


Abstract
Two types of cannabinoid receptor have been discovered so far, CB1 (2.1: CBD:1:CB1:), cloned in 1990, and CB2(2.1:CBD:2:CB2:), cloned in 1993. Distinction between these receptors is based on differences in their predicted amino acid sequence, signaling mechanisms, tissue distribution, and sensitivity to certain potent agonists and antagonists that show marked selectivity for one or the other receptor type. Cannabinoid receptors CB1 and CB2 exhibit 48% amino acid sequence identity. Both receptor types are coupled through G proteins to adenylyl cyclase and mitogen-activated protein kinase. CB1 receptors are also coupled through G proteins to several types of calcium and potassium channels. These receptors exist primarily on central and peripheral neurons, one of their functions being to inhibit neurotransmitter release. Indeed, endogenous CB1 agonists probably serve as retrograde synaptic messengers. CB2 receptors are present mainly on immune cells. Such cells also express CB1receptors, albeit to a lesser extent, with both receptor types exerting a broad spectrum of immune effects that includes modulation of cytokine release. Of several endogenous agonists for cannabinoid receptors identified thus far, the most notable are arachidonoylethanolamide, 2-arachidonoylglycerol, and 2-arachidonylglyceryl ether. It is unclear whether these eicosanoid molecules are the only, or primary, endogenous agonists. Hence, we consider it premature to rename cannabinoid receptors after an endogenous agonist as is recommended by the International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. Although pharmacological evidence for the existence of additional types of cannabinoid receptor is emerging, other kinds of supporting evidence are still lacking.
I. Introduction: Overview of the Cannabinoid Receptors
Cannabinoid receptors received their name as those receptors that respond to cannabinoid drugs, such as Δ9-tetrahydrocannabinol (Δ9-THC1; Fig. 1), derived from Cannabis sativa and its biologically active synthetic analogs. As detailed under Section II., synthetic agonists that bind to cannabinoid receptors include Δ9-THC-like analogs and aminoalkylindole compounds typified byR-(+)-WIN55212. Several endogenous ligands for cannabinoid receptors have also been identified, most notably arachidonoylethanolamide (anandamide), 2-arachidonoylglycerol, and 2-arachidonylglyceryl ether (noladin ether) (Section II.). However, because it is not yet clear whether these eicosanoid molecules are the only, or primary, endogenous agonists, we continue to call the receptors cannabinoid receptors rather than prematurely renaming them after an endogenous agonist as is recommended by the NC-IUPHAR. Cannabinoid receptor types are denoted by the abbreviation CB and numbered in the order of their discovery by a subscript (CB1, CB2). At present, two cannabinoid receptor types have been determined, the distinction between them being based on differences in their predicted amino acid sequence, their signaling mechanisms, and their tissue distribution. It has also proved possible to develop potent agonists and antagonists with marked selectivity for CB1 or CB2 receptors (Section II.) as well as CB1, CB2, and CB1/CB2 knockout mice (Section VI.).
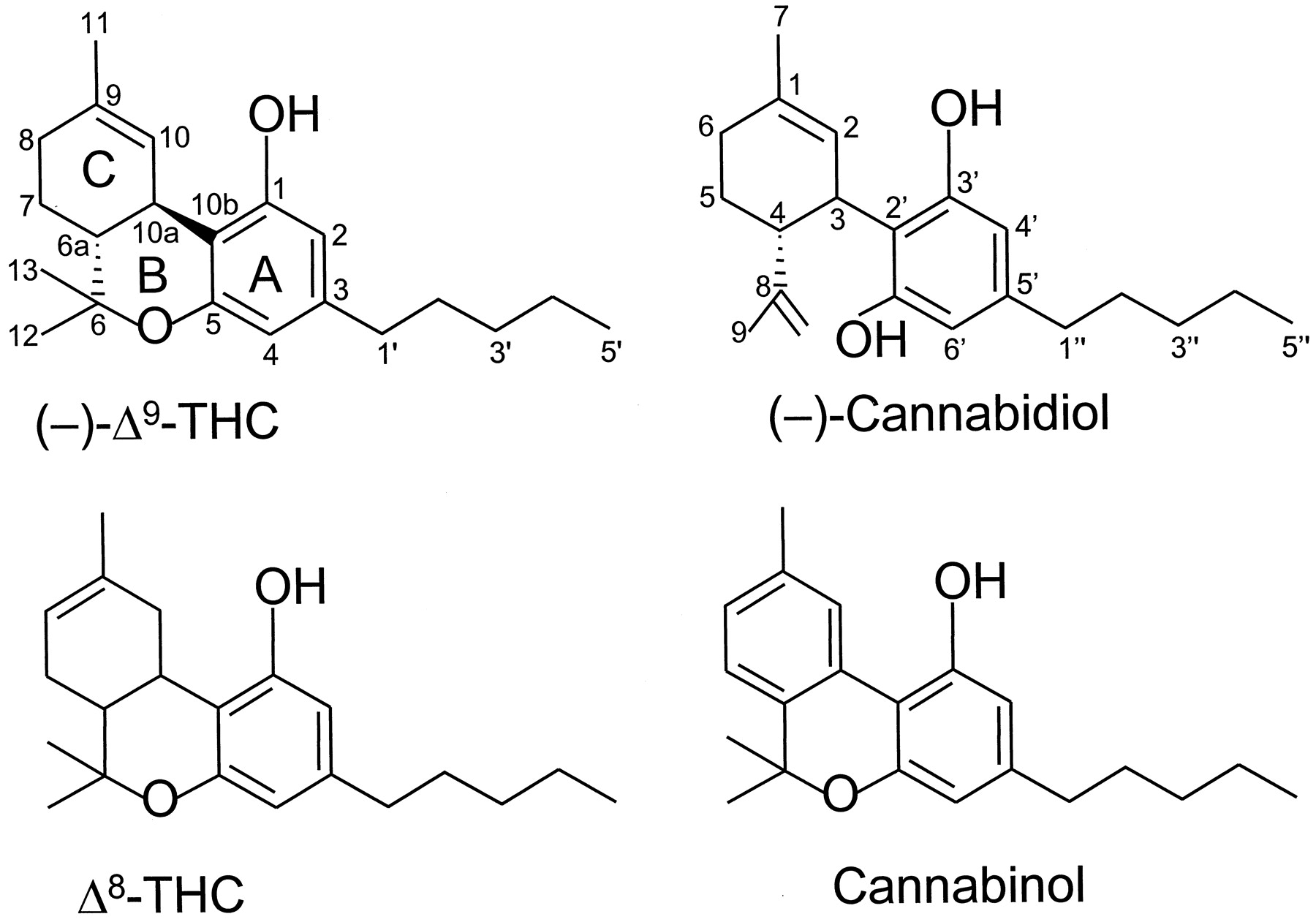
The structures of four constituents of cannabis: Δ9-THC, Δ8-THC, cannabinol, and cannabidiol.
The CB1 cannabinoid receptor (2.1:CBD:1:CB1:) has been cloned from rat, mouse, and human tissues and exhibits 97 to 99% amino acid sequence identity across species (Section V.). Its structure is that of a seven-transmembrane domain receptor, consistent with biochemical and cellular determinations of signal transduction via G proteins (Section IV.). CB1 receptor mRNA and protein are found primarily in brain and neuronal tissue (Section VII.). The CB2 cannabinoid receptor (2.1:CBD:2:CB2:) exhibits 48% homology with the CB1 cannabinoid receptor (Section V.). Expressed CB2receptor protein binds Δ9-THC-like, aminoalkylindole, and eicosanoid ligands (Section II.) and signals a response (Section IV.), thereby defining this receptor as being of the cannabinoid receptor class. The mouse CB2 receptor has been cloned and has an 82% sequence identity to the hCB2 receptor (Section V.). CB2 receptor mRNA is found primarily in immune tissue and is notably absent from normal nervous tissue (Section VII.). Any novel type(s) of cannabinoid receptor will be defined based on multiple criteria of primary structure homology, pharmacological characteristics in biological systems, and signal transduction mechanisms. Although some preliminary pharmacological evidence for the existence of additional types of cannabinoid receptor has already emerged (Section XI.), other kinds of evidence are still lacking.
The CB1 cannabinoid receptor has been extensively characterized for biological responses, and information about the structure-activity relationships of ligands for interaction with this receptor is extensive (Section II.). Claimed central nervous system responses to Δ9-THC and other cannabinoid receptor agonists include therapeutically beneficial effects of analgesia, attenuation of the nausea and vomiting in cancer chemotherapy, reduction of intraocular pressure, appetite stimulation in wasting syndromes, relief from muscle spasms/spasticity in multiple sclerosis, and decreased intestinal motility (for reviews, see Pertwee, 2000b; 2001a,b, 2002; Piomelli et al., 2000). Untoward side effects accompanying these therapeutic responses include alterations in cognition and memory, dysphoria/euphoria, and sedation (see Abood and Martin, 1992 for a review). Animal models that distinguish cannabinoid receptor activity include drug discrimination paradigms in rodents, pigeons, and nonhuman primates, a typical static ataxia in dogs, and a tetrad of responses in rodents (hypothermia, analgesia, hypoactivity, and catalepsy; reviewed under Section III.). Nerve-muscle tissue preparations (e.g., mouse vas deferens and guinea pig small intestine) respond to CB1 cannabinoid receptor agonists with an inhibition of electrically evoked contraction, believed to be the result of diminished release of neurotransmitter (Section III.). CB2mRNA has been found primarily in cells of the immune system (Sections VII. and IX.). However, because CB1 receptor transcripts have also been found in immune cells and tissues, it cannot be assumed that immune responses are solely regulated by the CB2 cannabinoid receptor. Therapeutic applications or untoward effects of cannabinoid receptor agonists in the immune system remain unclear. CB1 and CB2 cannabinoid receptors are both coupled to pertussis toxin-sensitive Gi/o proteins to inhibit adenylyl cyclase activity and to initiate the mitogen-activated protein kinase and immediate early gene signaling pathway(s) (Section IV.). In addition, CB1 receptors are coupled through Gi/o proteins to various types of potassium and calcium channels (Section IV.).
As to endogenous cannabinoid receptor agonists (endocannabinoids), it is likely that anandamide and 2-arachidonoylglycerol both function as neurotransmitters or neuromodulators and that one of their roles may be to serve as retrograde synaptic messengers (Section VIII.). Thus, there is evidence that they are synthesized by neurons “on demand”, that they can undergo depolarization-induced release from neurons, and that after their release, they are rapidly removed from the extracellular space by a membrane transport process yet to be fully characterized (Di Marzo et al., 1998; Maccarrone et al., 1998; Di Marzo, 1999; Piomelli et al., 1999; Hillard and Jarrahian, 2000). Once within the cell, anandamide is hydrolyzed to arachidonic acid and ethanolamine by the microsomal enzyme, fatty acid amide hydrolase (FAAH) (Di Marzo et al., 1998; Maccarrone et al., 1998; Di Marzo, 1999;Ueda et al., 2000). 2-Arachidonoylglycerol can also be hydrolyzed enzymically, both by FAAH and by other hydrolases yet to be characterized (Di Marzo et al., 1998; Di Marzo, 1999; Khanolkar and Makriyannis, 1999). Mechanisms underlying the release and fate of noladin ether remain to be identified.
This review summarizes the main features of the structure, pharmacology, and function of cannabinoid receptors that provide the basis for the classification of these receptors. Because it does not set out to be a comprehensive review of the literature, readers seeking more detail should refer to the many relevant reviews in the field (Table 1).
Recent reviews on cannabinoid receptors or endogenous cannabinoids
II. Classification of Ligands That Bind to Cannabinoid Receptors
A. Cannabinoid Receptor Agonists
1. Classical Cannabinoids.
This group of cannabinoids consists of ABC-tricyclic dibenzopyran derivatives that are either compounds occurring naturally in the plant, C. sativa, or synthetic analogs of these compounds. The most investigated of the classical cannabinoids have been Δ9-THC (Fig. 1), Δ8-THC (Fig. 1), 11-hydroxy-Δ8-THC-dimethylheptyl (HU-210) (Fig. 2), and desacetyl-l-nantradol (Fig. 2). Of these, Δ9-THC is the main psychotropic constituent of cannabis. Δ8-THC is also a psychotropic plant cannabinoid, whereas HU-210 and desacetyl-l-nantradol are synthetic cannabinoids. All these cannabinoids have been demonstrated to elicit cannabimimetic responses both in vivo and in vitro (Johnson and Melvin, 1986; Howlett et al., 1988; Martin et al., 1991; Martin et al., 1995; Pertwee, 1999).

The structures of the synthetic classical cannabinoid receptor agonists, HU-210 and desacetyl-l-nantradol, and of HU-211, the (+)-enantiomer of HU-210.
Δ9-THC was first isolated from C. sativa in pure form by Gaoni and Mechoulam (1964), who also elucidated its structure. Its absolute stereochemistry was subsequently shown to be (6aR,10aR) (Mechoulam and Gaoni, 1967). Δ9-THC undergoes significant binding to cannabinoid receptors at submicromolar concentrations, with similar affinities for CB1 and CB2receptors (Table 2). At CB1 receptors, it behaves as a partial agonist, the size of its maximal effect in several CB1receptor-containing systems falling well below that of cannabinoid receptor agonists with higher relative intrinsic activity, such as CP55940 and R-(+)-WIN55212 (Gérard et al., 1991;Breivogel et al., 1998; Griffin et al., 1998; Pertwee, 1999). The relative intrinsic activity of Δ9-THC at CB2 receptors is even less than its relative intrinsic activity at CB1 receptors (Bayewitch et al., 1996; Pertwee, 1999). Indeed, in one set of experiments with CHO cells transfected with hCB2 receptors, in which the cyclic AMP assay was used, Δ9-THC failed to show any agonist activity at all, behaving instead as a CB2 receptor antagonist (Bayewitch et al., 1996). Δ9-THC has also been reported to behave as an antagonist at CB1 receptors both in the [35S]GTPγS assay performed with rat cerebellar membranes (Sim et al., 1996; Griffin et al., 1998) and when the measured response was cannabinoid-induced inhibition of glutamatergic synaptic transmission in rat cultured hippocampal neurons (Shen and Thayer, 1999).
Ki values of certain ligands for the in vitro displacement of [3H]CP55940, [3H]R-(+)-WIN55212, or [3H]HU-243 from CB1- and CB2-specific binding sites
Δ8-THC has affinities for CB1 and CB2 receptors that are similar to those of Δ9-THC (Table 2) and also resembles Δ9-THC in behaving as a partial agonist at CB1 receptors (Matsuda et al., 1990;Gérard et al., 1991). However, its synthetic analog, HU-210, has relative intrinsic activities at CB1 and CB2 receptors that match those of the high-efficacy agonists, CP55940 and (+)-WIN55212 (Slipetz et al., 1995;Song and Bonner, 1996; Burkey et al., 1997; Griffin et al., 1998). HU-210 also has affinities for CB1 and CB2 receptors that exceed those of these other cannabinoids (Table 2). As a result, it is a particularly potent cannabinoid receptor agonist. Its pharmacological effects in vivo are also exceptionally long lasting. The enhanced affinity and relative intrinsic activity shown by HU-210 at cannabinoid receptors can be largely attributed to the replacement of the pentyl side chain of Δ8-THC with a dimethylheptyl group (see also below).
Like THC and HU-210, most classical cannabinoids that bind to CB1 have affinity for CB2as well, without major selectivity for either of these receptors. Thus, Δ9-THC-dimethylheptyl, 5′-F-Δ8-THC, 11-OH-cannabinol, 11-OH-cannabinol-dimethylheptyl, and cannabinol-dimethylheptyl-11-oic acid bind to both CB1 and CB2 receptors without major differences in theirK i values, although there are significant differential levels of potency between the various compounds (Showalter et al., 1996; Rhee et al., 1997). For example, theK i for Δ9-THC is about 40 nM for either receptor, whereas that for HU-210 is about 100 times lower (Showalter et al., 1996). Because binding values differ due to experimental conditions, data from different laboratories may vary considerably, but the general trend is apparently retained (Table2).
The first SAR determinations based on the Δ9-THC structure were summarized by Edery et al. (1971), and numerous reviews on this topic have since appeared (Mechoulam and Edery, 1973; Pars et al., 1977; Razdan, 1986; Mechoulam et al., 1987; Mechoulam et al., 1992; Martin et al., 1995). Most of the originally proposed SARs have withstood the erosion of time, although exceptions have been noted and certain refinements have had to be made. The SARs for classical cannabinoids at CB1receptors are summarized below (see Mechoulam et al., 1992 for references). They were established by animal experimentation (overt behavior in rhesus monkeys or baboons, dog static ataxia, the mouse ring test, spontaneous activity in rats and mice, and drug discrimination in THC-trained rats and pigeons, etc.; see Section III.). These tests are all presumed to involve CB1 receptor-mediated activity, and, indeed, a good correlation has been established between some of the above animal data and CB1 binding (Compton et al., 1993). However, since receptor binding is only the first step in a signal transduction pathway, lack of activation at some other point of the mechanistic cascade may result in a discrepancy between binding and activity. Thus, for example, Δ8-THC-11-oic-dimethylheptyl acid binds well to the CB1 receptor, but its inhibition of adenylyl cyclase is poor (Rhee et al., 1997). Current SAR information about classical cannabinoids is summarized below.
A dihydrobenzopyran-type structure with a hydroxyl group at the C-1 aromatic position and an alkyl group on the C-3 aromatic position seems to be a requirement. Opening of the pyran ring generally leads to complete loss of activity if both phenolic groups are present and are not substituted. Thus, (−)-cannabidiol (Fig. 1) has markedly less affinity for CB1 or CB2 receptors than Δ8- or Δ9-THC (Tables 2 and3).
The aromatic hydroxyl group has to be free or esterified for significant CB1 activity. Blocking of the hydroxyl group as an ether inactivates the molecule. It is possible that the esters are actually inactive but undergo hydrolysis to the free phenols in vivo. Thus, Δ9-THC acetate, when tested in vitro, shows negligible activity in biochemical reactions in which Δ9-THC is active (Banerjee et al., 1975).
The length of the chain on C-3 is of major importance. Some activity may be noted with propyl or butyl substitution; Δ9-THC has a pentyl group. A 1′,1′-dimethylheptyl or 1′,2′-dimethyl heptyl side chain strongly potentiates the cannabimimetic activity of compounds that have low activity in the n-pentyl series. An all carbon side chain on C-3 is not an absolute requirement. The side chain may contain an etheric oxygen (Loev et al., 1973).
11-Hydroxy THCs, which are major metabolites of classical cannabinoids, are potent cannabimimetics. Monohydroxylation on other positions of the terpene ring also usually leads to active derivatives. Dihydroxylation generally causes loss of activity. Further oxidation of the C-11 hydroxyl group to a carboxyl group causes inactivation.
Hydroxylation of C-1 of the side chain on C-3 abolishes activity. Hydroxylation at the other C-3 side chain carbons retains activity, with hydroxylation on C-3 of the side chain potentiating activity. Some of these hydroxylated compounds have been detected as major metabolites.
Alkylation of the C-2 aromatic position retains activity; alkylation on the C-4 position eliminates activity. Electronegative groups, such as carbonyl or carboxyl, at either C-2 or C-4 eliminate activity.
The methyl group on C-9 is not an absolute requirement for activity; 9-nor-Δ9-THC and 9-nor-Δ8-THC are active in the dog static ataxia test (Martin et al., 1975).
The double bond in the terpene ring is not essential for activity (Mechoulam and Edery, 1973; Mechoulam et al., 1980), and, indeed, this ring may be exchanged by some heterocyclic systems (Pars et al., 1977;Lee et al., 1983).
CB1 and CB2 Ki values of stereoisomers of cannabidiol and of two cannabidiol analogs
Changes in the stereochemistry at various carbons of THC-type molecules may cause significant changes in pharmacological activity. The following tentative SARs have been proposed (Mechoulam et al., 1992):
The stereochemistry at 6a,10a in the natural active cannabinoids is trans(6aR,10aR). A few cis isomers have been tested and have shown very low activity. However, ciscompounds have not been studied over a wide range of tests. (6aS,10aS) THCs are either completely inactive or show very low activity both in animal tests and in binding assays. Thus, although the 6aR,10aR analog HU-210 is a highly potent cannabinoid, its 6aS,10aSenantiomer (HU-211), when well purified, has been shown to be less active by more than three orders of magnitude (Järbe et al., 1989; Howlett et al., 1990; Mechoulam et al., 1991; Felder et al., 1992; Pertwee et al., 1992). With Δ8- and Δ9-THC, the picture is less clear. In the original publications, the synthetic (+)-enantiomers of these cannabinoids were apparently not completely separated from the corresponding (−)-enantiomers, such that activity was determined to be about 5 to 10% of the (−) compounds (Mechoulam et al., 1992). For Δ9-THC, careful purification led to a (+)-enantiomer with activity less than 1% of the (−)-enantiomer (Herkenham et al., 1990; Matsuda et al., 1990; Felder et al., 1992;Pertwee, 1997).
Reduction of Δ9-THC leads to hexahydrocannabinol epimers that are both active, the equatorial epimer being considerably more active than the axial one (Mechoulam and Edery, 1973; Mechoulam et al., 1980). The same relationship is observed with the 11-hydroxyhexahydrocannabinols (Mechoulam et al., 1991). Thus, it seems that an equatorial substitution (i.e., one in which the C-9 methyl or hydroxymethyl group is in the plane of the cyclohexane ring) is preferable to an axial one.
Several hydroxylated metabolites of Δ9-THC and Δ8-THC are known in both epimeric forms. For example, 8α- and 8β-hydroxy-Δ9-THC and 7α- and 7β-hydroxy-Δ8-THC have been identified as relatively minor metabolites, and slight differences in activity between the epimers in each pair have been observed (Mechoulam and Edery, 1973; Razdan, 1986).
Recent experiments have shown that stereochemical changes can also affect the pharmacological activity of cannabidiol-type molecules (Bisogno et al., 2001). More specifically, (+)-CBD, (+)-5′-dimethylheptyl-CBD, and (+)-7-OH-5′-dimethylheptyl-CBD each has significantly greater affinity for CB1 and CB2 receptors than its corresponding (−)-enantiomer (Table 3). Unexpectedly, these findings indicate that the stereochemical prerequisites for binding to CB1 and CB2 receptors are not the same in the cannabidiol series in which the (+) (3S,4S) enantiomers show the greater cannabinoid receptor affinity as in the THC series in which the (−) (6aR,10aR) enantiomers show the greater cannabinoid receptor affinity. It is also noteworthy that both (+)- and (−)-CBD behave as vanilloid receptor agonists. Interestingly, these two enantiomers are equipotent at vanilloid receptors, each having an EC50 in the low micromolar range (Bisogno et al., 2001).
Despite the lack of CB1/CB2selectivity shown by the first generation of classical cannabinoids, it has proved possible to develop CB2-selective agonists from this series by making relatively minor changes to the THC molecule (Gareau et al., 1996; Huffman et al., 1996; Hanus et al., 1999). More specifically, Huffman et al. (1996) discovered that removal of the phenolic OH group from HU-210 to form 1-deoxy-11-OH-Δ8-THC-dimethylheptyl (JWH-051; Fig. 3) greatly enhanced affinity for CB2 receptors without significantly affecting CB1 affinity (Table 2). More remarkable still is the high degree of CB2 selectivity shown in binding experiments by JWH-133, JWH-139, and HU-308 (Fig. 3) and by the Merck Frosst compounds L-759633 and L-759656 (Fig. 3) (Merck Frosst Canada Ltd., Kirkland, QC, Canada), all of which bind to CB2 receptors at concentrations in the low nanomolar range (Table 2). L-759633 and L-759656 are both equipotent and equiefficacious with the high relative intrinsic activity agonist CP55940 at inhibiting forskolin-stimulated cyclic AMP accumulation in CHO cells expressing recombinant CB2 receptors (Ross et al., 1999a). It has also been found that L-759656 (10 μM) is inactive at CB1 receptors and that L-759633 behaves as a weak agonist at these receptors, with an EC50 of about 10 μM (Ross et al., 1999a). Similarly, HU-308 and JWH-133 are much more potent inhibitors of forskolin-stimulated cyclic AMP production by CB2- than by CB1-transfected CHO cells (Hanus et al., 1999;Pertwee, 2000a).

The structures of the CB2-selective cannabinoid receptor agonists, HU-308, L-759633, L-759656, JWH-133, JWH-139, and JWH-051.
2. Nonclassical Cannabinoids.
During the course of their extensive SAR studies on the analgesic activity of classical cannabinoids, researchers at Pfizer synthesized new analogs lacking the dihydropyran ring of THC. CP47497 (Fig.4) represents the prototypical compound of this series of AC-bicyclic and ACD-tricyclic cannabinoid analogs (Melvin et al., 1984; Melvin et al., 1993). Further developments ultimately led to the bicyclic analog, CP55940 (Fig. 4), which has become one of the major cannabinoid agonists. Less lipophilic than THC, [3H]CP55940 has allowed the discovery and characterization of the CB1 cannabinoid receptor (Devane et al., 1988), and it is still the most used radiolabeled cannabinoid ligand. It binds to CB1and CB2 receptors with similar affinity (Table 2) and displays high activity in vivo as well, being 10 to 50 times more potent than Δ9-THC in the mouse tetrad model (Johnson and Melvin, 1986; Little et al., 1988). CP55940 behaves as a full agonist for both receptor types, its maximal effects in CB1 and CB2 receptor assay systems often matching or exceeding the maximal effects of several other cannabinoid receptor agonists (Pacheco et al., 1993; Slipetz et al., 1995; Burkey et al., 1997; Griffin et al., 1998; MacLennan et al., 1998; Pertwee, 1999). One potent ACD-tricyclic nonclassical cannabinoid is CP55244 (Fig. 4), which also displays signs of high affinity and high relative intrinsic activity, at least for CB1 receptors (Howlett et al., 1988; Little et al., 1988; Herkenham et al., 1990; Gérard et al., 1991; Griffin et al., 1998). Indeed, CP55244 seems to have even higher CB1 affinity and relative intrinsic activity than CP55940. It seems likely that other nonclassical cannabinoids share the ability of CP55940 to interact with CB2receptors; however, this remains to be established. Like classical cannabinoids, nonclassical cannabinoids with chiral centers exhibit significant stereoselectivity, those compounds with the same absolute stereochemistry as (−)-Δ9-THC at 6aand 10a (6aR,10aR) exhibiting the greater pharmacological activity (Little et al., 1988; Herkenham et al., 1990; Melvin et al., 1993).
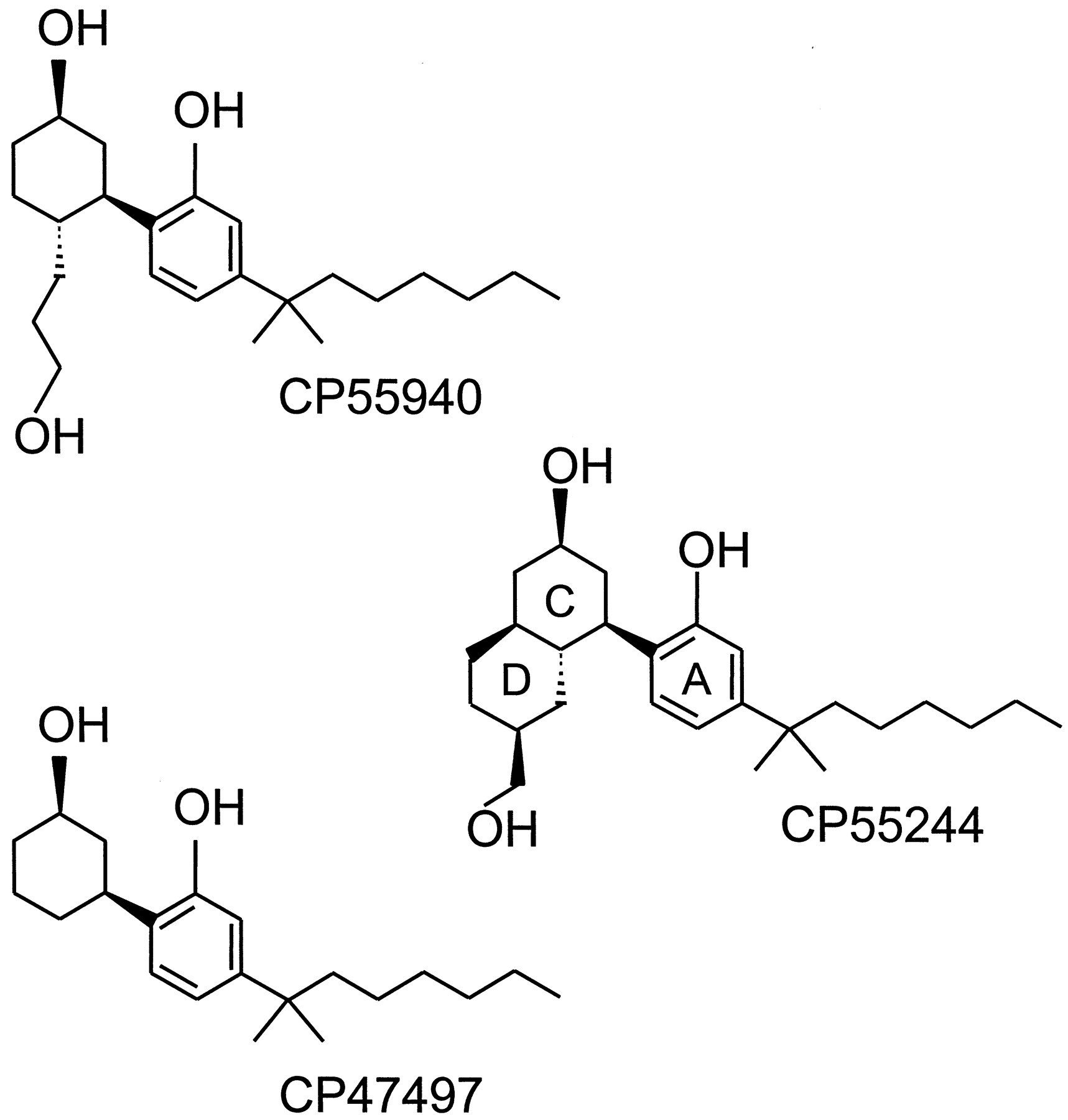
The structures of the (−)-enantiomers of three nonclassical cannabinoid receptor agonists: CP55940, CP47497, and CP55244.
3. Aminoalkylindoles.
Until the early 1990s, all the compounds known to act as cannabimimetics were structurally derived from THC. The situation changed when Sterling Winthrop researchers reported a new family of aminoalkylindoles possessing cannabimimetic properties. This discovery resulted from the development of structurally constrained analogs of pravadoline (Bell et al., 1991;Pacheco et al., 1991), a series of compounds with reduced ability to behave as nonsteroidal anti-inflammatory agents that inhibit cyclooxygenase but increased ability to bind to the CB1 receptor (D'Ambra et al., 1992; Eissenstat et al., 1995). R-(+)-WIN55212 (Fig.5) is the most highly studied, commercially available compound of the series. It displays high affinity for both cannabinoid receptors, with moderate selectivity in favor of the CB2 receptor (Table 2), and exhibits high relative intrinsic activity at both CB1 and CB2 receptors (Bouaboula et al., 1997; Griffin et al., 1998; Tao and Abood, 1998; Pertwee, 1999). In vivo, it produces the full spectrum of pharmacological effects of THC and substitutes totally for other cannabinoids in discriminative stimulus tests, whereas its S-(−)-enantiomer, WIN55212-3, lacks activity both in vivo and in vitro (Martin et al., 1991; Compton et al., 1992a;Pacheco et al., 1993; Slipetz et al., 1995; Wiley et al., 1995b;Pertwee, 1997; Pertwee, 1999). A [3H]R-(+)-WIN55212 assay has been developed, which has been used to characterize and map cannabinoid receptors in rat brain (Jansen et al., 1992; Kuster et al., 1993). There is evidence that R-(+)-WIN55212 binds differently to the CB1 receptor than classical or nonclassical cannabinoids, albeit in a manner that still permits displacement byR-(+)-WIN55212 of other known types of cannabinoid from CB1 binding sites (Petitet et al., 1996; Song and Bonner, 1996; Pertwee, 1997; Chin et al., 1998; Tao and Abood, 1998; see also Section V.).
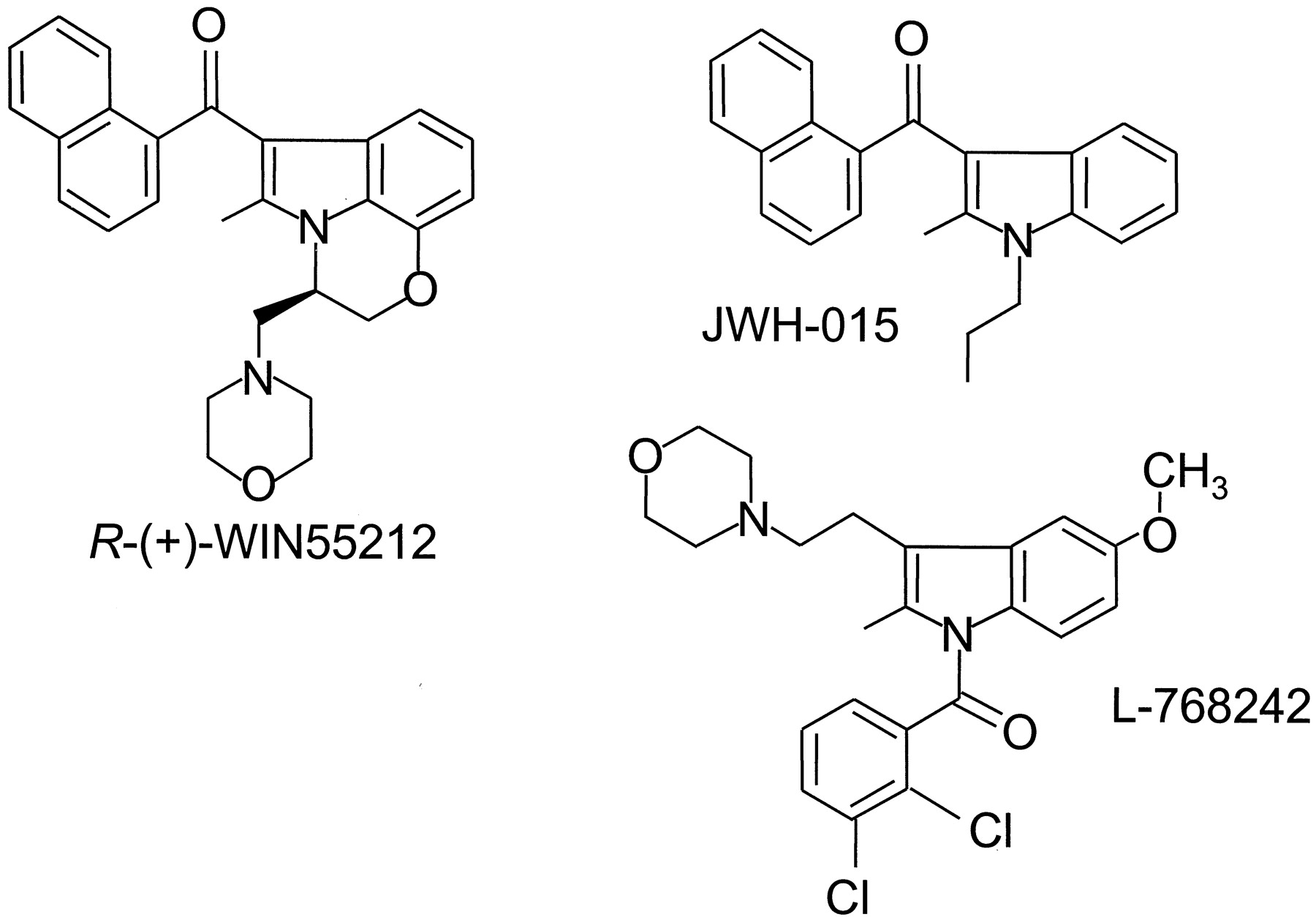
The structures of three aminoalkylindole cannabinoid receptor agonists: R-(+)-WIN55212, JWH-015, and L-768242.
A number of cannabinoid receptor agonists based on the aminoalkylindole structure have been prepared (see Huffman, 1999). As a result, it has been possible to demonstrate that activity is retained when the aminoalkyl substituent is replaced by simple n-alkyl chains (Huffman et al., 1994) or when the indole nucleus is replaced by a pyrrole ring (Lainton et al., 1995; Wiley et al., 1998) or an indene ring (Kumar et al., 1995). Interestingly, some of these newer aminoalkylindoles have been found to display significant selectivity for the CB2 receptor. Among these are JWH-015 (Fig. 5) and a series of Merck Frosst compounds that includes L-768242 (Fig. 5) (Gallant et al., 1996; Showalter et al., 1996) (see also Table 2).
4. Eicosanoids.
The prototypic member of the eicosanoid group of cannabinoid receptor agonists is anandamide, which belongs to the 20:4, n-6 series of fatty acid amides (Fig.6). This is the first of five endogenous cannabinoid receptor agonists to have been discovered in mammalian brain and certain other tissues (Devane et al., 1992b), the other compounds being homo-γ-linolenoylethanolamide and docosatetraenoylethanolamide (Hanus et al., 1993), 2-arachidonoylglycerol (Mechoulam et al., 1995; Sugiura et al., 1995), and noladin ether (Fig. 6) (Hanus et al., 2001). Of these endocannabinoids, the most investigated to date have been anandamide and 2-arachidonoylglycerol.

The structures of five endogenous cannabinoids.
Anandamide resembles Δ9-THC in behaving as a partial agonist at CB1 receptors and in exhibiting less relative intrinsic activity at CB2 than CB1 receptors (Bayewitch et al., 1995; Rinaldi-Carmona et al., 1996a; Griffin et al., 1998; Pertwee, 1999). In line with this classification as a CB2 receptor partial agonist, it shares the ability of Δ9-THC (Section II.A.1.) to attenuate CB2receptor-mediated responses to an agonist with higher relative intrinsic activity (2-arachidonoylglycerol) (Gonsiorek et al., 2000). The anandamide molecule does not contain any chiral centers; however, some of its synthetic analogs do, one example being methanandamide, theR-(+)-isomer, which has nine times greater affinity for CB1 receptors than the S-(−)-isomer (Abadji et al., 1994). Structural modification of the anandamide molecule, which itself displays marginally higher affinity for CB1 than CB2 receptors, has led to the development of the first generation of CB1-selective agonists. Notable examples areR-(+)-methanandamide (Khanolkar et al., 1996; Lin et al., 1998), arachidonyl-2′-chloroethylamide (ACEA), arachidonylcyclopropylamide (ACPA) (Hillard et al., 1999), and O-1812 (Fig. 7) (Di Marzo et al., 2001a). The CB1 selectivity ofR-(+)-methanandamide stems from the introduction of a methyl group on the 1′ carbon of anandamide, a structural change that also confers greater resistance to the hydrolytic action of FAAH. Neither ACEA nor ACPA show any sign of reduced susceptibility to enzymic hydrolysis by FAAH, presumably because they lack a methyl substituent. Indeed, the addition of a methyl group to the 1′-carbon of ACEA markedly decreases the susceptibility of this compound to FAAH-mediated hydrolysis (Jarrahian et al., 2000). However, another consequence of this addition is a reduction of about 14-fold in CB1 receptor affinity. O-1812 also possesses a 1′-methyl substituent, and it too appears to lack significant susceptibility to hydrolysis by FAAH (Di Marzo et al., 2001a). Compared with anandamide, O-1812 exhibits higher affinity for the CB1 receptor, greater CB1/CB2 selectivity, and higher in vivo potency as a CB1 receptor agonist.

The structures of the CB1-selective synthetic cannabinoid receptor agonists, methanandamide, ACEA, ACPA, and O-1812.
The following SARs have been proposed by Martin et al. (1999) for the production of CB1-like effects by the anandamide series of compounds (see Di Marzo et al., 1999; Palmer et al., 2000 for other recent reviews on the anandamide SAR).
Monosubstitution of the amide is a requirement for activity. Substitution by an alkyl, fluoroalkyl, or hydroxyalkyl increases activity, with a two- or three-carbon chain being optimal. Branching of the chain (methyl is optimal) retains activity.
Substitution of the hydroxyl in anandamide by a methyl ether, phenyl ether, or forming a phosphate derivative of anandamide decreases activity, whereas introduction of an amino or a carboxyl group eliminates activity.
Highest potencies are observed when structural changes are carried out in both the arachidonoyl and ethanolamide moieties of anandamide.
The introduction of an alkyl substituent (methyl is optimal) on the carbon α to the carbonyl or on the carbon adjacent to the nitrogen increases metabolic stability.
The SAR of the end pentyl chain (C-16 to C-20) in anandamide is very similar to that of classical cannabinoids; however, by branching the chain, the effect on pharmacological measures is not as dramatic in the anandamide series as in the classical series.
As a requirement for activity in the 20:x, n-6 series, x has to be three or four; however, activity is strongly reduced when n-6 is changed to n-3.
Activity is retained by increasing the chain length of anandamide by two methylenes (i.e., 22:4 and n-6) but is dramatically reduced or eliminated if the chain length is decreased by two methylenes.
Interpretation of SAR data for anandamide is complicated by evidence firstly, that this fatty acid amide is also an agonist for non-CB1, non-CB2 receptors, and secondly, that some of its metabolites also have pharmacological activity (Adams et al., 1998; Craib et al., 2001; Pertwee and Ross, 2002).
Turning now to 2-arachidonoylglycerol, there is evidence that this compound is an agonist for both CB1 and CB2 receptors (Stella et al., 1997; Sugiura et al., 1997b; Ben-Shabat et al., 1998) and that it exhibits higher relative intrinsic activity than anandamide at both CB1 and CB2 receptors (Pertwee, 1999; Gonsiorek et al., 2000; Savinainen et al., 2001). Like anandamide, 2-arachidonoylglycerol has marginally higher affinity for CB1 than CB2 receptors, its affinity for each of these receptors matching that of anandamide when the latter is protected from enzymic hydrolysis by phenylmethylsulfonyl fluoride (Table 2). Rather few structure-activity experiments have been performed with analogs of 2-arachidonoylglycerol thus far. The available data suggest that 1(3)-arachidonoylglycerol has similar CB1 and CB2 binding properties to 2-arachidonoylglycerol (Mechoulam et al., 1998) and that it is about three times more potent than 2-arachidonoylglycerol as a CB1 receptor agonist in vitro (Stella et al., 1997). There is also evidence that 2-palmitoylglycerol and 2-linoleoylglycerol lack significant affinity for CB1 or CB2 receptors (Mechoulam et al., 1995, 1998; Ben-Shabat et al., 1998) and that 1(3)-palmitoylglycerol and 1(3)-stearoylglycerol (10 μM) do not share the ability of 1(3)- and 2-arachidonoylglycerol to behave as CB1 receptor agonists in vitro (Stella et al., 1997).
As yet, few pharmacological experiments have been performed with noladin ether. These have generated data indicating that in contrast to anandamide and 2-arachidonoylglycerol, noladin ether has much higher affinity for CB1 receptors than for CB2 receptors (Hanus et al., 2001; Table 2). It also appears to have less relative intrinsic activity at CB1 receptors than 2-arachidonoylglycerol (Savinainen et al., 2001). As expected for a CB1receptor agonist, noladin ether produces hypokinesia, antinociception, catalepsy, and hypothermia in mice (Hanus et al., 2001).
B. Cannabinoid Receptor Antagonists/Inverse Agonists
1. Diarylpyrazoles.
The prototypic members of this series of compounds are the Sanofi compounds SR141716A, a potent CB1-selective ligand, and SR144528, a potent CB2-selective ligand (Fig.8). These ligands readily prevent or reverse effects mediated respectively by CB1 and CB2 receptors (Rinaldi-Carmona et al., 1994,1998). There are many reports that, by themselves, SR141716A and SR144528 can act on CB1 or CB2 receptors to produce effects that are converse to those produced by cannabinoid receptor agonists (Pertwee, 1999). Although these effects of the arylpyrazole antagonists may be attributable to the inhibition of endogenously produced agonists in the biological preparation, there is evidence that SR141716A and SR144528 can evoke inverse agonist responses (Bouaboula et al., 1997; MacLennan et al., 1998; Pan et al., 1998; Rinaldi-Carmona et al., 1998; Portier et al., 1999; Ross et al., 1999a; Coutts et al., 2000; Sim-Selley et al., 2001). This notion rests on the ability of the CB1 and CB2 receptors to exhibit signal transduction activity in the absence of endogenous or exogenous agonists (constitutive activity). As such, arylpyrazoles can behave as “inverse agonists” to reduce the constitutive activity of these signal transduction pathways. In some experiments, SR141716A has been found to be more potent in blocking the actions of CB1 receptor agonists than in eliciting inverse cannabimimetic responses by itself (Gessa et al., 1997, 1998a;Schlicker et al., 1997; Acquas et al., 2000; Sim-Selley et al., 2001).Sim-Selley et al. (2001) have obtained evidence that this may be because SR141716A binds with relatively low affinity to a site on the CB1 receptor that is distinct from the agonist binding site for which it has higher affinity. Their data also suggest that it is this lower affinity site that is responsible for the inverse agonist properties of SR141716A.

The structures of the cannabinoid receptor antagonists/inverse agonists, SR141716A, AM251, AM281, SR144528, and LY320135.
Two analogs of SR141716A that have also been used to block CB1 receptor-mediated effects are AM251 and AM281 (Fig. 8). AM281 has 350 times greater affinity for CB1 than CB2 receptors (Table 2), and both analogs share the ability of SR141716A to attenuate responses to established cannabinoid receptor agonists (Gifford et al., 1997b; Al-Hayani and Davies, 2000; Cosenza et al., 2000; Izzo et al., 2000; Huang et al., 2001; Maejima et al., 2001; Simoneau et al., 2001;Wilson and Nicoll, 2001). There are also reports that like SR141716A, AM281 behaves as an inverse agonist when administered alone (Gifford et al., 1997b; Cosenza et al., 2000; Izzo et al., 2000). Current information about the SARs for SR141716A-like compounds can be summarized as follows.
Disubstitution of the amide nitrogen of SR141716A strongly decreases CB1 affinity (Lan et al., 1999b).
Replacement of the amide function by ketone, alcohol, or ether also greatly decreases CB1 binding affinity (Wiley et al., 2001). Interestingly, some of the ether or alkylamide derivatives display partial agonist activity in mice in vivo. The highly hindered endo-fenchyl amide was used to design the CB2 receptor antagonist SR144528 (Rinaldi-Carmona et al., 1998).
Although the 2,4-dichlorophenyl substituent at the 1-position of the pyrazole ring seems to be optimal (Barth and Rinaldi-Carmona, 1999), its replacement by a 1-(5-isothiocyanato)-pentyl group decreases CB1 affinity only by a factor 4 (Howlett et al., 2000). The phenyl group has been replaced by a 4-methylbenzyl group in SR144528 (Rinaldi-Carmona et al., 1998).
In the 3-position of the pyrazole ring of SR141716A, replacement of theN-aminopiperidine substituent by the related 5- or 7-membered rings or by cyclohexyl does not alter CB1 binding affinity, whereas replacement by aminomorpholine or linear alkyl chains leads to a reduction in CB1 affinity (Lan et al., 1999b; Wiley et al., 2001).
Compounds with methyl, bromine, or iodine in the 4-position of the pyrazole ring are approximately equipotent, whereas replacement of methyl with hydrogen at this position results in a 12-fold decrease in CB1 affinity (Wiley et al., 2001). Methyl has been replaced by hydrogen at the 4-position of the pyrazole ring in SR144528.
In the 5-position of the pyrazole ring, replacement of the 4-chloro substituent of the phenyl group by other halogen or alkyl groups does not alter CB1 binding affinity (Thomas et al., 1998; Lan et al., 1999b). However, replacement by nitro or amino groups or displacement from the 4-(para) position to the 2-position of the phenyl group leads to poor CB1 receptor ligands, and replacement of the aromatic ring by alkyl groups abolishes CB1 affinity (Lan et al., 1999b).
A particularly potent compound in the SR141716A series is AM251 (Fig.8). This contains a para-iodophenyl group at the 5-position, a piperidinyl carboxamide at the 3-position, and a 2,4-dichlorophenyl group at the 1-position of the pyrazole ring (Lan et al., 1999b).
2. Other Chemical Series.
The most notable members of these series are the substituted benzofuran, LY320135, and the aminoalkylindole, 6-iodopravadoline (AM630) (Fig.9). LY320135, developed by Eli Lilly, shares the ability of SR141716A to bind with much higher affinity to CB1 than CB2 receptors (Table 2). However, it has less affinity for CB1receptors than SR141716A and, at concentrations in the low micromolar range, also binds to muscarinic and 5-HT2receptors (Felder et al., 1998). Like SR141716A, LY320135 not only blocks the effects of CB1 receptor agonists (Felder et al., 1998; Coruzzi et al., 1999; Holland et al., 1999;Molderings et al., 1999; Christopoulos et al., 2001) but also exhibits inverse agonist activity at some signal transduction pathways of the CB1 receptor (Felder et al., 1998; Christopoulos et al., 2001).
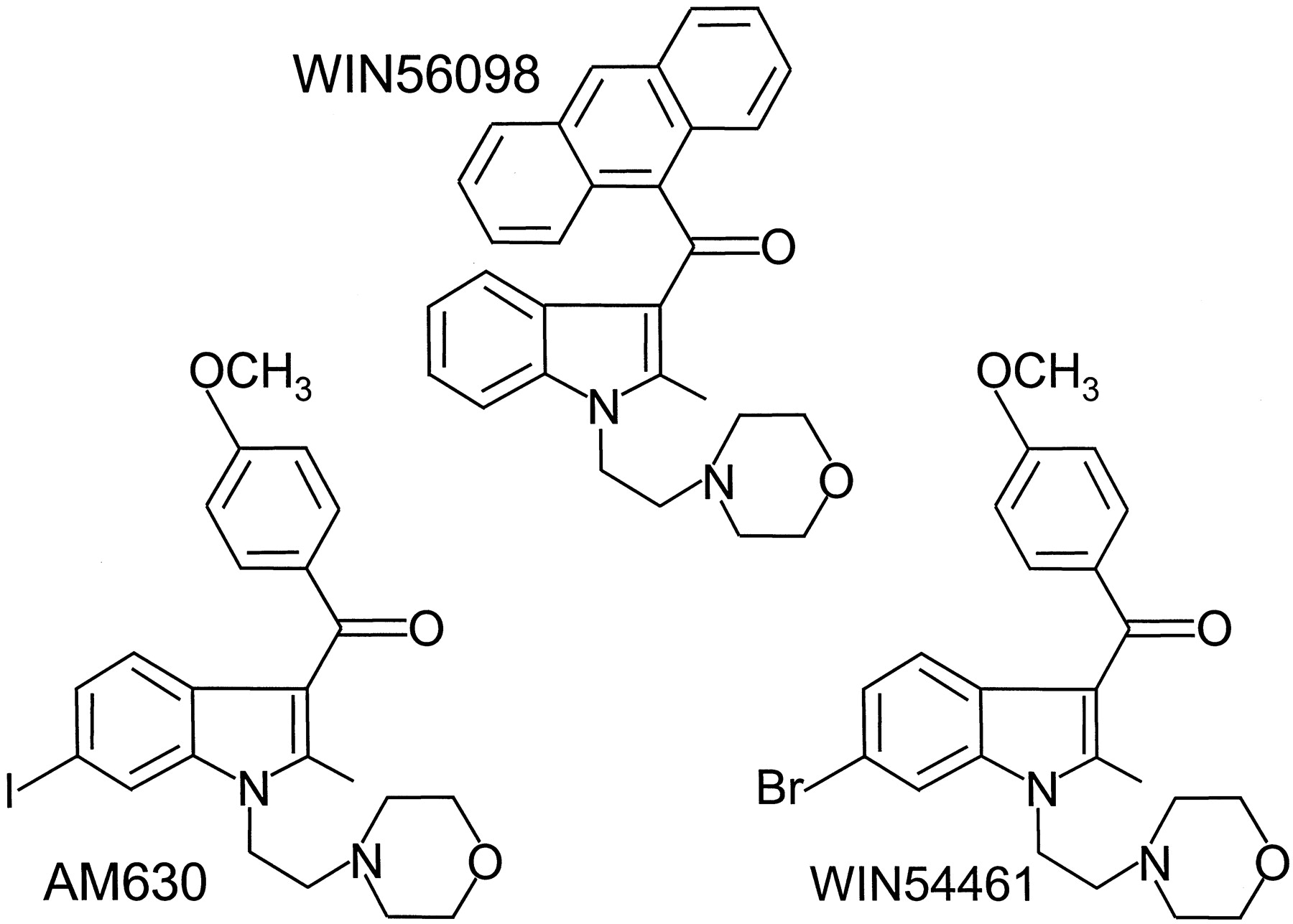
The structures of the pravadoline analogs, AM630, WIN56098, and WIN54461 (6-bromopravadoline).
AM630 is a CB2-selective antagonist/inverse agonist. Thus, experiments with hCB2-transfected CHO cell preparations have shown that it potently reverses CP55940-induced inhibition of forskolin-stimulated cyclic AMP production (EC50 = 128.6 nM) and that when administered by itself, it enhances forskolin-stimulated cyclic AMP production (EC50 = 230.4 nM) and inhibits [35S]GTPγS binding (EC50 = 76.6 nM) (Ross et al., 1999a). The inverse agonist activity of AM630 at CB2receptors appears to be less than that of SR144528 (Ross et al., 1999b). As to the ability of AM630 to interact with CB1 receptors, results from several investigations, when taken together, suggest that this ligand has mixed agonist-antagonist properties and that it is a low-affinity partial CB1 agonist (Pertwee et al., 1996; Hosohata et al., 1997a,b; Pertwee, 1999; Ross et al., 1999a). There is also one report that it can behave as a low-potency inverse agonist at CB1 receptors (Landsman et al., 1998). The ability of AM630 to behave as a cannabinoid receptor antagonist was first noted in experiments with the mouse isolated vas deferens, which yielded dissociation constant (K B) values for AM630 against Δ9-THC and CP55940 of 14.0 and 17.3 nM, respectively (Pertwee et al., 1995a). The pharmacological properties of AM630 in vivo have yet to be investigated. Two other aminoalkylindoles that have been found to attenuate responses to cannabinoids in the mouse isolated vas deferens are the Sterling Winthrop compounds, WIN56098 and WIN54461 (Fig.9). WIN56098 is the weaker antagonist, itsK B value for antagonism of Δ9-THC being 1.85 μM (Pacheco et al., 1991). Corresponding potency values for WIN54461 againstR-(+)-WIN55212 and Δ9-THC have been reported to be 159 and 251 nM, respectively (Eissenstat et al., 1995). The IC50 value of WIN54461 for displacement of [3H]R-(+)-WIN55212 from rat cerebellar membranes has been reported to be 515 nM by Eissenstat et al. (1995). However, they also found WIN54461 to lack detectable antagonist properties in vivo.
One compound that is close to being a CB1/CB2 receptor antagonist that lacks any agonist or inverse agonist activity is the classical cannabinoid 6′-azidohex-2′-yne-Δ8-THC (O-1184) (Fig. 10). In addition to a terminal N3 group, the C-3 alkyl side chain of this ligand contains a carbon-carbon triple bond, a structural modification that decreases relative intrinsic activity at CB1 and CB2 receptors without affecting CB1 or CB2 affinity (Ross et al., 1999b). At CB1 receptors, O-1184 behaves as a high-affinity, low-efficacy agonist, whereas at CB2 receptors, it behaves as a high-affinity, low-efficacy inverse agonist (Ross et al., 1998, 1999b). O-1238 (Fig.10), in which the carbon-carbon triple bond of O-1184 is replaced by a carbon-carbon double bond, has higher efficacy than O-1184 at CB1 receptors and behaves as a high-affinity, low-efficacy partial agonist at CB2 receptors (Ross et al., 1999b).
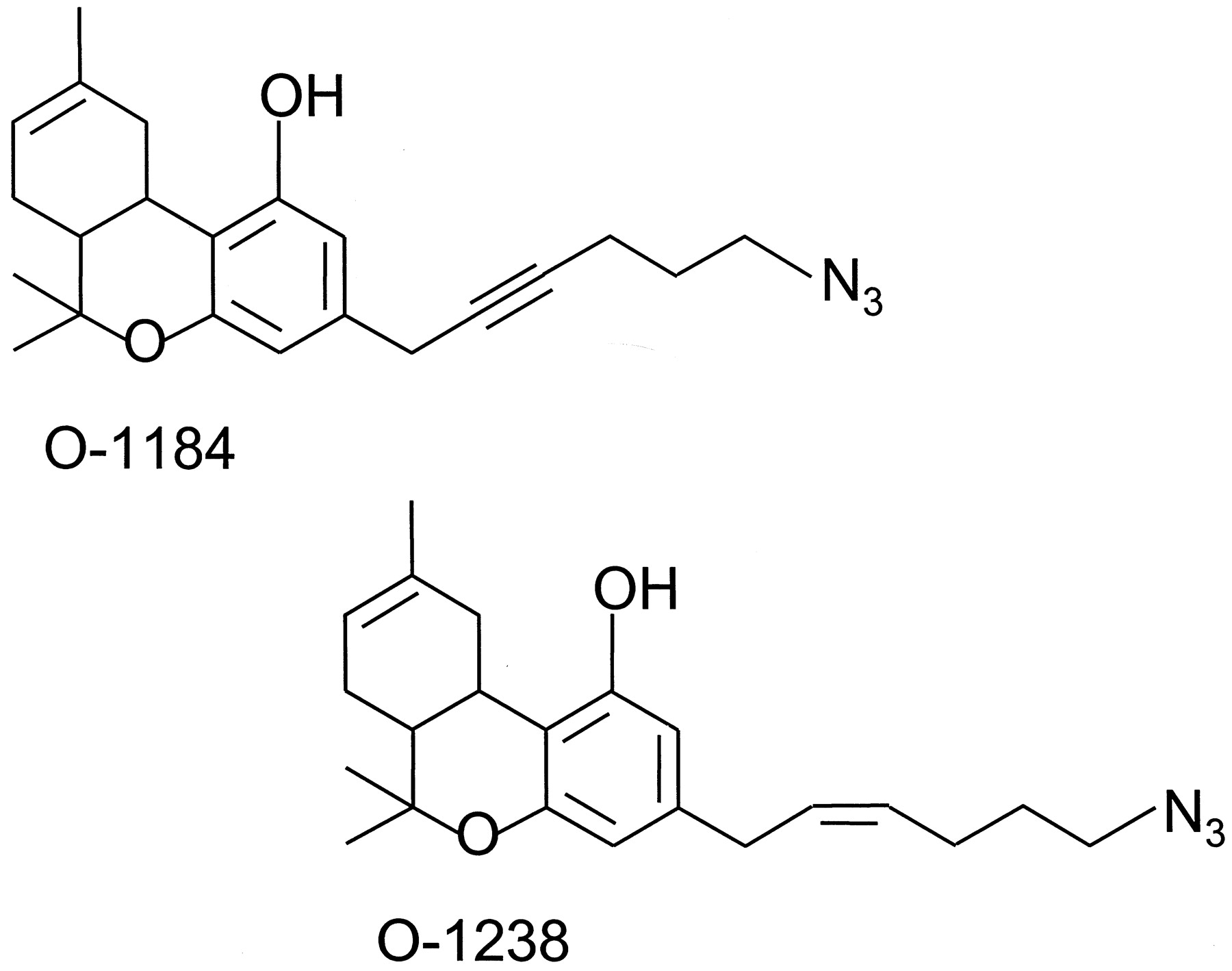
The structures of O-1184 and O-1238.
III. Bioassay
A. In Vivo Bioassay Systems
1. Introduction.
Cannabinoids produce a complex array of behavioral effects that have been characterized in numerous animal species as well as in humans. Although the diverse behavioral effects of cannabinoids provide ample opportunity for quantitating the pharmacological actions of this class of compounds, they provide a challenge to the elucidation of mechanism of action. A major focus of cannabinoid research has been the identification of pharmacological effects that are receptor-mediated. Until the recent development of a specific CB1 receptor antagonist, SARs provided the only in vivo approach for implicating receptor mechanisms. A major goal of cannabinoid research is elucidating the mechanisms responsible for the behavioral “high”. Of course, the psychotomimetic effects can only be assessed in humans, which imposes severe restrictions on SAR studies. Few cannabinoid analogs have sufficient toxicological histories to qualify for human experimentation. The difficulties with human studies have necessitated close examination of pharmacological effects in several animal species, many of which vary in their response to cannabinoids. However, it has now been established that numerous pharmacological effects are mediated via the cannabinoid receptor. There are several fundamental principles that have guided this undertaking. One of the most critical aspects of the choice is whether the pharmacological measure in animals is representative of cannabinoid effects in humans. Equally important is the characterization of behavioral effects that are unique to cannabinoids (i.e., mediated through cannabinoid receptors). Finally, there are the practical aspects of selecting pharmacological effects that can be quantitated and readily obtained. Using these criteria, several pharmacological effects in vivo can be attributed to the activation of cannabinoid receptors.
2. Dog Static Ataxia.
Walton et al. (1937) described the effects of cannabinoids in dogs, which represented one of the first animal models that was highly unique for this class of compounds. These effects include sedation, catalepsy, motor incoordination, and hyperexcitability; however, it is the combination of these effects that causes dogs to weave to and fro while remaining fixed in one spot that led to the somewhat anomalous term “static ataxia”. Again, the primary advantage of this model is that these behaviors describe a highly specific profile for cannabinoids that is not confused with that produced by other behaviorally active compounds. These behaviors can also be semiquantitated, and extensive SAR studies have revealed both dramatic changes in potency with modest changes in structure (Walton et al., 1937; Martin et al., 1975; Beardsley et al., 1987) and enantioselectivity (Dewey et al., 1984; Little et al., 1989). The strength of this model is that the results obtained correlate well with psychoactivity. These findings strongly suggest that cannabinoid-induced static ataxia is receptor-mediated. Moreover, the CB1 receptor antagonist, SR141716A, antagonizes the effects of Δ9-THC in this model, a finding that strongly supports CB1 involvement (Lichtman et al., 1998).
3. Overt Behavior in Monkeys.
Mechoulam and colleagues (Edery et al., 1971) synthesized a large number of cannabinoid analogs that allowed them to develop the first framework for describing the structural features that were critical for cannabinoid pharmacological activity. Their model was based on the gross observation of overt behavioral effects in monkeys. The cannabinoids produced sedation, ptosis, body sag, etc., which was reasonably selective for cannabinoids and could be rated in a semiquantitative fashion. They described a SAR that also included enantioselectivity (Edery et al., 1971); however, there have been no reports of reversal of these effects by the CB1 receptor antagonist, SR141716A.
4. Rat Drug Discrimination.
Drug discrimination is considered one of the most reliable means of predicting whether test drugs produce subjective effects similar to those of a known drug. Initially, an animal is trained to press a lever for food reward and then subsequently trained to press a specific lever for this reward when under the influence of Δ9-THC and another lever when any other drug is administered. Therefore, on test days, which lever the animal chooses tells the experimenter whether the test compound is perceived as THC-like or not. Much of the early rat drug discrimination literature for the cannabinoids was generated by Järbe's laboratory (Järbe and Ohlin, 1977; Järbe and McMillan, 1979, 1980; Järbe et al., 1989; Järbe and Mathis, 1992). Rats have also been trained to discriminate between CP55940, a potent cannabinoid agonist, and vehicle (Gold et al., 1992). These animals perceived Δ9-THC as being like CP55940. Furthermore, the Δ9-THC-discriminative cue has been shown to be selective for cannabinoids (Barrett et al., 1995).
SAR data have been obtained in drug discrimination experiments conducted with the aminoalkylindoles (Compton et al., 1992a), various other structurally dissimilar cannabinoids (Wiley et al., 1995b), and anandamide (Wiley et al., 1995a). The results from all of these studies are consistent with receptor affinity for the CB1receptor. In addition, SR141716A was shown to block the discriminative properties of rats trained on CP55940 (Wiley et al., 1995b) and on Δ9-THC (Wiley et al., 1995c). Therefore, the discriminative properties of cannabinoids appear to be mediated through CB1 receptors. More importantly, there is an excellent correlation between drugs that engender cannabinoid responding in the drug discrimination paradigm and psychoactivity in humans (Balster and Prescott, 1992).
5. Monkey Drug Discrimination.
The above description of drug discrimination in rats applies to monkeys; however, it has been argued that primates may provide a more accurate reflection of cannabinoid behavioral effects in humans. This model has provided reassuring data that novel cannabinoids, such as CP55940 (Gold et al., 1992),R-(+)-WIN55212 (Compton et al., 1992a), and the endogenous ligand anandamide (Wiley et al., 1997), are likely to produce cannabinoid behavioral effects in humans. Establishing this fact is particularly crucial since these compounds are being used widely as cannabinoid probes. As with the rat drug discrimination, SR141716A was shown to block the discriminative properties of Δ9-THC (Wiley et al., 1995c), thereby implicating CB1 receptors.
6. Mouse Tetrad Model.
As mentioned earlier, cannabinoids are known to produce a wide range of pharmacological effects that include hyperstimulation, sedation, catalepsy, and several other depressant properties. Individually, none of these effects can be considered unique for cannabinoids, since all of these properties are shared by numerous classes of centrally active agents. Several years ago, it was discovered that i.v. administration of cannabinoids in mice produced sedation, hypothermia, antinociception, and catalepsy in the same dose range and within the same time frame, so that all four behaviors could be determined in the same animal for each injection (Martin et al., 1987). Compounds active in this composite model also produce effects in models that we traditionally consider to be highly predictive of cannabinoid effects, such as drug discrimination (Compton et al., 1993). Furthermore, the SAR studies in the mouse tetrad model are consistent with affinity for the CB1 receptor for CP55940 and related analogs (Little et al., 1988; Compton et al., 1992b), enantiomers of dimethylheptyl analogs of THC (Little et al., 1989), aminoalkylindoles (Compton et al., 1992a; Huffman et al., 1994), and endocannabinoids (Adams et al., 1998). It has also been shown that SR141716A is highly effective in blocking the effects of most cannabinoid analogs in the mouse tetrad model (Rinaldi-Carmona et al., 1994; Compton et al., 1996), confirming the involvement of CB1 receptors. The one exception has been the endocannabinoids (Adams et al., 1998). Although SR141716A fails to block the effects of anandamide, it is capable of blocking the effects of metabolically stable anandamide analogs (Adams et al., 1998). However, some anandamide analogs are effective in the mouse tetrad and apparently bind with little affinity for the CB1receptor (Di Marzo et al., 2001a). There are several possible explanations for these discrepancies, one of which is that the mouse tetrad may not be selective for cannabinoids. If future studies reveal that false positives can occur in this model, then it will be necessary to verify the results in this model with antagonism studies using a CB1-selective antagonist.
7. Memory Models.
The naturally occurring cannabinoids, as well as a wide range of synthetic compounds, have been demonstrated to impair learning and memory in rodents (Carlini et al., 1970), nonhuman primates (Ferraro and Grilly, 1973), and humans (Abel, 1971). Δ9-THC has been found to disrupt memory as assessed in the delayed match-to-sample task (Heyser et al., 1993), Lashley III maze (Carlini et al., 1970), and the eight-arm radial maze (Nakamura et al., 1991). Δ9-THC, CP55940, andR-(+)-WIN55212 all impaired working memory in rats in the eight-arm radial maze and the delayed nonmatch-to-sample task. Lichtman and Martin (1996) also found that Δ9-THC, CP55940, and R-(+)-WIN55212, administered systemically, impaired spatial memory in rats as assessed by the eight-arm radial maze and retarded completion time. Direct injection of CP55940 into the hippocampus impaired memory, which appeared specific to cognition since no other pharmacological effects were produced (Lichtman et al., 1995). The effects of cannabinoid on memory in rats are also blocked by SR141716A, providing strong evidence that these effects are mediated through CB1 receptors (Lichtman and Martin, 1996). Furthermore, the eight-arm radial maze has also been modified to evaluate agents for their potential to enhance memory performance. Under these conditions, SR141716A administration improved the performance of rats (Lichtman, 2000). Another learning and memory paradigm that has become increasingly popular in recent years is the Morris water maze. Reference memory can be assessed by requiring a well trained rat or mouse to navigate to a hidden platform that always remains in the same location, whereas working memory is assessed by requiring the animal to learn a new platform location each session. In this model, Δ9-THC disrupts working memory at doses much lower than those required to interfere with reference memory (Varvel et al., 2001). Additionally, SR141716A reverses the effects of Δ9-THC, demonstrating CB1-mediated effects. This model is ideal for assessing the SARs of cannabinoid agonists and antagonists.
8. Human Assays.
Cannabinoids that have been evaluated in humans include the active constituents in marihuana, their metabolites, and some agents with therapeutic potential (Razdan, 1986). Some of the earlier studies demonstrated that SAR could be conducted in humans (Perez-Reyes et al., 1972; Hollister, 1974). These evaluations in humans provided the basis for correlating psychotomimetic potency to potency in animal models. For the more than 20 cannabinoids that have been evaluated in humans, an excellent correlation exists between the cannabinoid subjective effects in humans and drug discrimination in laboratory animals (Balster and Prescott, 1992). Since CB1 receptors have been implicated in mediating drug discrimination, as discussed above, it seems most plausible that the behavioral effects in humans are mediated through the CB1 receptor. More conclusive evidence came from recent studies demonstrating that SR141716A blocks cannabinoid subjective effects as well as cannabinoid-induced tachycardia in humans (Huestis et al., 2001).
B. In Vitro Bioassay Systems
1. Binding Assays.
As detailed elsewhere (Pertwee, 1997,1999), the most widely used radiolabeled cannabinoid receptor probe is [3H]CP55940. Because CP55940 has approximately equal affinity for CB1 and CB2 binding sites (Table 2), displacement assays with [3H]CP55940 that are directed at characterizing the binding properties of novel unlabeled ligands are generally performed with membranes that are known to contain either CB1 or CB2 receptors but not both receptor types. These membranes are often obtained from cells transfected with CB1 or CB2receptors. An alternative practice has been to use tissues that express dense populations of CB1 or CB2 receptors naturally, usually brain tissue for CB1 receptors and spleen tissue for CB2 receptors. However, although brain tissue is largely populated with CB1 receptors, some CB2 receptors may also be present on microglia (Kearn and Hillard, 1999; see also Section VII.B.). Similarly, although most cannabinoid receptors in the spleen are CB2, some CB1 receptors are expressed by this tissue as well (Bouaboula et al., 1993;Galiègue et al., 1995; Ishac et al., 1996). The possibility also exists that brain and/or spleen express types of cannabinoid receptor yet to be identified. Indeed, there is already some evidence that mammalian brain, spinal cord, and peripheral nervous system can express additional types of cannabinoid receptor (Section XI.).
Other commercially available probes with high affinity for cannabinoid receptors are [3H]SR141716A, which is CB1-selective (Rinaldi-Carmona et al., 1996b; Table 2), [3H]HU-243, which binds more or less equally well to both CB1 and CB2 receptor (Devane et al., 1992a; Bayewitch et al., 1995), and [3H]R-(+)-WIN55212, which has marginally greater affinity for CB2than CB1 binding sites (Slipetz et al., 1995;Song and Bonner, 1996; see also Pertwee, 1999). Tritiated 11-hydroxy-Δ9-THC-1′,1′-dimethylheptyl has also been synthesized and used in cannabinoid binding assays (Thomas et al., 1992). However, this ligand is not generally available. Three other radiolabeled ligands have been developed as potential probes for human single photon emission computed tomography or positron emission tomography experiments. These are 123I-labeled analogs of AM251 and AM281 (Lan et al., 1996; Gatley et al., 1997;Gatley et al., 1998) and an 18F-labeled analog of SR141716A (SR144385) (Barth, 1998). Particularly promising single photon emission computed tomography results have been obtained from animal experiments with [123I]AM281 (Gatley et al., 1998).
2. Inhibition of Cyclic AMP Production.
The ability of cannabinoid CB1 and CB2receptor agonists to inhibit basal or drug-induced cyclic AMP production is widely exploited for the quantitative, functional bioassay of cannabinoids in vitro (see Pertwee, 1997, 1999). Although many types of receptor are negatively coupled to adenylyl cyclase, it is still possible to achieve selectivity by using a CB1 or CB2 receptor antagonist or by performing assays with cells transfected with CB1 or CB2 receptors. Preparations that are particularly sensitive to the inhibitory effect of cannabinoids on cyclic AMP production are cultured cells transfected with CB1 or CB2 receptors, certain cultured cell lines that express CB1receptors naturally, and CB1 receptor-containing membrane preparations obtained from the brain (see Pertwee, 1997,1999). Cells expressing CB2 receptors naturally (e.g., mouse spleen cells and human lymphocytes) are relatively insensitive to cannabinoid-induced inhibition of cyclic AMP production (Pertwee, 1997).
3. [35S]Guanosine-5′-O-(3-thiotriphosphate) Binding Assay.
This bioassay exploits the coupling of CB1 and CB2 receptors to G proteins. It relies on the increase in G protein affinity for GTP (and hence [35S]GTPγS) that is triggered by the occupation by agonist molecules of CB1 or CB2 receptors, the measured response being net agonist-stimulated [35S]GTPγS binding to G protein. The assay can be performed with the same range of tissue preparations that are used for the cyclic AMP assay, again in the presence or absence of selective CB1 or CB2 antagonists. In addition, [35S]GTPγS is sometimes used in autoradiography experiments with tissue sections (Sim et al., 1995;Selley et al., 1996; Breivogel et al., 1997). To minimize [35S]GTPγS binding that occurs in the absence of the agonist and so maximize agonist-induced stimulation of binding, high amounts of GDP and sodium chloride are usually added to the bioassay system (Sim et al., 1995; Selley et al., 1996; Breivogel et al., 1998). Since GDP decreases basal binding of [35S]GTPγS to a greater extent than agonist-stimulated binding, the overall consequence of adding GDP is an increase in net agonist-stimulated [35S]GTPγS binding (Breivogel et al., 1998). The extent to which net agonist-stimulated [35S]GTPγS binding can be enhanced in this way is limited by the concentration-related inhibitory effect that GDP has on absolute levels of both basal and agonist-stimulated binding. Thus, as GDP concentrations are progressively raised, a point is eventually reached at which [35S]GTPγS binding has fallen to a level that is too low to be measured reproducibly (Selley et al., 1996). The optimal GDP concentration appears to be higher for the assay of agonists with high than with low relative intrinsic activities, such that the ability of an agonist with low relative intrinsic activity to increase [35S]GTPγS binding above basal levels may be completely abolished when the concentration of GDP is increased (Breivogel et al., 1998; Griffin et al., 1998).
The [35S]GTPγS assay is less sensitive than the cyclic AMP and isolated tissue assays described underSections III.B.2. or III.B.4. Presumably, this is because the measured responses in these other bioassays are located further along the signaling cascade than G protein, so that there is greater signal amplification. The [35S]GTPγS assay should be independent of any variations that may exist between tissues in the relative contribution made by different G protein-coupled effector mechanisms. This is because it provides a total measure of G protein-mediated cannabinoid receptor activation rather than a measure of the activation of just one particular cannabinoid receptor effector mechanism as in the cyclic AMP assay. However, the [35S]GTPγS assay will be affected by both the type and the relative abundance of G protein α subunits. For example, if more Goα is expressed than Giα, the Goα response will dominate. Also, some G protein α subunits, such as Gq/11, are difficult to detect in the [35S]GTPγS assay.
4. Inhibition of Electrically Evoked Contractions of Isolated Smooth Muscle Preparations.
Smooth muscle preparations most often used for the bioassay of cannabinoids are the mouse isolated vas deferens and the myenteric plexus-longitudinal muscle preparation of guinea pig small intestine. These bioassays, which are particularly sensitive, rely on the ability of cannabinoid receptor agonists to act through CB1 receptors to inhibit electrically evoked contractions (Pertwee et al., 1992; Pertwee, 1997, Pertwee, 2001a). The CB1 receptors are located on prejunctional neurons and mediate inhibition of electrically evoked contractile transmitter release (Coutts and Pertwee, 1997; Pertwee, 1997; Schlicker and Kathmann, 2001). It is also possible that CB2-like receptors (see Section XI.) share the ability of CB1receptors to mediate inhibition of evoked contractions of the mouse vas deferens (Griffin et al., 1997). Several types of noncannabinoid receptor can mediate inhibition of evoked contractions of the mouse vas deferens or myenteric plexus-longitudinal muscle preparation. Consequently, to achieve selectivity, it is necessary to establish the susceptibility of agonists to antagonism by a selective CB1 antagonist, such as SR141716A (Pertwee et al., 1995b, 1996).
C. Practical Difficulties
One practical difficulty associated with the bioassay of cannabinoids both in vivo and in vitro is the high lipophilicity and low water solubility of these compounds, as this necessitates the use of nonaqueous vehicles. Indeed, it was this difficulty that prompted the development of the water-soluble cannabinoid receptor agonist O-1057 (Pertwee et al., 2000). Commonly used vehicles for the in vivo or in vitro administration of cannabinoid receptor agonists and antagonists include ethanol, dimethyl sulfoxide, polyvinylpyrrolidone, Tween 80, Cremophor, Emulphor, and bovine serum albumin (BSA). These are used singly or in combination, either by themselves or mixed with water or saline. Results obtained using such vehicles should be interpreted with caution because the vehicles may themselves produce pharmacological changes, for example, by perturbing membrane phospholipids. Consequently, vehicle control experiments are vital. These vehicles may also affect the apparent potencies of cannabinoid receptor ligands. Indeed, as detailed elsewhere (Pertwee, 1997), there are reports that [3H]CP55940 binding to CB1-containing membranes can be markedly influenced by the concentration of BSA used for cannabinoid solubilization. For example, in binding experiments with rat brain sections, Herkenham et al. (1991) found the apparent dissociation constant of [3H]CP55940 to be 2.6 nM in the presence of 1% BSA but 15 nM in the presence of 5% BSA. For endocannabinoids, a second practical difficulty is that they are substrates both of membrane transporters and of hydrolytic enzymes such as FAAH (Section I.). It is for this reason that experiments with anandamide are often performed in the presence of a FAAH inhibitor, such as the general protease inhibitor phenylmethylsulfonyl fluoride (see Pertwee, 1997). Alternative strategies have been to perform experiments with FAAH−/− mice (Cravatt et al., 2001) or with analogs that are more resistant than anandamide to enzymic hydrolysis, for example, R-(+)-methanandamide (Section II.).
IV. Cellular Signal Transduction
Agonist stimulation of CB1 and CB2 cannabinoid receptors activates a number of signal transduction pathways via the Gi/o family of G proteins (see reviews by Howlett, 1995a; Pertwee, 1997, 1999). CB1 receptor signaling through G proteins has been demonstrated by [35S]GTPγS binding using rat brain membranes and brain slices (see Section III.B. for references). For CB1 receptor-stimulated [35S]GTPγS binding, anandamide andR-(+)-methanandamide are partial agonists compared withR-(+)-WIN55212, levonantradol, CP55940, 2-arachidonoylglycerol, and desacetyl-l-nantradol (see Howlett and Mukhopadhyay, 2000 for review and original references). In CHO cells expressing recombinant hCB2 receptors, [35S]GTPγS binding was stimulated by anandamide as a partial agonist compared with HU-210, whereas 2-arachidonoylglycerol was a full agonist (Hillard et al., 1999;Gonsiorek et al., 2000). Inverse agonist activity exhibited by SR141716A and analogs has been most clearly demonstrated by a decrement in [35S]GTPγS binding to G proteins in brain preparations (Landsman et al., 1997; Meschler et al., 2000).
Free Giα proteins regulate adenylyl cyclase, leading to an inhibition of cyclic AMP production. The consequent damping of phosphorylation by protein kinase A may modulate signaling pathways, such as that of ion channels and focal adhesion kinase. It is believed that free βγ dimers mediate the regulation of ion channels, mitogen-activated protein kinase (MAPK), and phosphatidylinositol-3-kinase (PI3K). However, it is not clear which Gi/oα subtypes might be associated with the βγ dimers in heterotrimers responsible for those responses. It should be noted that values of potency and relative intrinsic activity may differ for the various signal transduction pathways. The relative intrinsic activities of various cannabinoid receptor agonists to evoke a response via G proteins has been discussed by Breivogel et al. (1998)and Kearn et al. (1999). This section will summarize the most well characterized signaling pathways for cannabinoid receptors.
A. Regulation of Adenylyl Cyclase
Inhibition of adenylyl cyclase has been characterized in brain tissue and neuronal cells expressing CB1 and in human lymphocytes and mouse spleen cells expressing CB2 receptors (see Howlett and Mukhopadhyay, 2000and Pertwee, 1997, 1999 for review). The finding that cultured cell lines that express recombinant CB1 or CB2 receptors lead to inhibition of cyclic AMP production is supportive evidence that these receptor types can initiate this response (Matsuda et al., 1990; Felder et al., 1992;Vogel et al., 1993; Slipetz et al., 1995). CB1and CB2 receptor-mediated inhibition of adenylyl cyclase is a pertussis toxin-sensitive cellular event, indicating the requirement for Gi/o proteins (Howlett et al., 1986; Felder et al., 1992; Pacheco et al., 1993; Vogel et al., 1993). Adenylyl cyclase activity in N18TG2 membranes possessing endogenous CB1 receptors was inhibited by anandamide,R-(+)-methanandamide, and 2-arachidonoylglycerol, with relative intrinsic activities similar to desacetyl-l-nantradol, R-(+)-WIN55212, or CP55940 (Childers et al., 1994; Pinto et al., 1994; Howlett and Mukhopadhyay, 2000). In CHO cells expressing CB2receptors, anandamide and R-(+)-methanandamide partially inhibited forskolin-stimulated cyclic AMP accumulation at high concentrations (Felder et al., 1995; Hillard et al., 1999; Gonsiorek et al., 2000). The data suggest that anandamide is an agonist with low relative intrinsic activity for CB2 receptor- compared with CB1 receptor-mediated cyclic AMP production. 2-Arachidonoyl-glycerol has been found to behave as a full agonist when the measured effect is inhibition of forskolin-stimulated cyclic AMP accumulation in CHO cells expressing recombinant CB2 receptors (Gonsiorek et al., 2000).
Stimulation of adenylyl cyclase has been reported in pertussis toxin-treated cells, suggesting that in the absence of functional Gi/o coupling, the CB1receptor can activate Gs (Glass and Felder, 1997). The isoform of adenylyl cyclase expressed in cells is predicted to be a major determinant of the outcome of cannabinoid receptor activation, as demonstrated by studies in Vogel's laboratory (Rhee et al., 1998). These researchers found that expression of CB1 or CB2 cannabinoid receptors in a host cell coexpressing adenylyl cyclase isoforms 1, 3, 5, 6, or 8 resulted in inhibition of cyclic AMP accumulation. However, coexpression of either cannabinoid receptor type with adenylyl cyclase isoforms 2, 4, or 7 resulted in stimulation of cyclic AMP accumulation.
B. Regulation of Ion Channels
1. Ion Channel Modulation by Protein Kinase A.
CB1 cannabinoid receptors activate A-type potassium currents in rat hippocampal cells (Childers and Deadwyler, 1996). This response is due to the modulation of the intracellular cyclic AMP concentrations, thereby regulating the net phosphorylation of ion channel proteins by protein kinase A.
2. K+ Channel Activation.
Exogenously expressed CB1 receptors couple to the inwardly rectifying K ir channels in AtT-20 pituitary tumor cells in a pertussis toxin-sensitive manner, indicating that Gi/o proteins serve as transducers of the response (Henry and Chavkin, 1995; Mackie et al., 1995). Anandamide was a full agonist compared with R-(+)-WIN55212 in theK ir current activation in the AtT-20 cell model (Mackie et al., 1995); however, it was a partial agonist inXenopus laevis oocytes coexpressing the CB1 receptor and G protein-coupled inwardly rectifying potassium channel 1 and G protein-coupled inwardly rectifying potassium channel 4 channels (McAllister et al., 1999).
3. Inhibition of Voltage-Gated L, N, P, and Q Ca2+Channels.
L-type Ca2+ channels were inhibited by anandamide and R-(+)-WIN55212 in cat brain arterial smooth muscle cells, which express mRNA for the CB1 receptor (Gebremedhin et al., 1999). The cannabinoid-evoked inhibition of L-type Ca2+currents was blocked by pertussis toxin and SR141716A and was pharmacologically correlated with vascular relaxation in cat cerebral arterial rings (Gebremedhin et al., 1999).
The CB1 receptor inhibits N-type voltage-gated Ca2+ channels in neuronal cells through Gi/o protein (Caulfield and Brown, 1992; Mackie and Hille, 1992; Felder et al., 1993; Mackie et al., 1993; Pan et al., 1996). Anandamide was a partial agonist compared withR-(+)-WIN55212 or CP55940 (Mackie et al., 1993). 2-Arachidonoylglycerol and analogs inhibited the depolarization-evoked rise in intracellular Ca2+ as detected by Fura-2 in differentiated NG108-15 cells (Sugiura et al., 1997b). Anandamide was a partial agonist, and arachidonic acid was without effect.
R-(+)-WIN55212 and anandamide were both full agonists to inhibit Q-type Ca2+ currents in AtT-20 pituitary cells expressing recombinant CB1 receptors (Mackie et al., 1995). This response was pertussis toxin-sensitive, implicating Gi/o proteins as transducers. Anandamide inhibited P/Q-type Ca2+ fluxes (i.e., blocked by ω-agatoxin-IVa) as detected by Fura-2 fluorescence in rat cortical and cerebellar brain slices (Hampson et al., 1998). This response was blocked by SR141716A and pertussis toxin, indicating mediation by CB1 receptors and Gi/o proteins. Neither R-(+)-WIN55212 nor anandamide were able to inhibit Q-type Ca2+currents in AtT-20 cells expressing CB2receptors, indicating that the CB2 receptor fails to couple to this current (Felder et al., 1995).
C. Regulation of Intracellular Ca2+ Transients
Cannabinoid agonists evoked a rapid, transient increase in intracellular free Ca2+ in undifferentiated N18TG2 neuroblastoma and NG108-15 neuroblastoma-glioma hybrid cells (Sugiura et al., 1996, 1997a). This response was blocked by SR141716A, confirming mediation by the CB1receptor (Sugiura et al., 1996, 1999). For this response, HU-210, CP55940, Δ9-THC, anandamide, andR-(+)-methanandamide behaved as partial agonists compared with 2-arachidonoylglycerol or 1(3)-arachidonoylglycerol (Sugiura et al., 1996, 1997a, 1999). The 2-arachidonoylglycerol-evoked intracellular Ca2+ transient was blocked by pertussis toxin and by a phospholipase C inhibitor, suggesting a mechanism whereby a receptor-mediated release of Gi/o βγ subunits might activate phospholipase Cβ, leading to inositol-1,4,5-triphosphate (IP3) release (Sugiura et al., 1996, 1997a). An interaction between CB1 cannabinoid receptors and phospholipase C was shown in cultured cerebellar granule neurons, in which cannabinoid agonists augmented the Ca2+signal in response to NMDA receptor stimulation or K+ depolarization (Netzeband et al., 1999). The response was antagonized by SR141716A, pertussis toxin, and the phospholipase C inhibitor 1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione (Netzeband et al., 1999). The source of the released Ca2+ was a caffeine-sensitive and IP3 receptor-sensitive pool. In contrast, studies of CHO cells expressing recombinant CB1 or CB2 receptors were unable to detect release of IP3 or phosphatidic acid in response to anandamide or R-(+)-WIN55212, under conditions in which other exogenously expressed receptors coupled to phospholipases C could evoke IP3 release (Felder et al., 1992, 1995). This suggests that the cellular milieu may be a factor in this CB1 receptor signal transduction pathway.
D. Regulation of Focal Adhesion Kinase, Mitogen-Activated Protein Kinase, Phosphatidylinositol-3-Kinase, and Ceramide Metabolism
1. Signal Transduction via Focal Adhesion Kinase.
Cannabinoid agonists stimulatedtyr-phosphorylation of focal adhesion kinase (FAK) (pp125) in hippocampal slices (Derkinderen et al., 1996). The response could be blocked with SR141716A and pertussis toxin as evidence for mediation by CB1 receptors and Gi/o. Thetyr-phosphorylation of FAK in brain slices was reversed by 8-Br-cyclic AMP and mimicked by protein kinase A inhibitors, suggesting that Gi-mediated inhibition of adenylyl cyclase is integral to this pathway (Derkinderen et al., 1996). FAK is important for integrating cytoskeletal changes with signal transduction events, perhaps playing a role in synaptic plasticity.
2. Signal Transduction via Mitogen-Activated Protein Kinase and Phosphatidylinositol-3-Kinase.
MAPK (p38) was activated in CHO cells expressing recombinant CB1 receptors (Rueda et al., 2000) and in human umbilical vein endothelial cells possessing endogenous CB1 receptors (Liu et al., 2000). MAPK (p42/p44) was activated via CB1 receptors in U373MG astrocytic cells and in host cells expressing recombinant CB1 receptors (Bouaboula et al., 1995b). In C6 glioma and primary astrocyte cultures, Δ9-THC and HU-210 activated MAPK (p42/p44) (Sánchez et al., 1998;Guzmán and Sánchez, 1999). These effects were mediated by CB1 receptors and Gi/oproteins inasmuch as they were blocked by SR141716A and pertussis toxin. In WI-38 fibroblasts, anandamide promotedtyr-phosphorylation of extracellular signal-regulated kinase 2 and increased MAPK activity (Wartmann et al., 1995). In some cells, CB1 receptor signaling via MAPK was blocked by wortmannin (Bouaboula et al., 1995b; Wartmann et al., 1995), implicating PI3K as a mediator along this pathway. Δ9-THC promoted Raf-1 translocation to the membrane and phosphorylation in cortical astrocytes (Sánchez et al., 1998). From these studies, one could envisage a pathway whereby CB1 receptor-mediated Gi/orelease of βγ subunits leads to activation of PI3K, resulting in tyrosine phosphorylation and activation of Raf-1, and subsequent MAPK phosphorylation. Regarding functions regulated by the MAPK pathway, CP55940-stimulated MAPK activity led to activation of the Na+/H+ exchanger in CHO cells stably expressing the CB1 receptor (Bouaboula et al., 1999). Anandamide-stimulated MAPK activity was associated with phosphorylation of cytoplasmic phospholipase A2, release of [3H]arachidonic acid, and subsequent synthesis of prostaglandin E2 in WI-38 cells (Wartmann et al., 1995).
In C6 glioma and primary astrocyte cultures, Δ9-THC and HU-210 increased glucose metabolism and glycogen synthesis (Guzmán and Sánchez, 1999). The activation of Gi/o and PI3K by cannabinoid agonists led to activation of protein kinase B/Akt (isoform IB) in U373MG astrocytic cells and in CHO cells expressing recombinant CB1 receptors (Gómez del Pulgar et al., 2000). Protein kinase B phosphorylation and inhibition of glycogen synthase kinase-3 could account for increased glycogen synthase activity and increased glycolysis in responsive cells.
MAPK was activated in cultured human promyelocytic HL-60 cells possessing endogenous CB2 receptors and in CHO cells expressing recombinant CB2 receptors (Bouaboula et al., 1996). However, cannabinoid drugs failed to activate protein kinase B in HL-60 cells, suggesting that a PI3K mechanism may not be regulated by CB2 receptors in this model (Gómez del Pulgar et al., 2000).
3. Signal Transduction via Ceramide.
Studies with primary astrocyte cultures showed that anandamide, Δ9-THC, and HU-210 increased glucose metabolism, phospholipid synthesis, and glycogen synthesis via an SR141716A-inhibitable but pertussis toxin-resistant mechanism (see reviews by Guzmán and Sánchez, 1999 and Guzmán et al., 2001 for commentary and original references). Data supported a pathway that utilizes the adaptor protein Fan (factorassociated with neutral sphingomyelinase) to couple CB1 receptor stimulation to sphingomyelinase activation, release of ceramide, and subsequent activation of the Raf-1/MAPK cascade (Sánchez et al., 2001). In a second mechanism, ceramide activated carnitine palmitoyltransferase I within astrocyte mitochondrial membranes to stimulate ketogenesis and fatty acid oxidation (Blázquez et al., 1999).
Prolonged (days) elevation of intracellular ceramide has been associated with events leading to decreased proliferation and apoptosis in glioma cells (see Guzmán et al., 2001 for review). This response was initiated by chronic stimulation of both CB1 and CB2 receptors on a susceptible C6 glioma strain and involves increased ceramide synthesis via serine palmitoyltransferase, Raf-1 activation, and MAPK (p42/44) activation.
E. Immediate Early Gene Expression and Protein Synthesis Regulation
MAPK activation can be linked to expression of immediate early genes, as has been demonstrated for Krox-24 expression mediated by CB1 receptors in U373MG human astrocytoma cells (Bouaboula et al., 1995a). Krox-24 expression was stimulated via CB2 receptors in HL-60 promyelocytes (Bouaboula et al., 1996). Intracerebroventricular injection of anandamide evoked an increase in c-FOS immunoreactive protein in rat brain (Patel et al., 1998). Cannabinoid receptor agonists activated c-Jun N-terminal kinase (JNK1 and JNK2) in CHO cells expressing recombinant CB1 receptors (Rueda et al., 2000). The pathway for JNK activation involves Gi/o proteins, PI3K, and Ras (Rueda et al., 2000).
The suppression of prolactin receptor and trk nerve growth factor receptor synthesis by anandamide in human breast cancer MCF-7 cells may be due to a CB1 receptor-mediated decrease in protein kinase A and increase in MAPK activities (De Petrocellis et al., 1998; Melck et al., 1999). This CB1-mediated response ultimately led to an antiproliferative effect on the cells.
F. Regulation of Nitric Oxide Synthase
Nitric oxide (NO) production was stimulated by anandamide in rat median eminence fragments (Prevot et al., 1998) and by anandamide or CP55940 in leech or muscle ganglia (Stefano et al., 1997a,b; 1998). Responses in these tissues were blocked by SR141716A, implicating the involvement of a CB1-like receptor. Antagonism byN G-nitro-l-arginine methyl ester suggests that a signal transduction pathway must lead to regulation of NOS (Prevot et al., 1998). Because both anandamide and the NO-generating agentS-nitroso-N-acetyl-penicillamine could inhibit the release of preloaded radiolabeled dopamine from invertebrate ganglia, a role for NO in mediating the effects of anandamide on neurotransmitter release was implied (Stefano et al., 1997a).
Anandamide and HU-210 stimulated NO production in human saphenous vein segments (Stefano et al., 1998), cultured human arterial endothelial cells (Fimiani et al., 1999; Mombouli et al., 1999), cultured human umbilical vein endothelial cells (Maccarrone et al., 2000), and human monocytes (Stefano et al., 1996). These responses were blocked by SR141716A, implicating CB1 receptors. In cultured human arterial endothelial cells, NO generation was preceded by a rapid increase in intracellular Ca2+ concentration (Fimiani et al., 1999; Mombouli et al., 1999), consistent with the stimulation of a Ca2+-regulated constitutive NOS. In saphenous vein endothelia, the generation of NO required Ca2+ in the perfusate, suggesting that an extracellular source of Ca2+ might be required for NOS activation (Stefano et al., 1998). In human vein arterial cells, generation of NO and peroxynitrite was associated with activation of the anandamide transporter (Maccarrone et al., 2000).
Anandamide inhibited induction of inducible NOS (iNOS) by lipopolysaccharide plus interferon-γ in saphenous vein endothelium (Stefano et al., 1998) and neonatal mouse astrocytes (Molina-Holgado et al., 1997). The modulation of iNOS induction by anandamide required NO production, and this was blocked by SR141716A, implicating the CB1 receptor. The response could be mimicked byS-nitrosyl-N-acetyl-penicillamine, suggesting that transient NO production (presumably via a constitutive type of NOS) regulated the induction of iNOS (Stefano et al., 1998). Because both anandamide andS-nitrosyl-N-acetyl-penicillamine diminished the cyclic AMP accumulation evoked by lipopolysaccharide plus interferon-γ, these authors suggested that the mechanism for suppression of iNOS induction involved the inhibition of cyclic AMP production by NO (Stefano et al., 1998). It is well recognized that NO reversibly inhibits adenylyl cyclase isoforms 5 and 6 by acys-nitrosylation mechanism (Tao et al., 1998; McVey et al., 1999), providing a basis for postulating this mechanism.
The attenuation of iNOS induction by Δ9-THC in RAW 264.7 cells implicated the CB2 receptor and a mechanism involving a decrement in cyclic AMP (Jeon et al., 1996). In mouse peritoneal macrophages, the attenuation of iNOS induction by a series of cannabinoid drugs exhibited a relative order of potency that did not resemble the expected profile for CB1 or CB2 receptors (Coffey et al., 1996).
V. Molecular Biology of Cannabinoid Receptors
Although the existence of cannabinoid receptors was known before their cloning, the receptors presently known as CB1 and CB2 cannabinoid receptors were cloned as part of strategies based on conserved sequence motifs to clone G protein-coupled receptors in general rather than specifically trying to clone cannabinoid receptors. It was only after extensive screening of an expressed rat brain cDNA clone that it was identified as the CB1 cannabinoid receptor (Matsuda et al., 1990). Human (Gérard et al., 1990, 1991) and mouse homologues (Chakrabarti et al., 1995) have since been reported. They encode proteins of 472 (human) or 473 (rat, mouse) amino acids, including a rather long and well conserved amino terminal extracellular domain of 116 to 117 residues (Fig.11). Overall, these three receptors have 97 to 99% amino acid sequence identity. A recent sequence-based phylogenetic study of placental mammals (Murphy et al., 2001) included partial sequences from 60 placental mammals covering amino acids 53 to 381 of the rat or mouse sequence (i.e., from the middle of the amino terminal domain to the beginning of the seventh transmembrane domain). There are 24 positions of 329 where more than one sequence differs from the consensus (Table 4). Seven are highly variable positions (67–68, 75–79, and 94) where more than 25% of the sequences differ from the consensus, all of which occur in the amino terminal domain. Except for positions 75 to 79, where the variation is concentrated in Rodentia and Lagomorpha, these variations are broadly distributed across phylogenetic groups. Of potentially greater pharmacological significance are four positions (176, 187, 259, and 271) at which humans and three of the four most closely related primates share common alterations. Except for position 176, where there is a conservative isoleucine for valine substitution at the extracellular end of helix 1, these are highly nonconservative changes located in extracellular loops close to helices 3 to 5, where they might affect binding of large ligands.

Amino acid sequence alignment of human, rat, and mouse CB1 and CB2 receptors. Consensus matches are boxed and shaded with darker shading for identities and lighter shading for conservative substitutions. Numbering corresponds to the rat/mouse CB1 sequence. Underlines indicate the positions of the seven transmembrane helices. Helix 3 spans two lines as indicated by the arrowheads on the underline. The rat CB2sequence is a consensus of GenBank accession nos. AF286721 and AF176350together with edited trace data from the rat genome sequencing project (http://www.ncbi.nlm.nih.gov/genome/seq/RnBlast.html). The rat CB2 residue at alignment position 310 appears to be polymorphic [i.e., either Ala (as shown) or Thr].
Amino acid sequence variations in CB1 among 60 placental mammals
The CB1 coding sequence is contained in a single exon (see, for example, the human gene sequence in GenBank accession no. U73304), but the available cDNA sequences indicate that there must be at least one additional exon containing only 5′-untranslated sequence. However, an alternatively spliced form of the human receptor has been reported (Shire et al., 1995), in which a 167 base portion of the coding exon is spliced out of the human mRNA leading to the predicted substitution of a different 28-residue sequence for the first 90 amino acids. This shorter mRNA appears to be relatively rare by reverse transcription-polymerase chain reaction analysis: 1 to 20% of the message in most brain areas, according to the original report, although it now appears that these are substantial overestimates due to overexposure of the autoradiograms. Moreover, the invariant GT of the splice donor site becomes a GA in both the rat and mouse genes, which implies that this alternative splicing should not occur in these species. Although a similarly spliced form of the rat receptor was also reported (Shire et al., 1995), it now appears that it does not exist in either rat (Shire et al., 1996b) or mouse (Ho and Zhao, 1996). Most importantly, the short isoform is likely to be inefficiently translated because it initiates at the second AUG of the mRNA and has a T rather than the highly preferred A or G at the critical −3-position (i.e., three bases before the AUG) (Kozak, 1994). The question of whether the shorter protein is expressed in significant quantities is presently unanswered; however, if it were to be expressed in significant quantities, the guidelines of the International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification would dictate that the short isoform be referred to as CB1(b) and the major (i.e., larger) isoform should be CB1(a). To date, the short isoform has been referred to as CB1A (Shire et al., 1995). The CB1 mRNA is typically 5.5 to 6 kb, but an alternatively polyadenylated cDNA sequence was reported (Matsuda et al., 1990), which is 2.6-kb shorter in the rat. This species is not usually detected on Northern blots, but the predominant mRNA in human testis is only 4 kb and might represent a similar alternatively polyadenylated mRNA (T. I. Bonner, unpublished observations).
There was no substantial evidence for a second cannabinoid receptor until the hCB2 cDNA was cloned from HL-60 cells (Munro et al., 1993). Its 360-amino acid sequence is quite different from that of CB1, especially in its much shorter amino terminal domain where there is no significant conservation (Fig.11). Between transmembrane domains 1 and 7, the CB2 protein is only 48% identical to that of CB1, substantially less than the 70 to 80% usually seen between different types of G protein-coupled receptors, but enough to have led to its identification as a cannabinoid receptor. It is reported to be expressed primarily in spleen (Fig.12). The mouse CB2 gene has been cloned (Shire et al., 1996a) and is only 82% identical in amino acid sequence to the human receptor and is 13 amino acids shorter at the carboxyl terminal. The rat gene (Griffin et al., 2000) is similar to the mouse gene, except that it is 13 amino acids longer at the carboxyl terminal. It should be noted that this rat receptor is in fact a hybrid mouse-rat receptor with the first and last six amino acids derived from mouse sequence used as polymerase chain reaction primers. As with the CB1 gene, the coding sequence is contained in a single exon of the mouse gene (see GenBank accession no. U21681), but available cDNA sequence indicates that there is at least one additional exon containing only 5′-untranslated sequence.

Autoradiographs show cannabinoid receptor binding (a, f, g) and CB1 (b, d) and CB2 (c, e) mRNA expression in sections from the mouse (sagittal) and human brain (coronal) and mouse spleen (M. Herkenham and A. Hohmann, unpublished observations). Receptor binding of [3H]CP55940, a high-affinity agonist, shows high levels of receptors in the basal ganglia, cerebellum, hippocampus (hipp), and cerebral cortex (a). Cells expressing CB1 mRNA are shown in a similar plane of section (b). Lack of detectable CB2 expression in brain (c) indicates that the binding is to the CB1 type. In contrast, spleen has the opposite relative abundance of CB1 (d) versus CB2 mRNA (e) expression. The human brain has a distribution of cannabinoid receptors that closely matches that of the mouse, with high levels expressed in the basal ganglia, intermediate levels in the amygdala and hypothalamus, and low levels in the thalamus (f, g). The high levels of binding in many areas [cerebellar molecular layer, globus pallidus (GP, GPe), entopeduncular nucleus (Ep, GPi), substantia nigra pars reticulata (SNR), and dentate gyrus molecular layer] are on axons of cells expressing mRNA in afferent areas, such as the caudate putamen (CPu). Some cells in cortex and hippocampus express extremely high levels of CB1 message (arrows in b). Bars measure 1 mm for mouse and 1 cm for human.
Although the amino terminal domain of the CB1receptor is uncommonly long and well conserved, it appears to play no major role in ligand recognition, as deletion of the first 89 amino acids of the hCB1 receptor has no effect on CP55940 binding affinity (Rinaldi-Carmona et al., 1996a). Similarly, the altered amino terminal sequence presented by the CB1(b) isoform has little effect (0- to 3-fold) on the pharmacological properties of several agonists and only a 5- to 10-fold effect on the properties of the SR141716A antagonist.
Site-directed mutagenesis has only recently begun to define which residues constitute the agonist binding sites. Mutation of lysine 192 of the hCB1 receptor to an alanine demonstrated that this lysine is critical for the binding of several agonists (CP55940, HU-210, and anandamide), whereas the mutation has no appreciable effect on either binding or receptor activation byR-(+)-WIN55212 (Song and Bonner, 1996). Clearly, the agonist binding site is not precisely the same for all agonists. This lysine is located at the extracellular end of helix three in both the CB1 and CB2 receptors, a region commonly implicated in agonist binding in other G protein-coupled receptors. This result was extended (Chin et al., 1998) to show that the conservative substitution of an arginine for the lysine had little effect, whereas potentially much more disruptive substitutions of glutamine or glutamic acid eliminated binding of CP55940 but had little effect on binding of R-(+)-WIN55212. However, when the corresponding mutations of the hCB2 receptor at lysine 109 were tested, both the arginine and the alanine substitutions had little effect (Tao et al., 1999). Molecular modeling of the two alanine-substituted receptors (CB1K192A and CB2K109A) indicated that the CB2 receptor still could bind CP55940 via hydrogen bonds to serine 112 that were absent in CB1 at the corresponding residue, glycine 195. When the CB2K109A receptor was altered to also change Ser112 to Gly112, its properties recapitulated those of the CB1K192A receptor, thus confirming the modeling prediction. More recently, mutation of the CB1receptor to change Gly195 to Ser195, analogous to the CB2 receptor, has been shown to increase affinity for R-(+)-WIN55212 4-fold (Chin et al., 1999). Thus, there are two residues that are adjacent on the same face of helix 3, which play a critical role in binding of agonists other thanR-(+)-WIN55212 but a minor role in binding ofR-(+)-WIN55212. A complementary situation occurs in helix 5, where the corresponding residues Val282 in CB1and Phe197 in CB2 confer the selectivity ofR-(+)-WIN55212 for CB2 (Song et al., 1999). Substitution of phenylalanine for Val282 in CB1 results in an increase in affinity forR-(+)-WIN55212 to the CB2 value, whereas the converse mutation, replacing Phe197 of CB2 with a valine, results in a decrease ofR-(+)-WIN55212 affinity to the CB1value. Neither substitution affects affinities for CP55940, HU-210, or anandamide.
A number of other mutations have been reported that alter residues that are highly conserved throughout the rhodopsin family of G protein-coupled receptors, such as the aspartic acid in helix 2 (Tao and Abood, 1998; Roche et al., 1999), the DRY motif at the intracellular end of helix 3 (Rhee et al., 2000b), the tryptophan in the middle of helix 4 (Rhee et al., 2000a), and the tyrosine near the intracellular end of helix 7 (Feng and Song, 2001). These mutations generally give the same results as observed with the analogous mutations in other receptors. Given the highly conserved nature of these residues and their positions generally near the intracellular ends of the helices, it is likely that they are not so much a part of the agonist binding site as they are important for conformations that play a role in transmitting the binding signal to the G proteins. Of more interest for the agonist binding sites is the tryptophan at the extracellular end of helix 4. Conservative mutations of Trp172 in hCB2 to phenylalanine or tyrosine had little effect, but removal of the aromatic side chain by substitution of alanine or leucine eliminated binding of HU-210, CP55940, andR-(+)-WIN55212. The implications of these results are not clear, but it is worth noting that Trp172 is part of a GWNC motif shared (with some deviations from the G and N) by the sphingosine-1 phosphate and lysophosphatidic acid receptors and a small group of orphan receptors, GPR3, GPR6, and GPR12. All of these receptors have a cysteine at the extracellular end of helix 4 instead of the cysteine that is commonly found at the extracellular end of helix 3 and thought to participate in disulfide bonding that constrains the ends of helix 3 and 5. Similar loss of binding has been reported for the CB2 receptor when nearby Cys174 is replaced with serine (Shire et al., 1996a).
Studies with chimeric CB1/CB2 receptors (Shire et al., 1996a) demonstrate that the selectivity of the antagonist SR141716A for CB1 is provided about equally by the portions of the receptor on either side of the beginning of helix 5. Substitution of helices 4 through 5 of the CB2receptor into CB1 resulted in loss of SR141716A binding without altering CP55940 binding, which, together with chimeras substituting only the loop between the two helices, suggests that the specificity lies within helices 4 and 5. However, the critical chimera in which helices 4 and 5 from CB1 might have been expected to confer high-affinity antagonist binding on a CB2 receptor failed to bind either ligand. More recent mutations of the hCB2 receptor aimed at defining the selectivity of SR144528 for CB2identified three mutations in or adjacent to helix 4, S161A, S165A, and C175S, which eliminated SR144528 binding but had little effect on CP55940 or R-(+)-WIN55212 binding or activity (Gouldson et al., 2000). A molecular model was presented that accounted for the role of the two serine residues but did not account for the Cys175 residue. The complementary mutations of the CB1 receptor that might have been expected to gain SR144528 binding were not attempted. Nevertheless, this is yet another case where mutations have been identified that have dramatic effects on the binding of one ligand but not others.
No significant genetic polymorphism has been reported for the cannabinoid receptor genes. A silent mutation in the coding sequence of the CB1 gene, 1259G → A in codon 453 (Thr), has been reported (Gadzicki et al., 1999) to be common in the German population, but since this does not alter the amino acid sequence of the receptor, it is of little pharmacological significance. Another study that determined the coding sequence from 21 individuals, seven of whom exhibited extreme responses to cannabis, found no amino acid-changing mutations (Hoehe et al., 2000).
VI. Cannabinoid Receptor Knockout Mice
The relatively recent creation both of transgenic mice bearing a genetic deletion of the CB1 or CB2 receptor and of CB1/CB2 double knockouts has provided additional avenues for probing cannabinoid receptor function in both the CNS and periphery. Through gene targeting and homologous recombination in embryonic stem cells, two independent laboratories have generated CB1 receptor knockout mice (Ledent et al., 1999; Zimmer et al., 1999). After implantation in pseudopregnant females, homozygous offspring (CB1 −/−) lacked expression of the wild-type CB1 receptor both in the CNS and periphery. Using identical techniques, mice were bred lacking the CB2 receptor (CB2 −/−) (Buckley et al., 2000). CB1/CB2double-knockout mice have been obtained with the expected mendelian frequency by mating mice heterozygous for both receptors (CB1 +/−/CB2 +/−) (N. E. Buckley and A. Zimmer, personal communication).
CB1 knockout mice bred on a C57BL/6J background showed a variety of spontaneous phenotypes, including hypoactivity, reduced locomotion and rearing, supraspinal hypoalgesia, and increased mortality (Zimmer et al., 1999). Subsequent studies revealed a spontaneous reduction in feeding behavior (Di Marzo et al., 2001b) and change in male hormone balance (Paria et al., 2001). In contrast, mice bred on a CD1 background showed increased locomotor and exploratory activity when newly exposed to an arena but no change in supraspinal hypoalgesia or mortality (Ledent et al., 1999). CB1 null mice showed an increase in long-term potentiation (Böhme et al., 2000) and improvements in memory scores (Reibaud et al., 1999), supporting a role for this receptor in cognitive function. Both CB1 receptor knockout mouse lines demonstrated complete loss of cannabinoid agonist-induced behaviors, such as hypolocomotion, hypothermia, spinal and supraspinal analgesia, and bradycardia, consistent with a central role for CB1 receptors in these phenotypes. Moreover, these mice demonstrated less responsiveness to the reinforcing properties of opiates but not other drugs of dependence, suggesting a role for CB1 receptors in specific addictive behaviors (Ledent et al., 1999; Mascia et al., 1999; Cossu et al., 2001). For the most part, results observed in mice treated with selective CB1 receptor antagonists mimic the findings observed in the transgenic animals. However, developmental changes may have occurred in brain architecture to compensate for the lack of CB1 receptors, as has been suggested from studies of neuropeptide expression (Steiner et al., 1999). These findings suggest that studies with CB1 receptor knockout mice, as with other knockout mice, should be interpreted with caution and should be supported with pharmacological experiments.
One of the most promising uses of receptor knockout mice is to uncover new receptor types (see also Section XI.). Studies with CB1 receptor knockout mice have revealed non-CB1receptor-mediated responses to cannabinoid agonists in the CNS (see also Section XI.).R-(+)-WIN55212-mediated reduction in excitatory postsynaptic currents occurred in both wild-type and CB1receptor null mice, suggesting that the γ-aminobutyric acid (GABA)ergic currents are modulated by an unknown cannabinoid receptor (Hájos et al., 2001). Anandamide showed analgesic and hypolocomotor effects of similar magnitude in both wild-type and CB1 receptor knockout mice, again indicating the expression of an anandamide-sensitive non-CB1, non-CB2 receptor in brain tissue (Di Marzo et al., 2000b). Radioligand binding studies and functional GTPγS binding assays using anandamide and R-(+)-WIN55212 indicate the presence of a non-CB1 or -CB2 receptor in brain tissue (Breivogel et al., 2001). Similar non-CB1 receptor-mediated regulation of mesenteric vasodilation was observed in CB1, CB2, and CB1/CB2 double-knockout mice (Járai et al., 1999).
Few studies have revealed a role for the CB2receptors using the CB2 knockout mice. To date, one study has shown a role for CB2 receptors in cannabinoid agonist-mediated inhibition of helper T cell activation, in which the response was lost in CB2 null mice but not in their wild-type controls (Buckley et al., 2000). A study detailing the phenotype of the CB1/CB2 double receptor knockout mice has not been published to date.
VII. Tissue Distribution of Cannabinoid Receptors
A. Neuronal Distribution of Cannabinoid Receptors
The distribution of CB1 cannabinoid receptors has been investigated in considerable detail. Studies have used quantitative autoradiography, in situ hybridization, and immunocytochemistry, yielding complementary information. Investigations of CB2 cannabinoid receptor distribution are fewer. These indicate that this receptor is primarily localized on cells in structures associated with the immune system.
Autoradiographic studies of CB1 receptors are noteworthy for several reasons. They preceded the cloning of the receptor, indicated that the receptor was expressed in regions predicted from the behavioral effects of cannabinoids, and also established that cannabinoid receptors are expressed at high levels compared with other G protein-coupled receptors. Historically, autoradiography studies with [3H]CP55940 helped to establish the existence of a high-affinity cannabinoid receptor. As shown in Fig. 12, cannabinoid receptors were found to be particularly enriched in cerebral cortex, hippocampus, basal ganglia, and cerebellum, regions that were predicted from the behavioral effects of cannabinoids. Lower levels were found in hypothalamus and spinal cord. CB1 receptor binding was almost absent from the respiratory centers of the brainstem, consistent with the clinical observation of the low lethality of cannabis overdose (Robson, 2001).
Detailed autoradiographic studies have been conducted in several species, including human, monkey, and rat (Herkenham et al., 1990,1991; Glass et al., 1997). Qualitatively, all species have similar distributions; however, subtle differences are seen. For example, in humans, CB1 receptors are more highly expressed in amygdala and cingulate cortex compared with rat or monkey (Herkenham et al., 1990). Differences like these may explain interspecies differences in the behavioral effects of cannabinoids. In contrast to other anatomical techniques, the autoradiographic studies can give a quantitative measure of the density of cannabinoid receptors. These studies often found levels of expression greater than 1 pmol/mg tissue. These densities are greater than those of most other G protein-coupled receptors and are comparable with levels found for common ionotropic receptors (Greenamyre et al., 1984; Bowery et al., 1987). Comprehensive anatomical surveys have also been conducted with tritiatedR-(+)-WIN55212 and with SR141716A. These compounds gave a similar distribution as [3H]CP55940 (Jansen et al., 1992; Rinaldi-Carmona et al., 1996b). However, with the recent demonstration of physiological effects of R-(+)-WIN55212 in CB1 knockout mice (Section XI.), reexamination of these latter studies is in order.
Soon after the cloning of the CB1 receptor, several in situ hybridization studies were conducted (Mailleux et al., 1992; Matsuda et al., 1993). The results of these studies generally agreed with the results of the preceding autoradiographic studies, taking into account that in situ hybridization will identify CB1 receptor mRNA in cell bodies, whereas autoradiography will label receptors throughout the neuron. An important finding from the in situ studies was the corroboration of the impression from the autoradiographic studies that CB1 receptors are often found on axons and probably their terminals (Fig. 12). Another interesting finding from the in situ studies was that cannabinoid receptor expressing neurons have two general patterns of distribution (Mailleux et al., 1992;Matsuda et al., 1993). In some regions, they are expressed broadly and uniformly. For example, in cerebellum, almost all granule cells express CB1. In contrast, in the hippocampus, despite intense labeling of the pyramidal cell layer in the autoradiographic studies, most neurons do not express appreciable levels of CB1 mRNA. Instead, a few neurons express very high levels. A similar pattern is found in the cerebral cortex.
Once antibodies were developed against the CB1receptor, immunocytochemical studies were possible. Several of these have been conducted using distinct antibodies (Fig.13). Two comprehensive surveys of CB1 receptor expression in rat brain have been undertaken (Tsou et al., 1998a; Egertová and Elphick, 2000). In both of these studies, cannabinoid receptors were found in the regions predicted from the earlier autoradiographic and in situ hybridization studies. These surveys emphasized the high levels of CB1 receptor expressed on axonal fibers, especially at their terminals. Detailed electron microscope (EM) studies in rat and human hippocampus found that cell-surface CB1 receptors were found almost exclusively on presynaptic terminals (Hájos et al., 2000; Katona et al., 2000). EM gold studies suggest that hippocampal CB1receptors are expressed on the membrane of the entire presynaptic bouton, with the exception of the active zone. In contrast, EM studies in striatum suggest that CB1 receptors may be expressed more widely. This report found CB1labeling of postsynaptic elements and even perivascular astroglia (Rodrı́guez et al., 2001).
Electron micrograph of consecutive rat brain hippocampal sections stained with the C terminus-CB1antibody showing that inhibitory terminals presynaptically express CB1 cannabinoid receptors in the hippocampus. Serial sections have been cut through a CB1-immunoreactive axon terminal forming a symmetrical (GABAergic) synapse (thick arrow) on a dendrite in the stratum radiatum of the CA1 region. Gold particle labeling (small arrows) is restricted to the inner surface of the bouton, where the intracellular carboxy terminus epitope of CB1 is located. A small arrowhead indicates a dense core vesicle. In contrast, the complete lack of staining in axon terminals (★), forming an asymmetrical synapse (large arrowhead), suggests that glutamatergic axons do not contain CB1 receptors. Scale bar is 0.2 μm. Courtesy of I. Katona and T. F. Freund.
The anatomical localization of cannabinoid receptors has also given additional insight into their function. For example, CB1 receptors are often expressed on synaptic terminals that release both GABA and cholecystokinin (CCK) (Katona et al., 1999; Marsicano and Lutz, 1999; Tsou et al., 1999; see also Fig.13). Thus, inhibition of neurotransmission by CB1receptor activation will cause not only a decrease in GABA release but also a decrease in CCK release (Section VIII.). Another interesting feature is the reciprocal nature of the localization of CB1 receptors and the endocannabinoid hydrolyzing enzyme (FAAH). In at least some brain regions, CB1 receptors and FAAH appear to be localized on opposing neurons (Egertová et al., 1998; Tsou et al., 1998b). For example, hippocampal pyramidal neurons and cerebellar Purkinje neurons both express high levels of FAAH and few CB1receptors. Conversely, FAAH expression is low in hippocampal interneurons and cerebellar granule cells, which synapse onto pyramidal neurons and Purkinje neurons, respectively.
In addition to the CNS, CB1 receptors are widely expressed in the peripheral nervous system, both on sensory nerve fibers and in the autonomic nervous system (e.g., Pertwee et al., 1992). Although detailed comparative anatomical studies have not been conducted on CB1 receptor expression in the autonomic nervous system, the physiological experiments suggest significant interspecies differences (e.g., Benowitz et al., 1979; Lake et al., 1997). CB1 receptors are also found in moderate levels in the testis (Gérard et al., 1991; Wenger et al., 2001); their function there is unknown. CB1receptors are also expressed in some immune cells, but their level of expression is considerably lower than that of CB2receptors (Section VII.B.).
As discussed in greater detail elsewhere (Pertwee, 1997, 2001b), CB1 receptor expression levels are highest in the CNS, particularly in brain regions associated with higher cognitive functions. Functionally significant levels of CB1receptors are also expressed in pain pathways and the autonomic nervous system. Often, CB1 receptors are expressed on nerve terminals. One consequence of their activation is to decrease calcium entry through voltage-dependent calcium channels decreasing neurotransmitter release (Sections IV. andVIII.). As detailed in the next section, CB2 receptors are primarily found on immune cells, particularly mature B cells, and, to a lesser degree, on macrophages.
B. Immune Distribution of Cannabinoid Receptors
Current knowledge about the immune distribution of CB1 and CB2 cannabinoid receptors is summarized in Table 5. Cannabinoid CB1 receptor mRNA is found primarily in neural tissue but can be found to a lower extent in peripheral tissues, including the adrenal gland, bone marrow, heart, lung, prostate, testis, thymus, tonsils, and spleen (Kaminski et al., 1992;Bouaboula et al., 1993; Galiègue et al., 1995; Noe et al., 2000). Messenger RNA for CB1 can be found at low levels in neonatal rat brain cortical microglia (Waksman et al., 1999;Carlisle et al., 2002) and in select immune cell lines, including human THP-1 monocytic cells, human Raji B-cells, murine NKB61A2 natural killer-like cells, and murine CTLL2 IL-2-dependent T cells (Daaka et al., 1995).
Detection of cannabinoid receptors in immune cells and tissues
Both in situ hybridization studies and autoradiographic studies suggest expression of CB2 receptors in multiple lymphoid organs (Lynn and Herkenham, 1994; Buckley et al., 1998). Cannabinoid CB2 receptor mRNA is found in spleen (Fig. 12), thymus, tonsils, bone marrow, pancreas, splenic macrophage/monocyte preparations, mast cells, peripheral blood leukocytes, and in a variety of cultured immune cell models, including the myeloid cell line U937 and undifferentiated and differentiated granulocyte-like or macrophage-like HL-60 cells (Bouaboula et al., 1993; Munro et al., 1993; Facci et al., 1995; Galiègue et al., 1995; Condie et al., 1996; Pettit et al., 1996; Schatz et al., 1997). Valk et al. (1997)reported the presence of CB2 receptor mRNA in 45 of 51 cell lines of distinct hematopoietic lineages, including myeloid, macrophage, mast, B-lymphoid, T-lymphoid, and erythroid cells. In spleen and tonsils, CB2 mRNA content is equivalent to that of CB1 mRNA in the central nervous system. However, the distribution pattern of CB2 mRNA displays major variation in human blood cell populations, with a rank order of B lymphocytes > natural killer cells ≫ monocytes > polymorphonuclear neutrophils > T8 lymphocytes > T4 lymphocytes (Galiègue et al., 1995). A rank order for CB2 mRNA content similar to that noted for primary human cell types has been recorded for human cell lines belonging to the myeloid, monocytic, and lymphoid lineages (Galiègue et al., 1995). Lee et al. (2001) have reported a similar pattern of CB2 mRNA distribution in murine immune cell subpopulations. CB2 mRNA was most abundant in splenic B cells, followed by macrophages and T cells. Messenger RNA for CB2 has been identified also in neonatal rat brain cortical microglia maintained in vitro at levels that exceed those for CB1 (Carlisle et al., 2002).
Cannabinoid receptor protein has been localized in a variety of immune cell types and tissues. Ligand binding assays have allowed for the assessment of cannabinoid receptor protein in rat lymph nodes, Peyer's patches, and spleen (Lynn and Herkenham, 1994). Cannabinoid receptor binding was confined to B lymphocyte: enriched areas such as the marginal zone of the spleen, cortex of the lymph nodes, and nodular corona of Peyer's patches. Specific binding was absent in T lymphocyte-enriched areas, such as the thymus and periarteriolar lymphatic sheaths of the spleen, and certain macrophage-enriched areas, such as the liver and lung. Binding assay also has permitted quantitation of cannabinoid receptors on membranes of a variety of immune cell types and lines. Bouaboula et al. (1993) used [3H]CP55940 as a ligand for characterizing cannabinoid receptors in human myelomonocytic U937 cells. AK d of 0.1 nM and aB max of 525 fmol/mg protein was determined from Scatchard analysis for membranes of these cells.
In addition, CB1- and CB2-specific antibodies have been used to identify cannabinoid receptors in immune cells. Cannabinoid CB1 receptor protein has been identified in the human Jurkat T cell line (Daaka et al., 1996), in Daudi human B-lymphoblastoid cells and macrophage-like cells from rat brain tissue (Sinha et al., 1998), and in cortical microglia cultured from neonatal rat brain (Waksman et al., 1999). Galiègue et al. (1995) used an anti-hCB2 IgG to localize CB2 receptors within B lymphocyte-enriched areas of the mantle of secondary lymphoid follicles in sections of human tonsil. Carayon et al. (1998) employed immunopurified polyclonal antibody to investigate the expression of CB2receptors in leukocytes and showed that peripheral blood and tonsillar B cells were the leukocyte subsets expressing the highest amount of CB2 receptor proteins. Dual-color confocal microscopy performed on human tonsillar tissues demonstrated a marked expression of CB2 receptors in mantle zones of secondary follicles, whereas germinal centers were weakly stained, suggesting a modulation of this receptor during the differentiation stages from virgin B lymphocytes to memory B cells.
Changes in levels of cannabinoid receptors or their mRNAs after treatment with a variety of immune modulators or activators have been reported. Levels of CB2 mRNA have been detected in peritoneal macrophages at differential levels in relation to cell activation state. Lee et al. (2001) and Carlisle et al. (2002)determined that CB2 mRNA was present in thioglycollate-elicited murine peritoneal macrophages but not in resident peritoneal macrophages. In addition to these studies on receptor expression at basal activity, CB2 mRNA expression was studied following immune cell activation. Bacterial lipopolysaccharide stimulation down-regulated CB2mRNA expression in splenocyte cultures in a dose-response manner, whereas stimulation through cluster of differentiation 40 (CD40) using anti-CD40 antibody up-regulated the response and costimulation with IL-4 attenuated the anti-CD40 response. Daaka et al. (1995) have indicated that lipopolysaccharide-stimulated Raji and PMA-stimulated THP-1 human acute monocytic leukemia cell lines show increased levels of CB1 cannabinoid receptor mRNA. It was demonstrated also that increases in CB1 mRNA were linked to comparable increases in cognate protein expression. Mitogen activation of Jurkat cells showed an increase in specific binding of [3H]CP55940, and Western analysis indicated the presence of immunoreactive proteins on membranes from mitogen-activated Jurkat cells but not on membranes of unstimulated cells. Noe et al. (2000) reported that anti-CD40, anti-CD3, and IL-2 stimulation induced contrasting changes in CB1 mRNA expression in mouse splenocytes. Splenocytes stimulated with the T cell mitogens PMA/Io and anti-CD3 showed a decrease in CB1message, whereas cultures stimulated with the B-cell mitogen, anti-CD40 antibody, showed an increase in message. In addition, cotreatment with mitogens and IL-2 uniformly caused an increase in CB1 mRNA. These observations suggest that signaling pathways activated by T cell mitogens lead to decreased CB1 gene activation, whereas pathways activated by B-cell mitogens and IL-2 lead to increased CB1. Collectively, these reports suggest that cannabinoid receptors have biological relevance in lymphoid and myeloid cells during defined stages of cell activation.
Changes in levels of rat spleen cannabinoid receptors have been reported also after chronic cannabinoid administration. Massi et al. (1997) assessed the effect of chronic in vivo administration of CP55940 on the expression of cannabinoid receptors. Spleen coronal sections processed for receptor binding autoradiography with [3H]CP55940 in the absence or presence of unlabeled CP55940 and subjected to densitometric analysis of the autoradiograms showed significant loss of [3H]CP55940 binding for chronic cannabinoid-treated, tolerant rats.
VIII. Effects on Neurotransmission
As detailed in Table 6, there is good evidence that the activation of presynaptic CB1 receptors can lead to inhibition of the evoked release of a number of different excitatory or inhibitory neurotransmitters both in the brain and in the peripheral nervous system. This evidence has been obtained from experiments in which release has been monitored either through the direct measurement of transmitter levels in vivo or in vitro (acetylcholine, noradrenaline, dopamine, 5-hydroxytryptamine, d-aspartate, cholecystokinin, and GABA) or indirectly using electrophysiological techniques (glutamate, glycine, and GABA). R-(+)-WIN55212 and Δ9-THC have been reported to inhibit GABA uptake into tissue obtained from rat globus pallidus (Maneuf et al., 1996a,b) or substantia nigra (Romero et al., 1998), albeit at a rather high concentration (50 μM). Even so, the main effect of cannabinoids on GABAergic transmission in rat hippocampus seems to be inhibitory in nature (Paton et al., 1998; Hoffman and Lupica, 2000). Although there are some electrophysiological data that support CB1 receptor-mediated inhibition of GABA release in rat substantia nigra (Table 6), it has not proved possible to detect any cannabinoid-induced inhibition of spontaneous or evoked release of [3H]GABA from fragments of rat substantia nigra preloaded with this radioisotope (Romero et al., 1998) or, indeed, from slices of globus pallidus (Maneuf et al., 1996a). Although there is little doubt that CB1 receptors play a major role in modulating neurotransmitter release, evidence has recently emerged from experiments with CB1 knockout mice that inhibition of hippocampal glutamate release is mediated by presynaptic,R-(+)-WIN55212-sensitive, non-CB1receptors (Section XI.).
Cannabinoid-induced inhibition of central and peripheral neurotransmitter release
Although the primary effect of CB1 receptor agonists on neurotransmitter release seems to be one of inhibition, this may sometimes result in enhanced neurotransmitter release at some point downstream of the initial inhibitory effect. For example, there is evidence that cannabinoids enhance dynorphin release within the spinal cord and that this effect depends on CB1receptor-mediated inhibition of tonically active neurons that exert an inhibitory influence on dynorphinergic neurons (see Pertwee, 2001b). There is also evidence from experiments both with whole animals (Chen et al., 1990a,b; 1991; French, 1997; French et al., 1997; Tanda et al., 1997; Gessa et al., 1998b; Melis et al., 2000) and with brain slices (Cheer et al., 2000) that CB1 receptor agonists can stimulate dopamine release in the nucleus accumbens, and it is likely that this effect stems from a cannabinoid receptor-mediated inhibition of glutamate release from extrinsic glutamatergic fibers. These are large fibers that form synapses in the nucleus accumbens with GABAergic neurons that project to the ventral tegmental area to exert an inhibitory effect on dopaminergic mesoaccumbens neurons (Robbe et al., 2001). It is possible that cannabinoid receptor-mediated disinhibition of dopamine release in the nucleus accumbens gives rise to increases in acetylcholine release in the prefrontal cortex that have recently been observed in microdialysis experiments with rats in response to intravenous injections of low doses of Δ9-THC, HU-210, or R-(+)-WIN55212 (Acquas et al., 2000, 2001). Thus, GABAergic neurons project from the nucleus accumbens to the prefrontal cortex, and it is thought that dopamine released in the nucleus accumbens may act on these neurons to disinhibit acetylcholine release in the cortex (Moore et al., 1999). Results from microdialysis experiments with rats have indicated that at low doses, intravenously administered cannabinoids can also act through CB1 receptors to increase acetylcholine release in the hippocampus (Acquas et al., 2000, 2001), whereas data from in vivo electrophysiological experiments suggest that systemically administered cannabinoids can enhance dopamine release from mesoprefrontal cortical neurons that project from the ventral tegmental area to the prefrontal cortex (Diana et al., 1998). This stimulatory effect on cortical dopamine release may result from inhibition of GABA release mediated by CB1 receptors that are presumed to be located on the terminals of prefrontal cortical GABAergic interneurons that modulate the activity of pyramidal neurons (Pistis et al., 2001). These prefrontal cortical pyramidal neurons project to the ventral tegmental area, where they form excitatory synapses on mesoprefrontal dopaminergic neurons that release GABA from the prefrontal cortical GABAergic interneurons that have been postulated to express CB1 receptors.
One apparently anomalous finding, obtained from microdialysis experiments with unanaesthetized rats, is thatR-(+)-WIN55212 can act through cannabinoid receptors in the cerebral cortex to enhance calcium-dependent glutamate release (Ferraro et al., 2001). The same investigation also provided evidence thatR-(+)-WIN55212 can produce cannabinoid receptor-mediated increases in spontaneous, calcium-dependent glutamate release in primary cultures of rat cerebral cortex. The reason for the apparent discrepancy between these glutamate release data and previous electrophysiological data that indicate an inhibitory effect of cannabinoids on glutamate release (Table 6) remains to be elucidated. It is possible that when administered in vivo, CB1 receptor agonists have dose-dependent biphasic effects on cortical and hippocampal acetylcholine release: a stimulant effect at low doses and an inhibitory effect at higher doses. This hypothesis has been put forward by Acquas et al. (2001) to explain why, in some microdialysis experiments with rats, cannabinoids increase acetylcholine release in prefrontal cortex and hippocampus (Acquas et al., 2000, 2001), whereas in other microdialysis experiments, they decrease acetylcholine release in these same brain areas (Table 6).
Results from a number of recent investigations suggest that endocannabinoids may act through presynaptic cannabinoid receptors to function as fast retrograde synaptic messengers. More specifically, there is evidence to suggest that the biosynthesis and nonvesicular release of endocannabinoid molecules can be rapidly triggered by intense activity at glutamatergic synapses in the hippocampus and cerebellum. In the hippocampus, such release seems to take place from pyramidal cells (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). These cells receive synaptic inputs from both (excitatory) glutamatergic neurons and (inhibitory) GABAergic neurons. It has been proposed that pyramidal cells produce and release endocannabinoid molecules in response to elevations in intracellular calcium levels induced by the synaptic release of glutamate, and that the endocannabinoid molecules so produced then act through CB1 receptors on GABAergic neurons to inhibit calcium influx, thus decreasing GABA release onto the pyramidal cells (depolarization-induced suppression of inhibition). In the cerebellum, glutamate released onto Purkinje cells appears to be capable of triggering endocannabinoid production and release both by transiently increasing calcium levels within these cells and by acting on postsynaptic metabotropic glutamate receptors (mGluR subtype 1) to activate G proteins without producing any elevation of intracellular calcium (Kreitzer and Regehr, 2001a; Maejima et al., 2001). Once released from the Purkinje cells, the endocannabinoid molecules are thought to act through cannabinoid receptors that are present on the terminals of climbing fibers and of parallel fibers of cerebellar granule cells to inhibit the ongoing glutamate release (depolarization-induced suppression of excitation) (Kreitzer and Regehr, 2001a; Maejima et al., 2001). There is also evidence that cerebellar depolarization-induced suppression of inhibition results from the release of endocannabinoid molecules from Purkinje cells onto presynaptic CB1 receptors that are present on GABAergic basket and stellate cell terminals (Diana et al., 2002;Kreitzer and Regehr, 2001b). Although depolarization-induced suppression of excitation should provide a negative feedback mechanism for damping down high synaptic activity, depolarization-induced suppression of inhibition will have more complex effects. The identity of endocannabinoid(s) that serve as fast retrograde synaptic messengers remains to be established. In the meantime, it is noteworthy that results from experiments with primary cultures of rat cortical neurons have indicated that glutamate and NMDA stimulate the formation of 2-arachidonoylglycerol and that anandamide formation can be stimulated by the simultaneous activation of nicotinic and NMDA receptors with glutamate and carbachol although not by either of these agents alone (Stella and Piomelli, 2001). There are also reports firstly, that high-frequency in vivo electrical stimulation of rat Schaffer collaterals (excitatory hippocampal CA1 afferents) provokes increased calcium-dependent release of 2-arachidonoylglycerol but not of anandamide (Stella et al., 1997) and secondly, that striatal concentrations of anandamide but not of 2-arachidonoylglycerol can be increased in rats in vivo by local perfusion with a depolarizing concentration of potassium chloride or with the D2-like receptor agonist quinpirole (Giuffrida et al., 1999). In addition, it has been found that anandamide release in the periaqueductal gray area of rat brain can be induced both by direct electrical stimulation of this brain area and by subcutaneous injection of a chemical irritant into the hindpaw (Walker et al., 1999).
IX. Immunological Effects
The identification of peripheral cannabinoid receptor mRNA and protein in a variety of immune cell types, and the recognition that cannabinoids inhibit adenylyl cyclase in immune cells through a pertussis toxin-sensitive mode (Kaminski et al., 1992, 1994; Kaminski, 1998), suggest a role for cannabinoid receptors in the modulation of immune cell functions. Kaminski et al. (1992) demonstrated that suppression of the humoral immune response by cannabinoids was mediated partially through inhibition of adenylyl cyclase by a pertussis toxin-sensitive G protein-coupled mechanism. Δ9-THC and the synthetic nonclassical bicyclic cannabinoid CP55940 inhibited the lymphocyte proliferative and the sheep erythrocyte IgM antibody-forming cell responses of murine splenocytes to PMA plus the calcium ionophore ionomycin. More direct evidence for a functional linkage of cannabinoid receptors to modulation of immune functional activities has been obtained through the use of CB1- and CB2-selective antagonists.
Select functional activities of macrophages and macrophage-like cells have been reported to be affected by cannabinoids through cannabinoid receptors. McCoy et al. (1995, 1999) demonstrated that Δ9-THC modulated the capacity of macrophages to process antigens that are necessary for the activation of CD4+ T lymphocytes. Δ9-THC was reported to inhibit the processing of intact lysozyme in a dose-dependent fashion, and this inhibition was blocked by the CB2-selective antagonist SR144528, indicating that the inhibitory effect was mediated, at least in part, through the CB2receptor. The CB1-selective antagonist SR141716A did not reverse the suppression caused by Δ9-THC, consistent with no functional linkage of this receptor to this event. These observations were confirmed using CB2 receptor knockout mice (Buckley et al., 2000). Δ9-THC inhibited helper T cell activation through macrophages derived from wild type, but not from knockout mice, consistent with alterations in antigen processing being mediated by the CB2 receptor.
Sacerdote et al. (2000) reported that in vivo and in vitro treatment with the synthetic cannabinoid CP55940 decreased the in vitro migration of macrophages in the rat and that this effect involved both CB1 and CB2 receptors. Spontaneous migration and formyl-methionyl-leucine-phenylalanine-induced chemotaxis assessed by the use of Boyden-modified microchemotaxis chambers were affected. Both SR141716A and SR144528 were able to block the CP55940-induced inhibition of spontaneous migration, although the CB2 antagonist was more potent, and only the CB2 antagonist was able to reverse the effect of CP55940 on formyl-methionyl-leucine-phenylalanine-induced chemotaxis. The CB1 receptor has also been reported to mediate inhibition of iNOS production by neonatal rat microglial cells (Waksman et al., 1999). The potent cannabinoid agonist CP55940 effected a dose-dependent inhibition of iNOS that was reversed by SR141716A. However, no data were provided regarding a role for the CB2 receptor in this process. On the other hand,Stefano et al. (2000) have reported that the endocannabinoid 2-arachidonoylglycerol stimulated constitutive nitric oxide release from human monocytes and vascular tissues and immunocytes of the invertebrate Mytilus edulis and that this effect is mediated through the CB1 receptor in human cells and through an apparent cannabinoid receptor in the invertebrate immunocytes. Furthermore, in both the monocytes and the immunocytes, NO release elicited in response to 2-arachidonoylglycerol exposure was blocked by a CB1 antagonist but not by a CB2 antagonist. Inhibition of lipopolysaccharide-induced iNOS expression by murine RAW 264.7 macrophage-like cells by cannabinoids and the putative cannabinoid CB2-like receptor agonist palmitoylethanolamide (Section XI.) also has been reported (Gross et al., 2000). The inhibition of nitric oxide production byR-(+)-WIN55212 but not palmitoylethanolamide was attenuated significantly by the CB2 receptor antagonist SR144528. These results suggested that inhibition of RAW 264.7 cell lipopolysaccharide-induced iNOS expression byR-(+)-WIN55212, but not palmitoylethanolamide, is mediated by the CB2 receptor.
Gross et al. (2000) suggested an involvement of the CB1 cannabinoid receptor in infection of macrophages by the intracellular pathogen Brucella suis, a Gram-negative bacterium. The influence of the CB1and CB2 receptor antagonists, SR141716A and SR144528, and the nonselective CB1/CB2 cannabinoid receptor agonists, CP55940 and R-(+)-WIN55212, on macrophage infection by B. suis was examined. The intracellular multiplication of Brucella was dose-dependently inhibited in cells treated with SR141716A but not with SR144528, CP55940, orR-(+)-WIN55212. The agonists CP55940 andR-(+)-WIN55212 reversed the SR141716A-induced effect, implicating an involvement of the CB1 receptor in this process.
The involvement of both CB1 and CB2 receptors in Δ9-THC-induced inhibition of natural killer activity has been reported (Massi et al., 2000). In vivo administration of Δ9-THC to mice significantly inhibited natural killer cytolytic activity without affecting concanavalin A-induced splenocyte proliferation. Pretreatment with the CB1 and CB2 cannabinoid receptor antagonists SR141716 and SR144528 partially reversed the inhibition of natural killer cytolytic activity by Δ9-THC. However, the CB1receptor antagonist was more effective than the CB2 receptor antagonist. The parallel measurement of interferon γ (IFN-γ) revealed that Δ9-THC significantly reduced production of this cytokine. The CB1 and CB2receptor antagonists completely reversed the IFN-γ reduction induced by Δ9-THC. Thus, both cannabinoid receptor types were involved in the complex network mediating natural killer cytolytic activity.
Sugiura et al. (2000) examined the effect of 2-arachidonoylglycerol on the intracellular free Ca2+ concentrations in human HL-60 promyelocytic leukemia cells that express the CB2 receptor. It was found that 2-arachidonoylglycerol induced a rapid transient increase in intracellular free Ca2+ concentrations. The Ca2+ transient induced by 2-arachidonoylglycerol was blocked by pretreatment of the cells with the CB2 receptor-specific antagonist SR144528 but not with the CB1 receptor-specific antagonist SR141716A, indicating the involvement of the CB2receptor but not the CB1 receptor in this cellular response. Two other putative endogenous cannabinoid receptor ligands, anandamide and palmitoylethanolamide, were found to be a weak partial agonist and an inactive ligand, respectively.
Carayon et al. (1998) reported that CB2 receptor expression is down-regulated at the mRNA and protein levels during B-cell differentiation. The lowest expression was observed in germinal center proliferating centroblasts of tonsillar tissues. The cannabinoid agonist CP55940 enhanced CD40-mediated proliferation of both virgin and germinal center B-cell subsets. This enhancement was blocked by the CB2 receptor antagonist SR144528 but not by the CB1 receptor antagonist SR141716. It was also observed that CB2 receptors were up-regulated in both B-cell subsets during the first 24 h of CD40-mediated activation. In addition, SR144528 was shown to antagonize the stimulating effects of CP55940 on human tonsillar B-cell activation evoked by cross-linking of surface immunoglobulins (IC50 = 20 nM) (Rinaldi-Carmona et al., 1998). These results suggest a functional involvement of CB2 cannabinoid receptors during B-cell differentiation.
A possible explanation for the capacity of cannabinoids to act through cannabinoid receptors so as to exert a broad spectrum of immune function effects is that these compounds exert differential expression of cytokine profiles. Δ9-THC and other cannabinoid agonists have been reported to augment the expression of immune inhibitory Th2-type cytokines while inhibiting that of Th1-type immune stimulatory cytokines. Δ9-THC has been reported to inhibit antitumor immunity by a CB2receptor-mediated, cytokine-dependent pathway (Zhu et al., 2000). It suppressed host immune reactivity against lung cancer using two different weakly immunogenic murine lung cancer models. Δ9-THC decreased tumor immunogenicity, as indicated by the limited capacity for tumor-immunized, Δ9-THC-treated mice to withstand tumor rechallenge. The immune inhibitory Th2 cytokines, IL-10 and transforming growth factor, were augmented, whereas the immune stimulatory Th1 cytokine, IFN-γ, was down-regulated at both the tumor site and in the spleens of Δ9-THC-treated mice. In vivo administration of the CB2-selective antagonist SR144528 blocked the effects of Δ9-THC. These findings suggest the Δ9-THC promotes tumor growth by inhibiting antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. Δ9-THC treatment of BALB/c mice also suppressed immunity and early IFN-γ, IL-12, and IL-12 receptor β2 responses to Legionella pneumophila(Klein et al., 2000). Levels of IL-12 and IFN-γ, cytokines that promote the development of Th1 cells as well as resistance to a challenge infection, were suppressed by Δ9-THC. Results obtained with selective cannabinoid receptor antagonists indicated that both the CB1 and CB2 receptors were involved in this process.
X. Anandamide Is a Vanilloid Receptor Agonist
There are several reports that the endocannabinoid anandamide can act on rat or human vanilloid receptors transfected into cultured cells to produce membrane currents or increase intracellular calcium (Zygmunt et al., 1999; Smart et al., 2000, 2001; Ross et al., 2001). Anandamide also acts on naturally expressed vanilloid receptors in neonatal rat dorsal root ganglia to produce membrane currents (Tognetto et al., 2001) and in rat or guinea pig isolated arterial strips to trigger both release of calcitonin-gene-related peptide from perivascular sensory nerves and relaxation of precontracted tissues (Zygmunt et al., 1999). Results from experiments with transfected rat vanilloid receptors suggest that anandamide has markedly less relative intrinsic activity at these receptors than capsaicin (Ross et al., 2001). Methanandamide activates vanilloid receptors even less potently or effectively than anandamide (Zygmunt et al., 1999; Ralevic et al., 2000; Ross et al., 2001), whereas the CB1/CB2receptor agonists 2-arachidonoylglycerol and HU-210 lack significant activity at these receptors altogether (Zygmunt et al., 1999).
CB1 receptors are negatively coupled to calcium channels, whereas vanilloid receptors open cation channels. Consequently, some experiments have been directed at exploring the consequences of simultaneously activating both receptor types. These have been performed with rat cultured dorsal root ganglion neurons that are known to coexpress CB1 and vanilloid receptors to a very high degree (Ahluwalia et al., 2000). The results obtained indicate that capsaicin-induced increases in intracellular calcium can be opposed by CB1 receptor activation (Millns et al., 2001) and that CB1receptor-mediated inhibition of electrically evoked calcium mobilization and calcitonin-gene-related peptide release can be opposed by the activation of vanilloid receptors (Tognetto et al., 2001). Anandamide was found to be considerably more potent in inhibiting calcium mobilization than in activating vanilloid receptors. There is evidence that in the mouse isolated vas deferens, inhibition of electrically evoked contractions can be mediated both by presynaptic CB1 receptors through reduction of contractile transmitter release and by vanilloid receptors that trigger the release of neuropeptide molecules, which then presumably inhibit contractile transmitter release (Pertwee, 1997; Ross et al., 2001). Anandamide appears to act through both CB1 and vanilloid receptors to inhibit electrically evoked contractions of this tissue preparation, whereas the inhibitory effect of R-(+)-WIN55212 seems to be mediated solely by CB1 receptors (Ross et al., 2001).
The finding that anandamide is an agonist for both cannabinoid and vanilloid receptors prompted the development of the anandamide/capsaicin hybrid molecule, arvanil, which has anandamide-like CB1 affinity, less relative intrinsic activity than anandamide at CB1receptors, and greater potency than anandamide as a vanilloid receptor agonist (De Petrocellis et al., 2000; Di Marzo et al., 2000a). AM404 is another anandamide analog that activates vanilloid receptors (Jerman et al., 2000; Zygmunt et al., 2000; Ross et al., 2001), albeit at concentrations no higher than those at which it inhibits anandamide membrane transport (Beltramo et al., 1997; Piomelli et al., 1999).
XI. Preliminary Pharmacological Evidence for Non-CB1, Non-CB2 Cannabinoid Receptors
A. A Putative CB2-Like Cannabinoid Receptor
It has been found by Calignano et al. (1998, 2001) that the endogenous fatty acid amide, palmitoylethanolamide, induces antinociceptive effects that are attenuated by the CB2-selective antagonist SR144528 but not by the CB1-selective antagonist SR141716A. These results were obtained in the mouse formalin paw test after intraplantar injection of palmitoylethanolamide and in the mouse abdominal stretch test after intraperitoneal injection of this compound (Calignano et al., 1998, 2001). The same investigators also found that in these bioassays, anandamide can be antagonized by SR141716A but not SR144528, and that palmitoylethanolamide and anandamide act synergistically. Palmitoylethanolamide lacks significant affinity for CB1 or CB2 receptors (Devane et al., 1992b; Felder et al., 1993; Showalter et al., 1996;Sheskin et al., 1997; Lambert et al., 1999). Consequently, Calignano et al. (1998, 2001) have proposed the existence of an SR144528-sensitive, non-CB2 cannabinoid receptor (“CB2-like” receptor). This putative receptor is thought not to be a vanilloid receptor, because palmitoylethanolamide does not share the ability of anandamide or capsazepine to suppress paw-licking behavior when coadministered with capsaicin into mouse hindpaw (Calignano et al., 2001). Evidence for the existence of CB2-like receptors has also been obtained in experiments with the mouse vas deferens (Griffin et al., 1997). Unlike anandamide or other established CB1receptor agonists, palmitoylethanolamide does not show antinociceptive activity in the mouse hot plate test, suggesting that it does not interfere directly with neuronally mediated transmission of pain signals to the central nervous system (Calignano et al., 2001).
B. A Putative SR141716A-Sensitive, Non-CB1, Non-CB2 Cannabinoid Receptor
There is some evidence that mesenteric arteries of mice and rats express receptors that can be activated by anandamide and methanandamide but not by other established CB1/CB2 receptor agonists and that are both non-CB1, non-CB2, and nonvanilloid. More specifically, anandamide and methanandamide can both induce a concentration-related relaxation of rat or mouse precontracted mesenteric arteries, whereas Δ9-THC, HU-210,R-(+)-WIN55212, and 2-arachidonoylglycerol cannot (Járai et al., 1999; Wagner et al., 1999). Other agonists for this putative novel receptor are the cannabidiol analogs, abnormal cannabidiol and O-1602 (Fig. 14), neither of which exhibits significant affinity for rat brain CB1 receptors (Járai et al., 1999). Anandamide, methanandamide, and abnormal cannabidiol also relax precontracted mesenteric arteries obtained from CB1 receptor knockout (CB1 −/−) mice or from CB1 −/−/CB2 −/−double-knockout mice, confirming a lack of involvement of either CB1 or CB2 receptors in this effect (Járai et al., 1999).
The structures of abnormal cannabidiol and O-1602.
The proposed mesenteric non-CB1, non-CB2 receptors can be blocked by SR141716A, albeit less potently than CB1 receptors. Thus, the relaxant effects of anandamide, abnormal cannabidiol, and O-1602 in precontracted mesenteric arteries obtained from rats or from CB1 +/+ or CB1 −/− mice have been found to be attenuated by SR141716A at 0.5, 1, or 5 μM (Járai et al., 1999; Wagner et al., 1999). At 10 μM, the nonpsychotropic plant cannabinoid, cannabidiol (Fig. 1), also attenuates the relaxation of rat or CB1 −/− mouse precontracted mesenteric arteries induced by anandamide or abnormal cannabidiol (Járai et al., 1999; Wagner et al., 1999). This cannabinoid exhibits at least some degree of selectivity in that it does not attenuate relaxation induced in such vessels by acetylcholine, bradykinin, or sodium nitroprusside (Járai et al., 1999). The relaxant effect of abnormal cannabidiol in rat precontracted mesenteric arteries has been found to be unaffected by a concentration of capsazepine (5 μM) that can attenuate the relaxant effect of capsaicin, ruling out any major involvement of vanilloid receptors (Járai et al., 1999). SR141716A (1 μM) does not attenuate capsaicin-induced relaxation of rat precontracted mesenteric arteries (Járai et al., 1999).
Anandamide-induced vasorelaxation is detectable both in endothelium-intact and in endothelium-denuded precontracted mesenteric arteries of rats (Wagner et al., 1999; Kunos et al., 2000). However, SR141716A only attenuates this vasorelaxant effect of anandamide in the presence of endothelium, and the relaxant effects of abnormal cannabidiol and O-1602 in rat precontracted mesenteric arteries are also largely endothelium-dependent (Járai et al., 1999). It seems likely, therefore, that there are at least two mechanisms by which anandamide relaxes precontracted mesenteric arteries, and that the SR141716A-sensitive, non-CB1, non-CB2 receptors for anandamide proposed byKunos and colleagues (2000) are present on the endothelium but not on mesenteric smooth muscle.
C. A Putative Receptor for Anandamide and R-(+)-WIN55212
Evidence has emerged for the existence in mouse brain of a G protein-coupled receptor that can be activated by anandamide andR-(+)-WIN55212 but not by other CB1/CB2 agonists (Di Marzo et al., 2000b; Breivogel et al., 2001). More specifically, it has been found that [35S]GTPγS binding can be activated in brain membranes from CB1 −/− mice by anandamide (EC50 = 3.6 μM) and R-(+)-WIN55212 (EC50 = 1.8 μM) but not by Δ9-THC, HU-210, or CP55940. These properties of this possible new cannabinoid receptor distinguish it from the CB2 receptor for which Δ9-THC, HU-210, and CP55940 are all established agonists. They also distinguish it both from the SR141716A-sensitive, anandamide-sensitive, R-(+)-WIN55212-insensitive receptor that George Kunos' group has postulated to be present in mesenteric arteries (Kunos et al., 2000; Section XI.B.) and from the vanilloid receptor, which is not coupled to G proteins and is unresponsive to R-(+)-WIN55212 (Zygmunt et al., 1999). Activation of [35S]GTPγS binding by anandamide and R-(+)-WIN55212 was detected in membranes from CB1 −/− whole brain and from CB1 −/− cerebral cortex, midbrain, hippocampus, diencephalon, and brain stem but not in membranes from CB1 −/−caudate-putamen/globus pallidus or cerebellum, brain areas that are well populated with CB1 receptors in wild-type animals (Breivogel et al., 2001). Near maximal concentrations of anandamide and R-(+)-WIN55212 were not fully additive in their effects on [35S]GTPγS binding, supporting the hypothesis that these two agents act through a common mechanism (Breivogel et al., 2001). Membranes from CB1 −/− cerebral cortex, hippocampus, and brain stem were found to contain specific binding sites for [3H]R-(+)-WIN55212 but not [3H]CP55940 (Breivogel et al., 2001). However, neither of these tritiated ligands exhibited detectable specific binding in membranes from CB1 −/− diencephalon, midbrain, caudate-putamen/globus pallidus, cerebellum, or spinal cord. Membranes from some CB1 −/− brain areas (brain stem, cortex, midbrain, and spinal cord) but not others (basal ganglia, cerebellum, diencephalon, and hippocampus) also contained specific binding sites for [3H]SR141716A. Even so, it is unlikely that this compound is a ligand for the proposedR-(+)-WIN55212/anandamide receptor, as the distribution patterns of [3H]R-(+)-WIN55212 and [3H]SR141716A binding sites in CB1 −/− brain are different. Moreover, although concentrations of SR141716A above 1 μM were found to attenuate the stimulatory effects of anandamide andR-(+)-WIN55212 on [35S]GTPγS binding to CB1 −/− membranes, this attenuation could be attributed entirely to the inhibition of [35S]GTPγS binding that was produced by SR141716A in the same concentration range (Breivogel et al., 2001).
Other evidence for the presence of anR-(+)-WIN55212-sensitive non-CB1receptor in mouse brain was obtained recently by Hájos et al. (2001) in electrophysiological experiments with hippocampal slices obtained from CB1 −/− or wild-type mice. Their results suggest that althoughR-(+)-WIN55212 probably acts through presynaptic CB1 receptors in the CA1 region of the hippocampus to inhibit GABA release, it acts through presynaptic non-CB1 receptors to inhibit glutamate release in this brain region. This conclusion is consistent with previous reports that CB1 immunostaining cannot be reliably detected in hippocampal axon terminals forming glutamatergic synapses (Katona et al., 1999, 2000; Hájos et al., 2000). It is noteworthy that the inhibitory effect of R-(+)-WIN55212 on glutamatergic transmission observed by Hájos et al. (2001) in hippocampal tissue from CB1 −/−mice could be reversed by 1 μM SR141716A.
D. Other Putative Types of Mammalian Cannabinoid Receptor
Results obtained by Sandra Welch's group in experiments with rats and mice have prompted the hypothesis that there may be more than one subtype of CB1 receptor in the spinal cord. Thus,Welch et al. (1998) have found that the potency of intraperitoneal SR141716A against antinociception in the mouse tail-flick test induced by intrathecal administration of certain established cannabinoid receptor agonists is agonist-dependent. SR141716A was most potent against CP55940, less potent against Δ9-THC and Δ8-THC, and least potent against anandamide. As detailed elsewhere (Pertwee, 2001b), Welch's group also found that, in mice, intrathecal morphine interacts synergistically with intrathecal Δ9-THC but not with intrathecal anandamide or CP55940. In addition, there is some evidence for signaling differences between the mechanisms mediating the antinociceptive effects of intrathecal Δ9-THC and anandamide in mice (Welch et al., 1995; Pertwee, 2001b). There is also evidence from rat experiments that although intrathecal Δ9-THC triggers spinal release of dynorphins A and B, intrathecal CP55940 increases the release of dynorphin B but not dynorphin A and intrathecal anandamide fails to affect the release of either peptide (see Houser et al., 2000; Pertwee, 2001b). Signs of differences between cannabinoid receptor populations in mouse spinal cord and brain have also been reported by Welch's group (Pertwee, 2001b).
XII. Conclusions
Genes for two types of cannabinoid receptor, CB1 and CB2, have been characterized, and the existence of endogenous agonists for these receptors has also been conclusively demonstrated. The use of cloned receptors expressed in cell lines has greatly facilitated elucidation of the coupling characteristics of CB1 and CB2 receptors and the development and validation of selective ligands for these receptors. The availability of highly selective and potent CB1 and CB2 agonists and antagonists/inverse agonists has assisted in the characterization of the pharmacological properties of naturally expressed cannabinoid receptors, and the development of selective antibodies has allowed detailed localization of cannabinoid receptors, particularly of the CB1 receptor. Some CB1 receptors are present on nerve terminals, and these mediate inhibition of transmitter release when activated by agonists for these receptors that are either released endogenously or administered exogenously. Less is known about the physiological roles of CB2 receptors, which most likely include modulation of cytokine release from immune cells. There is some pharmacological evidence that supports the existence of additional types or subtypes of cannabinoid receptor, the characterization of which is being aided by the availability of CB1, CB2, and CB1/CB2 knockout mice. However, critical evidence in the form of genes encoding receptors with the appropriate pharmacology is currently lacking. Given the rather low sequence similarity between CB1 and CB2, it may be difficult to identify candidate receptors with more divergent pharmacology. If such genes are identified, it will be important to define their endogenous agonists fully to determine how broadly the cannabinoid receptor family should be defined.
Footnotes
Address correspondence to: Professor R. G. Pertwee, Co-Chair of the NC-IUPHAR Subcommittee on Cannabinoid Receptors, Department of Biomedical Sciences, Institute of Medical Sciences, University of Aberdeen, Foresterhill, Aberdeen AB25 2ZD, Scotland, UK. E-mail: rgp{at}aberdeen.ac.uk
Abbreviations
- Δ9-THC
- Δ9-tetrahydrocannabinol
- THC
- tetrahydrocannabinol
- NC-IUPHAR
- International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification
- ACEA
- arachidonyl-2′-chloroethylamide
- ACPA
- arachidonylcyclopropylamide
- anandamide
- arachidonoylethanolamide
- CBD
- cannabidiol
- CCK
- cholecystokinin
- CD40
- cluster of differentiation 40
- CHO
- Chinese hamster ovary
- FAAH
- fatty acid amide hydrolase
- FAK
- focal adhesion kinase
- GABA
- γ-aminobutyric acid
- HU-210
- 6aR,10aR analog of 11-hydroxy-Δ8-THC-dimethylheptyl
- HU-211
- 6aS,10aS analog of 11-hydroxy-Δ8-THC-dimethylheptyl
- IFN-γ
- interferon γ
- IL
- interleukin
- NOS
- nitric-oxide synthase
- iNOS
- inducible NOS
- IP3
- inositol-1,4,5-triphosphate
- MAPK
- mitogen-activated protein kinase
- NMDA
- N-methyl-d-aspartate
- NO
- nitric oxide
- PI3K
- phosphatidylinositol-3-kinase
- PMA
- phorbol 12-myristate 13-acetate
- PMA/Io
- PMA plus calcium ionophore
- R-(+)-WIN55212
- (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenyl-methanonemesylate (WIN55212-2)
- SAR
- structure-activity relationship
- [35S]GTPγS
- [35S]guanosine-5′-O-(3-thiotriphosphate)
- JWH-051
- 1-deoxy-11-OH-Δ8-THC-dimethylheptyl
- BSA
- bovine serum albumin
- CNS
- central nervous system
- EM
- electron microscope
- AM281
- N-(morpholin-4-yl)-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM251
- N-(piperidin-1-yl)-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- CP55940
- (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol
- CP55244
- (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxy-propyl)cyclohexan-1-ol
- AM630
- 6-iodo-2-methyl-1-[2-(4-morpholinyl) ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone (6-iodopravadoline)
- RT-PCR
- reverse transcription-polymerase chain reaction
- SR141716A
- N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
- 5-HT
- 5-hydroxytryptamine
- JNK
- c-Jun N-terminal kinase
- kb
- kilobase(s)
- L-759633
- (6aR,10aR)-3-(1,1-dimethylheptyl)-1-methoxy-6,6,9-trimethyl-6a,7,10,10a-tetrahydro-6H-benzo[c]chromene
- L-759656
- (6aR,10aR)-3-(1,1-dimethylheptyl)-1-methoxy-6,6-dimethyl-9-methylene-6a,7, 8,9,10,10a-hexahydro-6H-benzo[c]chromene
- JWH-015
- (2-methyl- 1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone
- JWH-133
- 3-(1,1-dimethylbutyl)-6,6,9-trimethyl-6a,7,10,10a-tetrahydro-6H-benzo[c]chromene
- JWH-139
- 3-(1,1-dimethylpropyl)-6,6,9-trimethyl-6a,7,10,10a-tetrahydro-6H-benzo[c]chromene
- HU-308
- {4-[4-(1,1-dimethylheptyl)-2,6-dimethoxy-phenyl]-6,6-dimethyl-bicyclo[3.1.1] hept-2-en-2-yl}-methanol
- CP47497
- 5-(1,1-dimethylheptyl)-2-(3-hydroxy-cyclohexyl)-phenol
- L-768242
- (2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone
- WIN54461
- 6-bromo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone
- WIN56098
- anthracen-9-yl-[2-methyl-1-(2-morpholin-4-yl-ethyl)-1H-indol-3-yl]-methanone
- U.S. Government
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





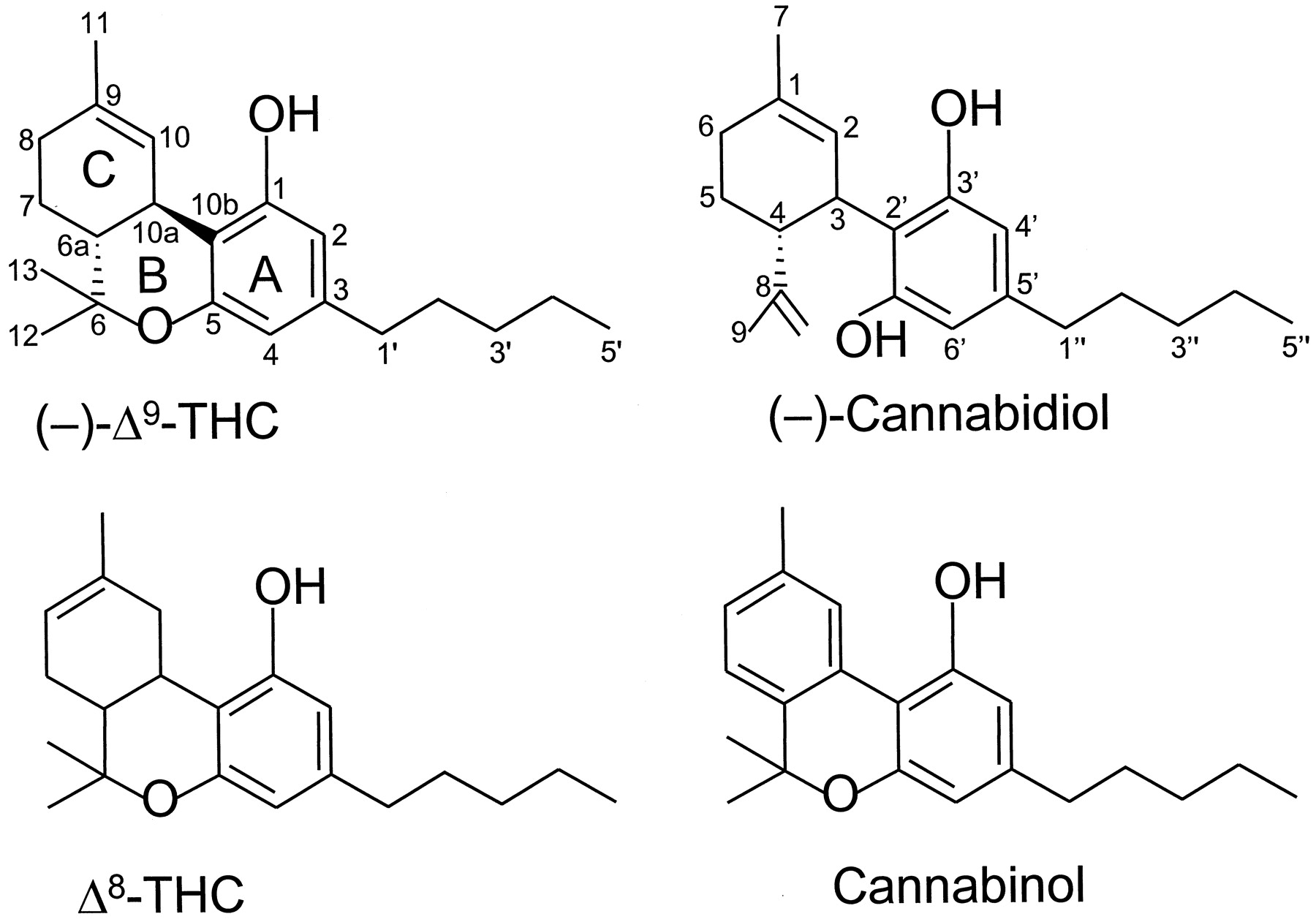{kind=link}
{kind=link}
{kind=link}
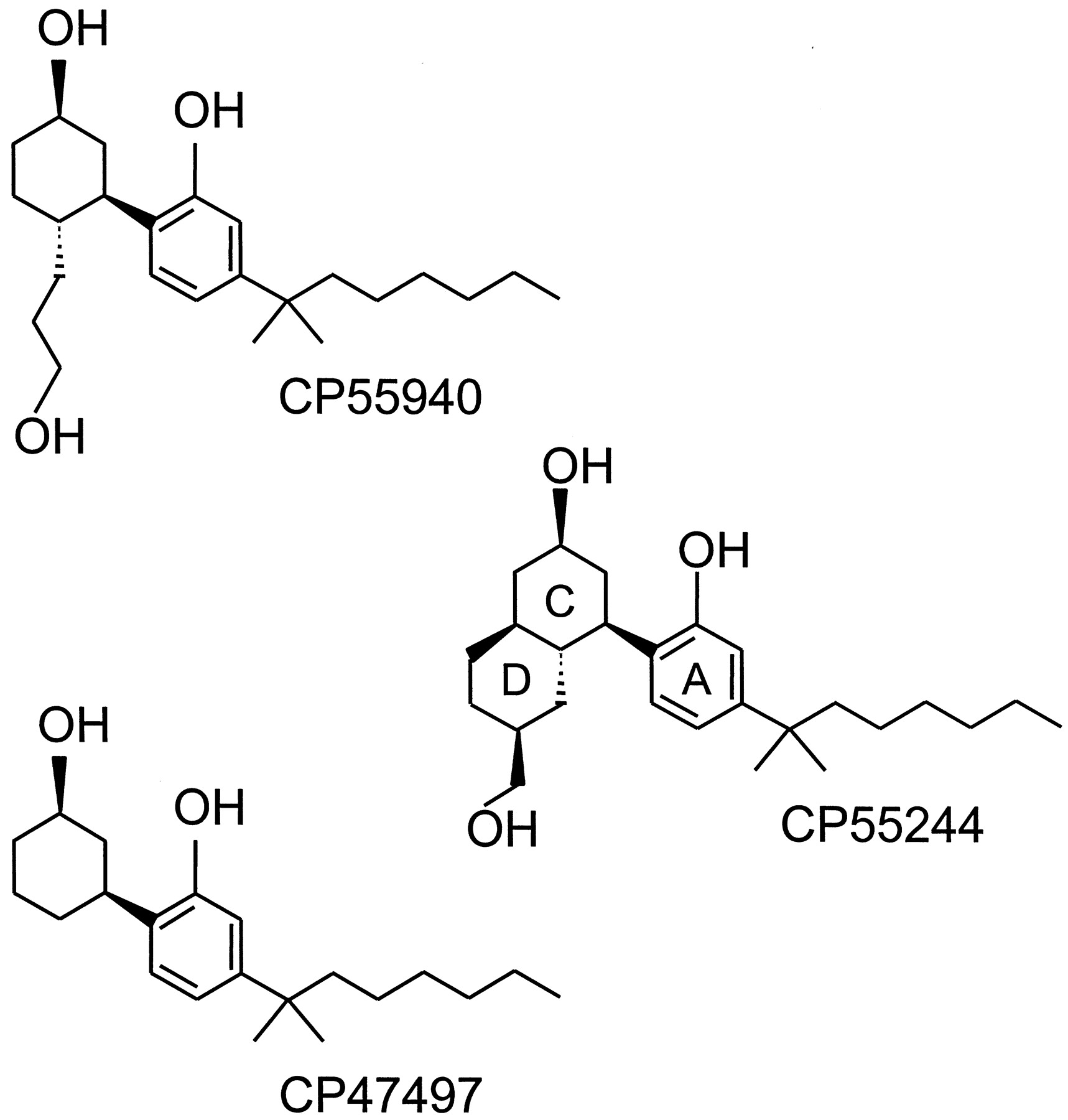{kind=link}
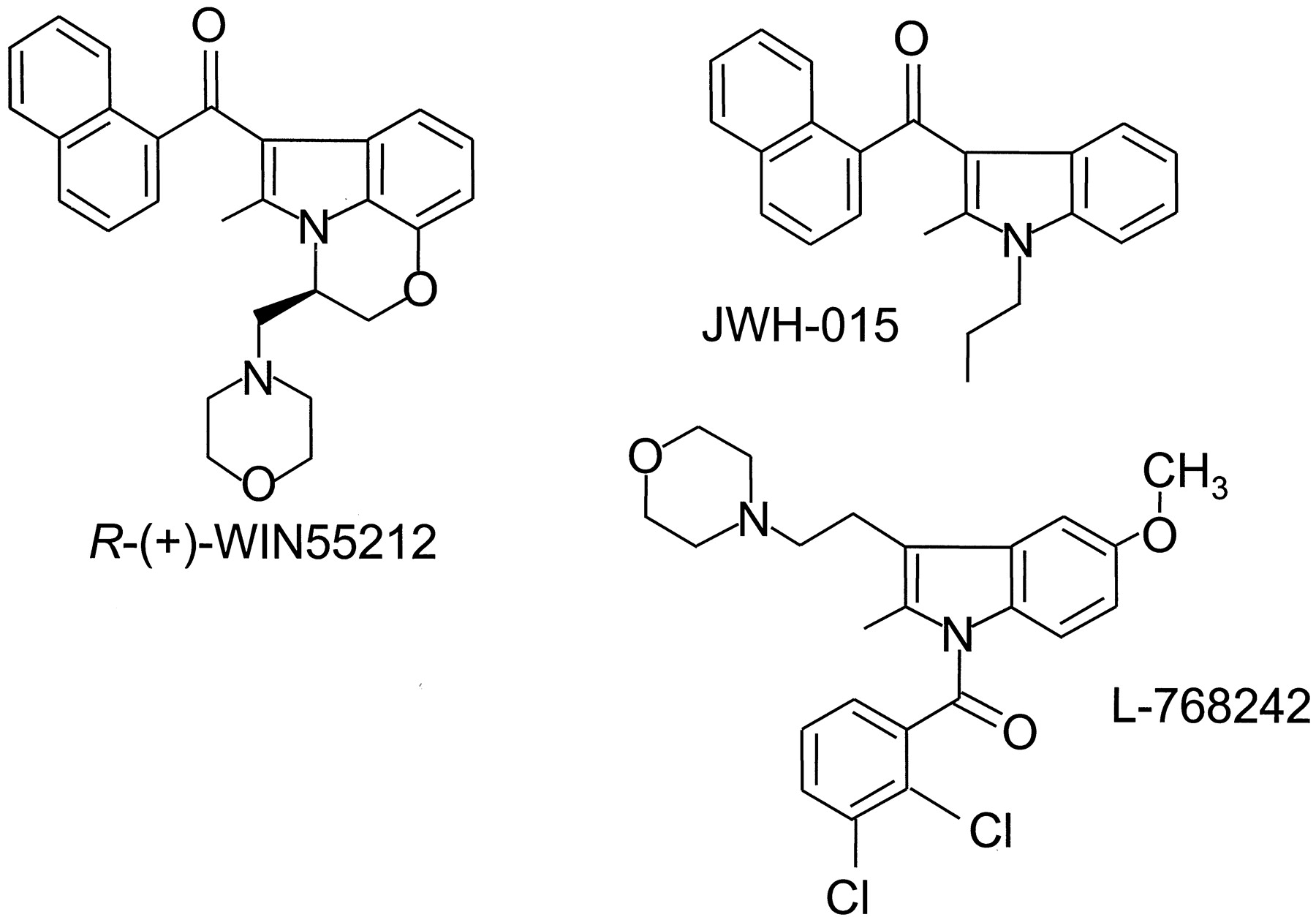{kind=link}
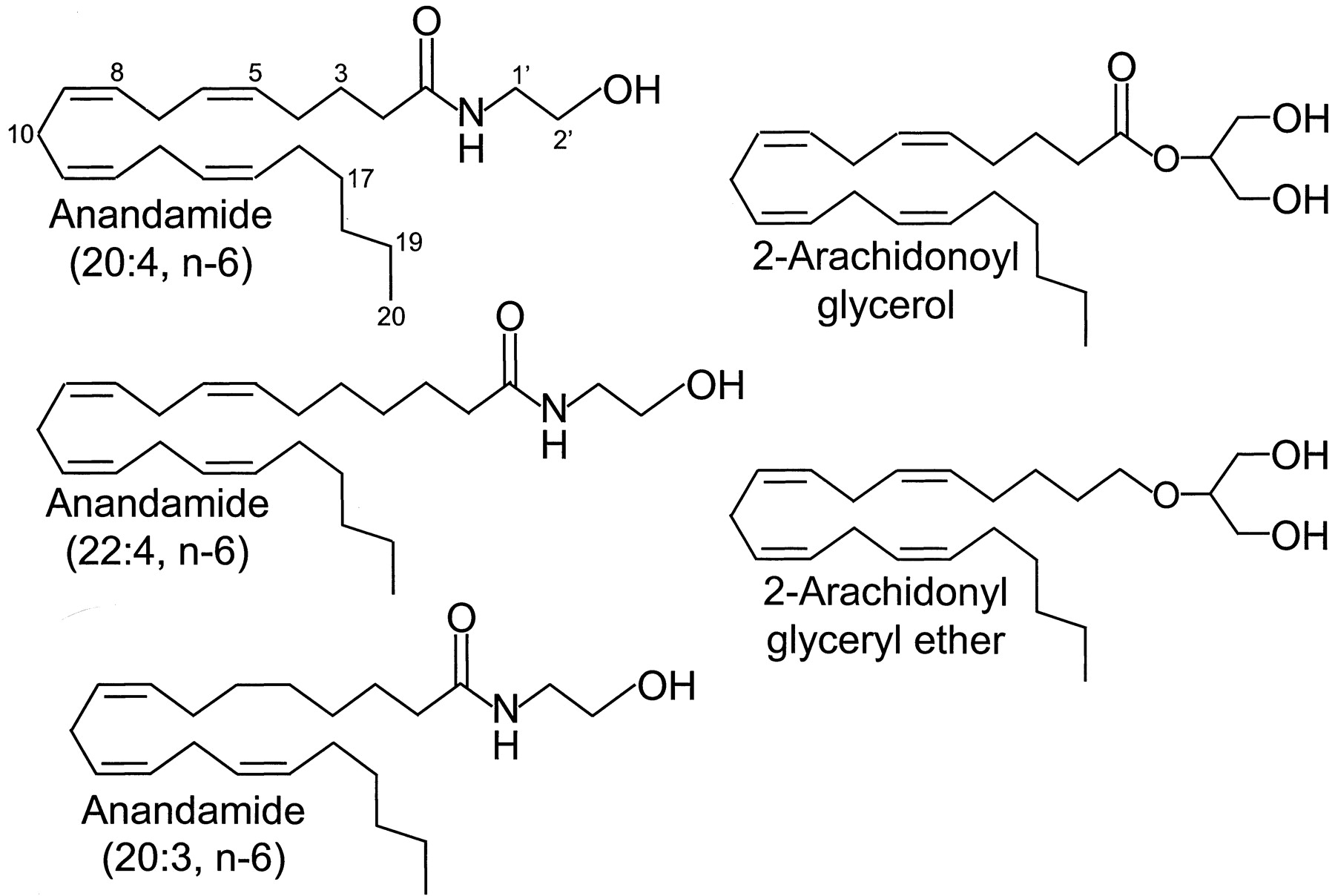{kind=link}
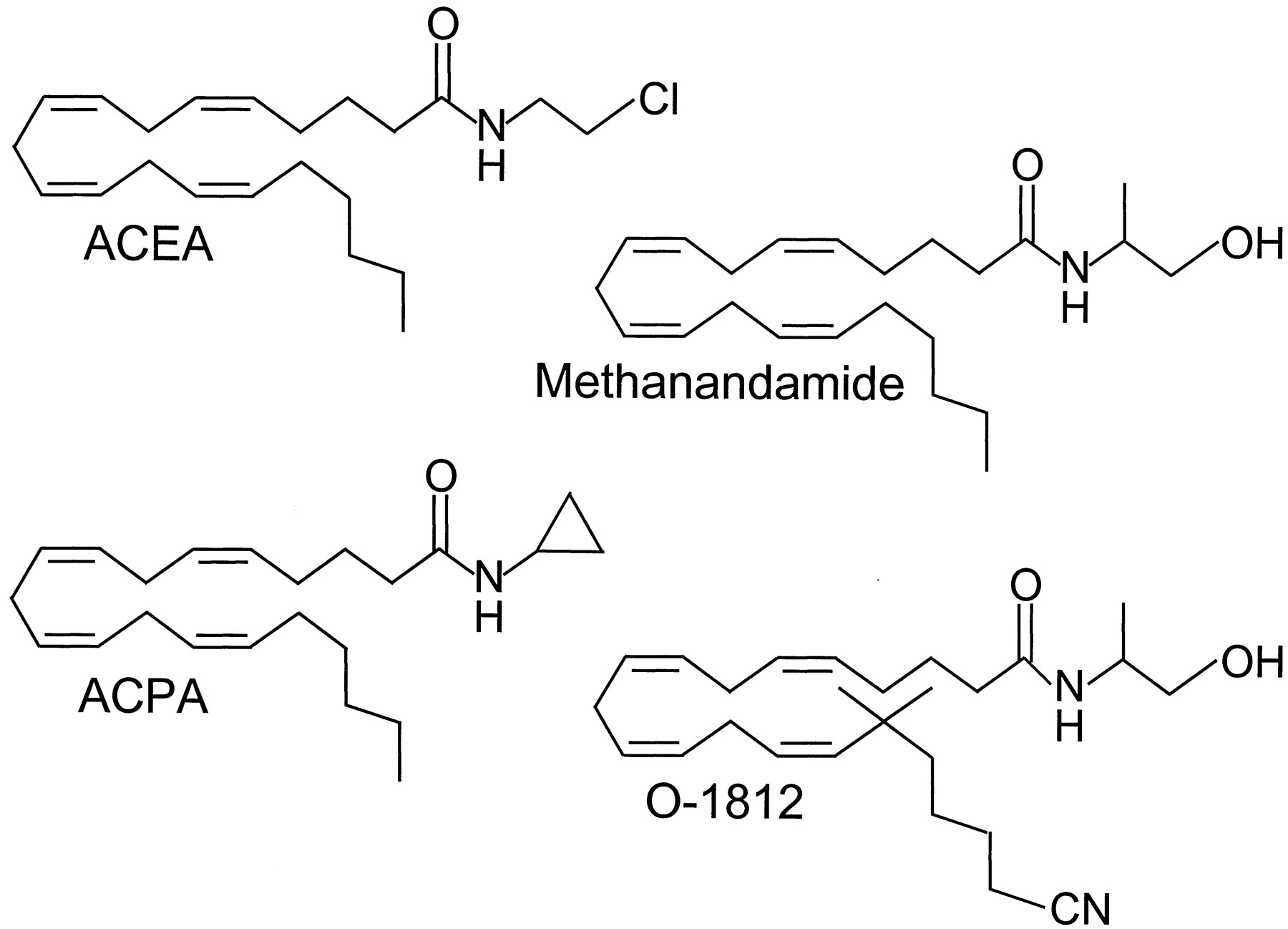{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction: Overview of the Cannabinoid Receptors
- II. Classification of Ligands That Bind to Cannabinoid Receptors
- III. Bioassay
- IV. Cellular Signal Transduction
- V. Molecular Biology of Cannabinoid Receptors
- VI. Cannabinoid Receptor Knockout Mice
- VII. Tissue Distribution of Cannabinoid Receptors
- VIII. Effects on Neurotransmission
- IX. Immunological Effects
- X. Anandamide Is a Vanilloid Receptor Agonist
- XI. Preliminary Pharmacological Evidence for Non-CB1, Non-CB2 Cannabinoid Receptors
- XII. Conclusions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters