewed by the above-mentioned committee and the muscarinic receptor subcommittee that the evidence presented in this review is sufficient to define the M5 receptor.
III. Molecular Definition of Subtypes
Cloning of complementary deoxyribonucleic acids for muscarinic receptor genes was spearheaded by the work of Numa and colleagues, who cloned the M1 and M2 genes (Kubo et al., 1986a,b), and was extended by the discovery of the M3, M4, and M5 genes (Bonner et al., 1987, 1988;Peralta et al., 1987). These five genes encode muscarinic receptor proteins (actually glycoproteins) which have the structural features of the seven transmembrane helix G-protein-coupled receptor family. Muscarinic receptor sequences have significant homologies with other members of this receptor superfamily (Hulme et al., 1990). The vertebrate receptor genes cloned so far are intronless within the coding regions and are notably similar across mammalian species (Hall et al., 1993; Eglen et al., 1996; table 2). The chromosomal localization of the human M1−M5 genes are reported to be 11q12–13, 7q35–36, 1q43–44, 11p12–11.2, and 15q26, respectively (Bonner et al., 1991).
Muscarinic receptor genes
In addition to being transfected into a variety of cells of mammalian/amphibian origin, either transiently (e.g., COS cells,xenopus oocytes) or stably (e.g., Chinese hamster ovary, A9L, Y1 cells), the receptors have also been expressed in insect (Sf9) cells (Vasudevan et al., 1995; Hepler et al., 1996), Dictyostelium (Voith and Dingermann, 1995), yeast (Payette et al., 1990; Huanget al., 1992), and Escherichia coli (Curtis and Hulme, 1997).
The apparent similarities between muscarinic receptor subtypes across species, at least at the amino acid level, are potentially misleading. Precedents from other types of receptors clearly show that a minor sequence difference between receptors in different species can have a major impact on their pharmacological profiles. For example, a single amino acid difference between the human and rat 5-hydroxytryptamine1B receptor is manifest as a major difference in ligand affinities (reviewed by Kenakin, 1996).
In muscarinic receptors the existing evidence is that the pharmacology is not significantly different between mammalian receptor homologs (insofar as it has been studied; see Hall et al., 1993, for example). However, the possibility cannot be excluded that novel ligands may make different interactions with regions of the receptor where there are sequence differences between species. Thus, there is a very good argument for using cloned human receptor genes expressed in cell lines to derive information about potential human therapeutic agents, rather than attempting to use a receptor system from a nonhuman species that provides the ‘best’ pharmacological match to the human profile.
In common with most members of the subgroup of G-protein-coupled receptor family whose ligand-recognition site binds small molecules, the major features of muscarinic receptor structure are:
-
The ligand recognition site is within the outer half of the membrane-embedded part of the protein.
-
The transmembrane segments are probably α-helices, three oriented approximately perpendicular to the membrane, four at a more acute angle (Baldwin et al., 1997).
-
There are two conserved cysteine residues that form a disulfide bond between the first and third extracellular loops (Kurtenbach et al., 1990; Savarese et al., 1992).
-
There is a conserved triplet of amino acids (Asp Arg Tyr) at the cytoplasmic interface of TMIII with the second intracellular loop, which is important for both the expression and function of the receptor (Zhu et al., 1994; Jones et al., 1995; Luet al., 1997).
-
The carboxy-terminus is on the intracellular side of the membrane because antibodies to the C-terminus sequences recognize cell-surface receptors only when cells are permeabilized (e.g., Lu et al., 1997). Palmitoylation of a cysteine residue at the carboxy-terminus of M2 receptors (C457) occurs in cells, but it is not an absolute requirement for the interaction with G-proteins even though function is enhanced (Hayashi and Haga, 1997).
-
There are one or more glycosylation sites on the N-terminus, but glycosylation apparently is not crucial for receptor expression and function, at least for the M2 receptor (Van Koppen and Nathanson, 1990). This study provides evidence for the extracellular location of the N-terminus.
In whole-cell studies, serine and threonine residues in the large postulated third intracellular loop of muscarinic receptors are phosphorylated by endogenous protein kinases (Pals-Rylaarsdam and Hosey, 1997), which indicates that the large i3 loop of the M2 receptor is indeed intracellular.
The projected three-dimensional structure of the receptors is expected to have more in common with that of rhodopsin, a G-protein-coupled receptor (Baldwin, 1993), than that of bacteriorhodopsin, a proton pump and another seven-transmembrane helix protein whose medium-resolution, three-dimensional structure is known (Henderson et al., 1990). The latter structure has been assumed to be an appropriate model for G-protein-coupled receptors in some studies.
As with the cationic amine receptors, all muscarinic receptors have an Asp residue in the distal N-terminal part of the third transmembrane domain which is thought to interact with the polar headgroup of amine ligands, including acetylcholine. This residue is alkylated specifically by the agonist, acetylcholine mustard, and the antagonist, propylbenzilylcholine mustard (Curtis et al., 1989; Spaldinget al., 1994). Uncharged muscarinic antagonists also will bind to muscarinic receptors but with lower affinity (e.g., Houet al., 1996). Residues important for the binding of these ligands have not been defined. Sites involved in binding different receptor-selective antagonists are probably quite diverse, depending on the antagonist (Wess et al., 1990, 1992; Matsui et al., 1995). It presently is difficult to identify amino acids that interact directly with the antagonists and to distinguish such residues from those which, when mutated, affect antagonist binding by indirectly changing the conformation of the binding site (or sites). Blümlet al. (1994) suggested that a conserved Asn residue in the sixth transmembrane segment is very important for the binding of certain subclasses of antagonists, notably atropine-like analogs.
An important molecular distinction between the different muscarinic receptor subtypes is the sequence divergence in the postulated third internal (i3) loops between the M1/M3/M5sequences compared with the M2/M4 sequences (Hulmeet al., 1990; Wess, 1996; Wess et al., 1997) that probably determines the quite specific coupling preferences of these two groups (Wess, 1993). More recently, further mutational studies have shown that coupling specificity of the M3receptor is determined by a small set of amino acids in the TMIII/i2 loop interface and in the membrane-proximal portions of the i3 loop (Blin et al., 1995). Similar studies with the M2 receptor have identified a four amino acid sequence (Val Thr Ile Leu) located at the interface of the i3 loop and TM VI which couples this receptor to its target G-proteins (Gαo/i) (Wess et al., 1997); this sequence is also present in other receptors with high coupling preference for these G-protein α subunits (Liu et al., 1995).
Muscarinic receptor-G-protein interactions and activation can occur in the absence of agonists. This can be demonstrated in binding studies carried out at low ionic strengths in the presence of Mg2+ (Hulme et al., 1981), as the existence of constitutive activity observed in functional studies by overexpression of the receptor (Vogel et al., 1995) or G-protein (Burstein et al., 1995), and also in a patch-clamp study in atrial cells (Soejima and Noma, 1984). Muscarinic antagonists regulate ‘basal’ activity [elevate cyclic adenosine monophosphate (cAMP) levels, inhibit inositol 1,4,5-trisphosphate production, or close atrial K+ channels, depending on subtype], but presently there is no evidence of differential effects of antagonists on their maximal effects. The existence of significant constitutive activity in vivo is not known, nor is there any therapeutic use of muscarinic antagonists acting as ‘inverse agonists.’
Another feature of muscarinic receptors is the presence of a specific binding site which, when occupied by ligands, can modify the binding and behavior of ligands binding to the acetylcholine recognition site. This allosteric site has been characterized by equilibrium and kinetic binding studies (Stockton et al., 1983; Ellis et al., 1991; Proska and Tucek, 1995; Lazareno and Birdsall, 1995) as well as in functional studies (Ehlert, 1988; Lazareno and Birdsall, 1995). Studies of chimeric and mutant receptors (Elliset al., 1993; Leppik et al., 1994; Matsuiet al., 1995) have begun to identify amino acids and receptor domains that may constitute the binding site.
IV. Pharmacological Definition of Subtypes
Pharmacological characterization of muscarinic receptor subtypes has long been dogged by a complete lack of agonists with any selectivity, and a lack of antagonists with very high selectivity for any single receptor subtype. Additionally, cells frequently coexpress more than one subtype, further adding to the difficulty of assigning a functional response to a single receptor subtype.
A. Antagonists
The definition of antagonist affinities for the five muscarinic receptors has been aided greatly by the use of radioligand binding techniques, with ligands such as [3H]pirenzepine and [3H]N-methylscopolamine, in combination with membrane preparations from cells transfected with the gene for a particular receptor, and thereby expressing a single receptor subtype. Affinity constants obtained from these experiments have been remarkably comparable with apparent affinity constants determined in functional experiments using Arunlakshana-Schild analysis (reviewed by Caulfield, 1993; table 3) or any of the acceptable variants (Lazareno and Birdsall, 1993a,b). Nevertheless, serious errors can be made in estimates of antagonist affinity constants by inappropriate design of radioligand binding experiments (Hulme and Birdsall, 1992). It also is possible to perturb grossly the antagonist structure-binding relationships by carrying out binding assays under conditions (particularly ionic strength, temperature, and solubilization in detergents) that differ substantially from those of the functional assays (Pedder et al., 1991). Similarly, the estimation of apparent affinity constants for antagonists in functional experiments can be flawed unless care is taken in experimental design (Caulfield, 1997).
Antagonist affinity constants (log affinity constants or pKBvalues) for mammalian muscarinic receptors3-a
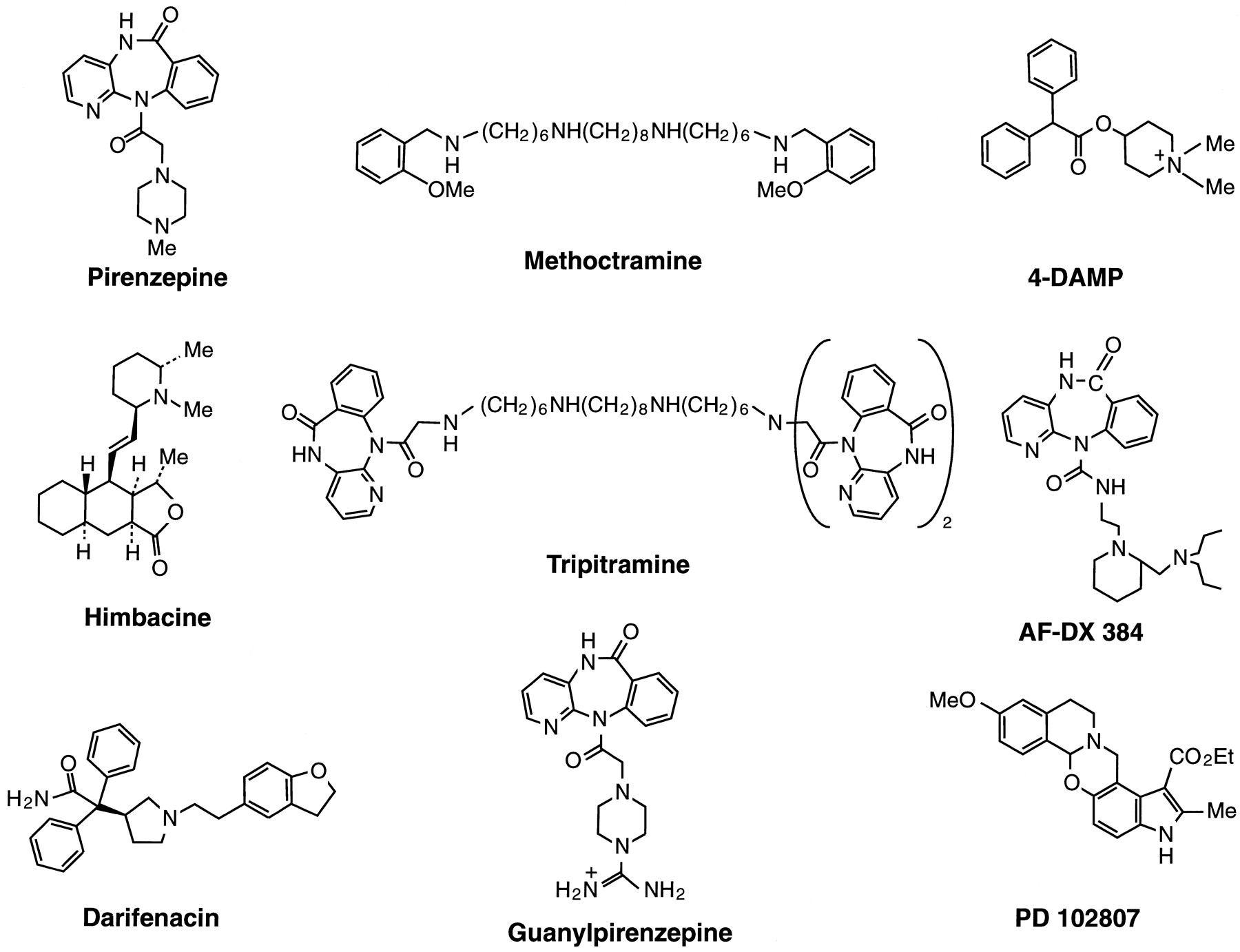
Table 3 summarizes the (log) affinity constants (and apparent affinity constants in functional studies) of atropine, some selective antagonists, and the two most selective muscarinic toxins for the different muscarinic receptor subtypes. The chemical structures of the selective nonpeptide antagonists are given in figure1. The data in table 3 include information from binding studies and functional studies (e.g., Lazareno and Birdsall, 1993a,b) on cloned receptors, and on natively expressed receptors (where the subtype involved has been defined satisfactorily; see Section IV.C.1–4).

Muscarinic antagonists.
The ranges of values represent the differences in values determined by different laboratories, and the inference is that when a value for a given antagonist lies outside the range for a given receptor, then that receptor is not detected (in a binding study) or does not mediate the response measured (in a functional study). Less well-characterized, but nevertheless potentially useful molecules are also included in table 3(e.g., guanylpirenzepine, darifenacin, and PD102807). Ranges of values for these antagonists are not given and caution should be taken in interpreting data obtained with these compounds until more information is available from further studies on cloned receptors and from a wider range of functional studies. These antagonists are of special interest because they have been reported to have a considerably higher affinity for one subtype over all other subtypes.
Also included in table 3 are two muscarinic snake toxins, MT3 and MT7. These toxins are two of several components of the venom of the green (Dendroaspis angusticeps) and black mamba (Dendroaspis polylepis) which have a high affinity for muscarinic receptors. These two toxins show great promise in that their apparently high subtype selectivity should be extremely useful in muscarinic receptor classification. However, whether they and the other muscarinic toxins have additional actions on nonmuscarinic systems has yet to be determined.
It is evident from table 3 that convincing evidence for the involvement of a particular receptor necessitates the use of more than one antagonist. It must be emphasized that, because of the lack of very high subtype selectivity of any single antagonist (except perhaps for the M1 receptor selectivity of MT7 toxin), it should not be acceptable to state that a particular receptor is involved when the experimental design involves procedures such as “block” of an agonist response by a fixed high concentration of antagonist, determination of IC50 values, “potency rank orders” of antagonists, or any other design that does not involve determination of affinity constants (or apparent affinity constants). Similarly, descriptions of compounds such as pirenzepine as “M1 receptor-selective” should be resisted. Most importantly, it is not advisable to use a “majority verdict” approach to receptor classification. Thus, if even one antagonist has a pKB that is significantly different from its pKB at one defined (preferably cloned) receptor, say M1, it is not acceptable to ignore that discrepancy and to classify the receptor as M1 without an experimental explanation for the discrepancy.
B. Allosteric Agents
Experiments with the allosteric compound gallamine (which acts as a selective allosteric antagonist at M2receptors) gave important early indications of muscarinic receptor heterogeneity (Riker and Wescoe, 1951; Clark and Mitchelson, 1976;Stockton et al., 1983). The effects of allosteric ligands can be detected in studies on cloned and expressed human muscarinic receptors, both in radioligand binding studies (e.g., Elliset al., 1991; Lazareno and Birdsall, 1995), and in functional studies (Lazareno and Birdsall, 1995). It is expected that the use of selective ligands acting at this site will be useful in muscarinic receptor classification, although the interpretation of their effects is complex (Lazareno and Birdsall, 1995).
Several allosteric ligands have been described (see e.g., Birdsallet al., 1987; Lee and El-Fakahany, 1991). Most ligands exhibit negative cooperativity with agonists and antagonists. Alcuronium and strychnine, however, are allosteric agents that are positively cooperative with the antagonist, N-methylscopolamine, at one or more muscarinic receptor subtypes (Tucek et al., 1990;Lazareno and Birdsall, 1995; Proska and Tucek, 1995), and brucine and certain analogs exhibit positive cooperativity with acetylcholine at specific receptor subtypes in both binding and functional studies (Birdsall et al., 1997; Jakubik et al., 1997;Lazareno et al., 1998)
C. Definition of Individual Receptor Subtypes
The affinity constants for the partially selective antagonists given in table 3 represent the basis for assigning a response or a binding site to a particular muscarinic receptor subtype. Definition of the selectivity of a novel muscarinic antagonist, or of the actions of a putative selective agonist, can be accomplished best using recombinant muscarinic receptors expressed in cell lines (Buckleyet al., 1989; Lazareno and Birdsall, 1993a; Dörjeet al., 1991b; Maggio et al., 1994). If the agent is intended to have therapeutic utility, it would seem logical to do these tests on cloned and expressed human receptors. Nevertheless, there have been many studies of new muscarinic agonists and antagonists on native receptors, often mediating functional responses, possibly because there is a view that a profile obtained with native functionally coupled receptors is “better” than similar data obtained either in binding or functional studies with cloned receptors. This might be true if there were reason to believe that native receptors behaved significantly differently from cloned receptors (e.g., because of posttranslational modifications). However, there is currently no evidence for such a difference (Caulfield, 1993). What is clear is that the behavior of agonists in particular (including their binding properties) will be determined by levels of expression of signal transduction proteins, including receptors, receptor kinases, G-proteins, RGS proteins, enzymes generating second-messengers, and other effectors, such as ion channels. For example, increasing the expression of Gαq increased the potency of agonists and induced constitutive activity (Burstein et al., 1995, 1997), which is what would be expected on theoretical grounds (Kenakin, 1996). A further complication that is likely to arise with high expression levels of receptors or other components of the signal transduction pathway is coupling to multiple effectors, especially with highly efficacious agonists (Kenakin, 1996). This has been observed in functional studies in membranes (Lazareno et al., 1993) and in whole-cell studies (see e.g., Ashkenazi et al., 1987;Gurwitz et al., 1994).
A true resolution of these problems will only be made when there is a full definition of the levels of each constituent of the signal transduction pathway involved in a given response, together with an understanding of how agonists work at the molecular level. Although there has been considerable progress in this regard, we are nevertheless a long way from this ideal state.
Notwithstanding this, many reports still use nonhuman functional systems to define the actions of agonists and antagonists at muscarinic receptor subtypes, often with a therapeutic end in mind. For this reason, we will outline briefly some model functional responses, which can be recorded by fairly simple techniques and which have been defined satisfactorily as involving a particular muscarinic receptor subtype.
1. M1 receptors.
a. Rat superior cervical ganglion.
Muscarinic agonist depolarization of rat isolated superior cervical ganglion, recorded extracellularly, is mediated by M1receptors (Brown et al., 1980). This is probably the result of inhibition of opening of the voltage-gated M-type K+ channels in these neurons (Marrion et al., 1989; Bernheim et al., 1992), although M1 receptors can modulate other conductances which could contribute to the depolarizing response (e.g., the protein kinase C-dependent Cl− current described byMarsh et al., 1995). The pharmacology of this system has not been investigated using the more recently discovered antagonists. However, the ablation of M-current inhibition in sympathetic ganglion neurons of the M1-knockout mouse argues convincingly for the linkage (Hamilton et. al., 1997.)
b. Canine saphenous vein.
Contraction of this preparation probably is mediated by M1 receptors, because the apparent pKB values of a range of partially selective antagonists is entirely consistent with an M1 receptor profile (O’Rourke and Vanhoutte, 1987; Sagrada et al., 1994; Watson et al., 1995). It should be noted that there is a low receptor reserve associated with the contraction. Most agonists, notably those of low intrinsic efficacy (including McN-A-343!), act as antagonists.
2. M2 receptors.
a. Guinea-pig heart.
Activation of muscarinic receptors in these preparations produces a reduction in force of contraction and (in nonpaced tissues) a decrease in the rate of beating. These effects are probably the consequence of inhibition of voltage-gated Ca2+ channels and activation of inwardly rectifying K+ channels, respectively. Extensive studies with many antagonists have defined this response as being mediated by the M2 receptor (reviewed byCaulfield, 1993).
M2 receptors can mediate both negative and positive inotropic responses in the left atrium of the reserpinized rat, that latter effect being insensitive to pertussis toxin (Kenakin and Boselli, 1990).
It also has been suggested that an M1 muscarinic receptor stimulates phospholipase C, and increases Ca2+ currents in pertussis toxin-treated guinea-pig and rat ventricular myocytes (see Sharma et al., 1997 for references). Supporting evidence for this contention was that subtype-specific antibodies detected M1 receptor protein in myocytes, and reverse transcriptase polymerase chain reaction detected significant m1 mRNA (Gallo et al., 1993;Sharma et al., 1997). However, one caveat to the report ofGallo et al. (1993) is that the effect of pirenzepine in antagonizing the muscarinic stimulation of phospholipase C extrapolated to an apparent pKB value of approximately 9.5, which is not consistent with any known muscarinic receptor.
3. M3 receptors.
a. Guinea-pig ileum.
The muscarinic receptors mediating contraction of guinea-pig ileum (and indeed of many other smooth muscle preparations) are defined pharmacologically as M3 (reviewed by Eglen et al., 1996). There is a large population of M2 receptors in many smooth muscles, and it seems likely that they are involved in antagonizing the relaxant effects of agents that elevate cAMP (Thomaset al., 1993; Eglen et al., 1994). M2 receptors in guinea-pig ileum also stimulate the opening of cation-selective channels that depolarize the muscle cells (Bolton and Zholos, 1997).
Several studies have indicated that the receptor mediating relaxation of vascular smooth muscle (via release of relaxing factors from endothelial cells) is M3 (reviewed by Eglen and Whiting, 1990; Caulfield, 1993; Van Zwieten and Doods, 1995) but there is also evidence, summarized in the reviews above, for differences in pKB values for selective antagonists in blocking the relaxant responses in some blood vessels.
There also have been suggestions that smooth muscle M3 receptors from different tissues may be heterogeneous. Thus, compounds such as zamifenacin, darifenacin, and p-F-HHSiD have been reported to distinguish between muscarinic agonist responses in tissues such as trachea, ileum, and urinary bladder (reviewed by Eglen et al., 1996). However, as pointed out byEglen et al. (1996), it seems premature to speculate about subtypes of M3 receptor (or a new receptor subtype) in the absence of supporting molecular evidence, either in the form of new genes or posttranslational modifications which change antagonist affinities.
4. M4 receptors.
a. Rabbit anococcygeus muscle.
In preparations in which the tone has been raised by histamine, muscarinic agonists relax the precontraction. This apparently is an exclusively presynaptic effect, involving the release of an inhibitory nonadrenergic noncholinergic neurotransmitter, probably nitric oxide (Gross et al., 1997). Muscarinic antagonists inhibit the relaxation, the pKB values indicating that M4 receptors mediate this response (Grosset al., 1997). There is a low receptor reserve for this response because agonist potencies are low and several agonists of low intrinsic efficacy act as antagonists.
b. NG108–15 cells.
The neuroblastoma-glioma hybrid cell line NG108–15 expresses M4 mRNA (Peralta et al., 1987) and M4 receptors can be detected readily in radioligand binding assays (Lazareno et al., 1990).
Inhibition of adenylyl cyclase activity by muscarinic agonists in rat corpus striatum probably is mediated by M4 receptors (Caulfield, 1993; Olianas et al., 1996). However, a 3- to 10-fold discrepant value for the pKB of methoctramine has been reported (Onali and Olianas, 1995).
5. M5 receptors.
Despite evidence for the presence of the M5 protein and its mRNA in the brain and periphery (Weiner et al., 1990; Flynn et al., 1997), it has not yet been possible to delineate a whole-tissue response whose location and pharmacology matches that predicted for the expressed gene product. There have been several studies of the function of the cloned M5 receptor, so this gene product does represent a functional receptor. However, it has been shown only recently that the pKB values for several selective antagonists in blocking function in cells transfected with the M5 gene agree with the binding affinities measured in membranes from the same cell line (Watson et al., 1998).Reever et al. (1997) have summarized the current knowledge about this somewhat ephemeral receptor subtype.
There is evidence that the A2058 human melanoma cell line expresses only M5 receptors (Kohn et al., 1996). This may provide a useful model for an endogenous M5 receptor in a human cell line, but it also should be noted that the coupling mechanisms in this cell line are somewhat unusual. Another potentially useful system is the eosinophilic leukemia cell line (EoL-1) where M5 (and M3) receptors can be induced on differentiation with interferon-γ (Mita et al., 1996).
The conclusion is that the characterization of the M5receptor is still incomplete but, based on the revised NC-IUPHAR guidelines, there is now sufficient evidence to warrant a M5 (rather than m5) nomenclature.
6. Functional systems whose classification is open to debate.
a. Rabbit vas deferens. In rabbit vas deferens, it has been argued that the inhibition of field stimulation-evoked twitch responses by muscarinic agonists such as McN-A-343 is mediated by M1 receptors, because the effect is antagonized by pirenzepine with an apparent pKB of 7.8 (Eltze, 1988, Grimm et al., 1994a). However, the pKB values obtained with some other antagonists are not consistent with this conclusion. Also, it previously has been suggested that the receptor mediating this functional response may be the M4 subtype, given the pKB value for himbacine (Caulfield, 1993). However, for a wider range of antagonists, comparison between pKB values obtained on this preparation with pKB values at cloned human receptors (or other well-defined systems where cloned receptor data are not available) clearly indicates that neither of these hypotheses can be true (fig.2). Thus, the rabbit vas deferens presynaptic muscarinic receptor subtype apparently is still not defined adequately. The possibility also remains that more than one muscarinic receptor subtype can couple to inhibit transmitter release in this preparation. A potentially further confounding factor is that most muscarinic agonistspotentiate the field-stimulated twitch response by activating a receptor that has a M2-like pharmacology (Eltze, 1988).

pKB(app) values for rabbit vas deferens functional assays versus pKB values on cloned muscarinic receptors. The functional (apparent) pKB values for six selective muscarinic antagonists obtained in Aruklashana-Schild-type experiments on rabbit vas deferens (Eltze, 1988; Sagrada et al., 1994; Grimm et al., 1994a; Waelbroeck et al., 1994) are compared with the log affinity values (pKB) from binding studies on cloned human receptors (Dörje et al., 1991b; Lambrechtet al., 1997). Where data are not available from work on cloned receptors, results are included from work on defined subtypes in nonhuman species (dicyclomine, Lazareno et al., 1990; O-methoxy silahexocyclium, Waelbroeck et al., 1994). Data have been included for those antagonists whose apparent pKB values on the rabbit vas deferens preparation deviate by more than 0.5 log units from the pKB values at either M1 or M4 receptors.
b. Rat duodenum.
McN-A-343 produces relaxation of this preparation, thought to be caused by stimulation of nonadrenergic, noncholinergic neurons (Micheletti et al., 1988), and it has been suggested, again on the basis of a pirenzepine pKB in the region of 8.0, that the receptor involved is an M1 receptor. This is not so, because the pKB values for 4-DAMP (Micheletti et al., 1990a), guanylpirenzepine (Micheletti et al., 1990b), and (S)-dimethindene (Pfaff et al., 1995) in this assay differ from the M1 binding affinities by approximately 1, 0.5, and 0.7 log units, respectively.
Both the above-mentioned systems involve presynaptic muscarinic receptors. The rabbit ear artery preparation is another presynaptic system in which the subtype that inhibits noradrenaline release has an atypical pharmacology (Darroch et al., 1992).
D. Agonists
There are no muscarinic agonists with a high selectivity for one particular subtype. Early studies of muscarinic receptors led to the suggestion that compounds such as McN-A-343 were selective for the M1 receptor, but this is not the case. In fact McN-A-343, if anything, may show a modest degree of M4 selectivity (Lazareno et al., 1993;Richards and Van Giersbergen, 1995). Extensive studies with functional systems involving both native and cloned receptors have demonstrated that the potency of an agonist is not a function of the receptor subtype, but rather is a function of the tissue or cell under study (reviewed by Caulfield, 1993, 1997; Eglen et al., 1996).
The concept of functional selectivity has been applied to the design of muscarinic agonists, which might, for example, be used in the treatment of the cognitive deficit in Alzheimer’s disease (e.g., Freedmanet al., 1993; Lambrecht et al., 1993; Ensingeret al., 1993; Jaen et al., 1995, Fisher et al., 1996). A selective action in such a disease is difficult to predict, however. The approach depends on the agonists having their greatest potency at the receptors in the target tissue. The effective receptor reserves in tissues cannot be manipulated, and there is no guarantee that an advantageous receptor reserve in the target tissue, relative to other tissues, can be maintained during prolonged agonist treatment or during the pathological progression of the disease. The new agonist (+)-3-(S)-3-[4-butylthio-1,2,5-thiadiazol-3-yl]-1-azabi- cyclo[2,2,2]octane (LY297802) is an antinociceptive agent at doses that do not produce parasympath mimetic effects. Its functional selectivity may be mediated via M4 receptors in the spinal cord (Shannonet al., 1997).
Although muscarinic agonists with receptor selectivity are certainly worthwhile targets, it will not be possible to define that selectivity until there is better control and understanding of the transduction processes mediating responses [including the nature of agonist-induced conformational change(s) at the receptor], and definition of the stoichiometry and nature of the G-proteins and other components of the transduction pathway.
V. Transduction Mechanisms and Functional Responses
It is well established that the “odd-numbered” muscarinic receptors (M1, M3, M5) typically couple via the α subunits of the Gq/11 family, whereas the “even-numbered” members (M2, M4) couple via Gi and Go α subunits. This preferential coupling resides at the molecular level mainly in the postulated membrane-proximal regions of the i2 and i3 loops of the different receptors, which are notably different between the “odd” and “even” receptor groups, and similar within the two groups. The coupling selectivity at the G-protein level is reflected in the generally, but not exclusively, observed downstream second-messenger pathways activated by the two groups of muscarinic receptors; phospholipase Cβ is activated by the “odd” receptors, whereas adenylyl cyclase is inhibited by the “even” receptors (reviewed byCaulfield, 1993; Felder, 1995). Thus, responses to activation of the latter group of receptors usually can be blocked by pertussis toxin-catalyzed adenosine diphosphate ribosylation of Gαi and/or Gαo.
Functional responses mediated by these major coupling pathways include the contraction of many smooth muscles and the stimulation of glandular secretion (by M3 receptors), and there is evidence that the M2 receptor-mediated inhibition of voltage-gated calcium channels in the heart is the result of adenylyl cyclase inhibition (see Méry et al., 1997 for references). The ablation of M-current inhibition in sympathetic ganglion neurons in the M1-knockout mouse provides direct evidence for this M1receptor-mediated transduction mechanism in this tissue (Hamiltonet al., 1997). Clearly, there are also many muscarinic responses that involve neither phospholipase Cβ nor adenylyl cyclase inhibition. Thus, the muscarinic activation of cardiac inward rectifier K+ channels (by M2receptors) results from a direct action of Gβγ subunits (released from the Gαiβγ heterotrimer) on the channels (see Wickman and Clapham, 1995). There are also several reports of muscarinic stimulation of adenylyl cyclase activity. This response is blocked by pertussis toxin pretreatment in membranes from the rat olfactory bulb and is thought to involve an indirect but synergistic interaction with Gs and a differential modulation of different isoforms of adenylyl cyclase (Onali and Olianas, 1995). In contrast Dittman et al. (1994)have provided evidence that M4 receptors can couple directly to Gs to activate adenylyl cyclase. Most unusually, it has been reported that M5 receptors in A2058 cells inhibit forskolin-stimulated cAMP production but do not stimulate inositol trisphosphate production (Kohn et al., 1996). The cAMP response is not sensitive to pertussis toxin pretreatment of the cell but depends on calcium influx.
Table 4 summarizes information on the preferred coupling mechanisms and the functional roles of the different muscarinic receptor subtypes. It also indicates some of the tissues where muscarinic receptors have been detected which are relevant to mediation of the functional response. Autoradiographic localization studies of muscarinic receptors have used selective radiolabeled antagonists, e.g., [3H]pirenzepine and [3H]AF-DX 384, but their relatively low subtype selectivity only results in preferential localization of one or more subtypes. A more discriminative method, originally developed byWaelbroeck et al. (1990), exploits the combination of the different kinetics of binding of [3H]N-methylscopolamine to the subtypes, together with the use of appropriate concentrations of combinations of selective antagonists, to allow a much more selective autoradiographic localization of M1 to M5receptors (Flynn et al., 1995). Subtype localization has been aided further by the development of subtype-selective antibodies, which have been useful in immunoprecipitation experiments and in immunocytochemical studies (e.g., Li et al., 1991; Wallet al., 1991a,b; Dörje et al., 1991a;Levey, 1993; Yasuda et al., 1993; Hersch and Levey, 1995;Rouse et al., 1997). These latter studies have allowed a comparison of the proportions of neurons expressing muscarinic receptor proteins and their mRNAs, as determined by in situ hybridization (Hersch and Levey, 1995; Weiner et al., 1990; Bernardet al., 1992; Vilaro et al., 1994). Immunocytochemical studies have been extended to the electron microscope level and have shown, for example, that the location of the M2 receptor protein is compatible with its acting as a presynaptic autoreceptor, a presynaptic heteroreceptor, and a postsynaptic receptor in the septohippocampal pathway (Rouse et al., 1997).
Muscarinic receptors: G-protein coupling, transduction pathways, localization, and functional responses
Coexpression of different receptor subtypes can be an important issue in classification of muscarinic receptor(s) mediating a functional response, especially where the response concerned (e.g., smooth muscle contraction) is the result of many potentially interacting steps in the transduction pathway. This is well illustrated by recent demonstrations of a function for M2 receptors in regulation of gut smooth muscle tone under certain circumstances (Thomas et al., 1993; Eglen et al., 1994). Clearly, the involvement of a mixture of subtypes will result in a confusing pharmacological profile, which may account for many of the controversies in the literature.
VI. Conclusions
Five muscarinic receptor genes have been characterized, and the understanding of their coupling characteristics is increasing, largely because of studies of cloned receptors expressed in cell lines. The use of selective antibodies has allowed the localization of muscarinic receptor subtype proteins. However, the paucity of highly selective antagonists, and the lack of any selective agonists has impeded the unambiguous identification of muscarinic receptor subtypes mediating many important responses. It is hoped that the discovery of compounds (and toxins) with greater receptor subtype selectivity will aid this process.
Acknowledgments
We are indebted to all the members of the NC-IUPHAR Committee on muscarinic acetylcholine receptors, and Tom Bonner, David Brown, Richard Eglen, Debbie Girdlestone, Ed Hulme, Pat Humphrey, Sebastian Lazareno, Ray Leppik, Michael Spedding, and Steve Watson for their critical reading of the manuscript and their constructive comments.
Footnotes
-
↵FNa Composition of the muscarinic receptor subcommittee of the International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification: N.J.M. Birdsall (Chair), Division of Physical Biochemistry, National Institute for Medical Research, Mill Hill, London NW7 1AA, UK; N.J. Buckley, Department of Pharmacology, Wellcome Laboratory of Molecular Pharmacology, University College London, Gower Street, London WC1E 6BT, UK; M.P. Caulfield, Department of Pharmacology, University of Dundee, Ninewells Hospital and Medical School, Dundee DD1 9SY, Scotland; R. Hammer, Drug Discovery, Boehringer Ingelheim KG, Binger Straβe 173,d-55216 Ingelheim/Rhein, Germany; H.J. Kilbinger, Pharmakologisches Institut, University of Mainz, Germany; G. Lambrecht, Department of Pharmacology, University of Frankfurt, Biocentre Niederursel, d-60439 Frankfurt, Germany; E. Mutschler, Department of Pharmacology, University of Frankfurt, Biocentre Niederursel, d-60439 Frankfurt, Germany; N.M. Nathanson, Department of Pharmacology, SJ-30, University of Washington, Seattle, WA 98195, USA; R.D. Schwarz, Parke-Davis Pharmacology Research Division, 2800 Plymouth Road, Ann Arbor, MI 48105, USA.
-
↵FNb Address for correspondence: Nigel J.M. Birdsall, Division of Physical Biochemistry, National Institute for Medical Research, Mill Hill, London NW7 1AA, UK. E-mail: n.birdsa{at}nimr.mrc.ac.uk.
- Abbreviations:
- cAMP
- cyclic adenosine monophosphate
- CNS
- central nervous system
- LY297802
- (+)−3-(S)−3-[4-butylthio-12,5-thiadiazol-3-yl]-1-azabicyclo [2,2,2]octane
- McN-A-343
- (4-Hydroxy-2-butynyl)−1-trimethylammonium-m-chlorocarbanilate chloride
- mRNA
- messenger ribonucleic acid
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}