


Visual Overview
Abstract
Hyperactivated Janus kinase (JAK) signaling is an appreciated drug target in human cancers. Numerous mutant JAK molecules as well as inherent and acquired drug resistance mechanisms limit the efficacy of JAK inhibitors (JAKi). There is accumulating evidence that epigenetic mechanisms control JAK-dependent signaling cascades. Like JAKs, epigenetic modifiers of the histone deacetylase (HDAC) family regulate the growth and development of cells and are often dysregulated in cancer cells. The notion that inhibitors of histone deacetylases (HDACi) abrogate oncogenic JAK-dependent signaling cascades illustrates an intricate crosstalk between JAKs and HDACs. Here, we summarize how structurally divergent, broad-acting as well as isoenzyme-specific HDACi, hybrid fusion pharmacophores containing JAKi and HDACi, and proteolysis targeting chimeras for JAKs inactivate the four JAK proteins JAK1, JAK2, JAK3, and tyrosine kinase-2. These agents suppress aberrant JAK activity through specific transcription-dependent processes and mechanisms that alter the phosphorylation and stability of JAKs. Pharmacological inhibition of HDACs abrogates allosteric activation of JAKs, overcomes limitations of ATP-competitive type 1 and type 2 JAKi, and interacts favorably with JAKi. Since such findings were collected in cultured cells, experimental animals, and cancer patients, we condense preclinical and translational relevance. We also discuss how future research on acetylation-dependent mechanisms that regulate JAKs might allow the rational design of improved treatments for cancer patients.
Significance Statement Reversible lysine-ɛ-N acetylation and deacetylation cycles control phosphorylation-dependent Janus kinase–signal transducer and activator of transcription signaling. The intricate crosstalk between these fundamental molecular mechanisms provides opportunities for pharmacological intervention strategies with modern small molecule inhibitors. This could help patients suffering from cancer.
I. Introduction
The Janus kinases (JAK) family consists of four cytosolic nonreceptor tyrosine kinases (TKs). These are tyrosine kinase-2 (TYK2), JAK1, JAK2, and JAK3 (Babon et al., 2014). These molecules consist of about 1150 amino acids and have a molecular mass of approximately 130 kDa. Genes encoding human JAKs locate in chromosomes 19p13.2 (TYK2), 1p31.3 (JAK1), 9p24.1 (JAK2), and 19p13.11 (JAK3) (Leonard and O’Shea, 1998).
All JAKs share a modular structure consisting of seven JAK homology (JH) domains that fall into four regions (Fig. 1). These are the N-terminal four-point-one, ezrin, radixin, moesin domain (FERM, JH5–JH7); the Src homology-2 (SH2)-like domain (JH3 and part of JH4); the inactive pseudokinase (JH2); and the catalytically active kinase (JH1) (Schindler et al., 2007) (Fig. 1). The four-point-one, ezrin, radixin, moesin domains mediate specific interactions of JAKs with plasma membrane–inserted receptors that bind extracellular signaling molecules (Funakoshi-Tago et al., 2006; Babon et al., 2014). The SH2 domains of JAKs contribute to their receptor binding and their dimerization for activating crossphosphorylation (Gorantla et al., 2010; Glassman et al., 2022). The pseudokinase domains of JAKs function as negative regulators of the tyrosine kinase domains by protein-protein interactions (Ungureanu et al., 2011; Lupardus et al., 2014).

Structural representation of the JAK family members JAK1–3 and TYK2. Shown is the organization of JAKs into the domains JH1–7 and frequent mutations of JAKs in tumor cells.
TYK2 received its name from its biochemical activity and from being the second identified tyrosine kinase that can phosphorylate tyrosine residues within proteins. Accordingly, TYK2 is the founding member of the JAK family and has been linked to cytokine and growth factor signaling since 1990 (Firmbach-Kraft et al., 1990; Krolewski et al., 1990; Verma et al., 2003).
JAKs were given the name of the ancient two-headed god Janus, who faces the past and the future (Darnell et al., 1994; Ihle et al., 1995; Pellegrini and Dusanter‐Fourt, 1997; Leonard and O’Shea, 1998). This two-headed functionality reflects their function as linkers between the receptors for cytokines and growth factors and the signal transducers and activators of transcription (STATs). These inducible transcription factors comprise STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Tyrosine-phosphorylated STATs enter the nucleus, where they bind to cognate STAT consensus motifs in genes and promote a recruitment of transcription machineries (Fig. 2). This consequently modulates gene expression patterns that crucially determine cell growth, proliferation, differentiation, and cell death. These vast biologic implications of JAK-STAT signaling can explain why dysregulation of this pathway is frequently associated with solid and hematologic malignancies, autoimmunity, and immunologic failures (Owen et al., 2019; Bharadwaj et al., 2020; Yang et al., 2020).

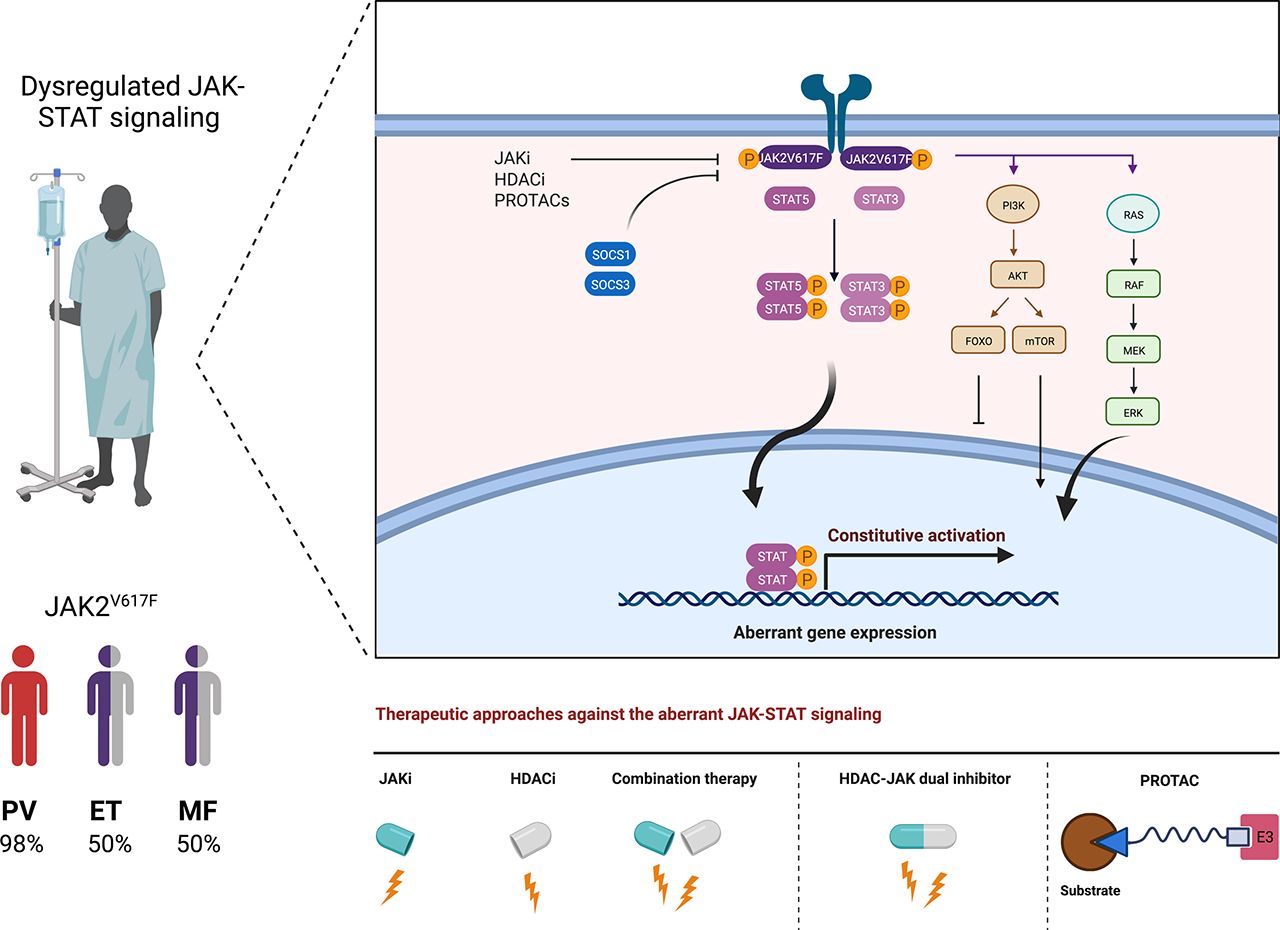
The diagram shows wild-type JAK-STAT (left) and mutant JAK2V617F-STAT (right) signaling. The binding of a cytokine, exemplified for erythropoietin (EPO), promotes the dimerization of its receptor and subsequently JAK2 dimerization and crossphosphorylation. Phosphorylation of the cytokine receptor creates a binding interface for the SH2 domains of preformed STAT dimers. These become phosphorylated by JAKs, form high-avidity dimers via SH2-pY interactions, and enter the nucleus to induce specific gene expression patterns. Cognate STAT consensus motifs in gene promoters are usually the palindromic γ-activated sequences and the interferon-stimulated response elements. Independent of receptor activation and due to structurally facilitated JAK2 dimer formation, JAK2V617F activates STATs. The resulting tonic gene activation promotes tumorigenesis. This article summarizes how HDACi can alter the molecules and mechanisms of these pathways. We exemplarily depict the suppressive effects of HDACi on JAK2V617F. These rely on the acetylation of STATs and their subsequent dephosphorylation and on the induction of SOCS proteins that directly inhibit and promote the degradation of JAKs. HDACi additionally decrease transcription of the JAK2 gene locus and accelerate the proteasomal degradation of JAK2V617F; see text for details. Created with biorender.com.
Histone deacetylases (HDACs) are epigenetic modifiers that fall into four classes. HDAC1, HDAC2, HDAC3, and HDAC8 form class I; HDAC4, HDAC5, HDAC7, and HDAC9 are class IIa; HDAC6 and HDAC10 represent class IIb; and HDAC11 is the sole member of class IV. The sirtuins (SIRTs) 1–7 represent class III (Table 1). HDAC classes I, II, and IV use Zn2+ to polarize H2O for a nucleophilic attack on the acetyl moiety in acetylated lysine residues (Krämer et al., 2009). SIRTs use NAD+ for their deacetylation reaction, which couples their catalytic activities to the energy state of cells (Stein and Imai, 2012; Cantó et al., 2015; Jiang et al., 2017b).
Human histone deacetylases, their cytogenetic and subcellular localization, and selected targets
Our review summarizes evidence on how structurally divergent histone deacetylase inhibitors (HDACi), derivatives thereof with tyrosine kinase inhibitor (TKi) activity, and TKi with a functional unit driving proteolysis of JAKs kill cancer cells. Moreover, we discuss how the extraction of JAK mutations and regulatory patterns in tumor cells can propel a rational development of new pharmacological approaches involving HDACi and TKi. We will first present how the four JAK proteins and the 11 Zn2+-dependent HDACs contribute to tumorigenesis.
II. Relevance of Janus Kinases for Tumorigenesis
A. Janus Kinase-1
JAK1 is frequently dysregulated in human hematologic malignancies. For example, 10%–27% of patients with T-cell acute lymphoblastic leukemia (T-ALL) and 1.5% of patients with B-cell acute lymphoblastic leukemia (B-ALL) have tumor cells with activating JAK1 mutations (Flex et al., 2008; Jeong et al., 2008; Mullighan et al., 2009b; Zhang et al., 2012; Li et al., 2017). Up to 2% of acute myeloid leukemia (AML) patients carry cells with JAK1 mutations (Tomasson et al., 2008; Xiang et al., 2008). In 8%–12.5% of T-cell prolymphocytic leukemia (T-PLL) cases, JAK1 mutations occur in the tumor cells (Bellanger et al., 2014; Springuel et al., 2015; Wahnschaffe et al., 2019).
Various residues in JAK1 are mutation hotspots (Fig. 1), and additional mutations are continuously updated in public databases, e.g., Genomic Data Commons of the National Cancer Institute (Grossman et al., 2016; Jensen et al., 2017), Catalogue of Somatic Mutations in Cancer (Tate et al., 2019), and cBioPortal for Cancer Genomics (Cerami et al., 2012; Gao et al., 2013).
Aberrant JAK1 signaling is linked to dysfunctional lymphopoiesis (Chen et al., 2012). For instance, V658F and S646P mutations occur in the JH2 domain of JAK1 (Fig. 1) and impair its autoinhibitory functions on the kinase domain. This leads to constitutively active STAT3 and MEK–extracellular signal-regulated kinases (ERK) signaling and allows a cytokine-independent proliferation of malignant B-ALL and T-ALL cells in vitro and in mice (Jeong et al., 2008; Hornakova et al., 2009; Mullighan et al., 2009a; Li et al., 2017). The finding that difficult-to-treat adult B-ALL cells are highly vulnerable toward the ATP-competitive JAK1/JAK2 inhibitor ruxolitinib verifies a disease relevance of JAK1S646P (Li et al., 2017).
Certain JAK1 mutations, such as A634D and R724H, are characteristic for T-ALL (Flex et al., 2008), whereas the V623A mutation occurs in AML (Xiang et al., 2008). Tumor cell–specific mutations of JAK1 are not only associated with tumorigenesis but also with therapy outcome. This is exemplified by patients with JAK1 mutations frequently showing resistance to chemotherapy (Li et al., 2017). These findings illustrate that JAK1 mutations contribute to both the development and the aggressiveness of blood disorders.
Solid tumors can also carry mutant JAK1 molecules. Fusions, rearrangements, missense mutations, nonsense mutations, silent mutations, frameshift deletions, and insertions in JAK1 occur in endometrial cancers, intestinal cancers, and stomach cancers (Albacker et al., 2017). Moreover, JAK1 is mutated in approximately 1.8% of patients suffering from breast carcinoma, uterine corpus neoplasm, colorectal adenocarcinoma, non–small-cell lung carcinoma, and prostate cancer (AACR Project GENIE Consortium, 2017), as well as in 3.8% of hepatocellular carcinoma cases and 11.5% of endometrial cancer patients (Cerami et al., 2012; Gao et al., 2013; Kan et al., 2013). Further research is required to comprehensively identify the therapeutic potential of such aberrations in these and other solid tumors (Qureshy et al., 2020).
B. Janus Kinase-2
In 2001 and updated in 2008, the World Health Organization categorized polycythemia vera (PV), essential thrombocythemia (ET), and idiopathic myelofibrosis (MF) together with chronic myeloid leukemia and some seldom leukemia subtypes into myeloproliferative neoplasms (MPNs) (Tefferi and Vardiman, 2008; Tefferi et al., 2009; Vannucchi et al., 2009; Vardiman et al., 2009; Helbig, 2018; Marcellino et al., 2020). PV, ET, and MF are clonal hematopoietic stem cell malignancies with increased self-renewal capacity and a lack of the leukemia fusion protein BCR-ABL, which defines chronic myeloid leukemia. The somatic point mutation JAK2V617F occurs in over 95% of patients with PV and in 50% of patients with ET or MF. Mutant JAK2 also occurs in 20% of other MPNs, such as refractory anemia with ring sideroblasts and thrombocytosis (Patnaik and Tefferi, 2015; Orazi et al., 2017). PV and ET patients have increased numbers of erythroid or megakaryocyte precursors, respectively. Fibrotic bone marrow, hemorrhage, splenomegaly, and enhanced cytokine-induced JAK1/JAK2 signaling are hallmarks of MF (Thiele et al., 2005; Barosi et al., 2007; Tefferi and Vardiman, 2008; Yönal et al., 2016). JAK2V617F manifests in ∼5% of AML and myelodysplastic syndrome cases (Renneville et al., 2006; Steensma et al., 2006; Verstovsek et al., 2006; Levine et al., 2007).
In 2005, JAK2V617F was discovered in tumor cells from patients with PV, ET, and MF (Fig. 1). This aberration stems from a guanine-to-thymidine substitution that replaces valine with phenylalanine at codon 617 within the JH2 domain of JAK2 (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005). Without a need for cognate receptor stimulation, dimeric JAK2V617F triggers tyrosine phosphorylation of the prosurvival JAK-STAT, phosphatidylinositol 3-kinase/serine-threonine protein kinase (AKT), and MAPK/ERK proteins as the root cause of aberrant cell growth and survival (James et al., 2005; Kralovics et al., 2005; Levine et al., 2005; Gorantla et al., 2010) (Fig. 2). Accordingly, ruxolitinib produces clinical responses in patients suffering from JAK2V617F-associated neoplasms (Barosi et al., 2014; Bose and Verstovsek, 2017b; Cervantes and Pereira, 2017).
Mutant JAK2 not only alters the initiation of cytoplasmic signaling cascades. JAK2V617F-positive cells show a high frequency of genomic instability, which is associated with an increased risk of transformation into aggressive AML (Theocharides et al., 2007; Beer et al., 2010; Aynardi et al., 2018). This serious adverse outcome is seen in 5% of patients with ET or PV (Finazzi et al., 2005; Wolanskyj et al., 2006) and in 15%–30% of primary MF patients (Dupriez et al., 1996). These incidences markedly increase upon genotoxic treatment (Finazzi et al., 2005; Björkholm et al., 2011). This may be due to the capacity of JAK2V617F to suppress cytotoxic programs that are triggered upon chemotherapy-induced DNA replication stress and DNA damage by an upregulation of DNA repair enzymes (Plo et al., 2008; Kurosu et al., 2013; Ueda et al., 2013; Chen et al., 2015; Karantanos and Moliterno, 2018). Thus, alternative therapies that early and effectively eliminate JAK2V617F could prevent aggressive disease progression.
Mutant JAK2 additionally regulates tumorigenesis via the phosphorylation of the residue tyrosine-41 in histone H3. This displaces heterochromatin protein-1α and abrogates its tumor-suppressive functions (Shi et al., 2006; Dawson et al., 2009; Griffiths et al., 2011). It needs to be considered, though, that other kinases, such as the cell cycle regulatory kinase WEE1 and activated CDC42-associated kinase-1 (ACK1), also catalyze the phosphorylation of histones and thereby modulate the transcription of multiple genes that are relevant to cellular homeostasis (Mahajan et al., 2012; Mahajan et al., 2017; Kim et al., 2020). For instance, the WEE1-mediated phosphorylation of histone H2B at tyrosine-37 during the S phase of the cell cycle suppresses the transcription of histone genes (Mahajan et al., 2012). ACK1 phosphorylates histone H4 at tyrosine-88 and consequently activates transcription of androgen receptor. This drives the progression of prostate cancer to a castration-resistant state (Mahajan et al., 2017; Sawant et al., 2022).
Tightly controlled JAK2 signaling is also relevant for metabolic integrity. JAK2V617F and the JAK2 exon 12 (N542-E543del) mutation, which drive increased erythropoiesis, create a systemically dysregulated metabolic profile with highly elevated mRNA and protein levels of the key glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-3. Accordingly, this enzyme is a druggable vulnerability of cells with mutant JAK2 (Rao et al., 2019).
JAK2 mutants are likewise involved in chronic myelomonocytic leukemia (CMML). The JAK2 inhibitor TG101209, which was developed to treat MPN cells with JAK2V617F and MPLW515L/K (Pardanani et al., 2007), halted the spontaneous growth of granulocyte-macrophage colony-forming units of cells from CMML patients (Geissler et al., 2016; Padron et al., 2016).
JAK2 mutations are not limited to myeloid leukemia but likewise occur in 8.5% of high-risk B-ALL and in 18%–28% of B-ALL cases associated with Down syndrome (Gaikwad et al., 2009; Kearney et al., 2009). A majority of JAK2 mutations in the lymphoid lineage affect the R683 residue in JH2 (Fig. 1). Overexpression of cytokine receptor-like factor-2/thymic stromal lymphopoietin receptor (CRLF2) is prominent in JAK2I682F and JAK2R683G cells of patients with clinically unfavorable types of acute lymphoblastic leukemia (ALL). The association between these JAK mutants and CRLF2 propels ligand-independent cell proliferation (Mullighan et al., 2009a; Savino et al., 2017; Chang et al., 2021). Below, we summarize that such therapeutically challenging lesions create a remarkable vulnerability to HDACi.
The disease relevance of JAK2-dependent signaling likewise became evident with the JAK2L611S mutation in the JH2 domain of JAK2. This mutation was identified in a child with B-ALL (Fig. 1) and not detected in the bone marrow upon remission, suggesting that JAK2L611S drove this cancer (Kratz et al., 2006). Therefore, extended screening for JAK2 mutations in childhood ALL may deliver insights into an actionable drug target.
Mutations in JAK2 are not limited to blood malignancies. JAK2 alterations occur in about 2.65% of all cancers, including non–small-cell lung cancer (NSCLC), breast carcinoma, and colorectal adenocarcinoma (AACR Project GENIE Consortium, 2017). Although JAK inhibitors (JAKi) are effective against cells from various solid tumors in preclinical studies, it is currently not solved if such drugs provide a benefit for patients with solid tumors (Qureshy et al., 2020).
C. Janus Kinase-3
JAK3 mutations occur in 12% of juvenile CMML (Sakaguchi et al., 2013; Springuel et al., 2015) and in 15% of acute megakaryoblastic leukemia cases (Walters et al., 2006; Malinge et al., 2008; Springuel et al., 2015) (Fig. 1). These aberrations lead to constitutively active JAK3 that confers growth factor–independent proliferation, being a hallmark of tumorigenesis (Walters et al., 2006; Malinge et al., 2008; Riera et al., 2011; Vainchenker and Constantinescu, 2013). Genomic profiling of a wide variety of T-cell neoplasms showed that up to one-third of patients had genomic aberrations that gave rise to gain-of-function (GOF) mutations in JAK1 and JAK3 (Springuel et al., 2015; Greenplate et al., 2018). JAK3 was found mutated in 10%–16% of T-ALL patients (Zhang et al., 2012; De Keersmaecker et al., 2013; Vicente et al., 2015; Li et al., 2016; Girardi et al., 2017; Liu et al., 2017; Degryse et al., 2018a; Greenplate et al., 2018) and in 36%–42% of T-PLL cases (Bellanger et al., 2014; Springuel et al., 2015; Wahnschaffe et al., 2019).
Further studies revealed JAK3 mutations in 1.8% of all cancers. NSCLC, colorectal adenocarcinoma, breast carcinoma, melanoma, and uterine corpus neoplasm have the greatest prevalence of genetic alterations in the JAK3 gene (AACR Project GENIE Consortium, 2017). Further research will define whether they are therapeutic targets.
D. Tyrosine Kinase-2
Similar to the other JAKs, activating point mutations at the TYK2 locus predominantly accumulate in the JH1 and JH2 domains (Leitner et al., 2017; Hammarén et al., 2019) (Fig. 1). The first reported GOF mutation of TYK2 was V678F. This aberration is homologous to JAK2V617F but has not yet been reported in patients (Staerk et al., 2005; Gakovic et al., 2008). This finding suggests that members of the JAK family have common as well as very individual functions in tumorigenesis. Such a lack of functional redundancy might explain the highly divergent mutation spectra of JAKs in cancer cells.
Two GOF TYK2 germline mutations (P760L and G761V) were identified in leukemic pediatric patients (Fig. 1). These mutations are in the JH2 pseudokinase domain of TYK2 and likely constrict its autoinhibitory function on the JH1 kinase domain (Waanders et al., 2017). Sequence analysis of 17 cell lines and 45 pediatric T-ALL patient samples identified further activating mutations of TYK2 (G36D, S47N, V731I, E957D, R1027H) (Fig. 1). Such aberrations can promote cell survival through the inducible transcription factors STAT1 and STAT3 and activating effects on the antiapoptotic BCL2 proteins (Sanda et al., 2013; Wingelhofer et al., 2018; Wöss et al., 2019).
Activation of wild-type TYK2-STAT1 signaling and ensuing expression of the antiapoptotic BCL2 protein as well as germline TYK2 mutations (A53T, A81V, R197H, V362F, G363S, I684S, R703W, A928V, A1016S, P1104V) are linked to the development of ALL and AML (Kaminker et al., 2007; Tomasson et al., 2008; Sanda et al., 2013). TYK2P1104A was detected in breast, liver, stomach, and poor-prognosis malignant peripheral nerve sheath tumors (Hirbe et al., 2017). Although TYK2P1104A might inactivate TYK2 (Kaminker et al., 2007), increased levels of TYK2 correlate with cell migration and metastasis of prostate cancer cells (Ide et al., 2008; Santos et al., 2015). These data illustrate context-dependent tumor-promoting activities of wild-type and mutant TYK2.
Taken together, the above reports impressively demonstrate that a failure to control JAK-STAT signaling is a hallmark feature of early and late stages of cancer development.
III. Histone Deacetylases Are Valid Pharmacological Targets in Janus Kinase Mutant Tumor Cells
Hematopoietic and solid tumor cells frequently display overactive and overexpressed HDAC levels when compared with normal tissues (Fig. 3A). Consequently, acetylation patterns of histones and nonhistone proteins, chromatin compaction, and gene expression are dysregulated. These processes determine cell proliferation and cell fate at multiple layers, including hallmarks of tumorigenesis (Fig. 3, B and C). For example, aberrant HDAC activity restricts the expression of genes encoding tumor suppressors and cell differentiation proteins (Krämer et al., 2014; Wagner et al., 2014; Ceccacci and Minucci, 2016; Imai et al., 2016).

HDACs are dysregulated in cancer and control chromatin remodeling with histone acetyl transferases (HATs). (A) Cancer cells display altered HDAC expression and activity. This creates a vulnerability of transformed cells to HDACi. (B) HATs catalyze the acetylation of lysine moieties of histone and nonhistone proteins using acetyl-CoA as a cosubstrate. The acetyl groups neutralize the positively charged histone tails and thereby weaken the electrostatic interactions between histones and the negatively charged phosphate backbone of DNA. This causes a relaxed state of chromatin. In a chromatin context-dependent manner, this can render DNA more accessible for transcription (depicted as accelerated speed). Elimination of the acetyl groups by HDAC activity reverses these processes. HDACs catalyze the removal of the acetyl group from the lysine residues, causing the release of the acetate molecule. These alterations in transcription critically affect key cellular processes. (C) Biochemistry of the acetylation/deacetylation reactions. Created with BioRender.com.
For decades, these important physiologic functions of HDACs have encouraged the development and testing of small molecules that target HDACs (Figs. 3A and 4A). Initial HDACi were identified by their ability to induce histone acetylation before the discovery of the first HDAC in 1996 (Taunton et al., 1996). These compounds of natural and microbial origins include butyrate, trapoxin, and trichostatin A (TSA) (Hagopian et al., 1977; Yoshida et al., 1990; Kijima et al., 1993; Krämer et al., 2001). Crystal structures of HDACs were determined from 1999 on (Finnin et al., 1999). These allowed the development of an increasingly specific arsenal of HDACi based on structure-activity-relationship.
In the 1990s, butyrate and phenylbutyrate were the first HDACi that were applied in compassionate protocols to patients suffering from leukemia (Krämer et al., 2001). The discovery that the antiepileptic, mood-stabilizing drug valproic acid inhibits class I HDACs in vitro and in experimental animals (Göttlicher et al., 2001) illustrated that an agent that increased protein acetylation could be given safely to patients for decades (Michaelis et al., 2007). The US and China Food and Drug Administration (FDA) agencies have approved HDACi for the treatment of poor-prognosis cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma. These drugs are vorinostat (SAHA), romidepsin (FK228), belinostat (PXD101), and chidamide/tucidinostat (Piekarz et al., 2009; Food and Drug Administration, 2010; Whittaker et al., 2010; Li et al., 2019; Cappellacci et al., 2020). Clinically validated HDACi target Zn2+-dependent HDACs (Nebbioso et al., 2017; Beyer et al., 2019; Cappellacci et al., 2020) (www.clinicaltrials.org).
HDACi can be broad and isoenzyme-specific inhibitors (Fig. 4A). For example, the hydroxamic acids vorinostat, belinostat, and panobinostat inhibit HDACs belonging to classes I, IIa/b, and IV (pan-HDACi) at nano- to low micromolar levels. The short-chain fatty acid valproic acid selectively inhibits class I HDACs, whereas butyrate and its derivatives inhibit preferentially classes I and IIa at millimolar levels. The benzamide entinostat (MS-275) blocks the class I HDACs HDAC1, HDAC2, and HDAC3 at low micromolar levels, and the nanomolar HDACi romidepsin additionally targets HDAC8 (Fig. 4A) (Bradner et al., 2010; Lechner et al., 2022). Current approaches often focus on hydroxamic acid–based compounds for an improved design of HDACi. This relies on the ability of hydroxamic acids to block HDACs at low concentrations in vitro and in cells as well as the clinical experience with these HDACi.

HDAC classes, HDACi, and mode of action. (A) Schematic representation of different classes of HDACs and their pharmacological inhibitors. (B) Targeting the Zn2+-dependent HDACs by HDACi. The structure of hydroxamic acids is shown as an example of the three typical parts of an HDACi. The cap group binds the HDAC at its rim, and the linker serves to position the Zn2+-binding part toward the coordinated metal in the catalytic cleft of the HDAC. (C) The double-hit approach uses JAK-HDAC dual inhibitors. The cap group provides possibilities to attach a TKi. The TKi must preserve its structures that compete with ATP and the HDACi must retain the Zn2+-binding moiety. Ac-K, acetylated lysine residue in a protein.
The pharmacological warhead of HDACi is a Zn2+-binding group (Krämer, 2009), as exemplified for hydroxamic acids in Fig. 4B. On the other end of the drug, a cap group has residues for interactions with the rim of the enzyme. A linker of variable length connects these two moieties (Fig. 4B). This structure makes HDACi amenable for poly-pharmacophore design. The cap group can provide residues to anchor additional pharmacological moieties and exit vectors to attach independent pharmacophores. This can be the attachment of TKi (Fig. 4C). In contrast, the Zn2+-binding group must be preserved to maintain inhibitory activity against HDACs.
Individual structures of the HDACs (length and depth of the catalytic cavity, amino acid side chains at the entrance site of the catalytic cavity) have allowed the design of selective and specific HDACi. This has been achieved by varying the cap group, the length of the linker, and the type of warhead (Krämer et al., 2014). For example, tetrahydro-β-carboline–based hydroxamic acid HDACi with large cap groups specifically block the broad and shallow catalytic groove of the second catalytic domain of HDAC6 (17.5 Å). These HDACi selectively inhibit HDAC6 at nanomolar concentrations (Sellmer et al., 2018) and poorly enter class I HDACs because of their narrower catalytic channel rim (∼12.5 Å) (Butler et al., 2010). Researchers highly appreciate these and other HDACi as pharmacological tools to define individual functions of HDACs in vitro and in vivo. In addition, specific HDACi are expected to kill certain tumor cells with less adverse effects on normal cells from the same tissue (Michaelis et al., 2007; Krämer, 2009; Juengel et al., 2011; Falkenberg and Johnstone, 2014; Krämer et al., 2014; Koeneke et al., 2015). It will be interesting to see the full clinical potential of such HDACi.
A. Janus Kinase–Signal Transducer and Activator of Transcription Pathways Control Cellular Susceptibility to Histone Deacetylase Inhibitors
JAK-STAT signaling is not only a paradigm for an intracellular signaling pathway but also a determinant for the cellular susceptibility to changes in protein acetylation patterns. In CTCL and diffuse large B-cell lymphoma (DLBCL) cell lines and ex vivo patient samples, aberrant activation of the JAK-STAT pathway confers resistance to HDACi (Fantin et al., 2008; Smith et al., 2010; McKinney et al., 2014; Cortes et al., 2021). A vorinostat-phase IIb clinical trial revealed by immunohistochemistry of skin biopsies that nonresponding CTCL patients carried higher expression levels and activation of STAT1, STAT3, and STAT5. Consistent herewith, RNA interference (RNAi) against STAT3 or STAT5 synergistically induced apoptosis when combined with vorinostat in cultured lymphoma cells (Fantin et al., 2008). These findings encouraged a synchronized inhibition of JAKs and HDACs. A combination of vorinostat with the broad-range, ATP-competitive JAKi pyridone-6 caused growth arrest and triggered apoptosis in CTCL cells with primary robustness to vorinostat due to activated STATs (Fantin et al., 2008). Similarly, inhibition of JAK1 and JAK2 using ruxolitinib or momelotinib overcame the limited response of a subset of CTCL cells toward romidepsin in vitro and in xenografts (Cortes et al., 2021). These data are in line with findings from DLBCL, where concurrent inhibition of JAK-STAT and HDACs using ruxolitinib and panobinostat showed obvious synergy and could tremendously reduce tumor growth in vitro as well as in a xenograft model (McKinney et al., 2014; Davis et al., 2018).
In JAK2V617F-positive MPN cells, concurrent treatment with ruxolitinib and vorinostat resulted in G1-phase cell cycle arrest, apoptotic cell death, and inhibition of colony-formation capacity. This was due to the attenuated expression of phosphorylated and total JAK2V617F and significantly reduced phosphorylation of STAT3 and AKT (Hao et al., 2020). These effects are linked to the suppressors of cytokine signaling (SOCS) proteins, which are negative regulators of JAK-STAT signaling (Jiang et al., 2017a) (Fig. 2). The inhibition of JAK2V617F by ruxolitinib and vorinostat upregulated SOCS3. Furthermore, HDACi suppressed the mRNA levels of the antiapoptotic STAT3 target genes BCL2, BIRC5, MYC, and the cell cycle regulator CCND1, which can explain the above-mentioned cell cycle arrest in G1. This data shows that ruxolitinib and vorinostat combinations promise better therapeutic outcomes for patients with MPN (Hao et al., 2020). Below, we discuss clinical trials using HDACi and TKi in combination.
Unlike in the above-named tumor types, combinations of ruxolitinib and the hydroxamic acid–based pan-HDACi givinostat (ITF2357) did not induce more apoptosis than givinostat alone in B-ALL cells with CRLF2 overexpression. This deviation may rely on the resistance of CRLF2/JAK2-mutated cells to type 1 (e.g., ruxolitinib, baricitinib; bind JAKs in the active conformation) and type 2 (CHZ-868; stabilizes the inactive conformation of JAKs) JAKi. Irrespective thereof, givinostat potently inhibited the growth and survival of such cells (Savino et al., 2017).
As in MPNs, JAKi and HDACi interact favorably against breast cancer cells. Vorinostat increased histone acetylation at the leukemia inhibitory factor receptor (LIFR) gene promoter. This subsequently recruited bromodomain-containing protein-4 (BRD4) to induce the expression of LIFR. This cytokine activates prosurvival JAK-STAT3 signaling in auto- and paracrine fashions. Inhibition of JAKs or BRD4 sensitized breast cancer to HDACi, indicating that JAKs and BRD4 promote an undesired cytoprotective effect in this setting. Thus, combinations of HDACi with inhibitors of JAKs or BRD4 appear as potential therapies for breast tumors (Zeng et al., 2016). Further studies will show whether JAK-STAT activation protects additional tumor types from HDACi.
Linear correlations between tonic activation of JAK-dependent STAT3 phosphorylation and HDACi resistance are, though, unlikely. For example, DLBCL cells with constitutively tyrosine-phosphorylated STAT3 were more sensitive to panobinostat than those cells lacking detectable STAT3 phosphorylation. In this system, chronic STAT3 phosphorylation was abrogated by HDACi-induced STAT3 hyperacetylation (Gupta et al., 2012), reminiscent of the inhibitory STAT1 phosphorylation-acetylation switch (Krämer et al., 2009). Furthermore, an unbiased screen with a panel of tumor cells demonstrated that PV cells with hyperactive JAK2V617F-dependent signaling cascades displayed the highest sensitivity to vorinostat (Liang et al., 2022). It is tempting to speculate that context-dependent high tonic levels of JAK-STAT signaling create a vulnerability for pharmacological approaches involving HDACi. Such considerations are relevant because clinically validated markers for the stratification of patients into responders and nonresponders to HDACi are not available.
B. Histone Deacetylase Inhibitors Can Correct Aberrant Janus Kinase–Signal Transducer and Activator of Transcription Signaling
In the following section, we sum up evidence on how HDACi decrease aberrant JAK signaling, the underlying molecular mechanisms for this, and potential therapeutic implications of the requirement of HDAC activity for JAK-dependent signaling.
1. Impact of Histone Deacetylase Inhibitors on Janus Kinase-1
In 2004, it was revealed that pretreatment of colon cancer cells with structurally divergent HDACi suppressed cytokine-induced JAK-STAT signaling. The carboxylic acid butyrate and the hydroxamic acids TSA and vorinostat abrogated the activating phosphorylation of JAK1 on Y1022/Y1023 upon treatment with the type II interferon (IFN)-γ (Klampfer et al., 2004). This impaired the IFN-induced phosphorylation of STAT1 at Y701/S727, subsequently caused a failure of STAT1 to translocate to the nucleus, and blunted IFNγ-dependent gene expression. Both overexpression of HDAC1, HDAC2, and HDAC3, as well as RNAi-mediated depletion of these enzymes, illustrated that they promote JAK-STAT activation and the expression of IFNγ target genes. These effects were not due to a caspase-dependent degradation of STAT1 (Klampfer et al., 2004; Owusu and Klampfer, 2017), which can occur in leukemic cells undergoing apoptosis in response to HDACi (Licht et al., 2014).
In contrast to this antagonistic relationship between HDACi and IFNγ, both drugs synergistically suppressed the survival of colon cancer cells and reduced the antiapoptotic BCL-XL protein (Klampfer et al., 2004). This finding may point to a therapeutic option based on HDACi and IFNs. Studies illustrating apoptosis induction by HDACi and IFNs in human melanoma cells support this idea (Krämer et al., 2006; Roos et al., 2011). Moreover, butyrate levels of up to ∼7 mM in the colon can be anti-inflammatory and chemopreventive due to a suppression of STAT1 signaling and prosurvival factors (Klampfer et al., 2004; Stempelj et al., 2007). Unlike normal cells, cancer cells often rely on glycolysis to gain energy, instead of the mitochondrial citrate cycle (Warburg effect). This makes cancer cells unable to fuel butyrate into this metabolic pathway. Consequently, butyrate accumulates and causes protein hyperacetylation and antiproliferative activities (Meyer et al., 2021).
2. Impact of Histone Deacetylase Inhibitors on Janus Kinase-2
The oncogenic role of JAK2V617F spurred the development of small-molecule inhibitors of hyperactive JAK2 (Zhao et al., 2016a; Musumeci et al., 2019). The success and survival benefit, along with its striking efficacy regarding spleen volume and symptom reduction, have made ruxolitinib a meaningful therapy in MPNs. Since 2011, ruxolitinib has become the main drug for the treatment of MPNs refractory to the ribonucleotide reductase inhibitor hydroxyurea (Bose and Verstovsek, 2017a). However, ruxolitinib is not fully curative in most cases. This particularly applies to MF patients, who suffer from short survival, and to patients with JAK2 mutant cells that are insensitive to JAKi (Barosi et al., 2014; Bose and Verstovsek, 2017b; Cervantes and Pereira, 2017; Chang et al., 2021). Accordingly, ongoing research strives to identify improved drugs for the treatment of MF patients.
a. Histone deacetylase inhibitors attenuate JAK2V617F in blood malignancies
An alternative, indirect route to target mutant JAK2 in patients suffering from MPNs relies on HDACi (Bose and Verstovsek, 2017b). Givinostat selectively reduced the protein levels of JAK2V617F but not wild-type JAK2 in cell lines and primary human MPN cells. Congruent herewith, JAK2 wild-type cells became the dominant population in heterogeneous patient samples containing wild-type and mutant JAK2 (Guerini et al., 2008). The HDACi-induced attenuation of JAK2V617F occurred by a yet unclear mechanism and despite unchanged mRNA expression (Guerini et al., 2008). Remarkably, givinostat produced promising results in a clinical trial with 29 patients suffering from JAK2V617F-positive blood disorders. Givinostat decreased the allelic burden of JAK2V617F, proving on-target activity against MPN cells (Rambaldi et al., 2010). The givinostat-induced loss of JAK2V617F tied in with anticancer activity in patients with MPNs that were unresponsive to the maximally tolerated dose of hydroxyurea (Finazzi et al., 2013). It similarly appears promising that in an ongoing 4-year study involving 54 MPN patients (51 PV cases; 42–80 years old; trial #NCT01761968), a maximally tolerated dose of 200 mg orally taken givinostat per day has produced over 80% overall response rates. This treatment has been well tolerated, with at most 10% of grade 3 events that have occurred in patients who have concomitantly received hydroxyurea (Rambaldi et al., 2021). These clinical data support the long-term use of givinostat in patients.
Like givinostat, vorinostat, panobinostat, and a further hydroxamic acid–based pan-HDACi, pracinostat (SB939) (Fig. 4A), produce antimyeloproliferative activities that are molecularly linked to reduced JAK2V617F. Vorinostat significantly hampered the aberrant phosphorylation of JAK2V617F and of its downstream targets STAT5, STAT3, AKT, and ERK1/ERK2 in murine and human PV cells (Akada et al., 2012). This finding agrees with remarkable vorinostat-evoked hematologic responses in a JAK2V617F knock-in mouse PV model and a significantly reduced mutant allele burden. Interestingly, vorinostat was more potent than pyridone-6 against erythroid progenitors in this model (Akada et al., 2012). In a phase II clinical trial involving patients with PV and ET, vorinostat demonstrated high efficacy against JAK2V617F-positive peripheral blood mononuclear cells (Andersen et al., 2013). Likewise, panobinostat was effective against MPNs alone or in combination with JAKi. Panobinostat showed clinical efficacy in patients with wild-type JAK2- or JAK2V617F-positive MF and decreased the JAK2V617F allele burden (DeAngelo et al., 2013a; Mascarenhas et al., 2013; Mascarenhas et al., 2017). Pracinostat decreased JAK2V617F, STAT5, and, more pronouncedly, their phosphorylation in cultured PV and ET cells. This was associated with apoptosis-related processing of poly-ADP-ribose polymerase-1 (PARP1). Both pracinostat and the dual JAK2/FMS-like tyrosine kinase-3 (FLT3) inhibitor pacritinib significantly reduced the growth of such tumor cells in mice. Combined application of pracinostat and pacritinib pronounced these effects against ET xenografts (Novotny-Diermayr et al., 2012).
An observation in clinical trials using HDACi is that lower drug doses over longer time are often well tolerated by patients. Moreover, patients with less advanced disease achieve better therapeutic responses. Another recurrent finding in the clinic is that subgroups of patients are particularly amenable to treatment with HDACi (DeAngelo et al., 2013a, 2013b; Mascarenhas et al., 2013; Mascarenhas et al., 2017). Unraveling the molecular features of their malignancies will show which patients can profit from HDACi.
Noteworthy, wild-type JAK2 status does not preclude leukemic cell killing by JAKi and HDACi. Concurrent treatment with low doses of ruxolitinib and vorinostat acted synergistically against six out of 12 cell lines from hematologic malignancies with no known mutations of JAK2. This drug combination triggered reactive oxygen species–mediated tumor cell death (Civallero et al., 2017).
B-ALL represents about one-third of pediatric leukemia, and among them, 7% have overexpressed CRLF2 due to its translocation to strong promoter elements. As mentioned above, B-ALL cells with increased CRLF2 expression and mutant JAK2 are insensitive to JAK2 inhibitors. Promisingly, givinostat was effective against permanent and fresh human B-ALL cells with mutations in CRLF2 and JAK2 (JAK2I682F and JAK2R683G) (Fig. 1) (Savino et al., 2017). Unlike in PV cells (Guerini et al., 2008), givinostat decreased JAK2V617F mRNA in CRLF2/JAK2 mutant human B-ALL cells. Moreover, givinostat increased the mRNA of PTPN1, which is the founding member of the protein tyrosine phosphatase family and an inhibitor of JAK-STAT signaling (Savino et al., 2017). Although the protein levels of mutant JAK2 and PTPN1 were not determined in this study, it illustrates that the B-ALL–associated cooccurrence of dysregulated CRLF2/JAK2 is a pharmacodynamic marker for the high efficiency of givinostat.
In contrast to the reported specific loss of mutant and not wild-type JAK2 in givinostat-treated leukemic cells (Guerini et al., 2008), the benzamide HDACi chidamide, which selectively inhibits HDAC1, HDAC2, HDAC3, and HDAC10 (Fig. 4A), decreased wild-type JAK2 and JAK2V617F mRNA and protein levels in PV and myelodysplastic syndrome cells. This loss of JAK2V617F was associated with decreased STAT3 phosphorylation, cell cycle arrest, apoptosis, and upregulation of SOCS3 mRNA and protein expression. The authors of this work concluded that SOCS3 accumulation, likely upon hyperacetylation of histones in its gene promoter, attenuated wild-type and mutant JAK2 (Zhao et al., 2016b). The possibility of eliminating JAK2V617F and wild-type JAK2-STAT3–dependent signaling with a class I HDACi might offer a clinically safer treatment due to fewer side effects than with pan-HDACi. It is yet unclear whether pharmacological inactivation of class I HDACs and/or the class IIb enzyme HDAC10 by chidamide decreased JAK2.
Studies with the clinically tested hydroxamic acid citarinostat, which has some preference for HDAC6 (Lechner et al., 2022), did not attenuate total and constitutively phosphorylated JAK2 in a panel of lymphoid tumor cells (Cosenza et al., 2020). This data suggests that JAK2 stability does not depend on the catalytic activity of HDAC6. Curiously, highly synergistic cytotoxic combinations of momelotinib and citarinostat decreased p-JAK2 but spared total JAK2 levels. We speculate that such HDACi/TKi combinations unleash yet undefined molecular mechanisms that selectively target a small pool of p-JAK2. The size of this pool varies between cells, and it is conceivable that only a few of these highly catalytically active kinase molecules can modulate biologic processes. Such a low stoichiometry of activation corresponds to strong biologic effects that small fractions of activated STAT1 can induce (Göder et al., 2021).
Although these data suggest exploiting HDACi for the treatment of preleukemic conditions and leukemia driven by JAK2V617F, additional research is necessary to fully establish HDACi as a valid pharmacological option to treat MPNs. For example, it is ill-defined through which mechanisms HDACi target JAK2V617F-transformed cells. Is it the preferential degradation of JAK2V617F, or are there further mechanisms involved? If it is the degradation of JAK2V617F, then how do HDACi discriminate between wild-type and mutant JAK2? Which factors regulate such processes? It is likewise unclear if the targeting of a subset of HDACs or even of a single member of the 18 HDAC family members in humans suffices to attack leukemic cells driven by JAK2V617F. Considering that JAK2V617F and HDACs control the replication stress and DNA damage responses of leukemia cells (Plo et al., 2008; Kurosu et al., 2013; Marty et al., 2013; Ueda et al., 2013; Chen et al., 2014; Roos and Krumm, 2016; Nikolova et al., 2017), the impact of HDACi on such mechanisms should additionally be investigated as antitumoral effects.
b. Histone deacetylase inhibitors modulate aberrant JAK2 signaling in solid tumor cells
Although JAK2V617F has never been detected in solid malignancies (Lee et al., 2006; Herreros-Villanueva et al., 2010), aberrant JAK2 signaling and constitutive STAT activation are malignant features of multiple solid tumors. Aberrant growth hormones, epidermal growth factors, and chemokines can trigger a hyperactivation of JAK2/STAT5 in prostate, breast, and colorectal cancers (Harry et al., 2012; Thomas et al., 2015; Buchert et al., 2016; Bharadwaj et al., 2020; Qureshy et al., 2020). In the following section, we summarize evidence that JAK2 is a druggable vulnerability of solid tumors.
In colon cancer cells, TSA induced cell cycle arrest and apoptosis. TSA upregulated SOCS1 and SOCS3 through the hyperacetylation of histones H3 and H4 in their gene promoters. This tied in with the reduction of both phosphorylated and total forms of JAK2 and STAT3 as well as their downstream targets BCL2, survivin, and the cell cycle regulator p16/INK4A (Xiong et al., 2012) (Fig. 2). Since JAK2 activation correlates with poor survival of patients with colorectal cancers or prostate cancers (Li et al., 2004; Mao et al., 2011), HDACi might be an option for particularly aggressive colon cancer types.
Additional cancer cells, such as the highly fatal pancreatic ductal adenocarcinoma (PDAC), are sensitive toward the JAK2/JAK3 inhibitor AZ-960 (Scholz et al., 2003; Corcoran et al., 2011). Since PDAC cells are also vulnerable to HDACi (Schneider et al., 2010; Laschanzky et al., 2019), inhibition of JAK2 signaling and HDACs might combine favorably against PDAC cells. This may allow a dose reduction of HDACi and JAKi and, consequently, reduced toxicity against normal tissues.
The rare and highly deadly pancreatic neuroendocrine tumors (PanNET) display epigenetic alterations, including HDAC overexpression. Clinically achievable nanomolar doses of panobinostat stalled the proliferation and caused apoptosis of primary human PanNET and corresponding rodent tumor cells. Global RNA analyses indicated that panobinostat dysregulated JAK-STAT signaling in PanNET cells. Panobinostat did not alter JAK2 and SOCS3 levels but augmented STAT3 phosphorylation. The authors of this study interpret this as a compensatory mechanism suppressing panobinostat-induced apoptosis (Schmitz et al., 2021). Perhaps such compensatory pathways create a susceptibility for cotreatment with HDACi and drugs blocking STAT3 (Bharadwaj et al., 2020). This hypothesis is supported by the above-described notion that breast cancer cells become vulnerable to HDACi when LIFR-induced JAK-STAT signaling is antagonized by small molecules against JAKs and BRD4 (Zeng et al., 2016).
Epigenetic dysregulations also occur in hepatocellular carcinoma (HCC) and might be a vulnerability of this most common and highly fatal liver cancer (Anestopoulos et al., 2015). Increased activation of JAK2-STAT3 signaling in HCC correlates with reduced SOCS3 expression due to transcriptionally silenced methylated CpG regions in the SOCS3 promoter. Treatment of HCC cells with the DNA hypomethylating agent 5-aza-2'-deoxycytidine/decitabine successfully restored SOCS3 expression, blocked STAT3 activity, and suppressed tumor cell growth. These findings suggest that epigenetic changes promote tumorigenesis through JAK2-STAT3 signaling (Niwa et al., 2005). Whether HDACi and hypomethylating drugs combine favorably in HCC through JAK-STAT inactivation is yet unresolved. A recent study compared the efficacy of equimolar concentrations of vorinostat and decitabine in HCC cell models. Both compounds induced apoptosis, and vorinostat was more potent than decitabine. Vorinostat decreased mRNAs encoding class I HDACs, JAK2, STAT3, and increased SOCS1 and SOCS3 expression (Sanaei et al., 2021). Additional work can show whether these changes are functionally relevant. Irrespective thereof, evidence collected in different cell systems shows that HDACi augment SOCS protein levels (Fig. 2).
3. Impact of Histone Deacetylase Inhibitors on Janus Kinase-3
Activating JAK3 mutations in T-ALL and other blood malignancies position dysregulated JAK3 signaling as a potential drug target (Zhang et al., 2012; De Keersmaecker et al., 2013; Vicente et al., 2015; Li et al., 2016; Girardi et al., 2017; Liu et al., 2017; Degryse et al., 2018a, 2018b) (Fig. 1). Unlike other JAKs, JAK3 can be inhibited specifically due to its cysteine 909 residue (Cys909). This gatekeeper position of JAK3 is not present in JAK1, JAK2, or TYK2. This inspired the development of irreversible, selective JAK3 inhibitors (Goedken et al., 2015; Telliez et al., 2016; Elwood et al., 2017; He et al., 2017; Kempson et al., 2017; He et al., 2018). Docking experiments with the JAK3 kinase domain led to the identification of ATP-competitive irreversible tricyclic covalent compounds that inhibit JAK3 in the low nanomolar range. These drugs contain chloroacetamide or acrylamide-derived Michael acceptors to irreversibly bind Cys909. Such molecules displayed high selectivity against the other JAKs (IC50 values above 1 µM) and potently abrogated the JAK3-dependent activation of STAT5 in lymphocytes (Goedken et al., 2015). The irreversible JAK3 inhibitor PF-06651600 blocked JAK3 activity while sparing JAK1-mediated signaling (Telliez et al., 2016). Another irreversible covalent JAK3 inhibitor potently inhibited JAK3 (0.15 nM) with 4300-fold selectivity toward JAK3 over JAK1 (Elwood et al., 2017). Additional covalent irreversible JAK3 inhibitors preferentially inhibited JAK3 (IC50 = 57 nM) over other JAKs (IC50 > 10 µM) (He et al., 2017). Likewise, the compound T29 demonstrated the highest potency and selectivity in picomolar doses for JAK3 (IC50 = 0.14 nM) over other JAKs (IC50 > 5 µM) and other kinases with a cysteine residue analogous to Cys909 in JAK3 (He et al., 2018). Further irreversible selective agents impair JAK3 (IC50 = 1 nM) with high selectivity over JAK1 (IC50 = 1100 nM), JAK2 (IC50 = 1300 nM), and TYK2 (IC50 > 5000 nM). These molecules contain an acrylamide scaffold as an electrophilic warhead (Kempson et al., 2017). The development and characterization of specific JAK3 inhibitors verified their usefulness in treating autoimmune disorders. The putative activity of such compounds against cancer cells is unexplored.
HDACi are a putative option to kill cancer cells expressing mutant JAK3. In CTCL, JAK3 repressed the tumor-suppressive microRNA miR-22 via tyrosine phosphorylation of STAT5, leading to its nuclear translocation and binding to the miR-22 gene promoter. This attenuates the validated miR-22 target genes MAX, MYCBP, HDAC6, CDK6, and PTEN in malignant T cells. STAT3 and STAT5 repress such genes through the recruitment of transcriptional corepressors such as EZH2 and HDACs. Vorinostat time- and concentration-dependently upregulated miR-22 in CTCL cells (Sibbesen et al., 2015). HDACi additionally abrogate the JAK3-STAT5–dependent expression of miR-21 being associated with CTCL (Lindahl et al., 2016). This impact of HDACi on miRNAs may represent an Achilles’ heel of CTCL.
Selective JAK3 inhibitors may favorably combine with HDACi against subtypes of lymphoma and potentially other malignancies that particularly rely on mutant JAK3. A key consideration with such combinatorial approaches is at which therapeutic levels the desired specificity for JAK3 disappears due to the impact of HDACi on JAK1, JAK2, and TYK2. Nonetheless, if given for short periods, JAK3 and HDACi may hit the cancer cells hard and simultaneously.
4. Impact of Histone Deacetylase Inhibitors on Tyrosine Kinase-2
Panobinostat abrogated the tyrosine phosphorylation of JAK2, TYK2, STAT3, and expression of MCL1. This consequently induced apoptosis in cell lines and tumor samples derived from DLBCL patients (Gupta et al., 2010; Gupta et al., 2012). This paved the way for phase I/II clinical trials (#NCT01261247 and #NCT01523834) that assessed the efficacy as well as the safety profile of panobinostat in patients with relapsed/refractory DLBCL. Patients tolerated panobinostat and showed a 21% overall response. Whether patients unresponsive to treatment had persistent JAK signaling is an intriguing but unexplored possibility. Truly, other targets of HDACi might also dictate resistance. For example, a recent study using single-cell sequencing illustrates that c-FOS antagonizes HDACi-induced apoptosis and is upregulated in DLBCL cells surviving HDACi (Krämer and Schneider, 2022; Wang et al., 2022).
Another strategy to combat TYK2 may rely on the HDAC-dependent control of the ubiquitin-proteasome system and the finding that cytokines not only activate TYK2 but can lead to its proteasomal degradation. Upon activation by IFNα, a STAT1-dependent induction of the E2 ubiquitin conjugase UBCH8 accelerates the turnover of TYK2 in complex with the E3 ubiquitin ligase seven-in-absentia-homologue-2 (SIAH2) in NSCLC cells. Accordingly, SIAH2 levels inversely correlate with STAT3 phosphorylation and metastatic gene expression in NSCLC patient samples (Müller et al., 2014). HDACi induce E2 ubiquitin conjugase UBCH8 and its cooperativity with SIAH1/SIAH2 to accelerate the proteasomal degradation of multiple oncogenes (Buchwald et al., 2010; Buchwald et al., 2013; Krämer et al., 2013). Eliminating TYK2 and its overactivity in certain tumors with HDACi might be a promising anticancer strategy.
5. Histone Deacetylase Inhibitors Modulate Janus Kinase–Signal Transducer and Activator of Transcription–Dependent Immune Functions
Recent reports demonstrate that the effects of HDACi on JAK-STAT–dependent immunologic tumor surveillance can stall tumor cell proliferation. The overexpression of B7 homologue-1, which is rather known as programmed cell death ligand-1 (PD-L1), is a key mechanism of immune evasion by tumor cells and hence a modern anticancer target (Deng et al., 2018). Conspicuously, HDACs sustain an IFNγ-mediated expression of PD-L1 in gastric cancer. Pretreatment with butyrate, vorinostat, or TSA, as well as a genetic elimination of HDAC1 and HDAC3 by RNAi, suppressed the IFNγ-induced acetylation of histone H3 at lysine residue 9 at the promoter of the B7 homologue-1/PD-L1 gene. This equally occurred upon depletion of JAK2, p-JAK1, p-JAK2, and p-STAT1 and a subsequently impaired nuclear translocation of STAT1. HDACi suppressed IFNγ signaling in gastric cancer cells, which hindered their ability to escape detection and elimination by cytotoxic CD8+ T cells (Deng et al., 2018). These findings suggest that the reported negative impact of HDACi on STAT signaling (Krämer et al., 2009; Krämer and Heinzel, 2010; Ginter et al., 2012; Wieczorek et al., 2012; Kaowinn et al., 2017; Kotla et al., 2017; Liu et al., 2020; Yang et al., 2021) and herein summarized negative effects of HDACi on JAKs (Fig. 2) are key immunoregulatory and tumor-suppressive mechanisms of such drugs.
These insights could not only improve the treatment of gastric cancers. In murine models with lung and renal carcinoma, entinostat improved the tumor-suppressive effects of an antibody neutralizing the receptor programmed death receptor-1 (PD-1) (Orillion et al., 2017). The class I/IV HDACi mocetinostat (Fig. 4A) similarly promoted tumor antigen presentation, decreased immune suppressive cell types, and augmented antitumor effects of anti–PD-L1 antibodies in colon carcinoma-bearing mice(Briere et al., 2018).
JAK2V617F, and consequently p-STAT3/p-STAT5–induced transcriptional induction of PD-L1, are higher in primary MPN patient-derived monocytes, megakaryocytes, and platelets than in cells from healthy individuals. Inhibition of JAK2 with ruxolitinib or the JAK2 inhibitor SD-1029 and anti–PD-1 or anti–PD-L1 antibodies prolonged the survival of mice xenografted with human peripheral blood mononuclear cells from MPN patients and of JAK2V617F knock-in mice (Prestipino et al., 2018). Targeting PD-L1 via the depletion of JAK2V617F by HDACi may disable the immune evasion of MPNs.
The above-summarized reports inform that HDACi combine favorably with inhibitors of immune checkpoint molecules. The ability of HDACi to target immune suppressive mechanisms is not restricted to the PD-1/PD-L1 axis. For example, entinostat disrupts immunosuppressive functions of regulatory T cells in the tumor microenvironment through the acetylation of STAT3 and an ensuing downregulation of Foxp3 expression in renal and prostate murine cancer models (Shen et al., 2012).
6. Advantages of Indirect Pan–Janus Kinase Inhibition by Histone Deacetylase Inhibitors
Based on current evidence, HDACi function as indirect pan-JAKi that cause a slow decline of JAK-STAT signaling through transcriptional effects and an impact on JAK protein stability. Although details on such mechanisms are still a subject of investigation, the ability of HDACs to interfere with multiple members of the JAK family seems to be therapeutically beneficial.
Due to the transcriptional and protein stability-centered mechanisms of HDACi (Fig. 2), they can attenuate JAK mutants irrespective of a particular lesion (Fig. 1). This should also hold for JAK2 resistance mutations in the ATP-binding site that are acquired during the treatment of B-ALL cells with type 1 and type 2 JAKi (Downes et al., 2021) and in case of a cooperation between concomitant activating mutations in JAK1 and JAK3. This acquired resistance to JAKi favors the selection of TKi-resistant tumor clones and consequently increased risk of relapse in patients suffering from T-ALL, T-PLL, and extranodal natural killer (NK)/T-cell lymphoma (Bains et al., 2012; Koo et al., 2012; Atak et al., 2013; Bellanger et al., 2014; Springuel et al., 2014). In MPN cell models and granulocytes from MPN patients, overexpression of JAK2V617F at the transcriptional and protein levels as well as heterodimer formation between mutant JAK2V617F with JAK1 and TYK2 increases their phosphorylation and TKi resistance (Koppikar et al., 2012). Elimination of multiple JAKs by HDACi could abrogate this adaptation to JAKi. One might envision that HDACi can also be effective against tumor-associated JAK fusion proteins resulting from genomic rearrangements.
In a similar fashion, inhibition of heat shock protein-90 (HSP90) attenuates JAK1 and JAK2V617F in MPN cells (Wang et al., 2009; Fiskus et al., 2011; Koppikar et al., 2012; Weigert et al., 2012). Hyperacetylation of HSP90 in response to HDACi was proposed to promote proteasomal degradation of mutant JAK2 (Wang et al., 2009; Fiskus et al., 2011; DeAngelo et al., 2013a). Recent evidence, though, illustrates that the number of acetylation sites in HSP90 and their stoichiometry and biologic relevance are more complex than anticipated (Krämer et al., 2014; Prus et al., 2019). An advantage of HDACi over HSP90 inhibitors is the FDA approval for HDACi (Food and Drug Administration, 2010; Whittaker et al., 2010; Cappellacci et al., 2020).
Moreover, HDACi-triggered mechanisms that eliminate JAK mutants are not susceptible to an observed persistence of phosphorylated JAK2 in the presence of type 1 TKi (Meyer et al., 2015; Wu et al., 2015). Hyperactive JAK2 even appears to sensitize cells to HDACi (Guerini et al., 2008; Savino et al., 2017). This could be particularly relevant considering the allosteric activation mechanism of JAK2 that shifts the molecule from an autoinhibited toward a molecularly open, uninhibited state. Increased complementarity and hydrophobicity of JAK2V617F and other JAK2 mutants allow their ligand-independent dimerization and activation. HDACi-induced elimination of such JAK2 mutants decreases the likelihood of their dimerization and thus abrogates allostery. This idea is supported by the notion that the specific allosteric TYK2 inhibitors BMS-066/BMS-986165 bind the JH1 pseudokinase domain of TYK2 and can freeze it in its inactive, autoinhibited conformation in normal blood cells (Tokarski et al., 2015; Burke et al., 2019; Glassman et al., 2022).
Another advantage of HDACi and a synergistic killing of MPN cells with JAKi/HDACi combinations may rely on a suppression of the coincidence of B-cell non-Hodgkin lymphomas during treatment of MPN patients with JAK1/JAK2 inhibitors (Porpaczy et al., 2018). Immunosuppressive effects of such drugs on cytotoxic T lymphocytes and NK cells facilitate the conversion of malignant preexisting B-cell clones into aggressive B-cell lymphoma (Low and Song, 2016; Khandelwal et al., 2017; Kim et al., 2017; Assouan et al., 2018). As discussed above, there are beneficial effects of HDACi on the PD1/PD-L1 system.
In the context of the here-discussed interplay between HDACs and JAK signaling, one should still consider that HDACi can attenuate oncogenic signaling at multiple levels. For example, pan-HDACi and class I HDACi reduce WEE1 and ACK1 expression in cells derived from blood malignancies and solid tumors (Cornago et al., 2014; Zhou et al., 2015; Mahendrarajah et al., 2016; Göder et al., 2018; Hu et al., 2020; Wachholz et al., 2022). In addition, HDACi might abrogate JAK-dependent crossdrug resistance mechanisms. For instance, JAK1V658F (Fig. 1) confers resistance of the FLT3-ITD–positive AML cells to the TKi midostaurin and sorafenib (Rummelt et al., 2021). Indirect inhibition of JAK1 signaling by HDACi (Klampfer et al., 2004; Owusu and Klampfer, 2017) may disable such resistance mechanisms. Furthermore, concomitant degradation of FLT3-ITD by HDACi (Buchwald et al., 2010; Pietschmann et al., 2012; Wachholz et al., 2022) could be a double-hit approach against such aggressive cancer cells.
IV. Chemically Coupled Multitargeting Drugs
Despite their wide use and clinical relevance, combination therapies face challenges and limitations. These include, but are not limited to, undefined drug-drug interactions, the need to establish effective dose regimens for two agents, higher costs of development, extra efforts for clinical trials, and complications of the pharmaceutical formulations (Peters, 2013). To prevail over these drawbacks, multipharmacophore therapeutics combining two or more different pharmacophores are developed as an alternative strategy (Zimmermann et al., 2007; Anighoro et al., 2014; Nepali et al., 2014; Fu et al., 2017). Such a chemical coupling allows hitting two cancer-relevant targets with one molecule at the same time. This might overcome drug-drug interactions, might exert a more potent killing effect, can prevent the advent of drug-resistant cells due to mutations in a single drug target, and offers more predictable pharmacokinetic profiles. In addition, a hybrid molecule can facilitate drug dosing to ensure patient compliance (Morphy et al., 2004; Morphy and Rankovic, 2005). A key condition for the success of such strategies is that both pharmacophores retain specific and highly potent activities against their targets. Hence, critical warheads must be present in the fusion molecules (Fig. 4C).
Kinases were one of the earliest molecules to be cotargeted with HDACs. Examples of such compounds resulted from chemical coupling of vorinostat and the macrocycle templates of zotiraciclib/pacritinib (SB1317/SB1518). The macrocycle templates of these TKi are replaced by the cap group of vorinostat (Ning et al., 2015). This preserves the hydroxamic acid group for complex formation with the catalytically active Zn2+ in HDACs (Fig. 4C). These hybrid structures displayed high potency against JAK2, FLT3, and HDACs. The multifunctional hybrid molecule 32 is a nanomolar inhibitor of JAK2, FLT3, and global HDAC activity (IC50 values 686, 87, and 87 nM, respectively), and exhibited remarkable anticancer activities against leukemic cells as well as solid tumors (Ning et al., 2015). An additional JAK-HDAC dual inhibitor termed 20a showed high activity against JAK2 (IC50 = 8 nM) and HDAC6 (IC50 = 46 nM). This agent caused apoptotic cell death of AML cells (Huang et al., 2018a).
Pyrimidine-2-amino-pyrazole/hydroxamate derivatives are dual-acting inhibitors that inactivate JAK2 and HDAC6 at nanomolar doses. A compound named 8m produced higher activities in vitro and in vivo than vorinostat and ruxolitinib against a panel of leukemic cell lines (Liang et al., 2019).
Solid tumor-derived cells are also susceptible to HDACi-TKi fusion molecules. Most recently, a series of pyrrolo-[2,3-day]-pyrimidine–based derivatives emerged as dual-acting JAKi/HDACi. Compounds 15d and 15h block JAK1,2,3 and HDAC1/HDAC6 in nanomolar doses, attenuated tyrosine-phosphorylated STAT3, and suppressed the growth of triple-negative human breast cancer in vitro and in mice (Liang et al., 2022). Despite their ability to induce LIFR expression, these multitargeting drugs combated LIFR-induced STAT3 phosphorylation by tumor-associated fibroblasts. Remarkably, 15d/15h were superior to vorinostat. Despite an equal accumulation of acetylated histones and tubulin, targets of HDAC1 or HDAC6 (Table 1), vorinostat activated STAT3 phosphorylation upon induction of LIFR in breast tumor cells (Liang et al., 2022). These data illustrate that combined inhibition of JAKs and HDACs favorably abrogate cytokine-induced phosphorylation events.
The hybrid molecule 24, a chemical coupling of ruxolitinib and vorinostat, inhibits HDAC1,2,3,6,10, and JAK1/JAK2 with >10-fold selectivity over TYK2, JAK3, and 93 other kinases (Yao et al., 2017). Compound 24 inhibited STAT3 tyrosine phosphorylation, induced an accumulation of acetylated histone H3 at 0.15–7.36 µM, apoptosis-related processing of the DNA repair enzyme PARP1, and growth inhibition in a panel of leukemic cells (ALL, AML, MPN, multiple myeloma) and solid tumor-derived cells (breast, colon, prostate) (Yao et al., 2017). Although compound 24 is a more potent inhibitor of HDAC6 than of HDAC1 (1.4 versus 6.9 nM), experiments with the HDAC6 inhibitor tubastatin A show that inhibition of HDAC6 cannot explain the processing of PARP1 (Yao et al., 2017). These data correspond to a strong biochemical activity of HDAC6 inhibitors without any antiproliferative or cytotoxic effects in several leukemic and solid tumor cells in vitro and in vivo (Noack et al., 2017; Schäfer et al., 2017; Leonhardt et al., 2018; Sellmer et al., 2018; Bandolik et al., 2019; Beyer et al., 2019; Depetter et al., 2019; Pons et al., 2019; Nawar et al., 2022). Presumably, compounds 15d/15h killed cells through inhibition of HDAC1 (Liang et al., 2022). Work on compound 24 was extended by the same group who developed it. Fusing the pharmacophores of vorinostat and ruxolitinib via a single nitrogen atom yielded subnanomolar inhibitors of JAK2 and HDAC6. These compounds had remarkable selectivity against HDAC1 and improved potency against the individual pathways. Compound 13b presented the highest potency and striking subnanomolar activity against JAK2 with an IC50 of 41 pM and an IC50 against HDAC6 of 200 pM (Yao et al., 2018). Thus, optimized JAK-HDAC hybrid inhibitors could improve cancer therapy.
Similarly, merging the pharmacophores of ruxolitinib and vorinostat created molecules with high potency against solid and hematologic malignancies. These agents attenuated JAK2 mRNA expression and increased proteasomal degradation of JAK2. This associated with reactive oxygen species accumulation and tumor growth inhibition by apoptosis (Civallero et al., 2017). These data hold for other JAKi-HDACi fusions. Agents resulting from a chemical coupling of the JAK2-selective inhibitors XL019 or pacritinib with vorinostat produced antiproliferative activity and triggered apoptosis in a panel of cancer cells from ALL, MPN, multiple myeloma, breast, colon, and prostate (Yang et al., 2016; Chu-Farseeva et al., 2018). These findings shed light on new leads for bifunctional molecules that block JAKs and HDACs. Furthermore, these results suggest that inhibiting JAK2 phosphorylation with TKi does not exclude its proteasomal degradation upon HDAC inhibition. From these insights, one can deduce that the preferential loss of mutant JAK2 over wild-type JAK2 is unlikely related to JAK2 phosphorylation (Guerini et al., 2008). It will be interesting to see if such fusion molecules overcome the limited clinical potential of XL019 in MPN (Verstovsek et al., 2014) and whether they are effective against other neoplastic diseases.
An additional dual-acting molecule, Roxyl-zhc-84, is a fusion of vorinostat and the CDK4/CDK6 inhibitor abemaciclib. This dual pharmacophore resolved the problem of limited responses of cells from aggressive breast cancer, high-grade serous ovarian cancer, and NSCLC toward traditional HDACi. The mechanism underlying this benefit is an overcoming of JAK1-STAT3-BCL2–mediated drug resistance. Roxyl-zhc-84 was superior to the combinatorial treatment with HDACi and JAKi. Moreover, Roxyl-zhc-84 had favorable pharmacokinetic profiles in a rat model and produced low toxicity in a mouse model (Huang et al., 2018b). The same group showed that conjugates of HDACi and CDK inhibitors overcame limited responses of oral squamous cell carcinoma (OSCC) toward HDACi monotherapies (Zhao et al., 2019). Roxyl-ZR, a highly Roxyl-zhc-84–related conjugate of vorinostat and abemaciclib, significantly suppressed the survival of OSCC cells through inhibition of the JAK1-STAT3-BCL2 axis and ensuing apoptosis. In mice, Roxyl-ZR mitigated OSCC growth at low toxicity (Zhao et al., 2019).
These results imply the therapeutic potential of hybrid molecules and provide valuable lead compounds for further assessment and structural drug optimization.
V. Targeting Aberrant Janus Kinase Signaling by Proteolysis Targeting Chimeras and Histone Deacetylase Inhibitors
Proteolysis targeting chimeras (PROTACs) are heterobifunctional chimeras that contain ligands for target proteins, ligands that dock to E3 ubiquitin ligases, and linkers to connect these ligands (Fig. 5A). The formation of a ternary complex leads to K48-linked poly-ubiquitination of the target proteins, which are then recognized and rapidly proteolyzed by proteasomes (Pettersson and Crews, 2019; Vogelmann et al., 2020).

Exploiting VHL-based PROTACs and HDACi to correct aberrant JAK-STAT signaling. The game-changing PROTAC technology, as well as the impact of HDACi on VHL expression, suggests a new, synergistically active therapeutic approach through proteolytic mechanisms and HDAC inhibition. (A) PROTACs can accelerate the proteasomal degradation of JAKs. Shown is a VHL-based PROTAC. (B) The expression of the VHL gene is suppressed by HDACs. HDACi augment the VHL mRNA and protein levels, which can increase the proteasomal degradation of JAKs by PROTACs. This principle may also apply to targets of VHL-PROTACs beyond JAKs. Created with BioRender.com.
Despite over 600 E3 ligases in the human genome, until now few candidate E3 ligases, including inhibitors of apoptosis (IAPs), von Hippel-Lindau (VHL), cereblon (CRBN), murine double minute-2 homolog, damage-specific DNA binding protein-1 and Cullin-4–associated factor-15/16, and ring finger protein-114 are employed for targeted protein degradation (Liu et al., 2019). This is due to the scarcity of small-molecule ligands that recruit E3s without impairing their functions. The identification of more compounds that recruit E3s is anticipated to improve and refine the PROTAC technology (Belcher et al., 2021).
A key advantage of PROTACs over low-molecular-mass inhibitors is the rapid and sustained elimination of their targets. This is frequently accompanied by high efficacy and, in some cases, increased selectivity for their targets (Nowak et al., 2018; Sun et al., 2018; Khan et al., 2019; Shah et al., 2020; Zhang et al., 2020; Alabi et al., 2021; Chang et al., 2021). Besides, PROTACs targeting receptor tyrosine kinases disable both enzymatic activities and scaffolding functions of these proteins. This is important because scaffolding kinase functions reactivate oncogenic signaling pathways and propel kinome rewiring supporting escape clones with drug resistance (Shah et al., 2018; Murtuza et al., 2019). Moreover, JAK-PROTACs can disengage the crossreactivation of specifically inhibited JAKs by the other JAK family members. This is a major resistance mechanism against JAKi (Koppikar et al., 2012).
Most recently, the ubiquitin-proteasome machinery was successfully hijacked with PROTACs that degrade JAKs through IAPs, VHL, and CRBN in a variety of leukemic cells (Shah et al., 2020; Chang et al., 2021; Alcock et al., 2022). Guided by the JAKi chemotypes pyrimidine and quinoxaline, IAP, VHL, and CRBN-recruiting PROTACs were designed and tested for their impact on wild-type JAK1/JAK2 levels in human AML cells. These PROTACs accelerated the ubiquitinylation and proteasomal degradation of JAK1 and JAK2 in micromolar doses (0.6–10 µM). In this setting, IAP-based PROTACs induced the degradation of JAK1, JAK2, and JAK3 more effectively than PROTACs recruiting VHL and CRBN (Shah et al., 2020).
The clinically used type 1 JAK1/JAK2 inhibitors ruxolitinib and baricitinib were used to rationally design a new series of CRBN-directed PROTACs (Chang et al., 2021). This was achieved by analysis of the crystal structures of the human JAK2 JH1 domain complexed with baricitinib or ruxolitinib at 1.7 to 1.8 Å resolution. It turned out that the pyrimidine rings of the drugs offer a C2-carbon that points away from the catalytic cleft of JAK2 toward the solvent. Introducing a 4-amino-benzamide allowed the attachment of a linker and the imides thalidomide/lenalidomide/pomalidomide as recruiter elements for CRBN. Some of the obtained PROTACs abrogated aberrant JAK-STAT signaling and killed ruxolitinib/baricitinib-resistant ALL cells with a combined overexpression of CRLF2 (due to the IGH-CRLF2 translocation) and JAK2I682F/JAK2R683G. PROTACs 5, 6, 7, and 8 effectively inhibited aberrant JAK-STAT signaling (IC50 < 100 nM) in a panel of ALL cell lines and patient-derived primary ALL xenografts in mice. An up to 70,000-fold higher activity of the PROTACs compared with their parent compounds was not due to increased inhibition of JAK2 phosphorylation but related to its degradation. Even though these efforts provide a new window to target JAK-mutated malignancies, specificity and pharmacokinetics of the existing JAK-directed PROTACs require optimization. The JAK-PROTACs lost the specificity of ruxolitinib and baricitinib for JAK1/JAK2. In addition, these PROTACs inhibited MAP3K2, MAP3K3, and YSK4. Irrespective thereof, compound 8, a fusion of baricitinib and thalidomide, eliminated mutant JAK2 specifically and dose-dependently in cultured cells. Cells with a genetic elimination of CRBN by CRISPR–Cas9 retained JAK2 in the presence of compound 8, verifying CRBN-induced proteasomal degradation. Proteasomal degradation of the tumor-promoting CRBN neo-substrate G1-to-S-phase-transition-1 (GSPT1) by CRBN ligands can cause false-positive effects of PROTACs. This can blur target-specific effects of PROTACs. Interestingly, compound 8 did not degrade GSPT1 in cultured B-ALL cells with a translocation of CRLF2 and JAK2I682F. Nonetheless, compound-8 attenuated wild-type JAK2 and GSPT1 in primary B-ALL cells with a translocation of CRLF2 (Chang et al., 2021).
Based on ruxolitinib and baricitinib, the same group expanded their work and successfully developed JAK2/JAK3-selective PROTACs using the phenyl glutarimide (PG) ligand as CRBN-recruiting warhead. PG has a phenyl group instead of the phthalimide present in the immunomodulatory imide-based PROTACs. The PG-based PROTAC 11 significantly reduced off-target degradation of GSPT1 and showed higher selectivity for JAK2/JAK3 over the other JAKs in CRLF2-rearranged ALL cell lines. PROTAC 11 was more effective than the parental JAK2 inhibitors and displayed the most potency in xenografted, patient-derived ALL cells harboring an ATF7IP-JAK2 fusion (IC50 = 24 nM). Higher PROTAC doses were needed to kill ALL cells carrying both JAK2 fusions and CRLF2 rearrangements (IC50 < 120 nM) (Alcock et al., 2022).
Phenotypically similar to the PROTAC-mediated degradation of mutant JAKs, HDACi suppress JAK signaling by genomic and nongenomic mechanisms (Guerini et al., 2008; Rambaldi et al., 2010; Akada et al., 2012; Andersen et al., 2013; Zhao et al., 2016b; Bose and Verstovsek, 2017b; Savino et al., 2017) (Fig. 2). Transcriptional effects of HDACi may augment the efficacy of VHL-based PROTACs. Romidepsin and panobinostat induce the expression of VHL in endothelial cells and B-cell lymphomas (Kwon et al., 2002; Kalac et al., 2011). This might potentiate the effects of VHL-based PROTACs through an upregulation of VHL (Fig. 5B). PROTACs targeting HDACs might also promote the effectiveness of VHL-based PROTACs by increasing the amount of available VHL. PROTACs that decrease HDAC1/HDAC2 (Smalley et al., 2022), HDAC3 (Cao et al., 2020; Xiao et al., 2020), HDAC6 (Wu et al., 2019), HDAC8 (Chotitumnavee et al., 2022), and SIRT2 (Schiedel et al., 2018) in cultured cells as well as in animal models were identified. This implies the potential of PROTACs as a comprehensive approach for cancer treatment.
Combinations of JAK-directed PROTACs with HDACi might likewise be useful from the viewpoint that both drugs decrease mutant JAK isoforms. Particularly, the increased sensitivity of JAK2V617F compared with wild-type JAK2 toward HDACi (Guerini et al., 2008) could preferentially target leukemic cells over healthy cells, allowing sustained immune functions and proper hematopoiesis. In addition, the choice of attracted E3 ubiquitin ligase can prevent damage to normal tissues. For example, VHL- and CRBN-based PROTACs spare thrombocytes and platelets due to their low expression of VHL and CRBN. This could prevent fatal thrombocytopenia being a common cause of chemotherapy-associated death (Khan et al., 2019; Zhang et al., 2020). On the other hand, patients suffering from diseases such as ET may not profit well from VHL- and CRBN-based PROTACs and should obtain PROTAC-based drugs recruiting other E3 ubiquitin ligases.
VI. Clinical Trials to Assess the Safety and Efficacy of Combined Pharmacological Targeting of Janus Kinases and Histone Deacetylases
The FDA has approved 10 JAKi, including ruxolitinib and the JAK1/JAK3 inhibitor tofacitinib, for the treatment of MPNs and rheumatoid arthritis (Qureshy et al., 2020). As mentioned above, the FDA has also authorized HDACi. Table 2 lists a selection of clinical trials using combinations of JAKi and HDACi.
Examples of clinical studies that have tested JAKi and HDACi for cancer therapy
Panobinostat was clinically active against JAK2V617F-positive MPNs as monotherapy or in combination with ruxolitinib (DeAngelo et al., 2013a, 2013b; Mascarenhas et al., 2013; Ribrag et al., 2013; Mascarenhas et al., 2020). Preclinical studies with panobinostat plus ruxolitinib revealed that such combinations effectively corrected JAK2V617F-associated symptoms, including splenomegaly, as well as bone marrow and spleen histology in a JAK2V617F-driven MPN mouse model. Concurrent administration of panobinostat and ruxolitinib disrupted the JAK2V617F-induced STAT5 phosphorylation and significantly inhibited the growth of MPN cells (Evrot et al., 2013). Similarly, combinations of panobinostat and the JAK2 inhibitor TG101209 significantly disrupted JAK2V617F-driven signaling and synergistically induced apoptosis of primary CD34+ MPN cells. These were more affected by such treatment than normal CD34+ hemopoietic progenitor cells. This signifies a specific antileukemic effect (Wang et al., 2009). It is tempting to speculate that particularly the mutant JAK2V617F expression created this vulnerability.
These insights inspired multiple clinical trials using panobinostat plus ruxolitinib. When applied at lower doses over time, such drug regimen significantly corrected splenomegaly and ameliorated bone marrow fibrosis (Mascarenhas et al., 2020). In a phase Ib clinical trial (#NCT01433445), the combinatorial therapy of ruxolitinib plus panobinostat demonstrated an appropriate safety profile, with low dose-limiting toxicities and modest rates of grade 3/4 anemia (34.2%) and thrombocytopenia (21.2%) (Ribrag et al., 2013).
The phase I/II clinical study (#NCT01693601) confirmed the efficacy and safety of ruxolitinib administered twice daily at 10–15 mg and panobinostat at 10–20 mg three times biweekly. Anemia occurred as the most common therapy-associated feature in 47% of patients (Ribrag et al., 2013; Harrison et al., 2015; Mascarenhas et al., 2020). The reduction in spleen size in this trial was similarly seen in patients who received ruxolitinib in the phase III trial Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment I and Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment II (#NCT00934544) (Harrison et al., 2016).
In MF patients, the combination of ruxolitinib and panobinostat was well tolerated and reduced splenomegaly in 57% and 39% of the patients at weeks 24 and 48, respectively. Of note, some patients experienced a reduction in JAK2V617F allele burden and obvious correction in bone marrow fibrosis. Adverse events were not higher than with ruxolitinib or panobinostat single-agent treatments (Harrison et al., 2015).
As a single, orally given agent, pracinostat exhibited modest potency in MF patients (Zorzi et al., 2013; Chu et al., 2015). In a study with 25 MPN patients, 20 received ruxolitinib and pracinostat (#NCT02267278) (Table 2). Ruxolitinib was administered for three cycles, whereas pracinostat was given afterward in the fourth cycle. Eighty percent of patients had objective responses, and 74% experienced spleen responses. The National Cancer Institute Common Terminology Criteria for Adverse Events reported that anemia and thrombocytopenia were the most frequent adverse events (Bose et al., 2019). The small patient number requires additional investigations on the clinical validity of ruxolitinib and pracinostat.
An ongoing phase I/II study #NCT03598959 is devoted to exploring the efficacy and evaluating the safety profile of tofacitinib (Nwaogu et al., 2020), combined with chidamide in patients with relapsed and refractory NK/T-cell lymphoma.
The molecular mechanisms behind the beneficial interaction of JAKi and HDACi are not entirely clear yet. In at least some cell systems, ruxolitinib and other type 1 JAKi stabilize the phosphorylated form of JAK2V617F (Meyer et al., 2015; Wu et al., 2015). This TKi-mediated accumulation of mutant JAK2 can cause rebound effects when the drug concentrations decline, and this can cause adaptive processes termed hormesis. Since JAK2V617F is more prone than the largely unphosphorylated wild-type JAK2 to HDACi-induced degradation (Guerini et al., 2008; Savino et al., 2017), the elimination of the inhibited mutant JAK2 by HDACi can prevent and disengage such undesired processes. Of course, other mechanism can also contribute to the beneficial outcome of JAKi and HDACi combinations. A topical issue is whether clinical-grade JAKi-HDACi fusions and JAK-directed PROTACs, when applied alone or with HDACi, are as potent or even superior to combined application schemes of JAKi and HDACi.
Despite such success, not all patients profit from HDACi and JAKi. It needs consideration that leukemic (stem) cells can reside in both the periphery and bone marrow. Coculture systems revealed that bone marrow stromal cells protected MPN-derived JAK2V617F-positive cell lines as well as primary bone marrow–derived post-PV AML cells from lethal effects of ruxolitinib and vorinostat. The cytoprotective effects of bone marrow stromal cells were mediated by the secretion of soluble factors that maintain cellular homeostasis via activation of JAK-STAT, phosphatidylinositol 3-kinase, c-JUN N-terminal kinase, MEK-ERK, and nuclear factor-κB (NF-κB) signaling pathways (Cardoso et al., 2015). Such findings shed light on mechanisms of tumor survival and pave the way to novel, targeted therapeutic approaches.
VII. Summary
Members of the HDAC family are valuable targets for novel pharmacological intervention strategies against tumor cells. Modern, clinically relevant, broad-acting and isoenzyme-specific inhibitors of Zn2+-dependent HDACs modulate the activity and expression of the four JAK proteins and consequently their tumor-relevant downstream signaling. ATP-competitive JAK inhibitors, structurally diverse HDACi, JAKi-HDACi fusion pharmacophores, and JAK-directed PROTACs combine favorably against malignant cells in culture, experimental animals, and cancer patients. Future research will fully define how HDACs and acetylation-dependent molecular mechanisms control oncogenic JAK signaling and how clinicians can use this knowledge.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Mustafa, Krämer.
Footnotes
- Received March 11, 2022.
- Revision received July 8, 2022.
- Accepted August 15, 2022.
We gratefully acknowledge that work done by A.-H.M.M. in the group of O.H.K. is funded by the German Research Foundation/Deutsche Forschungsgemeinschaft (DFG) grant KR2291/12-1, DFG project number 445785155 (to O.H.K.) and was initially made possible by the DAAD. Additional support to O.H.K. is from the DFG project number 393547839 - SFB 1361, subproject 11; KR2291/9-1, DFG project number 427404172; KR2291/14-1, DFG project number 469954457; KR2291/15-1, DFG project number 495271833; KR2291/16-1, DFG project number 496927074; KR2291/17-1, DFG project number 502534123; the Wilhelm Sander-Stiftung (grant 2019.086.1); the Brigitte und Dr. Konstanze Wegener-Stiftung (Projekt 65); and the Walter Schulz Stiftung. We thank all our group members for helpful discussions and input on this work.
No author has an actual or perceived conflict of interest with the contents of this article. O.H.K. declares the patents “Synthesis, pharmacology, and use of new and selective FMS-like tyrosine kinase 3 (FLT3) inhibitors, WO2019/034538”, “Novel HDAC6 inhibitors and their uses, WO2016020369A1”, and "The use of molecular markers for the preclinical and clinical profiling of inhibitors of enzymes having histone deacetylase activity, WO/2004/027418." These patents cover substance classes that are discussed in this work.
Abbreviations
- ACK1
- activated CDC42-associated kinase-1
- AKT
- serine-threonine protein kinase B
- ALL
- acute lymphoblastic leukemia
- AML
- acute myeloid leukemia
- B-ALL
- B-cell acute lymphoblastic leukemia
- BRD4
- bromodomain-containing protein-4
- CDK
- cyclin-dependent kinase
- CMML
- chronic myelomonocytic leukemia
- CRBN
- cereblon
- CRLF2
- cytokine receptor-like factor-2/thymic stromal lymphopoietin receptor
- CTCL
- cutaneous T-cell lymphoma
- Cys909
- cysteine 909
- DLBCL
- diffuse large B-cell lymphoma
- ERK
- extracellular signal-regulated kinases
- ET
- essential thrombocythemia
- FDA
- Food and Drug Administration
- FERM
- four-point-one, ezrin, radixin, moesin
- FLT3
- FMS-like tyrosine kinase-3
- GOF
- gain of function
- GSPT1
- G1-to-S-phase-transition-1
- HCC
- hepatocellular carcinoma
- HDAC
- histone deacetylase
- HDACi
- histone deacetylase inhibitor
- HSP90
- heat shock protein-90
- IAP
- inhibitor of apoptosis
- IFN
- interferon
- JAK
- Janus kinase
- JAKi
- JAK inhibitor
- JH
- JAK homology
- LIFR
- leukemia inhibitory factor receptor
- MF
- myelofibrosis
- MPN
- myeloproliferative neoplasm
- MYC
- myelocytomatosis oncogene
- NF-κB
- nuclear factor-κB
- NK
- natural killer
- NSCLC
- non–small-cell lung cancer
- OSCC
- oral squamous cell carcinoma
- PanNET
- pancreatic neuroendocrine tumors
- PARP1
- poly-ADP-ribose polymerase-1
- PD-1
- programmed death receptor-1
- PDAC
- pancreatic ducal adenocarcinoma
- PD-L1
- programmed cell death ligand-1
- PG
- phenyl glutarimide
- PMF
- primary myelofibrosis
- PROTAC
- proteolysis targeting chimera
- PV
- polycythemia vera
- RNAi
- RNA interference
- SH2
- Src homology-2
- SIAH2
- E3 ubiquitin ligase seven-in-absentia-homologue-2
- SIRT
- sirtuin
- SOCS
- suppressor of cytokine signaling
- STAT
- signal transducer and activator of transcription
- T-ALL
- T-cell acute lymphoblastic leukemia
- TK
- tyrosine kinase
- TKi
- tyrosine kinase inhibitor
- T-PLL
- T-cell prolymphocytic leukemia
- TSA
- trichostatin A
- TYK2
- tyrosine kinase-2
- VHL
- von Hippel-Lindau
- Copyright © 2023 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





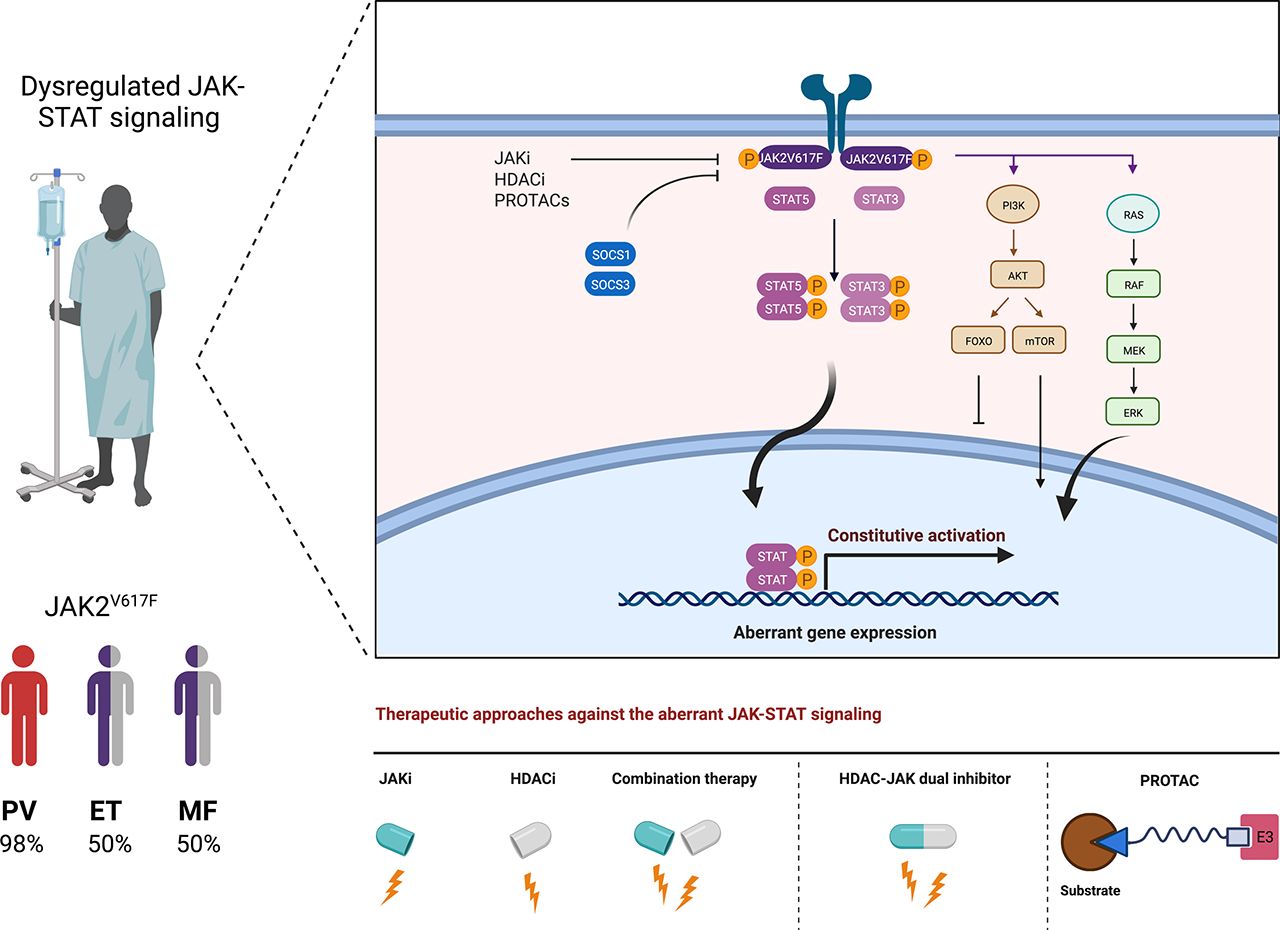{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- I. Introduction
- II. Relevance of Janus Kinases for Tumorigenesis
- III. Histone Deacetylases Are Valid Pharmacological Targets in Janus Kinase Mutant Tumor Cells
- IV. Chemically Coupled Multitargeting Drugs
- V. Targeting Aberrant Janus Kinase Signaling by Proteolysis Targeting Chimeras and Histone Deacetylase Inhibitors
- VI. Clinical Trials to Assess the Safety and Efficacy of Combined Pharmacological Targeting of Janus Kinases and Histone Deacetylases
- VII. Summary
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters