


Abstract
Over the last decade, hydrogen sulfide (H2S) has emerged as an important endogenous gasotransmitter in mammalian cells and tissues. Similar to the previously characterized gasotransmitters nitric oxide and carbon monoxide, H2S is produced by various enzymatic reactions and regulates a host of physiologic and pathophysiological processes in various cells and tissues. H2S levels are decreased in a number of conditions (e.g., diabetes mellitus, ischemia, and aging) and are increased in other states (e.g., inflammation, critical illness, and cancer). Over the last decades, multiple approaches have been identified for the therapeutic exploitation of H2S, either based on H2S donation or inhibition of H2S biosynthesis. H2S donation can be achieved through the inhalation of H2S gas and/or the parenteral or enteral administration of so-called fast-releasing H2S donors (salts of H2S such as NaHS and Na2S) or slow-releasing H2S donors (GYY4137 being the prototypical compound used in hundreds of studies in vitro and in vivo). Recent work also identifies various donors with regulated H2S release profiles, including oxidant-triggered donors, pH-dependent donors, esterase-activated donors, and organelle-targeted (e.g., mitochondrial) compounds. There are also approaches where existing, clinically approved drugs of various classes (e.g., nonsteroidal anti-inflammatories) are coupled with H2S-donating groups (the most advanced compound in clinical trials is ATB-346, an H2S-donating derivative of the non-steroidal anti-inflammatory compound naproxen). For pharmacological inhibition of H2S synthesis, there are now several small molecule compounds targeting each of the three H2S-producing enzymes cystathionine-β-synthase (CBS), cystathionine-γ-lyase, and 3-mercaptopyruvate sulfurtransferase. Although many of these compounds have their limitations (potency, selectivity), these molecules, especially in combination with genetic approaches, can be instrumental for the delineation of the biologic processes involving endogenous H2S production. Moreover, some of these compounds (e.g., cell-permeable prodrugs of the CBS inhibitor aminooxyacetate, or benserazide, a potentially repurposable CBS inhibitor) may serve as starting points for future clinical translation. The present article overviews the currently known H2S donors and H2S biosynthesis inhibitors, delineates their mode of action, and offers examples for their biologic effects and potential therapeutic utility.
I. Introduction
Over the last three decades, an unprecedented explosion occurred in the understanding of the biologic roles of the gaseous molecules nitric oxide (NO), carbon monoxide (CO), and—over the last decade—in the area of hydrogen sulfide (H2S), the “third gasotransmitter.” Enzyme systems producing these mediators have been discovered and characterized, and a multitude of scientific articles have been published on the metabolism, biologic roles, and the mechanisms of action of these three molecules. NO, CO, and H2S share many common properties: these rapidly diffusible gaseous molecules obey a different set of rules than most of the other classes of biologic mediators and pharmacological agents (reviewed in Wang, 2002; Szabo, 2010, 2016; Olson et al., 2012; Farrugia and Szurszewski, 2014). Each of the three gasotransmitter molecules can act as a vasodilator, cytoprotectant, and anti-inflammatory agent at lower concentrations, but they can also trigger cytotoxic and deleterious effects at higher concentrations.
Over the last decade, H2S has been the subject of intensive research and development efforts to understand its biologic roles in health and disease and to exploit its biologic pathways for therapeutic benefit. These efforts have resulted in a great number of innovative therapeutic approaches: they have produced pharmacological compounds and potential drug candidates that currently serve either as experimental tools (to characterize the biologic roles of H2S) and/or have advanced into clinical trials. After a brief overview of the biologic chemistry, physiology, and pathophysiology of H2S, the current article will present the state-of-the art on the various pharmacological approaches to donate H2S or to inhibit its biosynthesis.
II. The History of H2S as an Environmental Toxin
From a chemical standpoint, H2S is a colorless, flammable, water-soluble gas with the characteristic smell of rotten eggs. For a long time, H2S was viewed exclusively as a toxic gas and environmental hazard (often referred to as “swamp gas” or “sewer gas”). It is generated by various industrial sources (paper mills, tanneries, mining, petroleum refineries), and its toxicological profile has been extensively studied, both in experimental animals and humans, and in the context of environmental toxicology. A substantial body of toxicological literature (Beauchamp et al., 1984; Reiffenstein et al., 1992; Marshall et al., 2009; Haouzi, 2012) shows that increasing doses of H2S gas elicit various adverse effects, starting from eye irritation (at low doses), and, as the inhalation dose increases, extending into pulmonary injury and culminating, at high doses, in the characteristic “knockdown effect” (loss of consciousness, cardiopulmonary arrest, asphyxiation). Fatal effects occur in the range of approximately 1000 ppm (0.1%). Environmental toxicology recommendations typically specify the safely inhalable dose of H2S at 10–20 ppm. Inhaled H2S enters the blood stream through the lung (where it crosses from the alveolar space through the lung epithelial cells and then through the vascular endothelial cells and into the blood stream). The blood, in turn, carries it into all vascularized organs.
III. H2S, as an Endogenous Biologic Mediator: Physiologic Roles
The timeline of H2S research, and the transition from the status of H2S as a toxicological substance to an endogenous biological mediator, has recently been overviewed (Szabo, 2017a). Although it was originally described by DuVigneud in 1942 that liver homogenates, when incubated with sulfur-containing amino acids, produce H2S through an action of the transsulfuration pathway (Binkley and du Vigneaud, 1942), the biologic synthesis of H2S and its biologic roles had not received much attention until the last decade. Fifty years later, Kimura’s studies (Abe and Kimura, 1996), followed by a multitude of additional experiments, demonstrated that H2S is synthesized by mammalian tissues and serves as a biologic signaling molecule. According to our current knowledge, in most cells and tissues two pyridoxal-5′-phosphate-dependent enzymes responsible for metabolism of l-cysteine, cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE), and a third system, the combined action of 3-mercaptopyruvate sulfurtransferase (3-MST) and cysteine aminotransferase (CAT, also known as l-cysteine:2-oxoglutarate aminotransferase, aspartate aminotransferase, or aspartate/cysteine aminotransferase) are responsible for H2S biosynthesis. Additional details of H2S biosynthesis are covered in various review articles (Fiorucci et al., 2006; Lowicka and Bełtowski, 2007; Szabo, 2007; Li et al., 2011; Whiteman and Winyard, 2011; Predmore et al., 2012b; Kimura, 2014, 2015; Polhemus and Lefer, 2014; Huang and Moore, 2015; Papapetropoulos et al., 2015; Moore and Whiteman, 2015; Rose et al., 2017) (Fig. 1). The substrates of CBS and CSE (l-cysteine and l-homocysteine) are either of alimentary origin or can be liberated from endogenous proteins. In tissue homogenates, rates of H2S production are estimated to be in the range of 1–10 (pmol/s)/mg protein (Doeller et al., 2005); the relative contribution of CBS, CSE, and 3-MST to the total cell or tissue H2S output depends on the cell/organ studied as well as the experimental conditions.

Pathways of H2S generation in mammalian cells. Cystathionine-β-synthase (CBS; EC 4.2.1.22), cystathionine-γ-lyase (CSE; 4.4.1.1), and 3-mercaptopyruvate sulfurtransferase (3-MST; EC.2.8.1.2) are the three principal enzymes that contribute to the endogenous production of H2S. CBS and CSE are components of the reverse transsulfuration pathway, a biochemical pathway responsible for the conversion of methionine to cysteine, and catalyze a multitude of reactions that yield H2S, including the conversion of l-homocysteine to l-homolanthionine (by CSE), the conversion of l-homocysteine and l-cysteine to l-cystathionine (by CBS and CSE), the conversion of l-cystathionine to l-cysteine (by CSE), the conversion of l-cysteine to pyruvate and ammonia (by CSE), and the conversion of l-cysteine to l-serine and l-lanthinonine (by CBS). An additional pathway involves the CSE-dependent conversion of cystine to l-thiocystenine, which, in turn, produces H2S via thiol-dependent reactions. The third H2S-producing enzyme, 3-MST, is part of the cysteine catabolism pathway and uses 3-mercaptopyruvate (3-MP) as a substrate. 3-MST works in tandem with aspartate aminotransferase that also possesses cysteine aminotransferase activity (CAT) activity, generating 3-MP from cysteine via a series of reductions that first involve the generation of bound sulfane sulfur. 3-MP, in addition to acting as a substrate of 3-MST, can also produce H2S spontaneously. In some cells and tissues, d-cysteine can also be a significant substrate for H2S production; it is converted to 3-MP by d-amino acid oxidase (DAO). Pyridoxal 5′-phosphate (PLP) is a cofactor for CSE, CBS, and CAT.
Although the quantification of biologic H2S levels remains an intensively debated issue, it is generally estimated that mammalian cells and tissues are physiologically exposed to low micromolar H2S concentrations. Biologic H2S levels are dynamically regulated: they can be rapidly “consumed” and degraded by various mammalian tissues. The distribution and regulation of H2S producing enzymes is complex and is discussed in multiple review articles (Fiorucci et al., 2006; Szabo, 2007; Lowicka and Bełtowski, 2007; Qu et al., 2008, Li et al., 2011; Whiteman and Winyard, 2011; Predmore et al., 2012b; Polhemus and Lefer, 2014; Huang and Moore, 2015; Kimura, 2015; Papapetropoulos et al., 2015; Rose et al., 2016). Additional details of the enzymatic mechanisms responsible for H2S production by CBS, CSE, or 3-MST are covered in sections XXVI-XXVII.
The physiological roles of endogenous H2S are multiple and rapidly expanding. H2S plays an important physiological role as an endogenous modulator of vascular tone and blood pressure (Zhao et al., 2001, 2003; Ali et al., 2006; Xiao et al., 2006; Dawe et al., 2008; Yang et al., 2008), neurotransmission (Sen and Snyder, 2010; Kimura, 2013; Zhang and Bian, 2014; Kamat et al., 2015), angiogenesis (Wang et al., 2010a; Szabo and Papapetropoulos, 2011; Bibli et al., 2015a; Bibli et al., 2015b; Katsouda et al., 2016; Yuan and Kevil, 2016; Szabo, 2017b), nociception (Distrutti et al., 2006; Cunha et al., 2008; Smith, 2009; Linden, 2014), cardiac function (Predmore et al., 2012b; Polhemus and Lefer, 2014), various leukocytic functions (Zanardo et al., 2006; Dal-Secco et al., 2008; Wallace, 2010), penile erectile function (Srilatha et al., 2006; di Villa Bianca et al., 2015), and many others. On the basis of studies in Caenorhabditis elegans, H2S homeostasis affects thermotolerance and life span (Miller and Roth, 2007; Qabazard and Stürzenbaum, 2015).
IV. “H2S-Rich” and “H2S-Poor” Pathophysiological Conditions
H2S has been implicated in the pathogenesis of multiple diseases, as overviewed in review articles. These range from cardiovascular diseases (e.g., myocardial reperfusion injury, cardiac hypertrophy, heart failure, atherosclerosis, hypertension) (Predmore et al., 2012b; Polhemus and Lefer, 2014; Ahmad et al., 2015; Meng et al., 2015a, 2016; Shen et al., 2015; Wang et al., 2015a; Cao and Bian, 2016; van Goor et al, 2016; Kanagy et al., 2017; Greaney et al., 2017) to various neurologic diseases (e.g., stroke, neuroinflammation) (Wang et al., 2014a; Bhatia, 2015; Kida and Ichinose, 2015; Wallace et al., 2015; Sen, 2017) and metabolic diseases (e.g., diabetes mellitus) (Desai et al., 2011; Szabo, 2012; Okamoto et al., 2015; Carter and Morton, 2016) to various forms of local and systemic inflammation (e.g., hemorrhagic shock, septic shock, burn injury) (Wagner et al., 2009; Coletta and Szabo, 2013; McCook et al., 2014; Akter, 2016).
One can make initial attempts to classify the roles of H2S in various pathophysiological conditions. On one hand, there are disease states where local or systemic H2S deficiency exists - either due to inhibition of H2S biosynthesis and/or due to increased H2S consumption (e.g., reperfusion injury, asthma, diabetic vascular complications, acute and chronic cardiac diseases, aging). In these conditions, therapeutic H2S donation (replacement) may be warranted (e.g., Sun et al., 2007; Brancaleone et al., 2008; Wu et al., 2008; Whiteman et al., 2010a; Suzuki et al., 2011). On another hand, there are diseases where H2S biosynthesis is increased (due to upregulation of H2S-producing enzymes). Such diseases include various forms of critical illness and multiple forms of cancer (e.g., Mok et al., 2004; Collin et al., 2005; Jiang et al., 2005; Li et al., 2005; Zhang et al., 2006, 2007a,b; Bhatia et al., 2008a,b; Coletta and Szabo, 2013; McCook et al., 2014; Akter, 2016; Szabo, 2016). In these conditions inhibition of H2S biosynthesis may be therapeutically advantageous.1 However, due to the complex (often bell-shaped) pharmacological profile of H2S (Papapetropoulos et al., 2015; Szabo, 2016), the situation is much more complex. For example, in some conditions, H2S donors can be therapeutically beneficial, although the endogenous H2S levels are not diminished (e.g., antiviral effects of H2S). In other conditions, both H2S donors and H2S biosynthesis inhibitors can show efficacy (e.g., in cancer) (Szabo, 2016).
V. The Modes of H2S’s Biologic Actions
Similar to the other two gasotransmitters, NO and CO, H2S rapidly travels through cell membranes without using specific transporters (Cuevasanta et al., 2012; Riahi and Rowley, 2014). It is estimated that the sphere of action of endogenous H2S—as produced by a single cell—expands to involve more than 200 neighboring cells (Cuevasanta et al., 2012). H2S does not have one single “pathway” or “receptor”: it affects multiple cellular effectors in a cell-dependent, tissue-dependent, and species-dependent manner.
The physiological (generally, beneficial and cytoprotective) molecular mechanisms of H2S include antioxidant effects, either through direct chemical reactions with various oxidant species (Kimura and Kimura, 2004; Whiteman et al., 2004; Kimura et al., 2006; Esechie et al., 2008; Muzaffar et al., 2008) or through elevation of cellular glutathione levels by activation/expression of γ-glutamylcysteine synthase (Wei et al., 2008; Ansari and Kurian, 2016) or through the stimulation of various of intracellular antioxidant “master switches,” e.g., Nrf2 (Calvert et al., 2009; Hourihan et al., 2013; Peake et al., 2013; Xie et al., 2016a,b; Liu et al., 2016c). H2S also affects a variety of intracellular signal transduction processes, including the activation of the PI3K/Akt system (Cai et al., 2007; Hu et al., 2008; Sodha et al., 2008; Osipov et al., 2009, 2010; Papapetropoulos et al., 2009; Coletta et al., 2012; Kondo et al., 2013), the modulation of intracellular calcium homeostasis (Nagai et al., 2004), the modulation of various proinflammatory signal transduction mechanisms (e.g., nuclear factor-κB) (Anuar et al., 2006; Oh et al., 2006; Zhang et al., 2007a,b; Whiteman et al., 2010b; Li et al., 2011; Olas, 2015), and effects on many other systems including sirtuins (Hu et al., 2015; Xie et al., 2016b). The physiological effects of H2S include the opening of the ATP-sensitive potassium channels (KATP channels), an effect that occurs through the modification of critical regulatory cysteines in the channel via a process termed sulfhydration (also called persulfidation) (Zhao et al., 2001; Cheng et al., 2004; Tang et al., 2005; Mustafa et al., 2011; Iciek et al., 2016). In fact, a growing number of enzymes are subject to H2S-mediated sulfhydration, which can affect (either increase or decrease) their specific catalytic activity (reviewed in Iciek et al., 2015; Nagy, 2015).
Several lines of studies have demonstrated that H2S activates the transient receptor (potential cation channel), for example, in sensory neurons, urinary bladder, dorsal root ganglion, blood vessels, and other tissues, with important functional consequences (Kimura et al., 2013; Eberhardt et al., 2014; Terada and Kawabata, 2015; Hajna et al., 2016). Some of the effects of H2S occur at the level of cAMP and cGMP phosphodiesterases: H2S directly inhibits the catalytic activity of these enzymes, which, in turn, stimulates intracellular cAMP and cGMP levels, followed by the expected biologic responses (Bucci et al., 2010; Coletta et al., 2012; Modis et al., 2013c; Andreadou et al., 2015a,b; Bibli et al., 2015a,b). In the PI3K/Akt/eNOS system and the NO/cGMP system, the two gasotransmitters NO and H2S exhibit a remarkable degree of cooperative action and synergy (reviewed in Szabo, 2017b).
Recent work shows that H2S exerts a variety of effects in the mitochondria. At low concentrations, H2S can directly donate electrons into the mitochondrial electron transport chain through its action on the mitochondrial enzyme sulfide quinone oxidoreductase (reviewed in Szabo et al., 2014; Modis et al., 2014a). It can also support mitochondrial functions by inhibiting mitochondrial cAMP phosphodiesterases (Modis et al., 2013c), by exerting mitochondrial antioxidant effects (Pun et al., 2010; Suzuki et al., 2011; Xie et al., 2016), and by promoting mitochondrial DNA repair through direct interactions with mitochondrial DNA repair enzymes (e.g., sulfhydration of EndoG-like mitochondrial endo/exonuclease) (Szczesny et al., 2016). H2S can also directly stimulate the activity of mitochondrial ATP synthase (Complex V) through sulfhydration (Modis et al., 2016). On the other hand, at higher concentrations, H2S inhibits cellular respiration;2 this effect is primarily attributed to the inhibition of cytochrome c oxidase (i.e., mitochondrial Complex IV) by reacting with its copper center (Nicholls et al., 2013; Szabo et al., 2014). Cytochrome c oxidase is an essential component of the oxidative phosphorylation machinery within the cell that normally binds oxygen; if the function of this enzyme is inhibited, mitochondrial electron transport and ATP generation becomes impaired (Nicholls and Kim, 1982; Khan et al., 1990). The mechanism of the inhibitory effect of Complex IV by H2S was recently revisited by several investigators. It appears that the inhibitory action of lower and higher concentrations of H2S involves different molecular mechanisms, and the underlying reaction pattern is complex. Interestingly, the inhibitory effect is markedly enhanced at acidotic pH. For further mechanistic insight and discussions, see Collman et al., 2009; Nicholls et al., 2013; Szabo et al., 2014.
Although this inhibitory effect has been primarily linked to the toxic “side” of H2S (environmental toxicology, industrial exposures to H2S gas, etc.), there are some attempts to also explore this inhibitory action for potential therapeutic benefit. These approaches take advantage of the fact that the inhibition of Complex IV by H2S is reversible as opposed to the irreversible effect of cyanide on the same target. One such effort focuses on induction of reversible metabolic suppression (“hibernation”), most reproducibly achieved in mice and small rodents, to cope with the reduced oxygen availability to the tissues, for example, during lethal hypoxia or after severe blood loss (Blackstone et al., 2005; Blackstone and Roth, 2007; Aslami et al., 2009). Another application of the same concept may be the “on-demand,” reversible metabolic suppression of stored organs in an attempt to extend their storage life (Balaban et al., 2015; Lobb et al., 2015).
VI. H2S Delivery via Inhalation of H2S Gas
Since the natural form of H2S at room temperature and physiological pressure is the gas form, one may simply assume that the most convenient way of administering H2S to biologic systems is by inhalation.3 Similar to NO, H2S gas, upon inhalation, dissolves in the blood stream and “delivers” H2S to the tissues.
It is important from the standpoint of H2S donation to mention that in 2010 a bioequivalency study was conducted in rats that compared circulating H2S concentrations in response to H2S inhalation with the effect of infusion of the H2S donor NaHS, with the read-out being blood levels of biologically active H2S (quantified by reaction with monobromobimane). According to this study, 1 (mg/kg)/hour of intravenous sodium sulfide for 2 hours is approximately equivalent to 30 ppm of gaseous H2S inhalation for 2 hours (Wintner et al., 2010). Although the toxicological profile of H2S donors is determined by many factors (most importantly, its rate of H2S release), the above bioequivalency serves as a useful starting point when comparing toxicological and therapeutic doses of H2S.
Several H2S gas inhalation studies have been conducted in experimental animals. From the animal studies aimed at experimental therapeutic approaches using H2S, the study of Roth and colleagues (Blackstone et al., 2005) at the Fred Hutchinson Cancer Center received much attention. In mice, H2S inhalation was shown to induce a “hibernation-like state.” When placed in an atmosphere of 20–80 ppm H2S gas, mice exhibited dose-dependent reductions in core body temperature and metabolic rate (Blackstone et al., 2005). Over the course of several hours of H2S exposure, the animals’ metabolic rate continued to decrease as measured by their CO2 output (down to 10% of baseline). When the chamber of the animals was cooled, body temperature reached as low as 15°C. These effects were found reversible after resuscitation at room air and warming of the chambers. The original hibernation studies were subsequently repeated and suggested that some of the H2S-induced cardiovascular responses (e.g., decreased heart rate) may be consistent with the physiology of hibernation (Volpato et al., 2008; Seitz et al., 2012). The actions of H2S show some similarities with the effects of volatile anesthetics. For example, 250 ppm H2S and 0.9% isoflurane or halothane produce comparable (approximately 75%) decreases in CO2 production in mice; it has been, therefore, suggested that the decreased physical activity of the animals (and the consequently decreased skeletal muscle-related energy consumption) is a significant contributor to the hibernation-like effects of H2S inhalation in conscious mice (Li et al., 2012).4
Subsequent studies explored the potential benefit of H2S gas inhalation in various models of severe hypoxia and ischemia and found that H2S inhalation pretreatment extends the life of rodents subjected to severe hypoxia or severe hemorrhagic blood loss (Blackstone and Roth, 2007; Morrison et al., 2008). Follow-up studies in various rodent models of injury have demonstrated the beneficial effects of H2S inhalation. For instance, inhalation of H2S at 80 ppm for 6 hours protected against lung injury (including functional parameters, biochemical indices, histologic damage) in a ventilator-induced lung injury model, in an LPS-induced lung injury model, and in a cotton smoke inhalation model (Faller et al., 2010, 2012; Han et al., 2015b). Posttreatment with H2S (80 ppm, 6 hours) after challenge with a high dose of endotoxin (bacterial lipopolysaccharide, LPS) challenge exerted protective effects in a mouse model of endotoxic shock (Tokuda et al., 2012). In the above experiments, the mode of action of H2S did not require and did not involve hypothermia (Baumgart et al., 2010; Faller et al., 2010, 2012; Tokuda et al., 2012). Part of the protective effect of H2S inhalation against ventilator-induced lung injury may involve the activation of the Akt signaling pathway (Spassov et al., 2017).
In contrast to the beneficial effects of H2S inhalation in the above models, Zapol and colleagues (Francis et al., 2011) found no beneficial effect of H2S inhalation at 1 or 5 ppm in a lung injury model induced by high tidal ventilation, whereas a higher dose of H2S (60 ppm) exacerbated the injury. In contrast, intravenous administration of Na2S (0.55 mg/kg) exerted beneficial effects (reduction of pulmonary edema, suppression of inflammatory mediator expression) in the same study. Because the intravenous H2S dosing was efficacious, it is conceivable that the therapeutically effective dose of H2S inhalation was not reached in the above experiments; given the narrow and bell-shaped dose response, perhaps 1 and 5 ppm was too low, whereas 60 ppm was too high to produce therapeutic benefit. The dose-response relationships with inhaled H2S remain to be carefully explored in the various experimental models, taking into account the complex pharmacological properties of this gas.
Inhalation with either 40 or 80 ppm H2S protected rats in a ventricular fibrillation-induced cardiac arrest models (Wei et al., 2015; Geng et al., 2015). The potential benefit of H2S inhalation was even explored in models and diseases that are traditionally considered “chronic,” and not readily treatable by inhalation therapies, such as an MPTP model of neurodegeneration and movement disorder. Inhalation of 40 ppm H2S for 8 hours every day for 7 subsequent days prevented the MPTP-induced movement disorder and reduced the degree of tyrosine hydroxylase-containing neuron loss and attenuated neuronal apoptosis and gliosis in the nigrostriatal region after administration of MPTP (Kida et al., 2011; Faller et al., 2012). The neuroprotective effect of inhaled H2S in several models was associated (and possibly may be due to) the upregulation of genes encoding various antioxidant proteins, including heme oxygenase-1 and glutamate-cysteine ligase (Kida et al., 2011). In addition to concomitant H2S therapy or H2S pretreatment, various approaches of H2S “preconditioning” were also found to be effective in various models. In a study by Roviezzo et al. (2015), instead of breathing H2S gas, NaHS was aerosolized into the lungs (at a dose that corresponded to approximately 100 ppm H2S) or vehicle for up to 5 minutes daily for 2 weeks. This therapeutic regimen abrogated ovalbumin-induced bronchial hyperreactivity and the increase in lung resistance and prevented mast cell activity and fibroblast growth factor-2 and IL-13 upregulation (Roviezzo et al., 2015). In another study, breathing of H2S gas at 40 ppm for 8 hours every day for 7 days elicited a protective effect against a subsequent transient middle cerebral artery occlusion/reperfusion, for infarct size, functional outcome parameters (e.g., neurologic score), and biochemical parameters (oxidative stress, apoptotic markers) (Ji et al., 2016).
As discussed elsewhere (Lou et al., 2008; Haouzi, 2012; Asfar et al., 2014), the hibernation-inducing metabolic effects of H2S are easy to elicit in small animals (e.g., rodents) but not in larger animal species. Indeed, in anesthetized sheep, pigs, and piglets, H2S inhalation or infusion fails to slow down metabolic parameters (Li et al., 2008a; Haouzi et al., 2008; Satterly et al., 2015) or only has a slight effect (Simon et al., 2008). Nevertheless, beneficial effects of H2S have been reported in large animals subjected to various models of critical illness, suggesting that protective mechanisms other than metabolic suppression/hibernation are responsible for the therapeutic effects in large animal species.
The feasibility of another related approach of H2S gas delivery has been tested by Zapol and colleagues (Derwall et al., 2011). These investigators have delivered H2S gas into the circulation of sheep via extracorporeal membrane lung ventilation and tested its efficacy in a model of partial cardiopulmonary bypass. The extracorporeal membrane lung was alternately ventilated with air (control) or air containing 100, 200, or 300 ppm H2S for 1-hour intervals. H2S exerted significant hemodynamic effects (pulmonary vasoconstriction, and systemic vasodilatation, leading to a decrease in mean arterial pressure). In addition, exposure to 300 ppm H2S impaired arterial oxygenation. Overall, no systemic metabolic effects nor any improvement in the outcome of the cardiopulmonary bypass was noted. Overall, although based on a single study only, it appears that administration of H2S gas through extracorporeal membrane lung ventilation is not a promising approach for the experimental therapy of critical illness.
Induction of whole-body metabolic suppression may be difficult to achieve with systemic administration of H2S (via inhalation or even via infusion, see below), especially in larger animals. In contrast, reversible suppression of the metabolic activity of stored organs before transplantation has been successfully achieved in multiple studies. Most of these studies used H2S-donor containing solutions (reviewed in Modis et al., 2014a), but in some studies, H2S gas inhalation was tested in the donor animals before lung transplantation (i.e., during the “warm ischemia” phase). This approach (80 ppm H2S gas inhalation for 2 hours) produced an improvement of the mitochondrial structures, reduction in lactic acid levels, suppression of inflammation, oxidative stress, and apoptosis after transplantation (Meng et al., 2017).
For obvious safety reasons, the studies testing the effect of H2S inhalation in humans are limited to relatively short-term physiological experiments using very low doses of H2S. Starting from the 1980s, the effect of low-dose (5–10 ppm) H2S inhalation has also been investigated in a variety of physiological studies in human volunteers (Bhambhani and Singh, 1991; Bhambhani et al., 1996a,b, 1997; Fiedler et al., 2008). These studies, due to the low doses of H2S used, have demonstrated only mild or no significant effects on physical performance and various cardiac and respiratory parameters.
Although less rigorously documented in the scientific literature, human H2S delivery is commonly used in the context of balneotherapy, where H2S inhalation occurs as humans are soaking in H2S-containing thermal waters (where H2S delivery into the body probably occurs via inhalation and absorption through the skin), or, in some cases, are sitting in closed rooms with fountains of H2S-containing thermal water placed in the middle of the room, where the H2S concentration in the air of the room is regulated by a sensor/ventilation feedback system (e.g., Tabiano Spa in Italy). There are small-scale preclinical studies demonstrating the beneficial effects of H2S delivery via “Tabiano water” (e.g., Giuliani et al., 2013). In addition, exploratory clinical studies suggest anti-inflammatory effects of ultrasonic nebulization of sulfurous water in asthmatic patients (Strinati et al., 1999). The potential therapeutic effect of these approaches has not been studied in appropriately powered, randomized clinical trials.
One of the potential problems with all forms of H2S delivery, but especially with H2S inhalation, relates to the issue of potential overdosing and consequent intoxication. Although the inhibitory effect of H2S on Complex IV is reversible, and therefore supporting therapy can result in patient recovery in some cases (Guidotti, 2015; Mooyaart et al., 2016), there are currently no well-characterized pharmacological antidotes to H2S intoxication: the application of sodium nitrite and hyperbaric oxygen has been used in humans (Ravizza et al., 1982; Whitcraft et al., 1985; Hall and Rumack, 1997). In animal studies, hydroxycobalamin (vitamin B12a) (Smith et al., 1976; Truong et al., 2007) and its analog cobinamide (Jiang et al., 2016) have also been shown to be efficacious as H2S antidotes.
Although the current section focuses on H2S inhalation, we should also briefly mention that H2S can also be exhaled by the same processes working in reverse direction (blood stream to vascular endothelial cells in the lung to lung epithelial cells to alveolar space). This may be part of the physiological elimination process, but, more importantly, increased H2S levels in exhaled air have been demonstrated when animals or human volunteers were subjected to therapeutic doses of H2S donors (Insko et al., 2009; Toombs et al., 2010). Increased exhaled H2S has been demonstrated in asthmatic patients (Zhang et al., 2014, 2015) and in septic patients (Bee et al., 2017). Exhaled H2S measurements may be one potential future way to monitor exposure to H2S donating agents, with one of its benefits being the ability to obtain an immediate read-out (as opposed to methods using H2S derivatization of blood or plasma and subsequent biochemical detection).
Although inhalation of H2S gas has been successfully employed in many animal studies, this method of delivery is not ideal for a number of reasons. It requires specialized equipment and personnel to deal with storage and transportation (H2S gas tanks), mixing, and delivery (e.g., corrosiveness issues, specialized tubing, and masks). H2S concentrations and delivered H2S doses must be carefully monitored. In addition, H2S has a pungent odor (the nose of most mammals is sensitive to it down to the parts per billion levels), which may induce discomfort and vomiting in the patient, and is, at least, a nuisance (if not a safety risk) for bystander medical personnel. Finally, since inhaled H2S will first “meet” the lung alveolar epithelial cells (in which cells it will have its highest local concentration), adverse effects on lung epithelial cells are possible, as documented in a variety of environmental toxicology studies (Lopez et al., 1987; Khan et al., 1991; Dorman et al., 2004; Roberts et al., 2006, 2008). These issues have necessitated intensive research and development of pharmaceutically acceptable, oral, parenteral, and topical H2S donating molecules and formulations, as discussed in the sections VII-XXII.
VII. Sulfide Salts (“Rapid-Release H2S Donors”)
The most common way to generate H2S for pharmacological and biologic experiments is to use common salts such as Na2S and NaHS. Most frequently, aqueous solutions of NaHS.xH2O (sodium hydrogen sulfide) or the nanohydrate disodium salt Na2S.9H2O or their anhydrous forms are used (Fig. 2). These salts rapidly generate H2S, but the commonly used term “rapid H2S-releasing drugs,” is, in fact, technically incorrect, since they do not release H2S, but rather dissociate to yield H2S in an instantaneous and pH-dependent manner. In this type of concentration/time relationship of H2S, the “experience” of cells or animals is very different from the slow, steady-state production of H2S by endogenous sources (e.g., the three H2S-generating enzymes) and, therefore, on first principles, serves as a very poor approximation to study the biologic roles of H2S.

H2S delivery to cell in culture. H2S and HS− are immediately generated when rapid-release H2S donors (i.e., sulfide salts) are dissolved in aqueous stock solutions (1). Likewise, when H2S donors (e.g., GYY4137, AP39 etc.) are dissolved in solution, some H2S and HS− can already begin to form (the extent of which depends on the chemical properties of the donor) (2). When stock solutions are added to the cell culture medium, these species (H2S-donor molecules, H2S and HS−) are delivered, first into the medium (3,4) and from there into the cultured cells (5). Some donors themselves are hydrophilic and may not have high cell permeability; these donors are likely to remain extracellular, and the H2S produced from them will enter the cells. Other H2S donors may enter the cells more readily (some of them may be cell-compartment-specific, e.g., AP39 sequesters into the mitochondria and delivers H2S preferentially to the mitochondrial component). Intracellularly, production is via glutathione-dependent conversion mechanisms. Intracellularly, H2S will react with various molecules (proteins, thiols, nitric oxide, reactive oxygen species) to create a mixture of biologically active species (polysulfides, persulfides, hybrid S/N compounds). Some of these reactions, e.g., with proteins and thiols, will already occur extracellularly in the cell culture medium (not shown) (6). Thus the cellular effects of H2S donors are produced by a complex array of interactions and biological actions induced by multiple species. H2S decomposition products (sulfite, sulfate, thiosulfate) are also produced via enzymatic and nonenzymatic processes (7). Another way to deliver H2S is by bubbling H2S into aqueous solutions (for instance, the method was used to produce IK-1001) (8). This solution, then, can be added to cells the same way as the other H2S delivery approaches (3). One can also supply H2S gas into the cell culture headspace, which, in turn, dissolves in the culture medium (9, 10) and delivers H2S and HS− to the cells. As soon as the H2S donors are dissolved in the stock solution, H2S starts to escape through diffusion into the air (11). Loss of H2S will also occur through diffusion of H2S from the cells into the culture medium (12) and then into the headspace (13).
At physiological pH, approximately 85% of the sulfide delivered by the salts will be in the dissociated, hydrosulfide form (HS−), and 15% will be the dissolved gas form (H2S) (Fig. 2). Although the process of dissolving a white salt in phosphate-buffered saline or tissue culture medium appears to be a fairly easy task, we must emphasize early on that H2S above a certain concentration level exerts adverse effects and can be toxic, and these issues must be considered when working with the molecule. As overviewed by Hughes et al. (2009), H2S solutions in the laboratory should always be prepared and used in fume hoods. Since H2S is heavier than air, it will accumulate in low, unventilated areas. The human nose can detect H2S down to parts per billion levels (at which concentration H2S is not dangerous to human health). In fact, loss of ability to smell H2S is an early symptom of H2S toxicity (which usually occurs after prolonged exposure to 50 ppm or higher levels of H2S). In other words, paradoxically, if a laboratory worker works with H2S solutions and the smell appears to be disappearing, it should be taken as a warning sign. A full safety assessment (including input from a local safety officer) is essential when working with H2S in a laboratory environment. The risks are already considerable when making up large amounts of H2S salt solutions and become especially significant when working with H2S gas from a cylinder, with mass flow controllers, H2S gas chambers, and related equipment. Various H2S detectors (normally used in industrial and environmental toxicological applications) are commercially available and should be implemented as part of a general safety plan.
The generation process is instantaneous, which means that a rapid “peak” concentration of H2S will be generated, which will rapidly decline due to physical loss (outgassing into the headspace, first from the H2S stock solution into the tissue culture hood, which is why H2S stock solutions must always be made fresh and used immediately, and then from the cell culture plate’s tissue culture medium into the cell culture incubator), and will be degraded and consumed by various cellular processes (Fig. 2). In vitro, the half-life of H2S, generated from salts, ranges between 5 and 30 minutes, depending on the quality of the water used for the experiments (metal content of laboratory water can be a significant variable), as well as many other experimental conditions (Doeller et al., 2005; Suzuki et al., 2011; DeLeon et al., 2012; Papapetropoulos et al., 2015), including cell type,5 cell density, ratio of cell number versus the volume of the culture medium, shape of the tissue culture well, temperature, and other factors. Similarly, in vivo, injection of H2S salts results in a high initial concentration, which then rapidly (within minutes) declines (Wintner et al., 2010).
It has been suggested that this initial high concentration of H2S may exert a rapid “knockdown” type effect, perhaps because at these early time points the concentration of H2S may reach high enough levels to cause a transient inhibition of Complex IV, resulting in a transient inhibition of mitochondrial respiration (Bouillaud and Blachier, 2011). Even the vascular relaxant effect of H2S, which is generally viewed as a tightly regulated, physiological mechanism, can be associated with inhibition of vascular ATP generation (Kiss et al., 2008). One can speculate that such “induced chemical hypoxia,” on its own (i.e., largely independent of the actual chemical species that elicited it) can result in various adaptive responses in the cell, for example, the upregulation of antioxidant defenses, somewhat resembling the phenomena of ischemic preconditioning. In fact, multiple studies show that rapid H2S donors can induce preconditioning responses (both the early and the delayed, second-window forms) as well as postconditioning (Calvert et al., 2009; Pan et al., 2009; Yusof et al., 2009; Predmore and Lefer, 2011; Peake et al., 2013; Zhang et al., 2013; Andreadou et al., 2015a,b; Ji et al., 2016). Such preconditioning-type and early responses may, in part, explain some of the differential pharmacological and biologic effects observed with rapid-release H2S donors versus slow-release H2S donors (Bouillaud and Blachier, 2011; Olson, 2011).
Other issues often raised with rapid H2S donors relate to the often unknown purity of the material used (yellow discoloration is a telling sign of impurities; some of these impurities, e.g., sulfate, may be biologically inactive, whereas others, e.g., thiosulfate and, especially, polysulfides, have their own, distinct biologic effects).6 Polysulfides are now considered a separate class of signaling molecules, which work at substantially lower concentration than H2S and catalyze a qualitatively different set of chemical and biologic reactions, including a major role in protein sulfhydration (in contrast, H2S itself cannot directly react with thiols) (Nagy, 2015; Kimura, 2014, 2015; Park et al., 2015). Some groups have proposed washing the surface of Na2S crystals in redistilled argon-saturated water before preparing the solutions for biologic use (Nagy et al., 2015). However, it is likely that some amount of polysulfide will never be avoided completely in the stock solution (and even if one minimizes this external polysulfide “delivery,” as soon as the H2S makes contact with a biologic system, like a cell culture or an isolated organ, polysulfide generation will commence).
The fact that sulfide salts are hygroscopic will introduce a source of error when trying to calculate the exact H2S concentration or dose to be applied to the biologic system. The fact that sulfide salts also emit a pungent odor is not only an annoyance for experimenters in the laboratory environment, but it is a real problem when considering the use of these compounds for pharmaceutical and human therapeutic applications.
There are additional uncertainties of what concentration of H2S the cell will actually “see” and for how long (starting with the extent of outgassing from the stock solution: the variable time between making up the stock solution and applying it to biologic systems;7 as a rule, all sulfide donor solutions, especially sulfide salt solutions, must be made up freshly and must not be stored as frozen stock solutions) after a high concentration of a stock solution is injected into the tissue culture medium and the uncertainties related to the rapidly changing cellular concentrations. Some of these issues may be mitigated by using thoroughly deoxygenated solutions when dissolving the H2S salts.
One may also attempt to compensate for the decomposition of H2S by constantly “infusing” H2S into the culture medium (e.g., Porteus et al., 2014) or by repeating the H2S “dosing” several times in an attempt to maintain a steady concentration of H2S (e.g., Suzuki et al., 2011), but the vast majority of published studies do not attempt to compensate for the loss of H2S and apply a single “dosing” of the salt, followed by the observation of biologic effects (often much delayed compared with the H2S donor’s administration, e.g., 24 or 48 hours, i.e., at time points where the initial H2S “dose” has been long cleared from the biologic system).
In vivo, the dosing with H2S salts is also problematic; typical dosing regimens include intraperitoneal administration of the material, most commonly in a once-a-day regimen; only a small proportion of the studies use approaches that attempt to maintain a steady-state concentration of H2S, e.g., by using minipumps (Suzuki et al., 2011; Stubbert et al., 2014), an approach that also has its own problems, for example, due to potential local effects of the extreme pH of the stock solutions necessary to load the minipumps to provide sufficient H2S delivery for extended time periods. Although, surprisingly, the circulating or tissue H2S levels have not been documented in any of these studies, based on measurements of plasma levels of H2S in response to intravenous administration of H2S donor salts (Wintner et al., 2010), it is likely that once-a-day intraperitoneal administration of H2S-releasing salts must yield an initial high circulating concentration of H2S, followed by a decline, and will not provide a 24-hour “coverage” for H2S delivery in vivo. Many studies use oral administration of solutions of rapid H2S donors, either via gavage or simply dissolving it in the drinking water of the animals. Surprisingly, the oral bioavailability of H2S remains to be exactly quantified (in experimental animals as well as humans); due to the fact that the intestinal epithelium forms a strong barrier against H2S produced by bacterial microbiota, one can assume that most of the H2S administered orally will not absorb into the systemic circulation.
The multitude of technical, practical, and scientific issues discussed above and elsewhere (e.g., Olson, 2012; Olson et al., 2012; Wedmann et al., 2014; Papapetropoulos et al., 2015; DeLeon et al., 2016a,b) necessitated the development of various classes of controlled H2S donors (discussed in sections XVI-XXII). Nevertheless, one should emphasize that, even with the above-mentioned multitude of limitations and uncertainties, the “rapid-releasing H2S donors” (i.e., simple salts of sulfide) have been used in thousands of biologic studies over the last decade. In fact, the majority of the information on the biologic and pharmacological effects of H2S has been generated using these salts. PubMed searches identify approximately 2000 publications that use Na2S or NaHS (and rely on it solely, or, in a smaller percentage of studies, in combination with other H2S donors, or other H2S-generating approaches, e.g., using the cellular overexpression of H2S generating enzymes). These papers are too numerous to comprehensively overview them. One common theme that is important to emphasize is that in vitro studies often demonstrate a bell-shaped concentration response to sulfide salts. At lower concentrations, physiological (or beneficial) effects dominate, such as cytoprotection, stimulation of cellular bioenergetics, stimulation of cell proliferation, anti-inflammatory effects. In contrast, at higher concentrations, adverse (or pathophysiological) effects are common, such as cytotoxicity, inhibition of cell proliferation, and proinflammatory effects. In vivo, systemic administration of sulfide salts, at lower doses, have been shown to exert blood pressure-lowering effects, anti-inflammatory effects, protective effects against various forms of ischemia-reperfusion injury, neurotrauma, vascular injury (e.g., accelerated atherosclerosis) (reviewed in Szabo, 2007, Moore and Whiteman, 2015).
In 2016, Xu et al. (2016b) reported that ammonium tetrathiomolybdate [TTM, or (NH4)2MoS4], a compound clinically used in the treatment of copper intoxication (e.g., Wilson’s disease) in patients, acts as a water-soluble H2S donor, which probably releases H2S through a simple hydrolytic process, albeit with a relatively long (hours) half-life, releases more H2S under acidic conditions. TTM, at concentrations of 50–200 µM, exerts protective effects against oxidant-induced cell damage in vitro (Xu et al., 2016b). TTM has many different biologic effects, including inhibition of tumor cell proliferation (Chisholm et al., 2016). The contribution of H2S release (versus H2S-independent pharmacological effects of the molybdate moiety) to its biologic effects remains to be clarified in future studies.
Calcium sulfide is another sulfide salt, which can generate H2S via hydrolysis. It is used in various industrial processes, but it is rarely used in biologic studies, although there are occasional poisoning cases (Horowitz et al., 1997), and it is suggested that calcium sulfide may have some potential as an orally active, salt-based H2S donor (Li et al., 2009b).
Although H2S salts (“rapid-releasing H2S donors”) have been successfully employed in many cell-based and animal studies, unformulated sulfide salts obviously do not represent an optimal starting point for pharmaceutical development for a number of reasons, including their short half-life, rapid and uncontrolled release, and unpleasant odor. The last decade’s intensive research and development of pharmaceutically acceptable, controlled H2S donating molecules and formulations will be summarized in sections VIII-XXII. Ideally, an H2S-donating prodrug should have 1) a chemical composition that is biologically compatible, including the side products generated after the release of H2S; 2) a known, possibly tunable, or possibly biologically context-dependent, release profile of H2S, which should be definitely much slower onset than the rapid H2S generation by sulfide salts and should be matching the indication and the route of delivery of the compound; 3) water solubility, 4) suitable oral bioavailability for compounds intended for oral dosing; 5) chemical tractability of the prodrug itself, as well as its decomposition products; and, as the compound progresses from a pharmacological tool stage to a development candidate stage, 6) pharmaceutically acceptable synthetic route, purity (including a pharmaceutically acceptable impurity profile), stability (“shelf-life”), and biologic tolerability/safety/toxicity/metabolism profile that would make the compound suitable to progress through the investigational new drug-enabling studies mandated by the regulatory agencies. Although not an absolute requirement from an investigational new drug-enabling standpoint, with prodrugs, the use of acceptable control molecules (e.g., a similar chemical structure that does not have the ability of H2S release) can be very useful in preclinical efficacy and mode-of-action studies. As it will be shown in sections VIII-XXII, the unique chemical and pharmacological nature of H2S necessitated rethinking of some of the general pharmaceutical and drug development principles.
VIII. Sodium Polythionate (SG-1002)
An orally active H2S-releasing compound (SG-1002) was produced by Sulfagenix (Cleveland, OH) and characterized in multiple in vivo studies in the laboratory of Dr. David Lefer. The initial publication on the compound (Kondo et al., 2013) described the characterization of this material by powder X-ray diffraction and mass spectrometry and disclosed that the compound is, in fact, a mixture of various molecules. The main constituent is a circular eight-membered alpha-sulfur molecule (92%), with an additional 7% sodium sulfate and less than 1% each of sodium thiosulfate, sodium trithionate, tetrathionate, and pentathionate (Fig. 3). SG-1002, when administered in the diet of mice at a dose of 20 (mg/kg)/day, caused an increase in blood and tissue (myocardial) H2S levels, as well as sulfane sulfur levels (Kondo et al., 2013; Barr et al., 2015). The increase in circulating H2S and sulfane sulfur levels by SG-1002 was also demonstrated in a Yucatan minipig model (Donnarumma et al., 2016b). The relative contribution of the various constituents of SG-1002 to this increase has not been delineated.
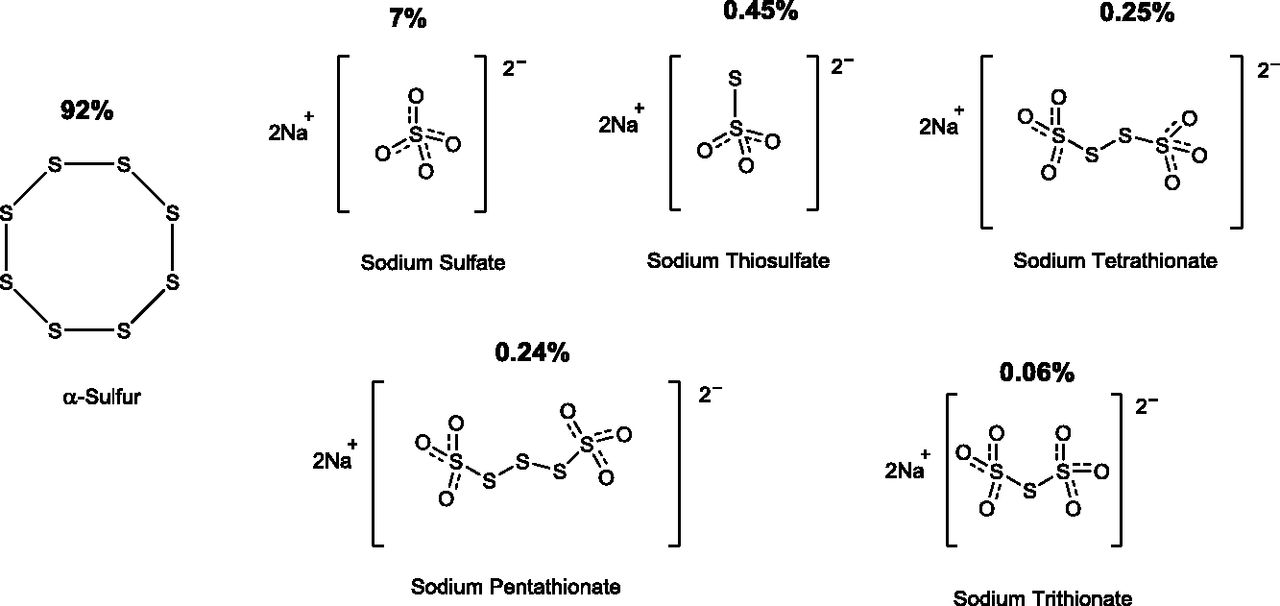
Chemical composition of SG-1002.
As far as preclinical efficacy studies, SG-1002 has been tested in a murine model of heart failure induced by transverse aortic constriction, where it was found efficacious against the development of myocardial hypertrophy and myocardial contractile dysfunction, and its effects were associated with reduction in oxidative stress parameters and stimulation of the Akt/eNOS signaling pathway (Kondo et al., 2013). It also exerted beneficial effects against myocardial hypertrophy and contractile dysfunction in a murine model of high-fat diet, both when it was administered in the beginning of the experiments, but also when the start of its administration was delayed to 12 weeks, a time when the animals started to exhibit signs of myocardial hypertrophy and dysfunction (Barr et al., 2015). The duration of SG-1002 was long in these studies (in some experimental groups up to 24 weeks) and was well tolerated. In addition to rodent models, the efficacy of SG-1002 was recently established in a pig model, as well. In Yucatan miniswine subjected to critical limb ischemia, treatment with SG-1002 (1600 mg/day orally) protected against the development of coronary artery endothelial dysfunction (Donnarumma et al., 2016b).
Despite the probable pharmaceutical and drug development challenges associated with the development of a material that contains multiple different active species, SG-1002 has now moved into the clinical development stage (designated as a “medicinal food”). In a Phase I clinical trial, its safety and its effects on H2S and NO bioavailability have been determined in a small number of healthy volunteers and in patients with heart failure (n = 7 or 8/group). Oral SG-1002 treatment (escalating dosages of 200, 400, and 800 mg twice daily for 7 days for each dose) was well tolerated and induced a significant increase in circulating levels of H2S at the two higher doses tested (Polhemus et al., 2015). There were also trends for increased blood sulfane sulfur levels, which, however, did not reach statistical significance. The elevation in free H2S plasma levels was more pronounced in healthy volunteers than in heart failure patients, most likely because the degradation of H2S is increased in the heart failure patients due to the oxidative stress associated with their condition. Importantly, serum brain natriuretic peptide levels (a marker of the severity of heart failure) were stabilized in the SG-1002-treated heart failure patients, whereas they tended to rise over time in the vehicle control group. However, due to the small patient number and low statistical power, additional studies are needed to confirm and extend these findings. According to the Sulfagenix website, a Phase II clinical trial (50 patients, randomized into a control and a SG-1002-treated group) is currently in the planning stages.
IX. IK-1001, a Pharmaceutically Acceptable, Parenteral Injectable Formulation of H2S
In 2007, the first report was published with IK-1001, a pharmaceutically acceptable formulation of H2S (“Sodium Sulfide for Injection”). This formulation was produced, under good manufacturing conditions by bubbling H2S gas into a physiologically balanced solution suitable for intravenous injection in humans. Many preclinical efficacy studies have been conducted with IK-1001, followed by the formal safety studies mandated before clinical trials. The preclinical studies demonstrated the efficacy of IK-1001 in various models, including rodent models of myocardial and hepatic ischemia-reperfusion (Elrod et al., 2007, Jha et al., 2008), cardiac arrest and resuscitation (Minamishima et al., 2009), various rodent and large animal models of myocardial infarction (Sodha et al., 2008, 2009; Osipov et al., 2009, 2010), and cardiopulmonary bypass (Simon et al., 2008, 2011; Szabo et al., 2011) and acute lung injury (Esechie et al., 2008, 2009). These protective effects require low doses of IK-1001 (e.g., 0.2 mg/kg bolus followed by 2 (mg/kg)/hour infusion), which are not associated with detectable physiological responses or any significant adverse effects. It is important that bolus administration of higher doses of IK-1001 (similar to the administration of sulfide salts discussed earlier) exerts a rapid hemodynamic effect, followed by a rapid decline in the concentration of H2S in the circulation (Wintner et al., 2010); therefore, the administration of IK-1001 is the safest and most effective when a low dose of initial bolus is followed by a constant infusion (Sodha et al., 2008; Osipov et al., 2009, 2010).
IK-1001 has successfully progressed through Phase I studies in healthy human volunteers, where its tolerability was monitored and its metabolism was evidenced by elevated thiosulfate plasma levels, and its elimination (exhalation) was documented through the lung. IK-1001 subsequently reached the Phase II trial stage (Leslie, 2008), at which point the sponsor company halted clinical development (Leslie, 2016), and two pending Phase II clinical trials (clintrials.gov identifier: NCT00858936 and NCT01007641) were terminated before the start of patient enrolment. To our best knowledge, the clinical program is no longer active with IK-1001.
X. Natural H2S Donors
In 2007 Benavides and colleagues (Benavides et al., 2007; Jacob et al., 2008) demonstrated that crude garlic extracts, as well as certain endogenous polysulfide compounds contained in garlic, release H2S in tissues. The release of H2S has been identified as the primary mechanism of the vasodilatory effect of garlic extracts (Benavides et al., 2007). Three compounds, diallyl sulfide or DAS (a weak H2S releaser), diallyl disulfide or DADS (an intermediate releaser of H2S, both in terms of net amount released and rate of release), and, the most active constituent of garlic, diallyl trisulfide (DATS), which releases the most amount of H2S and exhibits the fastest release rate (Liang et al., 2015), were proposed as the active H2S-donating principles of garlic (Fig. 4, A–C).
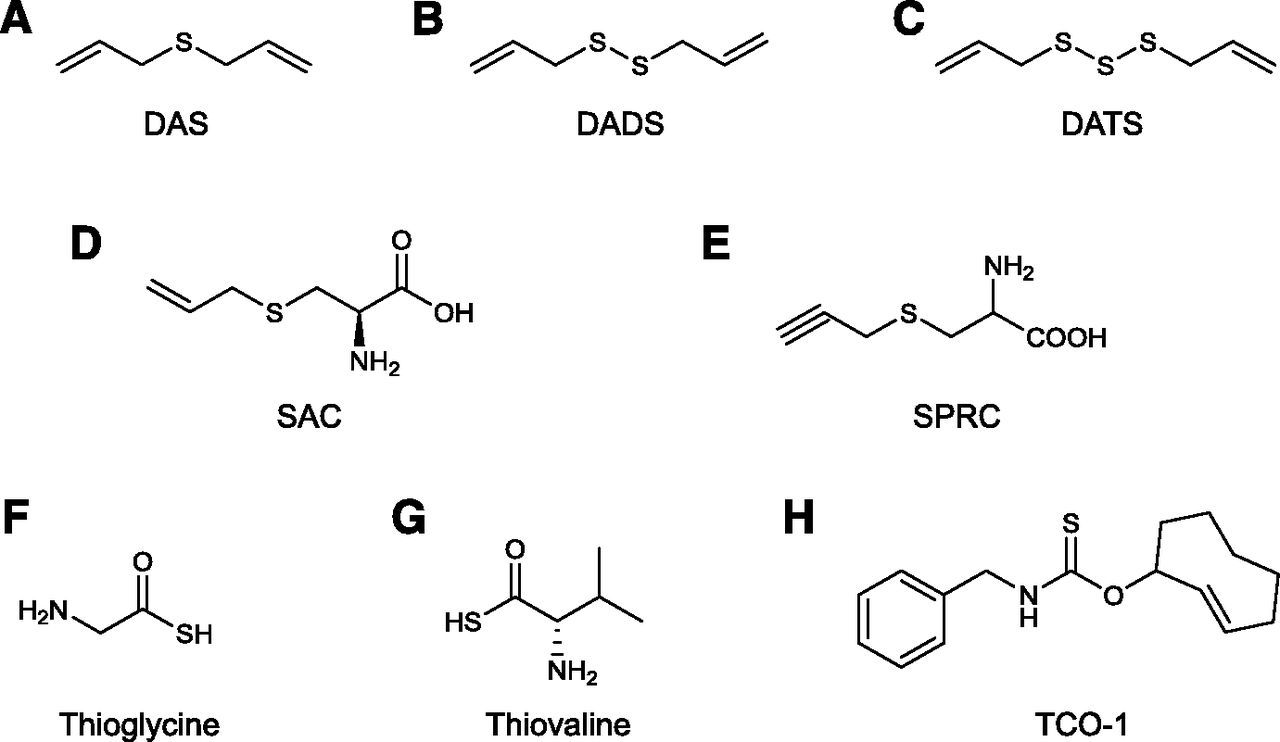
Structures of naturally occurring H2S donors and derivatives of naturally occurring compounds modified to release H2S. Diallyl sulfide (DAS; A), diallyl disulfide (DADS; B), diallyl trisulfide (DATS; C), S-allylcysteine (SAC; D), S-propargyl-l-cysteine (SPRC, also known as ZYZ802;E) thioglycine (TG; F), l-thiovaline (TV; G), thiocarbamate-functionalized carbonyl sulfide/H2S donor (TCO-1; G).
Cellular H2S release from DATS is dependent on its reaction with cellular glutathione. Briefly, the reaction of DATS with GSH produces the mixed disulfide allylglutathione and the low molecular weight hydropersulfide allylperthiol, from which H2S is released through a reaction with GSH. In turn, the reaction of DADS with GSH yields S-allyl-glutathione and allylperthiol, which reacts with GSH, thus releasing H2S (Benavides et al., 2007). Since these reactions occur in the intracellular environment, in the presence of various protein thiols, additional reactions may also occur, resulting in the covalent modification of proteins and formation of mixed disulfides. DATS can also directly transfer reactive sulfane sulfur to protein-SH groups, which results in the generation of protein hydropersulfides (Greiner et al., 2013). A variety of additional reactions have also been proposed that yield H2S or sulfane sulfur from various garlic-derived sulfur compounds (reviewed in Yagdi et al., 2016). The presence of l-cysteine in cell-free in vitro systems was found to significantly increase H2S release from DADS (Martelli et al., 2014).
In addition to direct chemical reactions, recent data indicate that garlic-derived polysulfides may also generate H2S via processes that involve various intracellular enzymes. As demonstrated in the kidney and liver tissues of mice, in vivo treatment of mice with DATS or DADS caused an increase in the activity of CSE in tissue homogenates (Iciek et al., 2012, 2016). Similar upregulation was also reported in cardiac myocytes exposed to DATS in vitro (Iciek et al., 2015, 2016; Tsai et al., 2015b). These findings may indicate that garlic-derived polysulfides produce H2S, at least in part, via CSE-dependent mechanisms. Alternatively, the upregulation of CSE and its “normal” physiologic function (conversion of cysteine and homocysteine) may also contribute to the elevation of H2S pools in various tissues after garlic-derived polysulfide treatment. Indeed, in H9c2 cells, siRNA-mediated silencing of CSE or treatment with the CSE inhibitor PAG attenuated the cytoprotective effects of DATS (Tsai et al., 2015b). The ability to induce CSE was also observed with other H2S donors (Meng et al., 2016), suggesting that CSE upregulation might be a common property among H2S generating compounds.8 Under some experimental conditions, not only CSE, but also CBS, has been reported to increase after DATS exposure (Chen et al., 2016a). Interestingly, DATS and DADS treatment also increased tissue rhodanese activity (Tsai et al., 2015b), perhaps as a compensatory mechanism to contribute to the elimination of the increased tissue H2S levels. Most recently, an additional mechanism, involving the oxidoreductase function of the antioxidant enzyme catalase, has also been demonstrated to contribute to the H2S release from DATS and other polysulfides (Olson et al., 2017). A final, indirect pathway that may also contribute to the enhancement of biologic H2S levels in response to garlic extracts or garlic-derived polysulfides may involve a generalized antioxidant action. Part of this action may involve a direct antioxidant effect. In addition, indirect effects may also contribute. Such indirect effects may involve the upregulation of various antioxidant pathways, which, in turn enhances the antioxidant status of cells. Potential mechanisms may involve 1) glutathione-S-transferase followed by elevation of intracellular glutathione levels, 2) activation of Nrf2 followed by the induction of various antioxidant pathways, and 3) an increase in the activity of the cysteine/glutamate antiporter and the cysteine transporter followed by increased intracellular accumulation of cysteine (Wu et al., 2001; Tsai et al., 2005; Kim et al., 2014; Kimura, 2015; Xu et al., 2015; DeLeon et al., 2016a). All of these responses would be expected to limit the oxidative degradation of H2S. However, the regulation of oxidative processes by garlic extracts and garlic-derived polysulfides is complex; under some conditions these species can exert not only antioxidant, but also pro-oxidant cellular effects (DeLeon et al., 2016a). The various potential mechanisms that may contribute to the elevation of biologic H2S levels in response to DATS are summarized in Fig. 5.
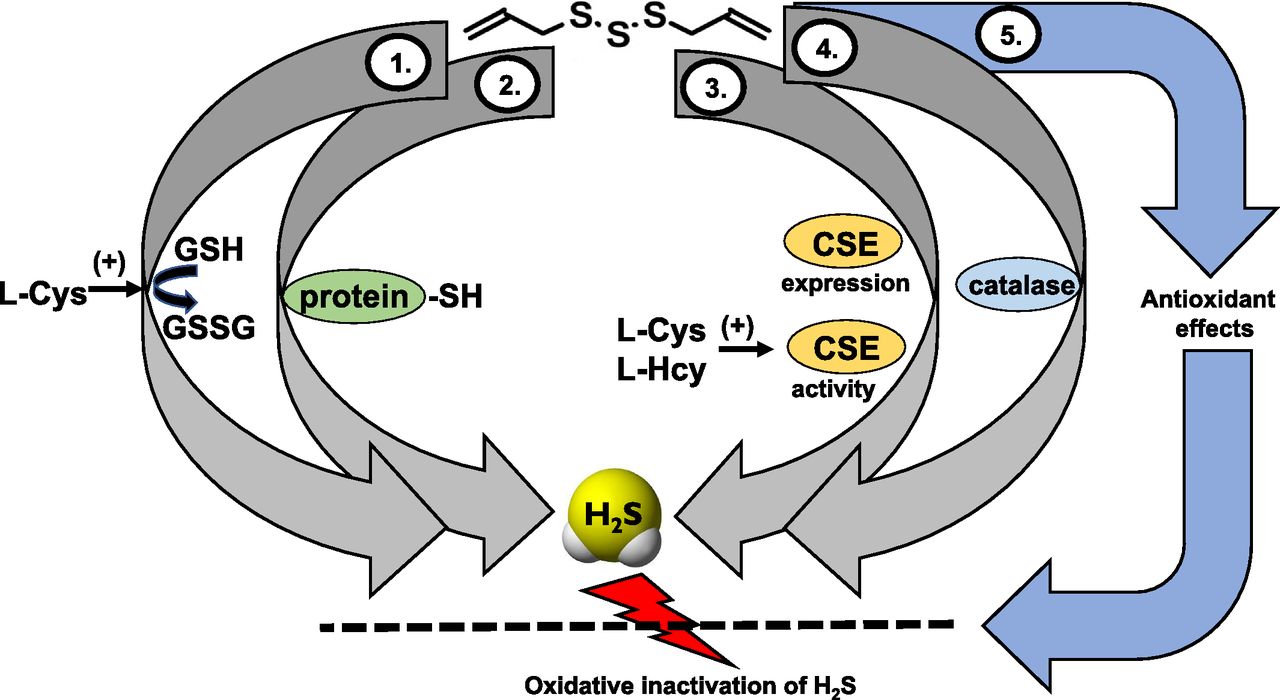
Pathways of H2S generation and mechanisms of action of polysulfide diallyl trisulfate (DATS) in mammalian cells. (1) H2S production via glutathione-dependent conversion mechanisms. This group of processes can involve several different mechanisms, including a carbon nucleophilic attack as well as various thiol-disulfide exchange reactions (not shown); (2) H2S production via reactions with protein-SH groups; (3) H2S production via upregulation of CSE and/or via stimulation of CSE activity. In these processes, H2S is produced from the endogenous substrates of CSE, l-cysteine/l-homocysteine, and DATS stimulates this reaction; (4) H2S production via the oxidoreductase function of catalase. An additional, indirect mechanism (5) involves redox mechanisms. DATS elevates the cell’s antioxidant pools, and this attenuates the oxidative degradation of H2S, in effect elevating the biologically available pools of H2S.
According to the most recent studies, in biologic contexts, the only relevant garlic-derived H2S donor is DATS; this compound, at concentrations of 100 µM, produces a clearly detectable increase in bioactive H2S in cellular systems (Liang et al., 2015). The previously reported H2S donating effect of DADS or DAS is likely attributable to DATS contamination of the samples.9 Although DATS is the fastest-releasing garlic-derived polysulfide, its H2S release rate is substantially slower than the H2S produced by the H2S-releasing salts NaHS and Na2S (Predmore et al., 2012a).
Although the initial product of garlic-derived polysulfides is H2S, in cells and tissues these molecules produce the most significant increases in the bound sulfane sulfur and polysulfide “pools”, rather than the free H2S levels (DeLeon et al., 2016a; Iciek et al., 2016).
Glutathionylated polysulfides, exemplified, for instance by the compound S-allylmercaptoglutathione, represent another species of garlic-derived slow-release H2S donors (Bhuiyan et al., 2015).
Pluth and colleagues (Cerda et al., 2017) recently reported on the synthesis of synthetic organic tetrasulfides, including bis(aryl) and bis(alkyl) tetrasulfides, as H2S donors, which release H2S in a first-order dependence on reduced glutathione (GSH) and release more H2S than the commonly used trisulfide DATS.
S-Allyl cysteine (SAC) (Fig. 4D) is another garlic-derived organosulfur-containing amino acid, which, however, appears to increase biologic H2S levels through a CSE-dependent mechanism (as opposed to releasing H2S directly or in cooperation with glutathione). This compound has been shown to exert protective effects in a rat model of myocardial reperfusion (Chuah et al., 2007; Wang et al., 2010b). However, in other studies, SAC (as opposed to DATS) did not exhibit significant inhibitory effects on inflammatory mediator production in LPS-stimulated microglial cells in vitro (Ho and Su, 2014). Although SAC clearly has beneficial effects in many models of disease (ranging from diabetic complications to hypertension) (Park et al., 2014; Denzer et al., 2016; Imai et al., 2016; Uzun et al., 2016; Brahmanaidu et al., 2017; Kattaia et al., 2017), it is likely that its pharmacological effects encompass multiple additional actions beyond H2S donation.
Systemic administration of garlic-derived polysulfides increases circulating H2S pools (both free H2S and sulfane sulfur) (Insko et al., 2009; Predmore et al., 2012a; Tsai et al., 2015b) and results in an increase in H2S exhalation (Insko et al., 2009). Garlic-derived polysulfides have been shown to exert cardioprotective and hepatoprotective effects via H2S release in several studies (Chuah et al., 2007; Shaik et al., 2008, Bradley et al., 2016). In a myocardial ischemia-reperfusion study, Lefer and colleagues (Predmore et al., 2012a) attributed the cardioprotective effect of DATS to H2S release, followed by activation of eNOS and elevation in circulating (cardioprotective) NO levels, but, in contrast to previous studies with fast-releasing H2S donors, the protection did not appear to involve the Nrf2 pathway. The question whether the many well-documented biologic effects of garlic, which include antioxidant effects, organ protective effects, radioprotective effects, anticancer effects, and many others (Belloir et al., 2006; Chuah et al., 2007; Herman-Antosiewicz et al., 2007; Münchberg et al., 2007; Pari et al., 2007; Sener et al., 2007; Amorati and Pedulli, 2008; Shaik et al., 2008; Predmore et al., 2012b; Yagdi et al., 2016), are related to H2S production remains to be clarified in future studies. It is clear, nevertheless, that DADS and DATS exert a wide range of pharmacological actions, many of which, according to our current knowledge, are probably unrelated to H2S release, including inhibition of histone deacetylase (Dashwood et al., 2006), inhibition of 3-hydroxy-3-methylglutaryl-coA (Rai et al., 2009), activation of metabolizing enzymes that detoxify carcinogens, modulation of regulation of cell-cycle arrest (Yi and Su, 2013), and, depending on the experimental conditions, either decreased or increased intracellular ROS production (Iciek et al., 2012; Smith et al., 2016).
In addition to garlic, numerous additional natural (in most cases plant derived) compounds have been characterized as H2S generators in vitro and, in some cases, have also been tested in vivo. Examples include lenthionine, isothiocyanate derivatives isolated from Brassicaceae species (Citi et al., 2014), shallots (Tocmo et al., 2014), and stinky bean (Parkia speciosa Hassk seeds) (Tocmo et al., 2016). The latter contains a rich collection of compounds that contain multiple sulfur groups and appear to generate H2S. These species include many cyclic compounds (2,4-trithiolane,1-3-5-trithiane, 1,2,3,5-tetrathiane, 1,2,3,5,6 penthathiane), as well as linear compounds such as dimethyl tetrasulfide (Tocmo et al., 2016).
XI. S-Propargyl-Cysteine
S-Propargyl-cysteine (SPRC, also termed ZYZ-802) (Fig. 4E), an analog of l-cysteine and a compound that is structurally closely related to SAC (a multifunctional molecule discussed in the section X), has been studied extensively in preclinical studies as an H2S donor (reviewed in Wen and Zhu, 2015). SPRC elevates H2S levels in biologic systems, an effect that presumably occurs either by direct H2S donation and/or by upregulation of H2S production through upregulation of endogenous CSE expression/activity and/or via CSE-dependent conversion of the compound to produce H2S (Wen and Zhu, 2015). The relative contribution of these potential actions remains to be further characterized. In endothelial cell proliferation and migration studies, the effect of SPRC was completely abrogated in the presence of the CSE inhibitor PAG, indicating that CSE stimulation (or CSE-mediated H2S production) may be a major component of its action (Tran et al., 2015). However, in a rat model of myocardial infarction, the beneficial effects of SPRC were only slightly reduced in the presence of PAG (Wang et al., 2009a), suggesting that the main mode of action of the compound does not (or does not always or does not necessarily) involve CSE.
As far as pharmacokinetic effects, in 2011, a report characterized the pharmacokinetics of SPRC (but, regrettably, not of its product H2S). The plasma half-life of SPRC was established as approximately 3 hours; its oral bioavailability was better than 95% (Zheng et al., 2011). A subsequent study also demonstrated the distribution, metabolism, and excretion of SPRC and showed the highest distribution of the compound to the kidney; heart and liver levels were also relatively high. SPRC exhibited low plasma binding. Its main metabolic route was identified as N-acetylation (Zheng et al., 2012). In in vivo studies, SPRC, at a dose of 50 mg/kg, induced only a slight, although statistically significant, elevation of circulating H2S levels (Wang et al., 2009a; Yang et al., 2015; Li et al., 2016).
In in vitro studies, SPRC (typically in the concentration range of 10–100 µM) stimulates cell proliferation and angiogenesis (Kan et al., 2014). SPRC also counteracts cell death induced by multiple insults, including ischemia-reoxygenation injury in cardiac myocytes (Wang et al., 2009a; Liang et al., 2015), high glucose-induced endothelial cell death and dysfunction (Yang et al., 2015), and doxorubicin-induced myocyte death (Wu et al., 2016c). SPRC was also shown to reduce tumor necrosis factor α (TNFα)-induced upregulation of adhesion molecules in endothelial cells (Pan et al., 2012) and IL-1β- or LPS-induced upregulation of multiple proinflammatory cytokines, adhesion molecules, and matrix metalloproteinases in various cell types (Pan et al., 2011; Wu et al., 2016c). Consistently with the bell-shaped concentration-response of H2S in cancer cells (reviewed in Szabo, 2016), very high concentrations (20–30 mM) of SPRC induce apoptosis in cancer cells (Ma et al., 2011).
Multiple studies have tested the efficacy of SPRC in various models of disease in vivo. Typically, the doses of SPRC are in the range of 10–50 mg/kg orally, once a day. In rat and mouse models of myocardial infarction induced by left anterior descending artery ligation, SPRC reduced myocardial infarct size, suppressed circulating markers of myocardial cell necrosis, improved survival, and stimulated postischemic angiogenesis (Wang et al., 2009a; Tran et al., 2015). In rat models of cognitive impairment induced either by intracerebroventricular administration of LPS or by β-amyloid, SPRC improved cognitive function and downregulated inflammatory mediator production (Gong et al., 2011a,b). In a mouse model of cerulein-induced pancreatitis, only a very minor effect of SPRC was noted on plasma amylase levels, but the compound protected against the histologic changes in the pancreas and downregulated the production of multiple inflammatory mediators (Sidhapuriwala et al., 2012). Curiously, in this model (which induces an increase in circulating H2S levels), SPRC did not cause any further increase in circulating H2S levels, but, rather, it caused a slight suppression of these levels (via a mechanism that remains to be explained). In a mouse model of hind limb ischemia, SPRC stimulated angiogenesis, resulting in an improved recovery and better blood flow responses (Tran et al., 2014). In a diabetes-induced kidney dysfunction model, SPRC inhibited the increase in plasma blood urea nitrogen and creatinine levels, reduced albuminuria, suppressed inflammation, and improved kidney histology (Qian et al., 2016). In an adjuvant-induced arthritis model, SPRC suppressed joint swelling and downregulated the production of multiple inflammatory mediators in the joint (Wu et al., 2016d). SPRC was also efficacious in a nonalcoholic liver disease model in mice (Li et al., 2016) and in a doxorubicin model of myocardial dysfunction in rats (Wu et al., 2016b). Consistently with the bell-shaped dose-response of H2S in cancer (reviewed in Szabo, 2016), higher doses of SPRC exerted inhibitory effects on tumor growth in tumor-bearing mice in vivo; for example, at 100 (mg/kg)/day, SPRC induced an approximately 50% inhibitory effect of the growth of gastric cancer cells implanted into nude mice (Ma et al., 2011).
The molecular and biochemical pathways associated with the effects of SPRC are multiple; they include the stimulation of Akt phosphorylation (Yang et al., 2015; Li et al., 2016), activation of the antioxidant “master switch” Nrf2 (Yang et al., 2015; Wu et al., 2016d), upregulation of CSE mRNA, CSE protein and CSE activity (Wu et al., 2009; Ma et al., 2011; Huang et al., 2013; Yang et al., 2015; Li et al., 2016), inhibition of nuclear factor-κB activation (Pan et al., 2012), vascular endothelial growth factor receptor activation followed by STAT3 activation (Kan et al., 2014; Wu et al., 2016d), upregulation of cellular antioxidant systems (superoxide dismutase, catalase, glutathione peroxidase, heme oxygenase-1) (Wu et al., 2016b; Li et al., 2016), elevation of intracellular glutathione levels (Wu et al., 2016b), reduction of cellular ROS levels (Pan et al., 2012; Li et al., 2016), and improvement of cellular calcium handling (Liang et al., 2015). In the context of cytotoxic effects of SPRC in cancer cells, at higher concentrations/doses, the compound was also found to increase bcl-2-like protein 4 and p53 expression (Ma et al., 2011).
In summary, the molecular mode of action of SPRC is complex and incompletely understood. Nevertheless, the compound elicits significant therapeutic effects in a variety of cell-based and animal models of disease and may be a candidate for future clinical translation. A recent study also reported in vivo efficacy with a controlled release form of the compound termed CR-SPRC (produced by solid dispersion technique with Eudragit RS30D as carrier) in an acute and chronic myocardial ischemia models (Huang et al., 2013; Tran et al., 2015). CR-SPRC [30 (mg/kg)/day] was reported to induce a very large, sixfold increase in plasma H2S levels over baseline in the report by Huang et al. (2013), whereas the unformulated SPRC [30 (mg/kg)/day] was reported to induce a threefold increase. These increases are substantially larger than all prior published reports with SPRC, where the increases in plasma levels were only in the range of 20%–50% above baseline. The reason for this discrepancy remains to be clarified.
The extensive pharmacokinetic and absorption, distribution, metabolism, and excretion studies published with SPRC (e.g., Zheng et al., 2011, 2012; Ma et al., 2015) as well as the recent efforts aimed at further optimization via pharmaceutical routes, suggest that clinical translation has been considered; however, to our knowledge, clinical trials have not yet been initiated.
XII. “Old School” Spontaneous H2S Generators: Thioacetamide and Lawesson’s Reagent
The fact that thioacetamide (CH3CSNH2) and Lawesson’s reagent (2,4-bis(4-methoxyphenyl)1,3,2,4-dithiaphosphetane-2,4-disulfide) release H2S has been known for over a century. The decomposition of thioacetamide is rapid upon its reaction with water (Lehrman and Schneider, 1955) (Fig. 6), and therefore, all the potential problems mentioned in section VII in relation to the sulfide salts apply to this compound as well. Thioacetamide is hepatotoxic and carcinogenic (Neal and Halpert, 1982). However, given the fact that these actions are not shared with other H2S donors, the mechanisms underlying these toxic effects are unrelated to the H2S-producing properties of thioacetamide. In some studies where thioacetamide is used to induce hepatic damage, the fact that thioacetamide also produces H2S is not always taken into account. For example, Wang et al. (2015b) used thioacetamide to generate liver damage in rats; unsurprisingly, circulating H2S levels were elevated in the thioacetamide group; also unsurprisingly, NaHS treatment of thioacetamide-treated rats exacerbated the liver damage, because presumably the total H2S generated by the two different approaches reached cytotoxic levels.
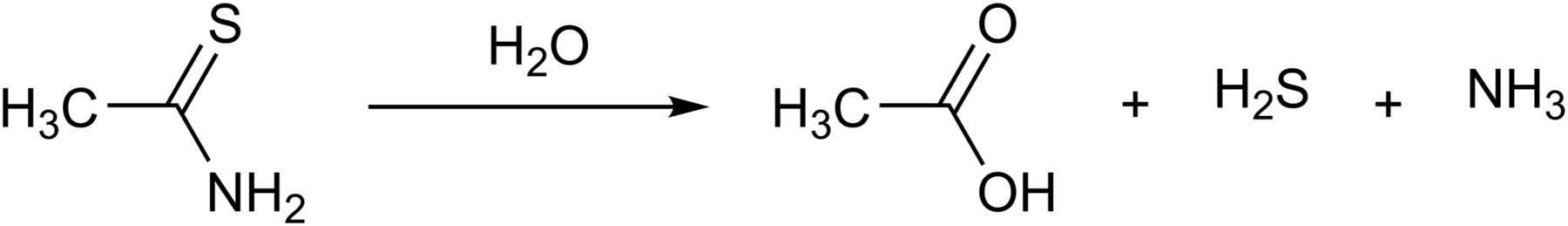
Thioacetimide releases H2S by hydrolysis.
Lawesson’s reagent (Fig. 7A), which contains a four-membered ring of alternate P and S atoms, has also been used in several biologic studies to generate H2S (Zanardo et al., 2006; Wallace et al., 2007a; Dal-Secco et al., 2008; Medeiros et al., 2009; Ekundi-Valentim et al., 2010; Spiller et al., 2010; Medeiros et al., 2012; Nicolau et al., 2013; Lucetti et al., 2017; Rodrigues et al., 2017). Dal-Secco et al. (2008) used Lawesson’s reagent to demonstrate its proinflammatory effects in some experimental settings and its anti-inflammatory effects in others (Ekundi-Valentim et al., 2010). The process of H2S release from Lawesson’s reagent involves the opening of the ring, followed by the generation of two molecules of dithiophosphine, R-PS2, which, in turn, decomposes to produce H2S. Similar to thiacetamide and the sulfide salts, H2S generation from Lawesson’s reagent is rapid and not controlled by cellular or biologic processes.

Structure of Lawesson’s reagent and structurally related compounds, including the slow-release H2S donor GYY4137. Lawesson’s reagent (A; 2,4-bis(4-methoxyphenyl)-2,4-dithioxo-1,3,2,4-dithiadiphosphetane), GYY4137 (B; P-(4-methoxyphenyl)-P-4-morpholinyl-phosphinodithioic acid), FW1256 (C; 3-dihydro-2-phenyl-2 sulfanylenebenzo[d] [1,3,2]oxazaphosphole).
Although dithiolethiones (1,2-dithiole-3-thiones) are primarily used as H2S-donating functional groups, typically coupled onto approved, clinically used drugs (see section XXIII), in a limited number of studies, these compounds, for instance, 1,2,dithiole-3-thione ADT-OH (5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione) or ADT (5-(4-methoxyphenyl)-3H-1,2-dithiole-3-thione) (Fig. 8), have also been used on their own as H2S donors. As expected from an H2S donor, these compounds exert various cytoprotective and anti-inflammatory effects (Li et al., 2007; Ozturk et al., 2007; Lee et al., 2010; Jia et al., 2013; Wang et al., 2014a; Liu et al., 2016a). The exact H2S release mechanism from these compounds probably occurs through a combination of a nonenzymatic process (hydrolysis) in aqueous solutions, as exemplified by the hydrolysis of ADT-OH (Fig. 9) as well as enzymatic events (e.g., through the action of esterases, as it is the likely case with dithothreitol) (Li et al., 2007; Qandil, 2012).

Commonly used compounds to deliver H2S. Structures shown are used either as stand-alone donors or are attached to known pharmacophores to give H2S-releasing properties in these structures, thus creating “combination donors.” DTT (A; 1,2,dithiole-3-thione), ADT-OH (B; 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione), or ADT (C; 5-(4-methoxyphenyl) -3H-1,2-dithiole-3-thione).
Spontaneous H2S release from ADT-OH (5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione). It has been suggested that the rates of H2S release from ADT-OH are increased when the compound is incubated with biologic material; however, the mechanisms responsible for the enhanced release remain uncharacterized.
The use of these “old-school” reagents to study H2S biology is not recommended, due to their nonspecific effects, as well as due to the availability of other, “cleaner” experimental tools to generate H2S in biologic systems. Their most acceptable use (for instance, as in Medeiros et al., 2009, 2012; Lucetti et al., 2017; Rodrigues et al., 2017) may be as a potential “add-on experimental group,” to confirm the biologic effects reported with multiple other classes of H2S donors.
XIII. GYY4137 and Other Phosphinodiothioate Derivatives
In 2008, Moore and colleagues (Li et al., 2008b) at the National University of Singapore reported on the synthesis of a water-soluble, slow-releasing H2S donor compound morpholin-4-ium 4 methoxyphenyl(morpholino) phosphinodithioate (GYY4137) (Fig. 7B). This novel compound, although its chemical structure bears obvious kinship with Lawesson’s reagent (Fig. 7A), is considered the first example of a new generation of H2S donor compounds specifically designed to mimic and model the slow rate of endogenous, physiological H2S production to supply (or perhaps replace missing pools of H2S) for biologic (therapeutic) effects. Immediate advantages of this compound over the “rapid-releasing” sulfide salts include 1) its slower release profile (see below), (2) its purity, i.e., better characterized chemical properties (i.e., lack of contamination with sulfide salts10), and 3) its water-soluble character, allowing the generation of stock solutions (although we should mention that some degradation takes place even in organic solvents) with higher initial concentration and less pH-dependent effects. GY4137, similarly to the sulfide salts, will immediately start releasing H2S when it comes in contact with water and, therefore, similar to the sulfide salts, it is best used when freshly prepared. The process of H2S release is increased at acidic pH (Li et al., 2008b). The mechanism of GYY4137 degradation has been investigated, in detail by Alexander et al. (2015) (Fig. 10). The process involves two distinct steps, the first of which involves a straightforward sulfur-oxygen exchange with water to give an arylphosphonamidothioate intermediate. However, this hydrolysis product is most likely not the most relevant byproduct of GYY4137, because of the second degradation step, which completes the hydrolysis to an arylphosphonate compound. Interestingly, the presence of l-cysteine in cell-free in vitro systems was found to significantly increase the H2S release rate of GYY4137 (Martelli et al., 2014), although the underlying chemistry has not been characterized.

GYY4137 releases H2S upon hydrolysis. Two molecules of H2S are released per molecule of GYY4137.
The initial report on GYY4137 focused on the vascular and cardiovascular effects of the compound and demonstrated that the compound exerts a slow-onset vasodilatory effects in isolated vascular rings and in perfused hearts and kidneys. In vivo, a single dose of the compound produced a significant sustained increase in circulating H2S levels, with better effects in response to intravenous (as opposed to intraperitoneal) administration and exerted antihypertensive effects in spontaneously hypertensive rats (Li et al., 2008b). The effects of GYY4137 (10–100 µM for 72 hours) were not associated with significant antiproliferative or cytotoxic effects in various cultured cell preparations (Li et al., 2008b).
Many investigators working in the biology of H2S have been using GYY4137 as a convenient tool to generate H2S in a controlled manner in vitro and in vivo. So far, over 100 publications have appeared using this compound in various experimental conditions (reviewed in Rose et al., 2015; Whiteman et al., 2015). Typically, the doses of the compound used in vivo are in the 50–100 mg/kg range (sometimes even higher); the relatively high doses are probably necessary because of the slow (perhaps too slow, see below) release of H2S from the compound. The major biologic effects included cytoprotection, i.e., improvement in cell viability, mitochondrial function, and overall cell survival in various cultured cells exposed to various reactive oxidant species, or to hypoxia, or to proinflammatory mediators. In vivo studies demonstrated the protective effect of GYY4137 in various models of ischemia-reperfusion and organ injury (myocardial, renal, hepatic) against early onset cell death and later-onset organ fibrosis (Lilyanna et al., 2015; Meng et al., 2015b,c; Zheng et al., 2015; Chatzianastasiou et al., 2016; Liu et al., 2016b; Karwi et al., 2016). GYY4137 was also found to attenuate both hyperoxic and aortic cross-clamping-induced lung injury (Madurga et al., 2014; Vadivel et al., 2014; Tang et al., 2017) and atherosclerosis and other forms of endothelial dysfunction and vascular remodeling (Liu et al., 2013; Wang et al., 2009b; Xu et al., 2014; Candela et al., 2016; van den Born et al., 2016; Nußbaum et al., 2017; Weber et al., 2017). GYY4137 was found to exert local and systemic anti-inflammatory effects, which are mediated by a combination of actions including inhibition of the production of various proinflammatory mediators, as well as antiplatelet and antithrombotic effects (Li et al., 2009a, 2013; Fox et al., 2012; Perry et al., 2011; Grambow et al., 2014, 2017; Chen et al., 2016b; Grambow et al., 2017; Rodrigues et al., 2017). GYY4137 administration prevented cardiac hypertrophy through SP1 sulfhydration and inhibition of KLF5 expression and activity (Meng et al., 2016). Recent studies have implicated H2S in the process of bone loss and demonstrated the efficacy of GYY4137 in its prevention (Grassi et al., 2016). GYY4137 was also found to inhibit viral replication and protect from viral airway inflammation (Li et al., 2015; Ivanciuc et al., 2016; Bazhanov et al., 2017). Another line of GYY4137-related research focused on cancer; GYY4137 was found to inhibit the proliferation rate of various cancer cells in vitro (Lee et al., 2011, 2014b; Lu et al., 2014) and reduced the growth of various transplanted murine models in vivo (Lee et al., 2011).
One of the caveats of working with GYY4137 (as well as with other H2S donors where there is a substantial “carrier” molecule) is that it is important to test for potential pharmacological or biologic effects of the “spent donor” molecule (the part of the molecule that is left behind after the H2S is released). This can easily be done by producing a “spent” GYY4137 stock solution by simply leaving it to decompose in aqueous solution for a few weeks.11 The issue is somewhat complicated by the fact that GYY4137 is produced as a dichloromethane complex, and dichloromethane can be decomposed to yield CO (another biologic gasotransmitter with significant effects, often cooperative or overlapping with those of H2S). Thus, several different experimental groups (e.g., freshly prepared GYY4137, spent GYY4137, freshly prepared dichloromethane) would be ideal to be included in well-designed studies using GYY4137. A simpler approach is to use the sodium salt (which does not contain the morpholine counter-ion or dichloromethane). However, these simple control groups, regrettably, are often absent from publications using GYY4137. Some investigators have suggested the compound ZYJ112 (morpholin-4-inum diphenylphosphinic acid, a non-sulfur compound that resembles GYY4137) as a control (Lee et al., 2011). Unfortunately, this compound is not sufficiently closely related to GYY4137 to be considered a satisfactory control. Moreover, it is not commercially available. At very high (millimolar) concentrations of GYY4137, one should even consider the use of osmotic controls.
Like with every other pharmacological agent, the higher the applied concentrations/doses of GYY4137, the more likely it is that some of the effects are due to nonspecific/secondary actions (in this case: actions other than H2S release). For instance, concentrations of GYY4137 of 10 mM or above (e.g., Grambow et al., 2014; Bala et al., 2014; Fitzgerald et al., 2014; Bazhanov et al., 2017) may be problematic and are not recommended to be used in biologic experiments, unless they are accompanied by the proper control experiments outlined above.
Several structural modifications of GYY4137 have been reported, many of them without any clear or obvious biologic difference between these novel compounds and the original donor GYY4137. For example, O-substituted 1,8-diazabicyclo[5.4.0]undec-7-ene salts behave similar to GYY4137 in terms of their H2S-releasing profile (Park et al., 2013; Park and Xian, 2015).
A group of investigators, which included some of the original scientists who created GYY4137, recently reported on the production of various GYY4137 derivatives, including ones with five-, six-, or seven-membered rings (Feng et al., 2015b, 2017; Huang et al., 2016). The compounds exhibited variable rates of H2S release and all of them were found to exert variable degree of antiproliferative effects in various cancer cell lines in vitro (Feng et al., 2017). One particular group of compounds, 2,3-dihydro-2-phenyl-2-sulfanylenebenzo[d][1,3,2]oxazaphospholes, of which the simplest member was designated as FW1256 (Fig. 7C), exerted significant cell-based biologic activity: inhibition of the production of various proinflammatory mediators in the 100 µM concentration range (Huang et al., 2016). This compound was also found to exert anti-inflammatory effects in mice challenged with LPS (Huang et al., 2016).
To vary the rate of H2S release, Whiteman et al. (2015) have also modified the phosphorodithioate core of GYY4137 and produced a series of compounds with faster release profile (e.g., AP105 > AP106 = GYY4137); some of these GYY4137-derivatives exhibit better anti-inflammatory and cytoprotective effects than equimolar concentrations of GYY4137.
One of the important biologic differences that relates to the rate of H2S release by various H2S donors is their differential ability to increase intracellular cGMP levels: slow H2S donors exert minimal or no activating effects on the cGMP system (i.e., do not raise cGMP levels), whereas “fast-releasing” donors (i.e., the sulfide salts) as well as some of the fast-releasing GYY4137-derivatives (e.g., AP72 and AP67) induce a significant elevation of cellular cGMP levels (Zhou et al., 2012; Bucci et al., 2012; Whiteman et al., 2015). These differences may become very important when assessing biologic effects; for example, in biologic conditions in which cGMP elevation is important for the beneficial or therapeutic effects, a slow-releasing donor may not perform as well as a faster-releasing molecule.12 Such differences in addition to important model-, cell-, and species-dependent differences are important in determining the extent to which the H2S and the NO system interact in various physiological and pathophysiological conditions.
XIV. Carbonyl Sulfide and its Prodrugs
Carbonyl sulfide (COS) is a sulfur compound that is present in the atmosphere in the concentration of 500 ppt. It is generated, for instance, by hot springs and volcanoes. In addition, COS can also be produced in biologic systems. A family of COS-synthesizing enzymes, thiocyanate hydrolases, has been localized in plants, as well as bacteria (but, up to this point, not in mammalian cells). COS has a short in vivo half-life, because it is rapidly hydrolyzed by carbonic anhydrase with the generation of H2S and CO2. COS donors have received little attention until recently, when Powell and colleagues (Steiger et al., 2016; Powell et al., 2017) hypothesized that thiocarbamates or N-thiocarboxyanhydrides (NTAs) may be useful as a platform from which to build various COS donors. Synthetic chemistry work followed by pharmacological characterization of a limited number of putative donors has demonstrated that thiocarbamates and NTAs indeed release H2S in the presence of carbonic anhydrase. The structure of the thiocarbamate-functionalized COS/H2S donor TCO-1 is shown in Fig. 4H. The rate of H2S production was found to be relatively fast (minutes). NTAs also served as a suitable platform for the generation of polymeric and nanotechnology-based H2S donors (to be discussed in section XXII). Another, very recent method to generate COS involves the inverse-electron demand Diels-Alder click reaction between a thiocarbamate-functionalized trans-cyclooctene and a tetrazine (Steiger et al., 2017b). These compounds, when incubated in biologic matrices that contain carbon anhydrase (e.g., red blood cells) release H2S over minutes to hours. Further biologic characterization of these compounds (in cell-based and animal-based systems) remains to be performed.
XV. Nonregulated, Nontargeted, Miscellaneous H2S Donors
Several groups have generated and (at least partially) characterized a variety of nontargeted and “nonregulated” H2S donors of various chemical classes. These donors will be reviewed in the current chapter, mostly based on a chronological order of their initial synthesis and disclosure.
In 2011, Zhao et al. (2011) reported on the synthesis of a series of H2S donors based on the N-(benzoylthio)benzamide template (Fig. 11A). These compounds were found to be stable in aqueous buffers; H2S generation required the presence of cysteine. In the presence of l-cysteine in vitro, or in the presence of plasma (which contains significant amounts of free cysteine), the compounds exhibited rapid-onset and sustained H2S-releasing profiles. Additional biologic characterization of these compounds has not been reported, but this overall concept yielded follow up thiol-activated compounds like NSHD-1 and NSHD-2 (see below).

Structures of thiol-activated H2S donors. N-(Benzoylthio)benzamide derivates (A), NSHD-1 (D), and NHSD-2 (E) are N-mercapto-based donors, Compound 8 is a perthiol-based donor (B), 4-carboxy-phenyl-isothiocyanate (4CPI; C), TAGDD-1 is a geminaldithiol-dithiol based compound (F). Cysteine-triggered H2S release from S-aroylthiooximes (G) have half-lives between 8 and 82 minutes depending on the substitution of the S-aroylthiohydroxylamine ring.
In 2012, the synthesis of FeIISH complexes, stabilized by an intramolecular N-H···S hydrogen bond, were reported (Galardon et al., 2012). These structures were found to act as H2S donors in solution (Fig. 12). Once again, further biologic characterization of these compounds has not been reported.

H2S release from FeIISH complexes.
In 2012, we took another, slightly different approach to design, synthesize, and pharmacologically characterize new water-soluble, slow-releasing H2S donors. We hypothesized that thioaminoacids could satisfy a number of criteria that would be beneficial for an H2S donor designed to be a general-purpose donor for in vivo use (e.g., lack of external thiol requirement for H2S release, no need for enzymatic conversion, and the generation of benign biologically acceptable degradation products after the H2S is released from the compound). Thioaminoacids are stable under acidic conditions, but at mildly alkaline pH, in the presence of bicarbonate, they spontaneously transform to the corresponding alpha-amino acid N-carboxyanhydrides by simultaneous liberation of H2S (a property known as the “bicarbonate effect”). Considering the bicarbonate concentration in blood, we hypothesized that thioaminoacids may serve as useful H2S donors in biologic systems. Two thioaminoacids (thioglycine and thiovaline) (Fig. 4, F and G) were synthesized and characterized (Zhou et al., 2012). The compounds produced a medium-fast release of H2S in biologic matrices. This included a sustained increase in circulating H2S levels that lasted for over 8 hours after a single intraperitoneal injection. The compounds also elicited marked increases in cellular cGMP levels in cell cultures in vitro, relaxed precontracted vascular rings, and protected cultured cells from oxidative injury. The effective in vitro concentration range was 0.01–0.1 µM. Moreover, at 4 µmol/kg, thiovaline significantly reduced infarct size in rodent models of myocardial infarction in vivo (Chatzianastasiou et al., 2016).
Another amino acid-related H2S donor is thiocysteine (or cysteine perthiol). This compound, in fact, can be considered a “natural” H2S donor, because it is involved in CSE-catalyzed H2S biosynthesis as an intermediate step (Stipanuk and Beck, 1982; Caliendo et al., 2010). In 2013, Zhao et al. (2013) reported on the synthesis and characterization of further perthiol-based H2S donors. One example is Compound 8a (Fig. 11B), which decomposes, in the presence of thiols, to yield H2S. Compound 8a generates H2S in cultured cells in vitro and reduces myocardial infarct size in vivo in a standard model of left anterior descending coronary artery occlusion/reperfusion (Zhao et al., 2013). Another class of cysteine-dependent H2S donors, the dithioperoxyanhydrides, produced in 2013 by Galardon and colleagues (Roger et al., 2013) also uses a similar, perthiol-based mechanism for the generation of H2S. Other than their vascular relaxant effects in isolated thoracic aortic rings, the biologic effects of the dithioperoxyanhydrides have not been characterized in detail.
In 2013 and 2014, Martelli et al. (2013, 2014) reported on the generation of several classes of arylthioamide compounds. The original report (Martelli et al., 2013) reported on p-hydroxybenzothiomide as the lead compound and made several structural modifications, including the replacement of the p-hydroxy group with an amido group and replacement of the phenyl ring with various heterocyclic ring structures. A subsequent report focused on five aryl-isothiocyanate-based H2S donors: aryl isothiocyanates (PhNCS, PhNCS–COOH, PhNCS–CH3, PhNCS–CF3, and PhNCS–iPr) and compared them with the reference donors NaHS, DADS, and GYY4137 (Martelli et al., 2014). The major finding of the study was that the arylthioamide compounds, similar to the N-(benzoylthio)benzamides discussed in the section XV, in cell-free conditions, do not release detectable amounts of H2S, unless they are incubated with l-cysteine. However, in biologic systems, where cysteine and other thiols are abundant, these compounds were expected to release H2S, which, indeed was proven to be the case. A carboxy-compound (PhNCS–COOH, also termed 4-carboxy-phenyl-isothiocyanate or 4CPI) (Fig. 11C) was reported as the most effective H2S donor. In biologic systems, PhNCS–COOH, typically in the concentration range of 10–300 µM or in the dose-range of 0.07–0.7 mg/kg, exerts “H2S-like” effects, such as vascular relaxation, inhibition of vasoconstriction, increase in coronary blood flow of perfused hearts, hyperpolarization of vascular smooth muscle cells, and a reduction of infarct size in a rat model of myocardial ischemia-reperfusion; some of these protective effects were also associated with a reduction of oxidative stress (Martelli et al., 2014; Testai et al., 2016).
In 2014, Zhao et al. (2014) reported on the synthesis of geminaldithiol (gem-dithiol)-based compounds (termed TAGDDs), which constitute another class of thiol-activated H2S donors that release H2S via the mechanism exemplified by the compound TAGDD-1 (Figs. 11F and 13). TAGDD-1, in the presence of cysteine of glutathione in vitro, releases H2S on the minutes-to-hours time scale. Cell-based release of H2S from TAGDD-1 was confirmed (Zhao et al., 2014), but this group of compounds still awaits further pharmacological and biologic characterization.

Mechanism of H2S release from TAGDD-1. The reaction is initiated by a reversible thiol exchange between TAGDD-1 and cysteine to generate S-acetyl cysteine and gem-dithiol. S-acetyl cysteine undergoes a fast S- to-N-acyl transfer to form N-acetylcysteine and drive the equilibrium. Meanwhile, the gem-dithiol releases H2S spontaneously in aqueous solution to yield benzaldehyde.
Bełtowski et al. (2014) characterized the synthetic nucleotide analogs adenosine- and guanosine 5′-monophosphorothioates (AMPS and GMPS) and demonstrated that these compounds, in the presence of thiols, convert to H2S and AMP or GMP, respectively. These compounds relax isolated kidney glomeruli in vitro and increases glomerular filtration rate in vivo.
A series of N-mercapto-based H2S donors, exemplified the compound NSHD-1 and NSHD-2 (Fig. 11, D and E) have also been generated and tested in recent years (Yang et al., 2014; Zhao et al., 2015). These compounds are thiol-activated and exhibit medium speed of H2S release (much faster than most of the GYY compounds but slower than the sulfide salts). NSHD-1 exhibits cytoprotective and antioxidant effects in vitro (Yang et al., 2014), whereas both NSHD-1 and NSHD-2 (100 µg/kg) reduce infarct size in a murine model of myocardial ischemia-reperfusion in vivo (Zhao et al., 2015).
Additional, relatively incompletely characterized classes of H2S donor compounds also include various S-aroylthiooximes (SATOs) (Fig. 11G), which release H2S in the presence of cysteine or glutathione via the scheme shown in Fig. 14 (Foster et al., 2014).

Mechanisms of H2S release from S-aroylthiooximes (SATOs). Two pathways could lead to H2S generation from SATOs. The first involving addition of the cysteine thiol to the SATO acyl group followed by rapid S→N-acyl transfer (Pathway A). The arylidenethiooxime would form, which could decompose to generate a ketone or aldehyde along with H2S and NH3. The second pathway involves a hydrolysis step to generate the S-aroylthiohydroxylamine and the ketone or aldehyde used to make the SATO (Pathway B). The fast reaction between SATOs and cysteine to yield H2S (t1/2 approximately 8−82 minutes) compared with the hydrolysis rate (t1/2 ranging from 45–250 hours) rules out pathway B as the mechanism of H2S release mechanism for most SATOs under the conditions tested in Foster et al., 2014.
Recently, Barresi and colleagues reported on the synthesis and partial characterization of a series of iminothioethers (N-benzylbenzothioamide and arylimidothioate derivatives). The compounds required the presence of thiols for H2S release. Various members of the series exhibited variable rates of H2S release, and — in line with the biological roles of H2S — produced relaxation in isolated vascular rings, increased coronary blood flow in perfused hearts, lowered blood pressure in rats and exerted hyperpolarizing and cGMP-increasing effects in cultured smooth muscle cells (Barresi et al., 2017).
XVI. pH-Controlled H2S Donors
As mentioned earlier, during the original characterization of GYY4137 it was noted that its H2S release rate is faster at low pH conditions (Li et al., 2008b). Using the core structure of GYY4137 and coupling it with a new activation strategy that involves the intracellular cyclization mechanism shown in Fig. 15, Kang et al. (2016) designed a new class of pH-controlled GYY4137-derivative H2S donors. This project hypothesized that the rate of cyclization will be pH dependent. An example of this class of compounds (JK-2) is shown in Fig. 16A. The compound JK-1 was found to be a donor that only releases H2S at only slightly acidic pH (5 and 6), but not at neutral (7.4) pH or at alkaline pH. In contrast, JK-2 (as well as the related compounds JK-3 and JK-4), exhibited slow and sustained H2S release profiles at pH 7.4 and pH 8, but the H2S generation from this compound was, once again, much faster at pH 5 or pH 6. Because ischemic tissues have a lower (acidic) pH in vivo, it was hypothesized that the low pH-dependent H2S release profile may be useful in conditions associated with local acidosis (for instance, myocardial ischemia-reperfusion injury). Thus, JK-1 and JK-2 were next compared in vivo, in a murine model of coronary artery ligation and reperfusion (a model, where previous studies have already demonstrated the cardioprotective effect of fast-releasing H2S donors, e.g., Elrod et al., 2007). The in vivo data demonstrated the protective effect of both compounds at the low doses of 50 and 100 µg/kg but did not show significant differences between the effect of the two compounds in terms of efficacy (Kang et al., 2016).

Mechanisms of H2S release from pH-controlled donors. Protonation of phosphonamidothioates at neutral or slightly acidic pH yields the corresponding phosphorothiols. This process facilitates the release of H2S if a nucleophilic carboxylate is presented at a suitable position. The formation of the five-membered ring could be the driving force for H2S release.

Controlled-release and targeted H2S donors. JK-2 (A) and JK-1 (C) are based on a phosphonamidothioate template and generate H2S in a pH-controlled manner, ΒW-HP-101 is an esterase cleavable H2S donor (B), SPD-1 (D) and SPD-2 (E) are photoactivatable H2S donors, Donors depicted in (F) are thiocarbamate-based donors that generate H2S after exposure to ROS (mainly H2O2). AP39 (G) is a mitochondria-targeted donor consisting of a mitochondria-targeting motif (triphenylphosphonium) coupled to an H2S-donating moiety (dithiolethione; DTT) by an aliphatic linker.
In a recent report, Feelisch, Singer, and colleagues (Dyson et al., 2017) reported on ammonium tetrathiomolybdate (ATTM), previously known as a copper chelator compound, as an H2S donor, where the action is time, pH, temperature, and thiol dependent. The molecule was tested in vivo: when given intravenously at myocardial or cerebral ischemia-reperfusion to rats, the donor significantly reduced infarct size following ischemia and also extended survival in a model of severe hemorrhage. Mechanistic studies (in vitro anoxia/reoxygenation) suggested that the donor, at least in part, exerts its action via mitochondrial effects (decreasing mitochondrial ROS production) (Dyson et al., 2017). It is interesting to note that this molecule, in fact, is a clinical-stage drug development candidate (in cancer, where its putative mode of action is copper chelation) (Chan et al., 2017).
XVII. Redox-Activated H2S Donors
Zhao and Pluth (2016) started out from the general principle that oxidative stress consumes H2S, and, therefore, H2S delivery should be targeted to biologic sites where ROS production is increased. They have, therefore, synthesized a new class of H2S donors, where H2S release is triggered by oxidative stress. Various thiocarbamate-based donors (peroxythiocarbamate: PeroxyTCM-1, PeroxyTCM-2, and PeroxyTCM-3) (Fig. 16F) were prepared and tested for H2S release in vitro in the presence of various oxidants. H2S release was most potently triggered by H2O2, but superoxide and peroxynitrite also induced H2S release. In contrast, a variety of other species (e.g., hypochlorite, hydroxyl radical, singlet oxygen, tert-butyl hydroperoxide, tert-butoxy radical, glutathione, NO, nitroxyl, nitrite, sulfate, or thiosulfate) did not trigger H2S release from these compounds (Zhao and Pluth, 2016). H2S release was also confirmed from these compounds in biologic systems; in cultured macrophages incubated with phorbol-12-myristate-13-acetate to induce endogenous ROS production, the compounds exhibited the expected preferential H2S release profile. Moreover, the compounds demonstrated a protective effect in HeLa cells challenged with cytotoxic concentrations of H2O2 (Zhao and Pluth, 2016). The potential therapeutic benefit of these compounds remains to be further investigated in vivo.
XVIII. Photoactivated H2S Donors
The first examples of photosensitive or photoactivatable H2S donors were provided by Devarie-Baez et al. (2013). The gem-dithiol-based release mechanism (discussed in section XV) was protected by the light-sensitive 2-nitrobenzyl group. The compounds liberate H2S in response to ultraviolet light as the gem-thiols become exposed and subsequently hydrolyzed. By employing the properties of the ketoprofenate photolabile protecting group, Fukushima et al. (2014, 2015) synthesized photo-controllable H2S donors (termed SPD-1 and SPD-2) (Fig. 16D, E) as research tools and, perhaps, as prototypes for future therapeutic applications (e.g., in the context of cancer therapy/photoirradiation). The compounds release H2S via the mechanism shown in Fig. 17; the H2S release with SPD-2 occurs at longer wavelengths than with SPD-1. H2S release from the SPD series of compounds was confirmed in both cell-free systems (Fukushima et al., 2014) and as HEK293 cells in vitro (Fukushima et al., 2015).

Photoactivated H2S release from SPD-2.
XIX. Esterase-Activated H2S Donors
Recent studies demonstrated the feasibility of producing esterase-activated H2S donors. Zheng et al. (2016) started out from prodrugs based on intramolecular cyclization and applied a lactonization prodrug system in which the nucleophilic hydroxy or amino group was masked as an ester or amide, and the drug H2S was conjugated to the carbonyl carbon in the form of a thioacid (Fig. 16B). After hydrolysis of the masking group, the nucleophile attacks the carbonyl group and undergoes a lactonization reaction, resulting in the release of H2S, as exemplified by the compound BW-HP-101 (Fig. 18). A series of donors were made based on the above principle, and all of them released H2S in the presence of esterase enzyme in phosphate-buffered saline over a period of approximately 60 minutes. The same group also synthesized BW-HP-105, where the above outlined H2S-releasing system was coupled to the clinically used NSAID naproxen. Another group synthesized pivaloyloxymethyl-based carbonothioates and carbamothioates that are activated by the esterase to generate carbonyl sulfide (COS), which is hydrolyzed to H2S via mechanisms outlined in section XIV (Chauhan et al., 2017).
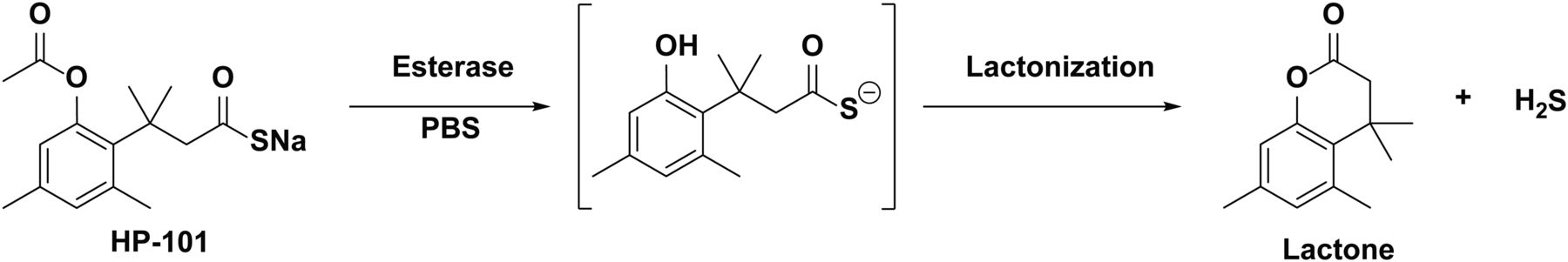
Mechanism of H2S release from esterase-cleaved prodrugs. Prodrugs of this category release H2S upon cleavage of an ester group, followed by lactonization. H2S release rates can be changed by 1) modifying the ester group (acyl moiety) and thus altering susceptibility to esterase and 2) altering structural features that are crucial for the lactonization rate. BW-HP-101 is shown as an example.
The original publications contained only very limited data in biologic systems, e.g., inhibition of TNFα production in cultured murine macrophages or antiproliferative effects in cancer cells (Zheng et al., 2016; Chauhan et al., 2017), but failed to investigate the standard issues related to esterase-activated prodrugs (e.g., their stability in plasma in the presence of plasma esterases, their pharmacokinetic properties and elaboration of H2S in plasma or in cell cultures or in vivo, relative conversion of the compounds by plasma versus intracellular esterases, in vivo H2S release profile and half-life, in vivo safety and tolerability, etc.). It is hoped that these issues will be delineated in follow-up studies. Without this information, it is difficult to ascertain whether esterase-convertible H2S releasing prodrugs, other than the lack of odor and their likely good stability upon storage, have any clear advantages.
XX. Mitochondrially Targeted H2S Donors
The H2S donors discussed in sections VI-XIX do not have any intended subcellular localization. It has been, therefore, assumed that these compounds either release their H2S “load” extracellularly (and then H2S diffuses into the cells and distributes into all cellular compartments), and/or the compounds themselves enter the cells (and, once again, enter all compartments without any particular targeting or specificity). Whether this was indeed the case, however, has not been directly tested until recently, when Montoya and Pluth (2016) produced a series of organelle-specific H2S detection probes using the SNAP-tag fusion protein methodology and tested subcellular localization of H2S after cells were exposed to various H2S donors. Indeed, NaHS, DATS, and GYY4137 produced an increase in all cellular compartments studied, including the mitochondria and the lysosomes.
Because of the multiple physiological and protective effects of low (physiological) concentrations of H2S on mitochondrial function (see above) and also because the mitochondria can produce high levels of ROS that can lead to a loss of biologic H2S levels, it makes sense to specifically target H2S delivery to the mitochondria. The first is an example in which mitochondrially targeted H2S donors were synthesized and characterized in vitro (Le Trionnaire et al., 2014; Szczesny et al., 2014). The compounds AP39 or (10-oxo-10-(4-(3-thioxo-3H-1,2-dithiol5yl)-phenoxy)decyl) triphenylphosphonium bromide (Fig. 16G) and a related compound AP123 combine the well-known mitochondrial targeting moiety triphenylphosphonium (TPP+) with a dithiolethione H2S delivery moiety. AP39 and AP123, as expected, selectively increase the H2S signal in the mitochondrial compartment of cultured cells (Le Trionnaire et al., 2014; Szczesny et al., 2014; Montoya and Pluth, 2016) and exert cytoprotective effects in various cell types (e.g., endothelial cells, epithelial cells, platelets, neurons, cardiac myocytes) against various forms of oxidative stress (H2O2, rotenone, glucose oxidase, elevated extracellular glucose, anoxia/reoxygenation) in vitro in high nanomolar/low micromolar concentrations (D'Araio et al., 2014 Le Trionnaire et al., 2014; Szczesny et al., 2014; Emerson et al., 2015; Sitek et al., 2015; Ahmad et al., 2016a; Chatzianastasiou et al., 2016; Gerő et al., 2016; Zhao et al., 2016a; Lobb et al., 2017). Currently, AP39, which appears to be a more effective H2S releaser than AP123 (Gerö et al., 2016), has been studied more extensively. Interestingly, and consistently with the positive bioenergetic effects of low concentrations of H2S, low concentrations of AP39 (25–100 nM) induced an elevation in basal mitochondrial activity (basal respiration and maximal respiration) of cultured endothelial cells and neurons, whereas at higher concentrations (250–300 nM) this effect is no longer detectable (Szczesny et al., 2014; Zhao et al., 2016a), consistent with the bell-shaped mitochondrial action of H2S. Mitochondrial H2S delivery with AP39 improved mitochondrial function in oxidatively stressed cells, at least in part due to the fact that it reduced mitochondrial ROS levels (Szczesny et al., 2014; Karwi et al., 2017). In addition, AP39 and AP123 also improved mitochondrial DNA repair in oxidatively stressed cells (Szczesny et al., 2014, Gerő et al., 2016). Recent work identified some of the cellular targets involved in this response and demonstrated that mitochondrial H2S promotes the assembly and activity of mitochondrial DNA repair complexes (Szczesny et al., 2016). Importantly, the intracellular signaling processes elicited by AP39 are different from the signaling induced by nontargeted H2S donors, consistent with the differential localization of the various cellular targets of H2S.
A potential advantage of mitochondrially targeted donors is that their effects are not dependent on the functional integrity of the NO system and do not involve activation of the cGMP/protein kinase G system (Tomasova et al., 2015; Chatzianastasiou et al., 2016; Karwi et al., 2017). However, actions through CaV3, RyR2, and Cl– cardiac membrane channels have been implicated in its pharmacological actions (Tomasova et al., 2015), perhaps related to the accumulation of AP39 in the nodal cells due to the high positive charge of the compound and high negative resting potential of the nodal cells. AP39 may also act as a mitochondrial permeability transition pore “desensitizer” and a blocker of the permeability pore (Chatzianastasiou et al., 2016; Karwi et al., 2017).
In vivo, AP39 recapitulates some of the physiological effects characteristic of H2S and other H2S donors (e.g., a decrease in blood pressure and heart rate) (Tomasova et al., 2015) and at low doses (approximately 1000-times lower doses than the typical doses of the “fast-releaser” salt-based H2S donors) exerts protective effects in various models, including acute cardiac arrest (Ikeda et al., 2015), renal ischemia-reperfusion induced by either renal artery ligation (Ahmad et al., 2016a) or storage and transplantation (Lobb et al., 2017), myocardial ischemia-reperfusion (Chatzianastasiou et al., 2016; Karwi et al., 2017), burn injury (Ahmad and Szabo, 2016), and neurodegeneration (Zhao et al., 2016a). Although the work with mitochondrially targeted H2S donors focuses primarily on cytoprotective actions, another line of studies indicates that the same compounds may also be used as an anticancer approach, e.g., in combination with methyl aminolevulinate-induced photodynamic therapy (Ferguson et al., 2014).
Although targeting H2S to the mitochondria appears to be an elegant and specific concept, a few complicating factors and potential concerns should also be mentioned. First, one of the key toxicological effects of H2S is the mitochondrial electron transport chain. Thus, delivery of H2S to the mitochondria at concentrations higher than desired is expected to induce a suppression of aerobic respiration and ATP generation; this is clearly shown by the bell-shaped dose-response of AP39 on cellular bioenergetics (Szczesny et al., 2014). Thus, the identification of the exact therapeutic concentrations/doses and avoidance of overdosing will be crucial with compounds of this type. Overdosing with the compound may also cause problems because the TPP moiety itself can also induce cytotoxicity (Guzman-Villanueva and Weissig, 2017). Second, at higher concentrations, AP39 will increase H2S levels not only in the mitochondria but also in cytoplasmatic compartments (Szczesny et al., 2014); thus, at higher doses/concentrations the compound is no longer mitochondrially selective. Third, when the accumulation of the compound in the mitochondrion relies on lipophilic cations such as the triphenyl phosphonium (TPP) moiety, there are limitations and caveats associated with this approach. The delivery of the molecule into mitochondria is highly dependent on the mitochondrial membrane potential; this means that in pathophysiological conditions associated with mitochondrial dysfunction, the mitochondrial targeting function of the molecule may be impaired. Indeed, in the presence of the mitochondrial uncoupler carbonyl cyanide-4-phenylhydrazone, AP39 no longer shows a preferentially mitochondrial increase H2S fluorescence (Szczesny et al., 2014). This means that in the very same diseases in which mitochondrial H2S delivery may be therapeutically warranted, the mitochondrial targeting of the H2S donor may become impaired. Finally, it should also be mentioned that, although over the last two decades the “TPP-coupling approach” has produced many useful experimental tools, e.g., targeting of various antioxidants to the mitochondria, as exemplified by mito-Q, mito-tempol, mito-E, etc. (Murphy, 2008; Reily et al., 2013) and some clinical trials (e.g., with mito-Q: Frantz and Wipf, 2010), it has not yet produced any clinically approved drugs.
XXI. NO/H2S Hybrid Donors
Starting out from the general principles that 1) in many diseases, NO and H2S homeostasis becomes impaired simultaneously and 2) H2S and NO often exert cooperative interactions, for instance on angiogenesis and vascular relaxation (Hosoki et al., 1997; Coletta et al., 2012, Altaany et al., 2013; Szabo, 2017b), several different groups of investigators have come up with the idea that simultaneous delivery of NO and H2S from the same molecule may be a potentially useful experimental therapeutic approach.
The compound ZYZ-803 (2-amino-3-propynylsulfanyl-propionic acid) was created when S-propyl-l-cysteine was combined with furoxan (a compound that releases NO in the presence of thiols) (Fig. 19A). In isolated aortic rings, ZYZ-803 (10–100 µM) produced a simultaneous elevation in NO and H2S levels (Wu et al., 2016a) and increased vascular cGMP levels in vitro and activated downstream signaling, evidenced by vasodilator-stimulated phosphoprotein phosphorylation (Wu et al., 2016a). It also stimulated endothelial cell angiogenesis in vitro (Hu et al., 2016). In vivo, intragastric administration of the compound induced the simultaneous elevation of plasma H2S and NO levels for several hours, and treatment with the compound [8.7 (mg/kg)/day for 14 days] improved angiogenesis in a hind limb ischemia model in mice. The results of concomitant control groups indicated that the efficacy of ZYZ-803 is higher than the effect of either the H2S or the NO-generating component of the molecule alone, as expected from the cooperative actions of the two mediators.
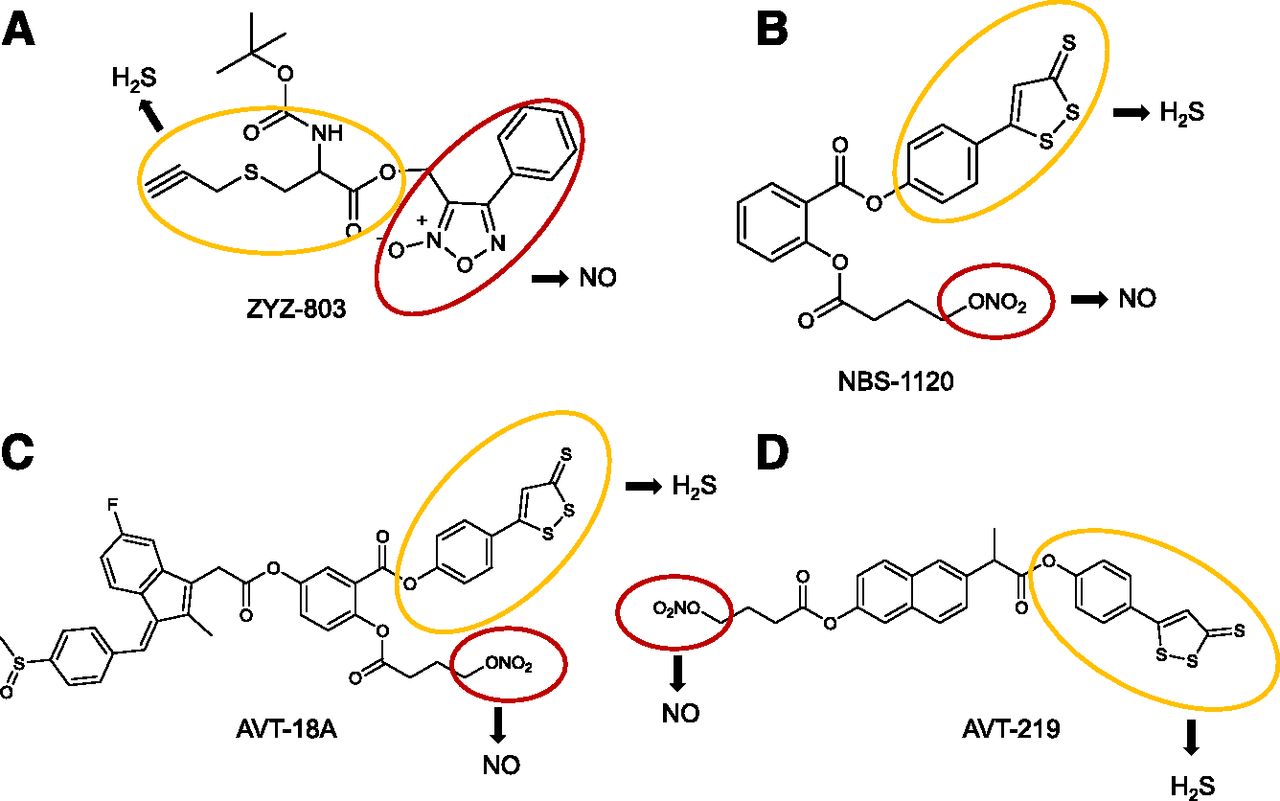
(A) ZYZ-803 is a derivative of S-propyl-l-cysteine and is thiol activated. (B) NBS-1120 is NOSH-aspirin (also designated as NOSH-1). (C) AVT-18A is a sulindac derivative. (D) AVT-219 is a naproxen derivative.
Another class of nitric oxide-hydrogen sulfide-releasing hybrids is based on a modified (S)-3-n-butylphthalide core (Wang et al., 2016d). From a series of molecules, compound NOSH-NBP-5 was found to release moderate amounts of NO and H2S and displayed significantly inhibitory effects on platelet aggregation in vitro. Further studies are needed to characterize the effects of this compound in vitro and in vivo.
In addition to H2S-donating versions of approved, clinically used drugs (as discussed below), several groups have also made combined NO/H2S donating versions of such drugs. The synthesis of several series of compounds, including NOSH-aspirin (NOSH-1 or NBS-1120), which subsequently has been subjected to detailed characterization (Fig. 19B) was reported in 2012 (Chattopadhyay et al., 2012a,b; Kodela et al., 2012). The release of both NO and H2S was confirmed from the compound in vitro. The first efficacy testing focused on proapoptotic and antiproliferative effects in cancer cell lines and in tumor-bearing mice in vivo. The compound exerted nanomolar inhibitory effects on cancer cell proliferation (which was markedly more potent than the effect of the parent compound or either the NO- or the H2S-releasing components on their own). In contrast to the remarkable in vitro potency, in vivo antitumor effects were reported only at one single, fairly high (although well tolerated), dose of the compound [100 (mg/kg)/day]. In the in vivo studies, the efficacy of NBS-1120 was not compared with the parent compound (aspirin) or to molecules that release either only H2S or NO; thus, the mode of the compound’s in vivo action remains to be clarified.
Subsequent work has synthesized meta-, ortho-, and para-isomers of NBS-1120 and found potent antiproliferative efficacy for all of them in vitro, with o-NOSH-ASA being approximately fivefold more potent than m-NOSH-ASA and 10-fold more potent than p-NOSH-ASA (Vannini et al., 2015a,b). Moreover, information disclosed in one of these follow-up reports revealed that part of the antitumor action of this series of compounds involves the induction of oxidative stress in the target tumor cells (Vannini et al., 2015a).
Subsequent work explored the anti-inflammatory efficacy of NBS-1120 in standard carrageenan models in vivo. Although in most tests (acetic acid-induced writhing responses, motor coordinance tests, carrageenan- and CFA-induced inflammatory hyperalgesia, carrageenan-induced neutrophil migration) the actions of NBS-1120 (150 µmol/kg) were comparable to the same dose of the parent compound (aspirin); in one of the assays (PGE2-induced hyperalgesia), NBS-1120 was significantly more potent than aspirin (Fonseca et al., 2015).
NOSH-sulindac (AVT-18A) (Fig. 19C) and NOSH-naproxen (AVT-219) (Fig. 19D) (Kodela et al., 2013), similar to the various NO-donating or H2S-donating NSAIDs synthesized earlier, were found to be safer in terms of gastric mucosal damage after oral administration than the parent compounds. AVT18-A exerted anti-inflammatory effects (reduction of prostaglandin production) to a similar degree than sulindac, but in contrast to sulindac, it did not induce the downregulation of superoxide dismutase activity in the stomach, and, once again, in contrast to sulindac, it did reduce tissue oxidative damage (tissue malon dialdehyde levels). ACT-18A maintained most of the anti-inflammatory, analgesic, antipyretic, and antiplatelet properties of its parent compound in standard murine carrageenan models (Kashfi et al., 2015). Moreover, AVT-18A and AVT-219 also exerted antiproliferative effects in various human cancer cell lines (colon cancer, Jurkat cells, breast cancer cells, pancreatic cancer cells) in vitro with substantially higher potency (mid-nanomolar IC50) than the parent compound. In contrast, the hybrid donor compounds had a less pronounced effect on the growth of nontransformed cell lines, and on these cells, the parent compounds and the hybrids had comparable (mid-micromolar IC50) potencies (Kodela et al., 2013; Kashfi et al., 2015).
XXII. H2S-Donating Polymers and Special Pharmaceutical Formulations
Targeted delivery and/or sustained release of H2S would be expected to confer significant advantages in the context of a variety of pharmaceutical applications (e.g., topical formulations, depot formulations).
Foster and Matson (2014) evaluated the effectiveness of thiooxime formation as a postpolymerization modification reaction for H2S release. Methacrylate polymers bearing pendant aldehyde functionality were prepared via reversible addition-fragmentation chain transfer polymerization of 2-(4-formylbenzoyloxy)ethyl methacrylate. Polymer side chain derivatizations with tert-butylhydrazide, O-benzylhydroxylamine, and S-aroylthiohydroxylamine were evaluated and were shown to form the corresponding hydrazone, oxime, and thiooxime molecules, respectively. According to the thiol functionality, S-aroylthiooximes decomposed to release H2S in the presence of thiols (Foster and Matson, 2014). Hasegawa and van der Vlies (2014) tested another approach and linked poly(ethylene) glycol with the H2S-releasing moiety ADT via an ether bond. The H2S-releasing activity of the resulting PEG−ADT conjugate was confirmed in murine macrophages. However, H2S release from the polymer was not detectable in the presence of serum proteins. The cell uptake of the PEG−ADT conjugate was mediated by the endocytic pathway. The molecules remained sequestered inside endolysosomes. PEG−ADT was capable of potentiating LPS-induced inflammation. Potentially therapeutically relevant (e.g., cytoprotective or anti-inflammatory) properties of this polymeric H2S donor were not reported; instead, the compound exerted a concentration-dependent inhibitory effect on cell viability and enhanced LPS-induced TNFα production in cultured macrophages (Hasegawa and van der Vlies, 2014).
PEG-conjugation has recently also been conducted in conjunction with garlic-inspired molecules by Ercole and colleagues (Ercole et al., 2017) A trisulfide linkage was incorporated into a conjugate comprising an mPEG tail and a cholesteryl head. The resulting compounds release H2S in a thiol-dependent manner. The compounds also release H2S in cultured HEK293 cells (without externally added thiols).
Starting out from thiol-activated H2S donors based on the S-aroylthiooxime (SATO) functional group (see above), Carter et al. (2015) reported on the preparation of a self-assembling peptide designed to form an H2S-releasing gel. Upon gelation, the SATO-containing aromatic peptide amphiphile appears to form at a β-sheet-type 3-dimensional structure forming a nanofiber. The material produced a time-dependent release of H2S, which was monitored over a 5-hour period in a cell-free system. Incubation of cultured endothelial cells with the gel also resulted in a detectable release of H2S, but only when 0.5 mM cysteine was also added to the culture medium (Carter et al., 2015).
Another approach explored the synthesis of macromolecular H2S donors based on copolymers having pendent oligo(ethylene glycol) and benzonitrile groups. The benzonitrile groups were subsequently transformed into primary aryl thioamide groups via thionation using sodium hydrosulfide. These thioamide moieties were, in turn, incorporated into a hydrophilic copolymer or a block copolymer structures (Ercole et al., 2016). The polymers exhibited rapid-onset, thiol-activated H2S release profiles. When added to the tissue culture medium of cultured H460 lung carcinoma cells, they activated extracellular signal-regulated kinase and protein kinase C signaling (Ercole et al., 2016).
Yet another approach produced H2S-releasing fibrous scaffold nanofibers via electrospinning of polycaprolactone solutions containing the H2S donor N-(benzoylthio)benzamide. Ultimately, the intended application of these fibers is wound dressing, based on the known stimulatory effect of H2S on angiogenesis and wound healing (reviewed in Szabo and Papapetropoulos, 2011). However, so far, all current studies with this fiber are in various in vitro models only. The nanofibers, in a cysteine-triggered manner, exhibited a sustained H2S release profile that lasted at least for 24 hours, induced H2S release, and afforded cytoprotection against oxidant-induced cytotoxicity in cell culture models. Moreover, the nanofibers were found to stimulate the proliferation of fibroblasts and induce the expression of various genes relevant for wound healing (e.g., collagen and smooth muscle actin) in cultured fibroblasts (Feng et al., 2015a).
An independent line of investigators explored a different approach for controlled, dermal delivery of H2S to accelerate wound healing. This approach started out with the pH-controllable H2S donor JK1 (discussed above) and incorporated it into H2S-releasing nanofibers through electrospinning of polycaprolactone. The resulting fibrous scaffold (designated as PCL-JK1) showed pH-dependent H2S-releasing properties similar to that of JK1 (higher H2S release rate at acidic pH), but overall the rate of H2S release was slower than H2S release from the parent JK1 in solution. In vitro studies demonstrated that that PCL-JK1 exhibits good cytocompatibility. PCL-JK1 was also tested in a cutaneous wound model in mice in vivo and found to enhance the wound repair and regeneration (including enhanced neovessel formation and increased collagen deposition), especially over the first week of the wound healing process (Wu et al., 2016c).
Recently, another H2S-releasing depot formulation (termed “NaHS@MPs”) has been produced for the therapy of wound healing (Lin et al., 2017). The formulation involves a microparticle system comprising of hydrophobic phase-change materials (1-tetradecanol and paraffin wax). The formulation provides an in situ depot for the sustained release of H2S. When wounds of diabetic mice were covered with the Tegaderm integrated with NaHS@MPs, re-epithelialization and wound closure was significantly accelerated, compared to the Tegaderm control group.
Recently, Wang et al. (2016c) produced mesoporous silica nanoparticles (MSNs) using the sol-gel method and loaded them with DATS. The resulting material, designated as DATS-MSN, exerted cytoprotective effects in endothelial cells and attenuated inflammatory mediator production and adhesion molecule expression in endothelial cells; it also enhanced their proliferation and migration. In addition, in vivo experiments were conducted to investigate the protective effects of DATS-MSNs on the endothelium of transplanted aortas. In these experiments, the survival and function of aortic segments either preserved in standard University of Wisconsin solution or University of Wisconsin solution supplemented with DATS-MSN were compared. Aortas were anastomosed onto the recipients’ abdominal aortas and evaluated 3 days later. DATS-MN storage reduced the number of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells in the grafts, indicative of cytoprotective effects (Wang et al., 2016c). In a follow-up study, DATS-MSN was also reported to exert cytoprotective and anti-inflammatory effects in rat myocardial ischemia-reperfusion models (Sun et al., 2017).
Another, natural H2S donor-containing nanoformulation, developed for antitumor indications, consists of DADS and α-linolenic acid as a protein-nanoemulsions (BAD-NEs). This formulation, as expected, exerts antioxidant effects and inhibits tumor cell proliferation (in the concentration range of 25–100 µM) (Ciocci et al., 2016).
One obvious area of long-acting H2S donor formulations is ocular applications (glaucoma), given the fact that intraocular administration of various H2S donors is known to have a significant lowering effect on the intraocular pressure (Salvi et al., 2016; Huang et al., 2017). Ali et al. (2014) at Teva Pharmaceuticals disclosed the preparation of a dozen sustained release H2S formulations (designated as “phase-sensitive smart polymer-based formulations”) by dissolving NaHS in polymer solutions, prepared by dissolving polymers (consisting of either polylactide or polylactide coglycolide, containing free carboxylic acid or capped allyl ester end group) in a mixture of benzyl benzoate and benzyl alcohol. The polymers contained 0.8% w/v NaHS, had physiological osmolarity, and their pH was adjusted to 7.4. Follow-up studies characterized the H2S-releasing properties of the formulations in artificial tear solutions. They had a variable degree (6%–27%) of initial H2S release (“burst release”), followed by a steady release of H2S, which was tracked over a period of 72 hours. It was concluded that controlled release of H2S (suitable for ocular indications where subconjunctival or subcutaneous administration would be the possible delivery routes) was achieved as a proof-of-principle by many of the disclosed formulations (Ali et al., 2014).
Most recently, sustained release formulations of GYY4137 (a compound that, as discussed above, is already very slow H2S releasing, even without any additional formulation) were reported by Patil et al. (2017). The delivery system (termed “phase-sensitive smart polymer-based in situ gelling delivery system containing GYY 4137”) exhibited sustained H2S release prepared by dissolving GYY4137 in poly lactide-coglycolide polymer (Resomer RG 502H) solution, which was prepared by dissolving polymer in a mixture of benzyl alcohol and benzyl benzoate in a ratio of 7:3, respectively. The formulation exhibited acceptable parameters for syringeability/injectability, pH and tonicity, moisture content, GYY 4137 degradation, and in vitro cytotoxicity in a retinoblastoma cell line. H2S release from the formulation was monitored over a 72-hour period; the formulation released H2S at a rate that is approximately 50% lower than the H2S release from unformulated GYY4137 solutions (Patil et al., 2017).
XXIII. Combined (or Hybrid) Molecules: H2S-Donating Derivatives of Clinically Used Drugs
To improve the therapeutic profile (safety and/or efficacy) of clinically used drugs, H2S-releasing versions of many clinically used drugs have been synthesized and characterized. Most of these compounds couple the H2S-donating group 4-hydroxy-thiobenzamide to the parent molecule. The molecule that progressed the most into clinical development (with Phase 2 trials completed) is the H2S-releasing derivative of naproxen (ATB-346 or [2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester]), which will be first discussed, in detail, in this section, followed by the various other, clinical or preclinical-stage combined/hybrid molecules.
The first report in the scientific literature on ATB-346 (Fig. 20A) was published in 2010 (Wallace et al., 2010). The selection of naproxen as the “base drug” in ATB-346 was made because it is considered the most cardiovascular-safe of the NSAIDs. Initial studies with ATB-346 (30 µmol/kg, orally) evaluated its anti-inflammatory effect in comparison with an equimolar dose of naproxen in standard zymosan mouse air pouch models. Zymosan injection into the air pouch induces a marked influx of leukocytes, coupled with a massive increase in exudate concentrations of PGE2 and an increase in blood TXB2 levels. Exudate leukocyte and PGE2 levels were significantly reduced by naproxen and its H2S-releasing derivative to a comparable degree. With respect to PGE2 levels, a significantly more pronounced inhibition was observed with ATB-346 than with naproxen, perhaps suggestive that the addition of the H2S-releasing group to naproxen, in addition to conferring a H2S-releasing property to the molecule, may increase the potency of the compound as a COX inhibitor. Whole blood TXB2 synthesis was almost completely suppressed by both naproxen and ATB-346. In contrast to naproxen (which induced the expected gastric damage), ATB-346 exerted no adverse effect on the integrity of the gastric epithelium (Wallace et al., 2010). Studies in an adjuvant arthritis-associated paw swelling model demonstrated the efficacy of ATB-346 in terms of inhibition or edema; at some time points, once again, ATB-346 appeared to be more efficacious than naproxen (Wallace et al., 2010). In a study in zymosan-induced arthritis in rats, once again, ATB-346 was more effective than naproxen in terms of various nociceptive responses and multiple inflammatory cellular and biochemical parameters (Dief et al., 2015), whereas in rat models of carrageenan arthritis, ATB-346 and naproxen were found to be comparably effective (Ekundi-Valentim et al., 2013). Despite these multiple suggestions that ATB-346 may not only be a safer version of naproxen, but, in some cases, may also be more potent or more efficacious, subsequent studies have generally focused on the H2S-donating properties of this compound, until the issue of potency difference has re-emerged in the human clinical trial stage (see below).

Combined (hybrid) H2S-donating derivatives of clinically used drugs. ATB-346 naproxen-benzamide conjugate (Α), ATB-429 is a mesalamine-ADT conjugate (Β), GIC-1001 is trimebutine 3-thiocarbamoylbenzene-sulfonate (C), ATB-337 (also known as ACS15) is a diclophenac-benzamide conjugate (D), ACS-6 is a sildenafil-ADT conjugate (E), ACS14 is an aspirin-ADT conjugate (F), ACS67 is a latanoprost-ADT conjugate (G) and ACS84 is an l-DOPA-ADT conjugate (H), compound 4 is an analog of adenosine (I).
A structurally related, but non-H2S-releasing analog of ATB-346 (naproxen-4-hydroxybenzamide) has also been produced and was tested in some of the initial studies (where it showed no gastric-sparing effects) (Gemici and Wallace, 2015) but has rarely been used in subsequent experiments.
Follow-on studies with ATB-346 have evaluated the efficacy and safety of this compound in a diverse array of models, typically in rats or mice. It has been demonstrated that ATB-346 does not induce gastric damage even at extremely high doses that are approximately 10-times higher than the doses required for its anti-inflammatory effects. ATB-346 was found also to be safe in models in which the gastric defense was impaired by systemic inhibition of nitric oxide synthesis. In addition, in contrast to naproxen, which caused significant damage to the small intestinal epithelium, ATB-346 was safe in this regard as well (Wallace et al., 2010). Finally, ATB-346 (in contrast to naproxen, which exerted a slight, but significant hypertensive effect in rodents) did not cause any significant change in blood pressure (Wallace et al., 2010).
In a follow-up study, ATB-346 (14.5 mg/kg) was evaluated in healthy, arthritic, obese, and hypertensive rats and in aged (19 months old) rats and in rats treated with low-dose aspirin and/or the proton pump inhibitor omeprazole. In all of these models (except hypertension), naproxen induced more severe gastric and/or intestinal damage than the corresponding effect in healthy rats. In addition, celecoxib-induced damage was significantly increased when the compound was coadministered with low-dose aspirin and/or omeprazole. ATB-346, at doses that were as effective as naproxen and celecoxib in reducing inflammation and inhibiting cyclooxygenase activity, exerted no adverse effects on the integrity of the gastric or intestinal epithelium, indicating that the gastrointestinal-sparing properties of ATB-346 are maintained even in the presence of various comorbidities (Blackler et al., 2012).
Part of the reason for the differential efficacy of ATB-346 than naproxen may be related to its differential absorption, metabolism, or excretion. Measurement of plasma naproxen levels after oral administration of ATB-346 or naproxen (16 and 10 mg/kg, respectively) to male Wistar rats found substantially higher naproxen levels in the naproxen-treated group than in the ATB-346-treated group (Ekundi-Valentim et al., 2013), indicative of slow conversion/metabolism of ATB-346 to naproxen in vivo. Similar findings (lower naproxen levels with ATB-346 than with naproxen) were reported in a second paper (Gemici et al., 2015). In contrast, an independent study in rats using a similar experimental design reported almost overlapping naproxen concentration/time curves after dosing at 86 µmol/kg (Elsheikh et al., 2014). A study in rats demonstrated that bile levels of naproxen (and associated bile-induced cytotoxicity) is higher in naproxen-treated animals than in animals treated with equimolar doses of ATB-346 (Blackler et al., 2015).
Additional experimental models where ATB-346 exerted beneficial effects include a mouse model of spinal cord injury (Campolo et al., 2013); a mouse model of traumatic brain injury (Campolo et al., 2014); a rat model of stress ulcers (Fomenko et al., 2014); a rat model of nonerosive esophagitis (Khyrivska et al., 2014); a rat model of ligature-induced periodontitis (Herrera et al., 2015); two different mouse models of intestinal carcinogenesis (Elsheikh et al., 2014; Paul-Clark et al., 2016); and a streptozotocin-induced cognitive impairment, neuroinflammation, and oxidative stress model in rats (Mostafa et al., 2016). In addition, at higher concentrations (100 µM) in vitro, ATB-346 induced melanoma cell apoptosis, whereas at a dose of [43 (µmol/kg)/day)] in vivo, ATB-346 significantly reduced melanoma development in vivo in tumor-bearing mice (De Cicco et al., 2016). Since these indications are unrelated to the current clinical development direction, the results of these studies are not discussed here in detail. However, from a mechanistic point of view, it should be mentioned that some of these reports unveiled pharmacological effects of the compound, which are not known if they are related to the H2S-donating action of the compound or are secondary to its anti-inflammatory effects (thereby possibly interrupting various positive feedforward cycles of injury) or independent pharmacological effects of the compound through mechanisms to be explored. Such actions include an inhibitory effect on the expression (not activity) of COX-2, an effect not shared with the parent compound naproxen (Campolo et al., 2013); an upregulation of the antiapoptotic protein bcl2 (an effect that was much more pronounced with ATB-346 than with naproxen) (Campolo et al., 2013); and the upregulation of glial cell-derived neurotrophic factor, GNF, vascular endothelial growth factor, and eNOS (Campolo et al., 2014). There is also a differential effect of ATB-346 and naproxen on the makeup of the intestinal microbiota (Blackler et al., 2015), an observation that needs to be further investigated for both its mechanism and its potential therapeutic relevance.
One of the unresolved issues with respect to ATB-346 (as well as many of the other hybrid H2S-releasing drugs discussed in the following paragraphs) is the mechanistic understanding of its H2S-releasing character. There is only a very limited amount of information on the rate and magnitude of H2S release from ATB compound in aqueous solutions or in plasma or in cells or cell extracts. When ATB-337 was incubated in potassium phosphate buffer, H2S generation, as detected from a H2S-sensitive electrode, was estimated as 12 nmol/min (while the control compound ADT-OH only released negligible amounts of H2S). On the other hand, both ATB-337 and ADT-OH released approximately three times higher amounts of H2S in liver homogenates (Wallace et al., 2007a). These data suggest a part spontaneous, part biologically catalyzed mechanism for H2S release. Based on claims contained in the patent literature it is suggested that 1) ATB-346 releases H2S spontaneously (in buffer, and a higher amount in the presence of tissue) via a two-step process proposed by Gemici et al. (2015) (Fig. 21);13 2) the release of H2S occurs independent of the activity of the two main enzymes for endogenous synthesis of H2S (CBS, CSE), 3) the concentration of H2S produced from 1 mM compound is in the 10–20 µM range, and 4) in vivo, the ATB-337 and ATB-346 produces detectable, but often modest and/or short-lived increases in plasma H2S concentrations (Wallace et al., 2007, 2008).

Proposed mechanism of H2S release and breakdown of ATB-346. The release of H2S from ATB-346 occurs at a very low pace when the drug is dissolved in an aqueous solution. The rate of H2S release is enhanced in the presence of tissue or in the presence of reducing agents (dithiothreitol, l-cysteine, or glutathione).
Importantly, in inflammation studies, separate administration of naproxen and the H2S-releasing group 4-hydroxy-thiobenzamide failed to induce the gastric protective effect seen with the compound (Wallace et al., 2010), suggesting that spatial proximity of the H2S-donating group to the site of naproxen’s action is probably critical for the beneficial pharmacological effects of ATB-346, although the mechanism of this action remains to be further characterized. In a melanoma study, where ATB-346 induced apoptosis, the parent compound naproxen did not (De Cicco et al., 2016), and therefore we can assume that the effects are related to the release of high local concentrations of H2S to the tumor cells. Likewise (and surprisingly) plasma levels of H2S after ATB-346 treatment in vivo are rarely reported. In the study by Dief et al. (2015), plasma levels of total sulfide are reported in arthritic animals with or without naproxen or ATB-346 treatment. Arthritis, on its own, fails to affect plasma H2S levels. In arthritic animals treated with naproxen [10 (mg/kg)/day for 5 days], plasma H2S levels are decreased below baseline by approximately 30%, whereas in arthritic animals treated with ATB-346 [15.9 (mg/kg)/day for 5 days], plasma H2S levels increased above baseline by approximately 30% (Dief et al., 2015). However, since no data were presented with either naproxen or ATB-346 in control (nonarthritic) animals, it is hard to evaluate the meaning of these data. Nevertheless, this is to our knowledge the only publication so far that reports the effect of ATB-346 on H2S levels in vivo.
Although some of the mechanistic issues related to ATB-346 remain to be clarified, it is clear from the above sections that the compound exerts anti-inflammatory effects in a multitude of models and exerts gastric protective effects. These properties justify clinical development, given the fact that the currently used NSAIDs leave plenty of room for improvement, both for safety and for efficacy. The status of the ATB-346 clinical program was recently reviewed by Wallace et al. (2017). In the phase 1 clinical trial, ATB-346 did not have any significant effects on blood pressure compared with placebo, in line with the preclinical data. Similarly, in the Phase 2 clinical trial in osteoarthritis patients (see below), systolic or diastolic pressure remained unaffected by the compound. ATB-346 was safe and well tolerated in the single-ascending-dose portion of the Phase 1 clinical trial, where ATB-346 was administered at increasing doses (from 25 to 2000 mg). Plasma naproxen levels increased in a dose-dependent manner after ATB-346 administration but remained lower than what would be expected from naproxen dosing. However, the plasma half-life of naproxen was longer in the ATB-346-treated healthy human volunteers than what would be expected from the administration of naproxen itself, either suggesting a slow conversion or perhaps a slower elimination of the compound. The multiple-ascending-dose portion of the Phase 1 trial showed similar patterns and the compound remained well tolerated, and the pharmacokinetics of the compound were as expected until the ATB-346 (750 mg twice daily) dosing group was initiated, where markedly higher naproxen levels started to accumulate, a finding that was not expected based on the results of the previous cohorts. At this dose, there were three patients with increased liver enzymes (consistent with the high plasma levels of naproxen) and one adverse event also occurred (marked elevation in liver enzymes), which led to discontinuation of a subject’s participation. Subsequent examination revealed that this patient should not have been enrolled into the trials in the first place (gallstone and undisclosed previous history of hepatitis).
These findings necessitated further examination of the pharmacokinetic and pharmacodynamic properties of ATB-346 before continuation of the clinical trial program. These studies revealed that, in patients, ATB-346 is a much more rapid onset and much more prolonged suppressor of COX) activity (as evidenced by prostaglandin E2 and thromboxane B2 levels) than the parent compound. The most likely explanation for this finding is that ATB-346 itself (or, perhaps a metabolite of ATB-346), is a potent COX inhibitor in vivo. These observations resulted in a substantial lowering of the dose of ATB-346 in the Phase 2 clinical trials. In these trials, ATB-346 was given once daily to patients with osteoarthritis of the knee at a dose of 250 mg for 10 days. This dose was selected because it was safe and well tolerated in the preceding Phase 1 trials and had a complete suppressive effect on COX activity. The trial, although it was open label and not placebo controlled, closed with demonstration of safety (no elevations of liver enzymes or other adverse effect) and with a suggestion for efficacy. Patients treated with ATB-346 exhibited improvements on the WOMAC (Western Ontario-McMaster University Arthritis Index) score (decreases of 4–7 on the score), and the inhibitory effect of ATB-346 on COX activity was confirmed by a marked and sustained inhibition of whole blood COX activity (Wallace et al., 2017). The original “strong point” ATB-346 in the preclinical studies was its gastrointestinal safety compared with naproxen. However, a 10-day-long arthritis trial is not suitable to assess gastrointestinal safety, because an effective dose of naproxen would have been also well tolerated in most patients for a 10-day study period (and, of course, based on historic data, naproxen would also have been efficacious in the same trial). Nevertheless, the suggestion for the efficacy of ATB-346 and the demonstration of its safety and tolerability are good starting points for further clinical testing of ATB-346.
The second clinical-stage combined/hybrid H2S donor molecule is GIC-1001 (trimebutine 3-thiocarbamoylbenzene-sulfonate) (Fig. 20C), an H2S-donating version of trimebutine maleate, a noncompetitive spasmolytic agent, with moderate affinity to µ- and κ-opioid receptors. The intended development indication is to use the compound as an alternative to intravenous sedation in patients undergoing colonoscopy. This direction is based on the hypothesis that the spasmolytic and colonic peripheral opioid agonistic activity of trimebutine combined with antinociceptive effects of H2S would be beneficial in clinical indications such as adjunct therapy of colonoscopy. Although the number of publications in the scientific literature with GIC-1001 is limited, in a murine study oral trimebutine maleate exerted only a slight inhibitory effect on the nociceptive response to increasing pressures of colorectal distension, whereas equimolar doses of orally administered GIC-1001 (30–60 mg/kg) significantly reduced nociceptive responses (Cenac et al., 2016). In Phase I single- and multiple-ascending dose clinical trials, GIC-1001 was orally administered to 80 healthy male and female subjects. The single-ascending dose studies demonstrated that levels of 125–1000 mg were tolerated. The multiple ascending dosing portion used three times daily doses of 125–500 mg over 7 days (19 consecutive doses), which were also tolerated, with adverse events including headache, somnolence, and nausea (Paquette et al., 2014) Surprisingly, there was no significant increase in total plasma H2S levels, which indicates that the H2S-release profile of the molecule must be very slow. According the clintrials database, in Trial NCT01926444, 240 patients were enrolled in a Phase 2 clinical trial, which has been completed. No further information is publicly available on the outcome of the trial or follow-on clinical development.
In addition to ATB-346 and GIC-1001, which are clinical-stage drug development candidates, a variety of additional H2S-releasing derivatives of clinically used drugs have been reported in the scientific literature. However, these compounds have not progressed into clinical trials. One such compound is the H2S-donating derivative of mesalamine (ATB-429 or 5-amino-2-hydroxybenzoic acid 4-(5-thioxo-5H-[1,2]dithiol-3yl)-phenyl ester) (Fig. 20B). The first report on this compound demonstrated superior anti-inflammatory and antinociceptive efficacy compared with the base mesalamine molecule in a rodent experimental model of colitis-associated colorectal distension (Distrutti et al., 2006), and additional studies demonstrated its efficacy and safety in animal models of colitis (Fiorucci et al., 2007). Most recently, a series of combined NO/H2S-releasing ATB-429 derivatives have been synthesized, with antitumor effects in vitro (Wang et al., 2016a).
Li et al. (2007) reported on the biologic effects of a H2S-releasing diclofenac derivative (ACS15, or S-diclofenac (2-[(2,6-dichlorophenyl)amino]benzene acetic acid 4-(3H-1,2,dithiol-3-thione-5-yl)phenyl ester) (Fig. 20D). The same compound is also made by Wallace’s group and is referred to as ATB-337 (Wallace et al., 2007b). The compound was effective in endotoxin-induced inflammation models in rodents and exhibited higher potency and lower gastric toxicity than the parent molecule. The compound also reduced IL-1β production and upregulated the anti-inflammatory cytokine IL-10 and exerted anti-inflammatory effects in rodent models of inflammation, myocardial reperfusion, and acute lung injury without adverse cardiovascular effects (Bhatia et al., 2008a,b; Sidhapuriwala et al., 2007; Rossoni et al., 2008). In vitro studies have demonstrated that the compound exerts anti-inflammatory effects (Lee et al., 2010) and is an inhibitor of angiogenesis and cell proliferation (Isenberg et al., 2007; Baskar et al., 2008). With an eye for potential use in chemoprevention, a series of in vitro studies evaluated the effect of ACS15 in human hepatoma HepG2 and human colonic adenocarcinoma LS180 cells on the activity and expression of the carcinogen activating enzymes, cytochromes P-450 (CYP) CYP1A1, CYP1B1, and CYP1A2 and reported inhibitory effects (Bass et al., 2009). An independent line of studies performed in cell culture and in mouse calvaria organ cultures demonstrated that ACS15 inhibits osteoclast formation and activity, suppresses breast cancer cell support for osteoclastogenesis, and prevents osteolysis; most of these effects were more pronounced with ACS15 than the corresponding effects of the parent compound diclofenac (Frantzias et al., 2012). In an in vivo study, ACS15 (25 and 50 mmol/kg, i.p.) significantly attenuated doxorubicin-related cardiac injury and cardiac dysfunction and improved the survival rate of mice with doxorubicin-induced cardiomyopathy, whereas the parent compound diclofenac was without significant effects in the same model (Zhang et al., 2011). The compound also exerted a reduced capacity to induce gastric toxicity and leukocyte adherence than the parent molecule diclofenac, although ATB-337 and diclofenac were comparable in their ability to inhibit cyclooxygenase (Wallace et al., 2007a). On the basis of these data, H2S-donating diclofenac derivatives may have multiple potential indications, not only as anti-inflammatory agents but possibly in bone diseases and cancer as well.
Additional, more recently described H2S-containing hybrid drugs include H2S-donating sildenafil (ACS6) (Fig. 20E), which, in cultured pulmonary endothelial cells, was shown to inhibit the upregulation of NADPH oxidases and suppressed the formation of superoxide (Muzaffar et al., 2008). ACS6 elicited a dose-dependent relaxation of isolated rabbit cavernosal strips but was not different in this regard than the parent compound sildenafil (Shukla et al., 2009). However, ACS6 was a more potent inhibitor of the upregulation of NOX and PDE5 in response to various risk factors for erectile dysfunction, and it has been hypothesized that these additional properties may confer therapeutic benefits to the compound beyond the effects of sildenafil (Shukla et al., 2009). However, this remains to be directly tested in future studies. In the most recent report with ACS6, the compound exerted protective effects against homocysteine-induced neurotoxicity in vitro (Tang et al., 2013).
An H2S-releasing aspirin derivative (ACS14) (Fig. 20F) has also been synthesized (Sparatore et al., 2009; Rossoni et al., 2010) and characterized in a number of in vitro and in vivo models. This compound has gastric mucosa sparing properties (Sparatore et al., 2009) as well as antihypertensive, vascular protective effects, and protective effects against myocardial ischemia-reperfusion in rodent models (Rossoni et al., 2010). ACS14 also protects microglial cells from amyloid-β-peptide-induced cytotoxicity (Liu et al., 2011b), protects endothelial cells against methylglyoxal-induced dysfunction (Huang et al., 2014), and inhibits platelet aggregation and exerts antithrombotic effects in vitro and in vivo (Pircher et al., 2012; Gao et al., 2015). One of the problems with the available literature with ACS14 is that head-to-head comparisons with aspirin are sometimes missing (e.g., Giustarini et al., 2010) or sometimes the comparisons are difficult to evaluate. For instance, in an in vivo study, ACS14 [50 (mg/kg)/day] or equimolar dose of aspirin [i.e., 23 (mg/kg)/day] both reduced platelet aggregation, but ACS14 was more potent in some respects (against ADP-induced aggregations) and was less potent against others (arachinoid acid- or thromboxane-induced aggregation) (Pircher et al., 2012). Data of this type make it difficult to ascertain whether ACS14 has sufficient pharmacological advantage over aspirin to justify clinical development.
ACS67 (Fig. 20G), a hydrogen sulfide-releasing derivative of latanoprost acid, has been synthesized for potential ocular indications. The compound has been shown to attenuate retinal ischemia and oxidative stress to RGC-5 cells in culture (Osborne et al., 2010), and in an in vivo follow-up study it was shown to reduce intraocular pressure in rabbits (Salvi et al., 2016). Once again, head-to-head comparison with the parent compound latanoprost was missing from the report.
A H2S-releasing derivative of l-DOPA methyl ester has also been synthesized and designated as ACS84 (Fig. 20H). The compound was shown to protect microglial cells from amyloid-β-peptide induced cytotoxicity (Liu et al., 2011a), exert cytoprotective effects against oxidative stress induced by 6-OHDA in SH-SY5Y cells (Xie et al., 2013), and in an in vivo study, it ameliorated the 6-OHDA-induced neuronal loss in rats, an effect that resulted in functional improvements as well (improved rotational behavior in unilateral 6-OHDA-lesioned rats) (Xie et al., 2013). Yet again, head-to-head comparison of ACS84 with the parent compound (l-DOPA methyl ester) are, unfortunately, missing from the publications.
Using the ADT-OH H2S-releasing group, Kashfi and coworkers (Chattopadhyay et al., 2012a,b; Kodela et al., 2015a,b) also synthesized and tested several H2S-releasing NSAID-derivatives (HS-NSAIDs), including HS-sulindac, HS-ibuprofen, HS-naproxen, and HS-aspirin, primarily focusing on antiproliferative and cytotoxic effects in various cancer cell lines. Although the parent compounds failed to inhibit the proliferation of the cancer cells, the H2S-releasing derivatives exerted inhibitory effects, with IC50 values in the single-digit micromolar concentration range (Chattopadhyay et al., 2012a,b). A follow-up study reported on additional in vitro and in vivo antitumor effects of HS-naproxen: the compound inhibited colon cancer growth with an IC50 of 72 µM (while the parent’s IC50 was established as 2800 µM). In vivo, however, in a colon cancer-bearing nude mice model, the efficacy of HS-naproxen was only reported at a single (and rather high) dose of 100 (mg/kg)/day (Kodela et al., 2015b). Some of the H2S-releasing NSAID-derivatives discussed above have been additionally modified to incorporate both an NO- and a H2S-releasing group, exemplified by the compound NOSH-sulindac (AVT-18A) discussed in section XIII.
A recent report evaluated the cardioprotective potential of H2S-releasing adenosine derivatives by coupling adenosine to 4-hydroxythiobenzamide or ADT-OH H2S-releasing groups with the idea that H2S and adenosine both activate separate, distinct cyto- and cardioprotective pathways, and their combination may be of therapeutic potential (Lougiakis et al., 2016). The slow H2S release from the compounds was confirmed in vitro, as well as the ability of the compounds to elevate cGMP levels in cultured cells. An example is Compound #4 (Fig. 20I). Several of the adenosine derivatives were also tested in vivo in a rabbit model of myocardial ischemia-reperfusion, where it was confirmed that new hybrid derivatives result in synergistic cardioprotective activity through the combination of the molecular pathways of adenosine and H2S (both of which trigger cardioprotection) (Lougiakis et al., 2016).
The ADT-OH H2S-releasing group has also been used to derivatize a variety of additional clinically used drugs including the N-methyl-d-aspartate receptor antagonists memantin and amantadine (Marutani et al., 2014), the antistroke compound 3-n-butylphthalide (Wang et al., 2014; Yin et al., 2016), doxorubicin (Chegaev et al., 2016), olenoic acid (Xu et al., 2016a), glycyrrhetic acid (Song et al., 2016), nicotinic acid (Sun et al., 2016b), naproxen, as an amide derivative resistant to hydrolysis (Hammers et al., 2016), valproic acid (Hammers et al., 2016), losartan (Martelli et al., 2012), and the Chinese traditional compound Danshensu [β-(3, 4-dihydroxyphenyl)lactic acid] (Yan et al., 2017). Most of these compounds remain to be further characterized with respect to their pharmacological effects in vitro and in vivo.
Most recently, a H2S-releasing derivative of the clinically used amino-biophosphonate compound, alendronate has been synthesized for the experimental therapy of osteoporosis. The compound (DM-22), releases H2S in an L-cysteine-dependent manner, inhibits osteoclast differentiation and increases mineralization in osteogenic human mesenchymal stem cells (Rapposelli et al., 2017).
XXIV. Alternative Means to Increase Biologic H2S Levels
There are a number of possibilities to elevate biologic H2S levels that do not involve direct H2S donation (Table 1). The most obvious method to increase H2S levels in the body, of course, is by “feeding” the endogenous H2S producing enzymes with its substrates: l-cysteine and l-homocysteine for CSE and CBS, 3-mercaptopyruvate for 3-MST, and α-ketoglutarate for CAT (which, in turn, produces 3-MP). Although it is generally assumed that cells contain saturating concentrations of these substrates and, therefore, exogenous supplementation would not have a marked effect, in fact, in vitro and in vivo studies demonstrate that these three substrates can increase H2S levels, in a fashion that is reduced by inhibitors of the respective H2S-producing enzymes (e.g., Kartha et al., 2012; Ahn et al., 2017; Tan et al., 2017), and induce biologic effects that are consistent with elevation of H2S production, such as smooth muscle relaxation (d’Emmanuele di Villa Bianca et al., 2009; Flannigan et al., 2013; Yamane et al., 2015; Prieto-Lloret and Aaronson, 2015; Kuo et al., 2016; Yetik-Anacak et al., 2016), cell proliferation and angiogenesis (Wang et al., 2013c; Coletta et al., 2015), organ protection (Elsey et al., 2010; Magierowski et al., 2017) and bell-shaped effects on mitochondrial function (stimulation at lower concentrations and inhibition at higher concentrations) (Modis et al., 2013a,b). In some tissues, d-cysteine can also yield H2S, through actions on the d-amino acid oxidase, with beneficial/cytoprotective results (Shibuya et al., 2013; Shibuya and Kimura, 2013; Souza et al., 2017). However, these substrate-based approaches are not ideal as potential therapies, for several reasons. First, only l-cysteine, 3-mercaptopyruvate, and α-ketoglutarate are benign enough to be considered, because l-homocysteine exerts various adverse vascular effects (as discussed in Lai and Kan, 2015 and Ganguly and Alam, 2015). Second, the concentrations/doses to be used are rather high; for example in vitro, in EA.hy926 or A10 cells, l-cysteine increased H2S levels in the concentration range of 1–5 mM (Kartha et al., 2012). Third, l-cysteine, 3-mercaptopyruvate, and α-ketoglutarate have multiple additional pharmacological actions. Fourth, application of these substrates may or may not increase H2S levels (if the endogenous substrate level of a given cell or tissue is sufficient to produce saturating substrate concentrations, further supply of the substrate is not expected to induce any further increase in H2S production.) Fifth, they may become less effective when the endogenous H2S-producing enzymes are inhibited or downregulated. In fact, several pathophysiological states (e.g., Aminzadeh and Vaziri, 2012; Guo et al., 2012; Holwerda et al., 2012; Wang et al., 2014a; Huang et al., 2017; Merz et al., 2017) as well as various pharmacological agents, e.g., dexamethasone (d'Emmanuele di Villa Bianca et al., 2015), can induce the downregulation of CBS or CSE, while 3-MST is oxidation-prone and can be inactivated in various pathophysiological states, for instance diabetes mellitus (Coletta et al., 2015). Sixth, these molecules are not novel structures nor proprietary, therefore they are of limited interest to develop them commercially.
Alternative means to elevate of suppress biologic H2S levels
N-Acetylcysteine (NAC), most widely known as a as a ROS scavenger, a clinically used drug in patients with cystic fibrosis, acetaminophen poisoning, and many other conditions, may also increase H2S levels in biologic systems. Direct measurements in various cells in culture demonstrated that NAC increases cellular H2S levels in millimolar concentrations (Kartha et al., 2012). The underlying mechanisms are multiple. First of all, NAC scavenges ROS, which, in turn, attenuates the ROS-mediated inactivation of H2S, resulting in increased H2S levels. Second, NAC undergoes deacetylation, and the resulting l-cysteine is metabolized via CBS and CSE. Third, increased intracellular cysteine levels in response to NAC may also drive the conversion of homocysteine to cystathionine, an effect that has been suggested to explain the clinically observed, well-established homocysteine-lowering effects of NAC (Chen et al., 2004). Fourth, NAC, in the presence of nitric oxide, may also undergo a series of complex chemical reactions that can yield various reactive nitrogen and sulfur species including thionitrite, the smallest S-nitrosated thiol (Bertova et al., 2010; Tsikas and Böhmer, 2017). Fifth, in vivo, NAC can also upregulate CBS, CSE, and 3-MST expression (Tai et al., 2016; Tain et al., 2016), which, in turn, would be expected to indirectly increase biologic H2S levels. A cell-permeable, ethyl ester analog of NAC was recently generated, and this compound was shown to elevate circulating H2S levels in rats; in contrast, NAC, at the same dose (50 mg/kg), did not elicit a significant elevation in plasma H2S levels (Giustarini et al., 2012).
A related approach may be the boosting of H2S production by activating the various H2S-producing enzymes by their various cofactors. The best characterized approach relates to the activation of CBS by its cofactor S-adenosyl-methionine (SAM). The mechanism of the stimulatory effect of SAM on CBS is subject to specialized articles (Meier et al., 2003; Jhee and Kruger, 2005; Ereño-Orbea et al., 2014; Hellmich et al., 2015; Majtan et al., 2016); what is relevant for the current review is that in various cell-based systems and in vivo experiments, SAM (as well as SAM analogs) have been shown to increase H2S levels and exert biologic effects that are consistent with the roles of H2S (e.g., vascular relaxation and biphasic effects on mitochondrial function) (Jensen et al., 2011; Módis et al., 2014a). A similar approach involves the stabilization, stimulation, and reactivation of 3-MST with reducing agents (including α-lipoic acid, which is a clinically approved drug for diabetic complications) (Coletta et al., 2015).
The final approach related to boosting biologic H2S levels via the modulation of H2S-producing enzymes relates to the upregulation of their enzyme levels. Although this is a standard approach in biologic experiments (i.e., overexpression of these enzymes by various methods of gene transfer) (e.g., Papapetropoulos et al., 2009; Jacobs et al., 2011; Sen et al., 2007, 2011, 2012; Coletta et al., 2012; Regnier et al., 2012; Duan et al., 2015; Panza et al., 2015) and is sometimes also used in in vivo efficacy studies (e.g., Weilan et al., 2017), it is not viewed as a clinically translatable approach. Nevertheless, a recent study on taurine should be mentioned. In this report, taurine supplementation was found to increase plasma H2S, concentrations via mechanisms that involve, at least in part, the upregulation of CBS/CSE expression. The functional results of taurine supplementation included significantly improved endothelium-dependent and endothelium-independent vasodilatory responses (Sun et al., 2016a).
Nonmolecular approaches to increase CSE expression include physical exercise (Tang et al., 2016). The sex hormone testosterone has also been shown to increase the expression of CSE, and this effect may be, at least in part, responsible for a sex difference in circulating H2S levels (males having higher levels than females) (Bucci et al., 2009; Brancaleone et al., 2015). However, the situation appears to be complex, because in several studies estrogens were also reported to increase CBS and CSE expression (Zhu et al., 2013; Lechuga et al., 2015; Li et al., 2017).
A related approach is the upregulation of H2S-producing enzyme levels through inhibition of their degradation. Inhibition of protease-mediated degradation appears to be a pathophysiological mechanism responsible for the upregulation and mitochondrial accumulation of CBS (Teng et al., 2013). Treatment with proteasome inhibitors has been demonstrated to boost CBS protein levels, which exerts beneficial effects in mice with CBS mutations where CBS levels need to be elevated for a therapeutic effect (Gupta et al., 2013). Although it was not measured in this study, one can assume that the same therapy is also effective in increasing H2S production.
The next obvious approach to increase biologic H2S levels is the inhibition of its degradation. One more general approach may involve targeting ROS. Although, in cell-free biochemical systems (e.g., stopped flow assays) the reaction rate between H2S and various ROS species has been quantified as slow (and often suggested to be irrelevant) (Cuevasanta et al., 2015; Trujillo et al., 2016; Cuevasanta et al., 2017), many biologic studies show that increased ROS levels can, in fact, lead to decreased H2S levels and inhibition of cellular ROS production or scavenging cellular ROS can, in fact, elevate H2S levels (Pun et al., 2010; Hancock and Whiteman, 2016; Olas, 2017). Obviously, reduction of oxidative stress, in addition to boosting H2S levels, will have a myriad of additional pharmacological actions. A second, more specific approach may involve the pharmacological targeting specific H2S-degrading enzymes. The biologic degradation and elimination of H2S involves multiple mechanisms to yield sulfite, sulfate, and thiosulfate (overviewed in Li et al., 2011; Stein and Bailey, 2013; Mishanina et al., 2015; Rose et al., 2015, 2017). The enzymes traditionally listed in H2S catabolism include rhodanese (also known as thiosulfate sulfurtransferase), sulfide quinone oxidoreductase, a sulfur dioxygenase enzyme encoded by the gene ethylmalonic encephalopathy protein 1. In addition, endogenous H2S levels are also regulated by cysteine dioxygenase (Roman et al., 2013) and Coenzyme Q. Inactivation of these enzymes has been demonstrated to produce elevations in cell, tissue, and systemic H2S levels (Tiranti et al., 2009; Jurkowska et al., 2014; Rose et al., 2015, 2017; Morton et al., 2016; Luna-Sanchez et al., 2017; Ziosi et al., 2017); it is therefore logical to propose that controlled inhibition of some of these degradation enzymes may be a potential way to boost H2S levels. However, to our knowledge, this approach has not yet been tested pharmacologically. The degree of inhibition of these H2S-degrading enzymes would have to be partial and well-regulated to prevent elevation of H2S levels above the desired levels. Since these enzymes may also serve other important biologic functions, this approach may also come with a variety of adverse effects.
Until recently, thiosulfate has been viewed as an inactive byproduct of H2S catabolism. However, studies over the last 5 years have indicated that cells have the ability to reduce thiosulfate and regenerate H2S from it in a cell- and tissue-dependent fashion and, at least in part, via mechanisms that involve 3-MST and rhodanese (Mikami et al., 2011; Olson et al., 2013; Libiad et al., 2015). In vitro, thiosulfate has cellular effects in the high micromolar (100–500 µM) concentration range (Bijarnia et al., 2015; Marutani et al., 2015; Lee et al., 2016), with no apparent toxicity even at concentrations as high as 20 mM (Bijarnia et al., 2015). The cellular uptake of sodium thiosulfate involves the sodium sulfate cotransporter 2 (Marutani et al., 2015). In vivo, its therapeutic efficacy in rodent models of LPS-induced lung injury (Sakaguchi et al., 2014), galactosamine/LPS-induced acute liver failure (Shirozu et al., 2014), angiotensin-induced hypertension and hypertensive heart disease (Snijder et al., 2014; Snijder et al., 2015), global cerebral ischemia (Marutani et al., 2015), vascular calcification (Subhash et al., 2015), and hyperoxaluric renal dysfunction (Bijarnia et al., 2015) can be seen at doses ranging from 10 to 2000 (mg/kg)/day. In clinical observational studies, plasma thiosulfate levels show positive correlations, with improved clinical outcomes in renal transplantation (van den Berg et al., 2014; Frenay et al., 2016). On the mechanistic side, one must keep in mind that thiosulfate is likely to have multiple pharmacological actions in addition to being a generator of H2S. On the practical side, however, given the fact that sodium thiosulfate (STS) is a safe clinically used drug that is approved for the therapy of sodium nitroprusside/cyanide intoxication (Baskin et al., 1992; Hall and Guest, 1992); the clinical and experimental therapeutic opportunities around STS are substantial and there may be various opportunities to repurpose STS for the experimental therapy of a variety of diseases that may benefit from H2S supplementation.
Several clinically used drugs may also involve elevated H2S mechanisms as part of their action. It has been occasionally reported that certain sulfur-containing drugs (among others, phenazopyridine, dapsone, metoclopramide with acetylcysteine, dermal application of dimethylsulfoxide) in some patients can produce sulfhemoglobinemia, which may be, in part, related to the release of H2S and/or other labile, reactive sulfur species in these subjects (Lambert et al., 1982; Hansen et al., 1994; Burgess et al., 1998; Noor and Beutler, 1998; Langford and Sheikh, 1999; Gopalachar et al., 2005). The antischistosomal and cancer chemopreventive agent oltipraz (a pyrrolopyrazine thione) also contains a dithiolethione group and therefore may be a potential H2S donor, although this has not yet been directly investigated. Interestingly, oltipraz has been shown to inhibit cytochrome c, followed by an increase in mitochondrial ROS production (Velayutham et al., 2007), effects that are similar to the mitochondrial effects of high concentrations of authentic H2S. Its ability to activate Nrf2 (Yu et al., 2011) and its ability to enhance wound healing (Noorafshan et al., 2014) are other pharmacological action that mimic the known pharmacological actions of authentic H2S. Anathole trithione (Christen, 1995) is another dithiolethione compound, which, once again, may be a H2S releaser. Although the approved indication of this drug is xerostomia, this compound also has been tested in clinical trials for chemopreventive indications (Lam et al., 2002) and possesses multiple pharmacological actions, such as modulation of cellular thiol homeostasis (Dringen et al., 1998; Giustarini et al., 2014), which may be potentially linked to H2S, although this has not yet been tested directly. Additional clinically used drugs that can induce increases in H2S levels in biologic systems include aspirin (in female, but not male mice) (Srebro et al., 2006), sildenafil (Fusco et al., 2012), and cinaciguat (Salloum et al., 2012). Although the mechanism of sildenafil’s effect remains to be characterized, the effect of aspirin may be related to an enhancement of H2S release from sulfane sulfur pools (Bilska-Wilkosz et al., 2013). The effect of cinaciguat may be, at least in part, related to the protein kinase G-mediated upregulation of CSE (Das et al., 2015). Finally, recent studies by Cirino and colleagues (Bucci et al., 2014) demonstrated that zofenoprilat, a sulfur-containing angiotensin converting enzyme inhibitor, also generates H2S spontaneously (an effect that is enhanced in the presence of thiols). Multiple lines of in vitro and in vivo studies have suggested (Bucci et al., 2014; Terzuoli et al., 2015; Donnarumma et al., 2016a; Monti et al., 2016) that the H2S-releasing property contributes to the therapeutic effect of this drug, independently of its effects as an angiotensin converting enzyme inhibitor.
XXV. A Brief Overview of Endogenous H2S Sources
H2S can be produced endogenously through both enzymatically catalyzed reactions and nonenzymatic routes. Cystathionine β-synthase (CBS; EC 4.2.1.22), cystathionine γ-lyase (CSE; 4.4.1.1), and 3-mercaptopyruvate sulfurtransferase (3-MST; EC.2.8.1.2) contribute to the endogenous production of H2S (Szabo, 2007; Kabil and Banerjee, 2014; Kimura, 2014, 2015; Papapetropoulos et al., 2015; also overviewed in the IUPHAR/BPS Guide to Pharmacology: (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=279). In addition, thiosulfate and sulfite have been proposed to yield H2S through reductive chemistry (Kolluru et al., 2013). Additional sulfane sulfur pools include inorganic polysulfides and protein persulfides that could yield H2S nonenzymatically or via the action of thioredoxin (Kabil and Banerjee, 2014; Wedmann et al., 2016). It should be noted that the exact contribution of enzymatic versus nonenzymatic sources to H2S levels has never been determined in a biologic system.
CBS and CSE in mammalian cells operate within the reverse transsulfuration pathway, a biochemical pathway responsible for the conversion of methionine to cysteine, and catalyze a multitude of reactions that yield H2S (Wang, 2012; Kabil and Banerjee, 2014; Kimura, 2014, 2015). This is due to a relaxed substrate specificity of these enzymes and the ability of CSE to act at both β- and γ-carbons of its substrates (Kabil and Banerjee, 2010; Singh and Banerjee, 2011). The third H2S-producing enzyme, 3-MST, is part of the cysteine catabolism pathway and uses 3-MP as a substrate (Nagahara, 2013). 3-MST works in tandem with aspartate aminotransferase that also possesses cysteine aminotransferase activity (CAT) activity, generating 3-MP from cysteine. It should be mentioned that 3-MP, in addition to acting as a substrate of 3-MST, can also produce H2S spontaneously (i.e., in solution, in the absence of any cellular components or enzymes) (Coletta et al., 2015). Moreover, lipoic acid, in addition to being a cofactor of the 3-MST reaction, has also been shown recently to release H2S nonenzymatically (Bilska-Wilkosz et al., 2017). These actions should be considered when interpreting the pharmacological effects of 3-MST, especially when substrates or cofactors are used at higher (micromolar) concentrations.
Unlike the generation of NO from NOS isoforms, where d-arginine was used as a negative control for l-arginine in biologic experiments (Moncada et al., 1991), d-cysteine is a substrate for H2S production. d-Cysteine is converted to 3-MP by d-amino acid oxidase (an enzyme that is preferentially expressed in peroxisomes) that then yields H2S (Shibuya et al., 2013). The d-cysteine pathway appears to be prominent only in a limited number of tissues (e.g., the cerebellum and the kidney; in the latter tissue, H2S production is approximately 80 times higher from d-cysteine than from l-cysteine) and, so far, its biologic significance has only been shown for ischemia/reperfusion injury in the kidney (Shibuya et al., 2013).
All three enzymatic pathways that lead to H2S production need, directly or indirectly, pyridoxal 5′-phosphate (PLP) for their activity (Singh and Banerjee, 2011; Wang, 2012; Kabil and Banerjee, 2014; Kimura, 2014, 2015). PLP is a cofactor for CSE and CBS. In contrast, 3-MST-does not itself need PLP to generate H2S; however, CAT, which supplies the 3-MST substrate 3-MP, is also PLP dependent (Fig. 1).
XXVI. Pharmacological Inhibitors of Cystathionine-γ-lyase
CSE (also abbreviated as CGL or CTH) is a major source of H2S in peripheral tissues, while it minimally contributes to H2S production in the central nervous system (De Luca et al., 1974; Vitvitsky et al., 2006; Yang et al., 2008; Kabil et al., 2011; Kimura 2014, 2015). CSE is readily detected at the protein level in the cardiovascular and respiratory system (Bucci et al., 2012, 2014; Kondo et al., 2013), the kidney, liver, uterus, and other organs (Kabil et al., 2011; Wang 2012; Kimura 2014, 2015). In line with its widespread presence in peripheral organs, serum levels of H2S in CSE−/− mouse are reduced by 50% (Yang et al., 2008). The contribution of CSE to H2S production varies significantly among tissues: free H2S in the heart is reduced by approximately 80%, whereas sulfane sulfur is less drastically affected (Yang et al., 2008; King et al., 2014). In the liver, CSE has been proposed to account for >95% of the H2S production, at least in mice (Kabil et al., 2011). Under resting conditions, CSE is a present in the cytosol. Although CSE has the capacity to translocate to the mitochondria upon prolonged increases in intracellular calcium triggered by a calcium ionophore (Fu et al., 2012), no physiological stimulus is known to alter the subcellular distribution of CSE to date.
CSE exists as a homotetramer with a subunit molecular mass of 45 kDa and is capable of catalyzing several H2S-generating reactions due to low stringency in substrate recognition (Kabil and Banerjee, 2014; Kimura, 2014). CSE can catalyze both β- and γ-replacement reactions using cysteine alone or cysteine and homocysteine as substrates (Singh et al., 2009; Kabil and Banerjee, 2010; Singh and Banerjee, 2011). The β-lyase reaction is approximately 20 times slower than the cleavage of the C-γ-S bond in the conversion of l-cystathionine to l-cysteine (Sun et al., 2009). In the presence of high concentrations of homocysteine, the γ-replacement reaction between two molecules of homocysteine becomes dominant in the production of H2S (Kabil et al., 2011). In some experimental conditions, CSE activity can be slightly stimulated by Ca2+/calmodulin (Yang et al., 2008; Coletta et al., 2012), whereas in others CSE activity is unaffected by Ca2+/calmodulin (Mikami et al., 2013; Kabil and Banerjee, 2014). As mentioned above, catalytic activity of CSE is known to depend on PLP. Lys212 (hCSE numbering) binds covalently to the PLP cofactor via the formation of a Schiff base between the amino group of the Lys212 side chain and the carbonyl group of PLP (Sun et al., 2009). Transition from the open apo-hCSE form to the closed PLP-bound form is accompanied by significant conformational changes.
Pharmacological inhibitors of CSE have been critical for our understanding of the biologic functions of H2S. The most commonly used compounds include dl-propargyl glycine (PAG; also known as PGG) and β-cyano-l-alanine (BCA) (Szabo, 2007; Whiteman et al., 2011; Wang, 2012). PAG is a stereoselective compound; it is the L-isomer that inhibits CSE, whereas the R-isomer is inactive (Huang et al., 1998); therefore, the L-isomer (and not the racemate form) should be used for CSE inhibition. Although both PAG and BCA have been shown to inhibit additional PLP-dependent enzymes, they can preferentially inhibit H2S generation from CSE versus CBS (Asimakopoulou et al., 2013) (Table 2). BCA, but not PAG, inhibits recombinant CBS at high concentrations (>1 mM). The IC50 for PAG using purified CSE and cysteine as a substrate in two different studies was reported to be 40 (Asimakopoulou et al., 2013) and 20 μΜ (Sun et al., 2009), whereas that of BCA was shown to be 14 μΜ (Asimakopoulou et al., 2013). However, these compounds are typically used at much higher concentrations (1–10 mM) to block H2S production in cell-based assays, suggesting that they poorly cross the cell membrane (Pan et al., 2006; Brancaleone et al., 2008; Bucci et al., 2009; Papapetropoulos et al., 2009; Schleifenbaum et al., 2010; Wang et al., 2013a,b; Lee et al., 2014a; Potenza et al., 2014; Martinez-Cutillas et al., 2015; Testai et al., 2015; Tsai et al., 2015a; Yang et al., 2015; Krause et al., 2016). A significant difference between the PAG and BCA is that the former has an irreversible mode of action, acting as a suicide inhibitor (Abeles and Walsh, 1973; Steegborn et al., 1999). hCSE has been cocrystalized with PAG, yielding significant information about the mode of inhibition of this compound (Sun et al., 2009) (Fig. 22). PAG does not directly bind PLP, and the entire structure and active region of the hCSE/PLP/PAG complex is identical to that of the PLP-bound enzyme in the absence of the inhibitor. Lys212 is still covalently bound to C4′ of PLP, and Tyr114 mediates π-stacking interactions with PLP. The inhibitor PAG is covalently bound to Tyr114 as a vinylether, whereas the amino and carboxyl groups of PAG form hydrogen bonds with Glu339 and with Arg119 and Arg62 from the adjacent monomer, respectively. The inhibitor occupies the space of the side chain of the substrate, sterically hindering accessibility of substrate molecules to the active site. Additionally, PAG by covalently binding and trapping Tyr114, interferes with the release of the substrate (Clausen et al., 1998).
Inhibitors of H2S synthesizing enzymes
For CBS and CSE that catalyze multiple reactions reported IC50 values correspond to reactions generating H2S. L-cysteine was used as a substrate for CSE and L-cysteine/homocysteine were used for CBS. SAM was sometimes included in CBS activity assays. The recombinant CBS and CSE enzymes used were human in the majority of the cases; the enzymes carried tags for purification purposes or were modified (truncated) to increase yield/activity. For most of the 3-MST measurements the murine enzyme was used.
Stereoview of PAG-hCSE active site (A) and superimposed PAG complexes (B). PAG, PLP, and nitrate ion are shown in a thick line. hCSE⋅PAG, methionine γ‐lyase⋅PAG, and CsdB⋅PAG are colored green, gray, and pink, respectively. Residues interacting with PAGs and nitrate ion are shown. (C): Stereoview of the 2Fo ‐ Fc simulated annealing omit map of PAG, Tyr114 from hCSE⋅PLP⋅PAG. All atoms within 3.5 Å of PAG and Tyr114 were omitted prior to refinement. The map was contoured at a level of 1.0σ. Reproduced with permission from Sun et al., 2009.
In addition to PAG, another glycine analog and natural toxin, aminoethoxyvinylglycine (AVG) was found to block hCSE (Steegborn et al., 1999; Asimakopoulou et al., 2013) but not hCBS (Asimakopoulou et al., 2013); AVG is more potent compared with PAG, with an IC50 of 1 μM (Asimakopoulou et al., 2013). Despite its higher potency, AVG suffers from the same selectivity drawbacks as BCA and PAG, inhibiting additional PLP-dependent enzymes (Clausen et al., 1997; Huai et al., 2001; Eliot and Kirsch 2004; Whiteman et al., 2011). In addition, AVG has not been used in cellular or in vivo assays so far to measure the contribution of H2S to the biologic response, so its usefulness as a pharmacological tool remains unknown.
On the basis of its structural similarity with cysteine, d-penicillamine, a clinically used drug, was tested for its ability to modify CSE activity (Brancaleone et al., 2016). By using recombinant human enzymes, d-penicillamine was found to be 30-fold more selective for CSE versus CBS, whereas in tissue homogenates d-penicillamine was 30-fold more potent than PAG in inhibiting H2S production. In addition, d-penicillamine reduced cysteine-induced relaxations and exacerbated the TNFα-induced vascular inflammation, in line with the vasorelaxant and anti-inflammatory actions of CSE.
By using a fragment-based design approach, Corvino et al. (2016) fused cysteine derivatives already used as substitutes with pharmacophore structures of known CSE inhibitors. The compound that showed the most promising results was an oxothiazolidine derivative (2a) that was 100-fold more potent than PAG in inhibiting cysteine-stimulated H2S production in tissue homogenates. In contrast to PAG, this new compound inhibited CSE in a competitive manner. Additional differences between PAG and 2a exist as they differentially affected the catalytic activity of CSE. PAG and 2a both inhibited the production of pyruvate, ammonia and hydrogen sulfide from l-cysteine. However, although 2a blocked CSE from converting l-cysteine to lanthionine, CSE in the presence of PAG lost the ability to generate H2S but was still active forming cystine. This observation raises the possibility that other known CSE and CBS inhibitors selectively inhibit some, but not all, of the reactions catalyzed these enzymes.
The above information, taken together, suggests that PAG, despite its shortcomings, still remains the drug of choice to pharmacologically inhibit CSE. Before CSE knockout mice became available, but also after their widespread use, PAG is used to investigate the role of CSE in a variety of physiological conditions and in disease models and to confirm the observed CSE knockout phenotype. Pharmacological inhibition of CSE with PAG demonstrated the importance of this enzyme for angiogenesis (Papapetropoulos et al., 2009), vasorelaxation (Zhao et al., 2001; Bucci et al., 2010; Al-Magableh and Hart, 2011), and erectile function (d'Emmanuele di Villa Bianca et al., 2009). Inhibition of CSE elevated mean arterial blood pressure (Yan et al., 2004; Roy et al., 2012), enhanced cardiac damage (Pan et al., 2006; Sivarajah et al., 2006; Zhu et al., 2007), increased ischemia/reperfusion injury in various organs (Fu et al., 2008; Tripatara et al., 2008; Han et al., 2015a), aggravated the severity of ulcerative colitis (Wallace et al., 2009) and atherosclerosis (Wang et al., 2009b), exacerbated gastric injury (Fiorucci et al., 2005), increased mortality in sepsis (Spiller et al., 2010), and lead to symptoms of preeclampsia (Wang et al., 2013a). Despite the usefulness of PAG, the limitations associated with its lack of selectivity (discussed above) remain. Studies aiming to investigate the potential involvement of CSE in biologic responses should use a combination of approaches: pharmacological inhibitors, genetically modified animals, and silencing of CSE through siRNA, shRNA, or CRISPR/Cas9 technologies.
XXVII. Pharmacological Inhibitors of Cystathionine-β-synthase
CBS is abundantly present in the central nervous system but has also been found in various other tissues, including the cardiovascular and the respiratory system and the gastrointestinal tract (Whiteman et al., 2011; Bucci et al., 2014; Kimura, 2014, 2015). CBS is mainly regarded as a cytosolic enzyme, but its presence has been documented in the mitochondria of normal and tumor cells (Szabo et al., 2013; Teng et al., 2013). Stimuli such as hypoxia increase mitochondrial CBS content, at least in the liver, through inhibition of Lon protease (Teng et al., 2013). Evidence has been presented that CBS is subject to sumoylation, a posttranslational modification that regulates nuclear localization (Kabil et al., 2006; Agrawal and Banerjee, 2008). While mitochondrial CBS impacts cellular bioenergetics (Szabo et al., 2013), the importance of nuclear CBS remains to be defined in future studies.
CBS expression can be traced in evolution, all the way back to single-cell organisms (Majtan et al., 2014). Despite its broad taxonomic distribution, the quaternary structure and regulatory mechanisms of CBS enzymes are not conserved across phyla. Human CBS is a homotetramer, with each subunit exhibiting a size of 63 kDa (Miles and Kraus, 2004). CBS is unique within the 140 members of the PLP-dependent family of enzymes in that it contains heme as a prosthetic group (Alessio et al., 2007; Singh and Banerjee, 2011). Heme is not required for its enzymatic activity, but rather serves as a redox sensor and aids in proper folding (Meier et al., 2001; Banerjee and Zou, 2005; Majtan et al., 2008). CBS is organized into an N-terminal heme-binding domain, a central catalytic core that harbors PLP, and a C-terminal 140-amino acid regulatory region that houses a tandem pair of CBS domains (Miles and Kraus, 2004; Wang, 2012; Kabil and Banerjee, 2014). CBS domains are structural motifs that bind adenosine nucleotides, thus regulating protein function (Baykov et al., 2011). In the case of CBS, CBS motifs bind the allosteric activator S-adenosylmethionine (SAM) and result in an up to fivefold increase (Shan and Kruger, 1998; Majtan and Kraus, 2012). In the basal state of hCBS, the CBS motif pair, also known as “Bateman module,” is placed just above the entrance of the catalytic cavity of the complementary subunit, restricting substrate access to the active site and lowering enzymatic activity (Ereño-Orbea et al., 2014). Binding of SAM to the CBS motif induces a conformational change that weakens the interaction between the regulatory domain and the catalytic core; the Bateman module is shifted away from the pore, facilitating substrate diffusion to the catalytic center. Removal of the regulatory domain of CBS results in a truncated constitutively active enzyme that organizes into dimers (Kery et al., 1998; Jhee et al., 2000).
CBS can catalyze multiple reactions involving serine, cysteine, and homocysteine (Kabil and Banerjee, 2010; Kabil and Banerjee, 2014). Based on substrate affinities and concentrations, the favored reaction for CBS is the condensation of homocysteine and serine to cystathionine that is then converted to cysteine by CSE (Taoka et al., 1998; Singh et al., 2009). However, CBS also uses cysteine as a substrate to yield H2S in at least three distinct reactions: β-replacement of cysteine by homocysteine, by a second cysteine, or water to form cystathionine, lanthionine, and serine, respectively. From these reactions, kinetically the most efficient one is the β-replacement of cysteine with homocysteine (Singh et al., 2009). It is still unknown what influences the transsulfuration pathway to alter its preference from cysteine synthesis to H2S generation. In a recent study, Kabil et al. (2016) provided a paradigm for how H2S synthesis might be favored. Under endoplasmic reticulum stress conditions, heme oxygenase-1 is upregulated. The increased CO produced by heme oxygenase-1 inhibits CBS, deviating homocysteine metabolism from the canonical reactions that yield cystathionine to the production of H2S by CSE. In addition, increasing the cysteine/serine ratio shifts CBS from catalyzing the canonical reaction between homocysteine and serine to catalyzing the condensation of cysteine and homocysteine that yields H2S (Majtan et al., 2017). This observation makes it easier to rationalize why although tissues are not depleted of cysteine, when additional cysteine is provided to biological systems H2S production is increased.
Missense pathogenic mutations in the cbs gene constitute the most common inherited disorder of sulfur amino acid metabolism (Mudd et al., 2011). A deficiency in CBS activity results in homocystinuria that clinically manifests by defects in the connective tissue, mental retardation, and thromboembolism. Mild elevations of plasma homocysteine constitute an independent risk factor for cardiovascular diseases, osteoporosis, and age-related dementia.
CBS is located on chromosome 21, of which 3 copies exist in patients with Down syndrome, leading to increased CBS expression. As elevated CBS expression would predict, increased levels of H2S have been found in individuals with Down syndrome, evidenced by the increase in urinary excretion of its degradation product thiosulfate (Belardinelli et al., 2001; Kamoun et al., 2003). Increased production of H2S in Down syndrome was also inferred from the increase in erythrocyte sulfhemoglobin content (Kamoun et al., 2003). In a different cohort of patients, H2S was measured in plasma using the methylene blue method; patients with Down syndrome were found to have 50% higher levels than controls (Abdel-Salam et al., 2013). Chronic elevations of H2S in Down syndrome patients have been postulated to contribute to the neurological deficits associated with the disease. CBS inhibitors have, thus, been proposed as candidates to improve, at least partially, cognitive functions of Down syndrome patients (Charre et al., 2013).
Despite its low potency that requires millimolar concentrations in cell-based assays, aminooxyacetic acid (AOAA), also known as (carboxymethoxy)amine hemihydrochloride (CHH) or hydroxylamine-O-acetic acid hemihydrochloride (Table 2), was extensively used for years to inhibit CBS not only in vitro but also in vivo (e.g., Mudd et al., 2011; Szabo et al., 2013). Interestingly, a recent human study using local acetylcholine perfusion and measurement of blood flow by laser Doppler flowmetry found that AOAA reduces acetylcholine-induced vascular relaxations, implicating H2S as a contributor to the regulation of vascular tone (Greaney et al., 2017).
In a study using purified, recombinant CBS and CSE, we demonstrated that AOAA can inhibit both enzymes of the transsulfuration pathway (Asimakopoulou et al., 2013); surprisingly AOAA exhibited greater potency against CSE compared with CBS (IC50 1.1 vs. 8.5 μM, respectively). In addition to its inability to discriminate between CSE and CBS, AOAA is known to inhibit several other PLP-dependent enzymes, including aspartate transaminase (also known as GOT1), 4-aminobutyrate aminotransferase (GABA-T), alanine transaminase, and possibly a number of the other aminotransferases (Wallach, 1961; Cornell et al., 1984; Sherry et al., 1998; Kurozumi et al., 1999; Dever and Elfarra, 2008) and CAT (Flannigan et al., 2013), the enzyme that supplies 3-MP for 3-MST. Thus, in a cellular context, AOAA will suppress H2S generation from all three major enzymatic sources (direct inhibition of CBS and CSE and indirect inhibition of 3-MST through inhibition of CAT). The various cellular targets of AOAA are illustrated in Fig. 23 in the context of the effects of AOAA in a cancer cell. In this context, some of these additional effects of AOAA (e.g., on GOT) may be therapeutically beneficial (and additive to the inhibitory effect of AOAA on CBS), because GOT contributes to the bioenergetic homeostasis of cancer cells (Módis et al., 2014a).

Multiple modes of AOAA’s action in cancer cells. By directly inhibiting CBS and CSE activity, by suppressing H2S formation through the 3-MST pathway via inhibition of CAT, and by inhibiting a variety of transaminases (including GOT1, a key enzyme of the malate/aspartate shuttle), AOAA acts as an inducer of “synthetic lethality” in cancer cells. CBS-derived and 3-MST-derived H2S supports mitochondrial electron transport and cancer cell bioenergetics by donating electrons at complex II, by stimulating ATP synthase, and by inhibiting intramitochondrial adenyl cyclase (this latter effect is not shown on this scheme). By inhibiting CBS and CAT, AOAA suppresses this bioenergetic pathway. The malate-aspartate shuttle translocates electrons that are produced in glycolysis across the semipermeable inner membrane of the mitochondrion to support oxidative phosphorylation. These electrons enter the electron transport chain at complex I. The shuttle system is required because the mitochondrial inner membrane is impermeable to NADH (a primary reducing equivalent of the electron transport chain). In humans, the cytoplasmic enzyme (GOT1) is one of the key enzymes in the malate shuttle: it functions to catalyze the interconversion of aspartate and α-ketoglutarate to oxaloacetate and glutamate using pyridoxal phosphate as a cofactor. By inhibiting GOT, AOAA reduces the transfer of electron donors to the mitochondria, thereby suppressing cancer cell bioenergetics. By the simultaneous inhibition of H2S production and various transaminases, AOAA interferes with key pathways of cancer cell mitochondrial function.
On the molecular level, the inhibitory effect of AOAA on CBS activity, and most likely on other B6-dependent enzymes, is believed to be due to an attack of the Schiff base linkage between PLP and the enzyme to form oxime-type complexes (Beeler and Churchich, 1976) (Fig. 24). AOAA remains the most potent CBS inhibitor known, because trifluoroalanine and hydroxylamine that had been used before to block CBS-derived H2S are even weaker inhibitors (IC50 = 66 and 278 µM, respectively) (Asimakopoulou et al., 2013). AOAA has low lipophilicity, which makes it difficult for it to enter cells, an effect that is likely to account for its low cell-based potency (millimolar), although its potency on the enzyme is higher (low micromolar) (Chao et al., 2016). Once it reaches the intracellular space, however, AOAA inhibits H2S production in all cellular compartments (Montoya and Pluth, 2016). The low cell uptake of AOAA lead the synthesis of various AOAA prodrugs that are more lipophilic and therefore are more cell permeable. This approach has been exemplified by YD0171 (AOAA methyl ester), which is cleaved by intracellular esterases and which exerts an approximately 10-fold increase in cell-based potency as well as in vivo efficacy compared with AOAA (Chao et al., 2016) (see below for more pathophysiological context).

Inhibition of CBS by AOAA. Docking simulation of PLP in the active center of CBS free (blue) or bound to AOAA (red) (left). Catalytic residues Tyr223 and Gly307 are also shown. Part of heme is visible in the lower right-hand side in dark blue. Residues in proximity to the AOAA-PLP complex are shown (right).
The need for better CBS inhibitors led several laboratories to embark on screening efforts to identify hits that could lead to compounds with improved selectivity and potency. In one such screen, Thorson et al., 2013 identified 12 compounds that significantly inhibited H2S at 150 μΜ. After excluding those compounds that scavenged H2S or quenched the azido-coumarin fluorescence that was used to detect H2S, the authors proposed the diuretic amiloride and the DOPA-dexarboxylase inhibitor benserazide as CBS inhibitors, with an IC50 of 89 μM and an IC25 125 μM, respectively. Τhe same group, after screening a library consisting of marine natural products and synthetic derivatives reported that their best hits were synthetic compounds derived from the polyandrocarpamines A and B; the potency of these derivatives was in the 100 μΜ range (Thorson et al., 2015). In another high-throughput assay, Zhou et al. (2013) identified 1,6-dimethyl-pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione (NSC67078) as a CBS inhibitor with a modest (threefold) selectivity over CSE. When docking analysis was used to tentatively identify the binding site this compound to hCBS, it was concluded that its binding site was different from that of PLP (Zhou et al., 2013).
It should be noted, however, that NSC67078 appears to have several additional pharmacological targets in addition to CBS; moreover, in the fluorescent assay used to estimate its potency, it also inhibits the H2S signal elicited by GYY4137, indicative of additional pharmacological actions beyond inhibition of the catalytic activity of CBS (e.g., H2S scavenging and potent inhibition of the β-catenin pathway (as discussed in Druzhyna et al., 2016). The most recently published effort to identify new CBS inhibitors through high-throughput screening used a natural compound library, yielding 11 hits with IC50 below 20 μΜ. The most potent among them was hypericin, one of the major active components of St. John’s Wort (Niu et al., 2017). However, as with most natural products, the specificity of hypericin against a single target is likely low.
We used a pool of 8871 well-annotated pharmacological compounds and clinically used drugs that included the LOPAC Library, the Food and Drug Administration Approved Drug Library, the National Institutes of Health Clinical Collection, the New Prestwick Chemical Library, the US Drug Collection, the International Drug Collection, the “Killer Plates” collection, and a small custom compilation of PLP-dependent enzyme inhibitors (Druzhyna et al., 2016). After using two counterscreens, the hit list was narrowed down to four compounds, hexachlorophene, tannic acid, aurin tricarboxylic acid, and benserazide, all of which were less potent than AOAA. In line with the known ability of copper to inhibit CBS (Matsuo and Greenberg 1958; Bar-Or et al., 2005), several copper-containing compounds contained in the libraries emerged as CBS inhibitors. The activity of these compounds was confirmed to be due to the inhibitory effect of copper ions themselves.14 It should be noted that benserazide was more potent an inhibitor for CBS (IC50 approximately 30 μΜ compared with the 125 μΜ IC25 reported by (Thorson et al., 2013). The lower inhibitor potency reported before either reflects differences due to the experimental conditions used or is due to degradation of the compound in the plate, a common source of errors in high-throughput screening assays. When benserazide was tested against the other two H2S-producing enzymes, it was found to only weakly inhibit CSE and 3-MST activity (16% and 35% at 100 μM, respectively). Moreover, the major benserazide metabolite 2,3,4-trihydroxybenzylhydrazine also inhibited CBS activity (Druzhyna et al., 2016). By using in silico docking simulations, we proposed that the mechanism of action of benserazide results from binding in the active site of the enzyme and reacting with the PLP cofactor, leading to the formation of a Schiff base-like adduct with the formyl moiety of pyridoxal (Druzhyna et al., 2016).
It is clear that despite intense efforts from several research teams to identify CBS inhibitors with an improved pharmacological profile, no truly selective new compounds have been found. However, the need to develop CBS inhibitors does not only stem from the desire to better understand the role of CBS in cell biology and its contribution to disease development and progression, but also because CBS has been proposed to be an important drug target for cancer (Hellmich et al., 2015) and stroke (Chan et al., 2015). In addition, inhibiting the CBS homolog present in bacteria, and other bacterial H2S producing enzymes renders bacterial pathogens highly sensitive to a multitude of antibiotics (Shatalin et al., 2011).
We recently discovered that CBS is highly expressed in colon cancer cell lines, including HCT116, LoVo, and HT29 (Szabo et al., 2013). Importantly, CBS levels are much higher in colon cancer biopsies compared with the surrounding normal mucosa. CBS in tumor cells produces high levels of H2S that serves as an alternative substrate for tumor bioenergetics, supporting tumor cell proliferation and driving angiogenesis (Szabo et al., 2013; Hellmich et al., 2015). Silencing of CBS or pharmacological inhibition with AOAA inhibited cancer cell line and tumor xenograft growth in vivo, validating CBS as an anticancer drug target (Szabo et al., 2013; Szabo, 2016). Increased expression of CBS was also noted in breast, ovarian, and bladder cancers (Bhattacharyya et al., 2013; Sen et al., 2015; Gai et al., 2016). Inhibiting CBS in a cisplatin-resistant orthotopic ovarian cancer model reduced nodule formation and sensitized tumor cells to chemotherapeutic treatment (Bhattacharyya et al., 2013).
AOAA has been used in human clinical trials in the 80s and 90s as a treatment of Huntington’s disease and tinnitus (Perry et al., 1980; Guth et al., 1990; reviewed in Hellmich et al., 2015). The observed therapeutic efficacy for these indications was not encouraging enough to proceed with clinical development of AOAA; however, the compound displayed acceptable tolerability. When comparing the potency of AOAA in recombinant CBS assays to the potency to inhibit HCT116 cancer cell proliferation, we noted more than 100-fold difference in the IC50 (Asimakopoulou et al., 2013; Szabo et al., 2013). We thus hypothesized that the markedly lower potency of AOAA in the cell-based assays was due to its limited cell membrane permeability. We synthesized a number of derivatives to increase potency but discovered that AOAA did not tolerate derivatization either on the amine group or the linker at the α-carbon position (Chao et al., 2016). Despite the greatly reduced potency of AOAA analogs against recombinant CBS, the AOAA optimization effort yielded a prodrug compound (YD0171) with superior potency in cell-based assays (Chao et al., 2016). The prodrug approach has been widely used in improving the pharmacological properties of various drugs or drug development candidates (Rautio et al., 2008). Coupling the active principle with a group that increases cellular penetration/uptake improves potency and efficacy. Methyl- or ethyl-esters are some of the most common prodrugs in existence, and clinical examples of such prodrugs include enalapril, oseltamivir, clopidogrel, famciclovir, and pivampicillin (Rautio et al., 2008). Indeed, generating a methyl ester of AOAA (YD0171) increased the water/octanol coefficient from 0.0019 (AOAA) to 0.1210 (for YD0171), indicating higher lipophilicity of the prodrug. Cleavage of the ester bond could be documented by measuring increased concentrations of methanol in cell homogenates (Chao et al., 2016). In vivo, YD0171 reduced growth of tumor xenografts in athymic mice with approximately a 20-fold increased potency over AOAA. Moreover, YD0717 induced the regression of established HCT116 tumors in vivo (Chao et al., 2016).
Taken together, although various screening efforts have identified many different CBS inhibitors, for most pharmacological studies (both in vitro and in vivo) AOAA remains the compound of choice; it has been used in a large number of publications over the last decade. The basic physiological papers include mechanistic studies investigating the role of endogenous H2S on various channels and cellular processes (e.g., Donovan et al., 2011; Gil et al., 2011; Roy et al., 2012; Martinez-Cutillas et al., 2015; Rios et al., 2015; Xiao et al., 2015; Krause et al., 2016; Liu et al., 2016d; Rios et al., 2016; Yan et al., 2016; Yetik-Anacak et al., 2016). Many of these reports are in the area of cancer, where they show the antiproliferative effects of this compound and as additive or synergistic antitumor effects in combination with various chemotherapeutic agents (Szabo et al., 2013; Bhattacharyya et al., 2013; Módis et al., 2014b; Szczesny et al., 2016). Additional pathophysiological conditions associated with H2S overproduction where AOAA has been shown to be of therapeutic benefit include oxygen-induced retinopathy (Gersztenkorn et al., 2016), stroke (Hadadha et al., 2015) and various forms of circulatory shock and burn injury (Chen et al., 2011; Ahmad and Szabo, 2016). Although, for the lack of better alternatives, we continue to recommend using AOAA as a CBS inhibitor (or as a combined CBS/CSE inhibitor), studies using AOAA (without various independent control experiments, e.g., CBS silencing or CBS−/− systems) should be regarded with caution, given the issues regarding the selectivity of this compound.
XXVIII. Pharmacological Inhibitors of 3-Mercaptopyruvate Sulfurtransferase
Consistently with its GC-rich and TATAless promoter that is characteristic of housekeeping genes (Nagahara et al., 2004), 3-MST has been shown to be present in all mammalian tissues (Kimura, 2015). However, 3-MST expression levels vary among tissues; brain, liver, kidneys, testes, large intestine, and endocrine organs in the mouse contain the highest amounts (Shibuya et al., 2013; Tomita et al., 2016). 3-MST is a 33-kDa Zn-dependent enzyme that exhibits a monomer-dimer equilibrium, with the monomer being the active form Nagahara, 2013) (Fig. 25). Two surface exposed cysteines (Cys154 and Cys263) are involved in intermolecular disulfide formation, determining enzyme activity (Nagahara et al., 2007). In addition to regulating cysteine degradation, 3-MST detoxifies cyanide (Nagahara et al., 2007). 3-MST is also designated as TUM1 (tRNA thiouridin modification protein 1), which is known to thiolate cytosolic tRNAs. Two TUM1 splice variants have been identified, showing similar kinetic behavior and comparable pH and temperature dependence, but the two variants differ in their cellular localization (Fräsdorf et al., 2014). TUM1-Iso1 is only present in the cytosol, whereas TUM1-Iso2 exhibits dual localization in the cytosol and mitochondria. Earlier studies also reported that 3-MST and CAT are found in both the cytosol and in the mitochondria (Shibuya et al., 2009). The mechanism of 3-MST catalysis involves transfer of the sulfur from 3-MP to a nucleophilic cysteine (Cys247) in its active site; the protein persulfide then yields H2S in the presence of some reductants (Mikami et al., 2011) or is released by specific enzymes (Nagahara, 2013; Kabil and Banerjee, 2014) (Fig. 25).
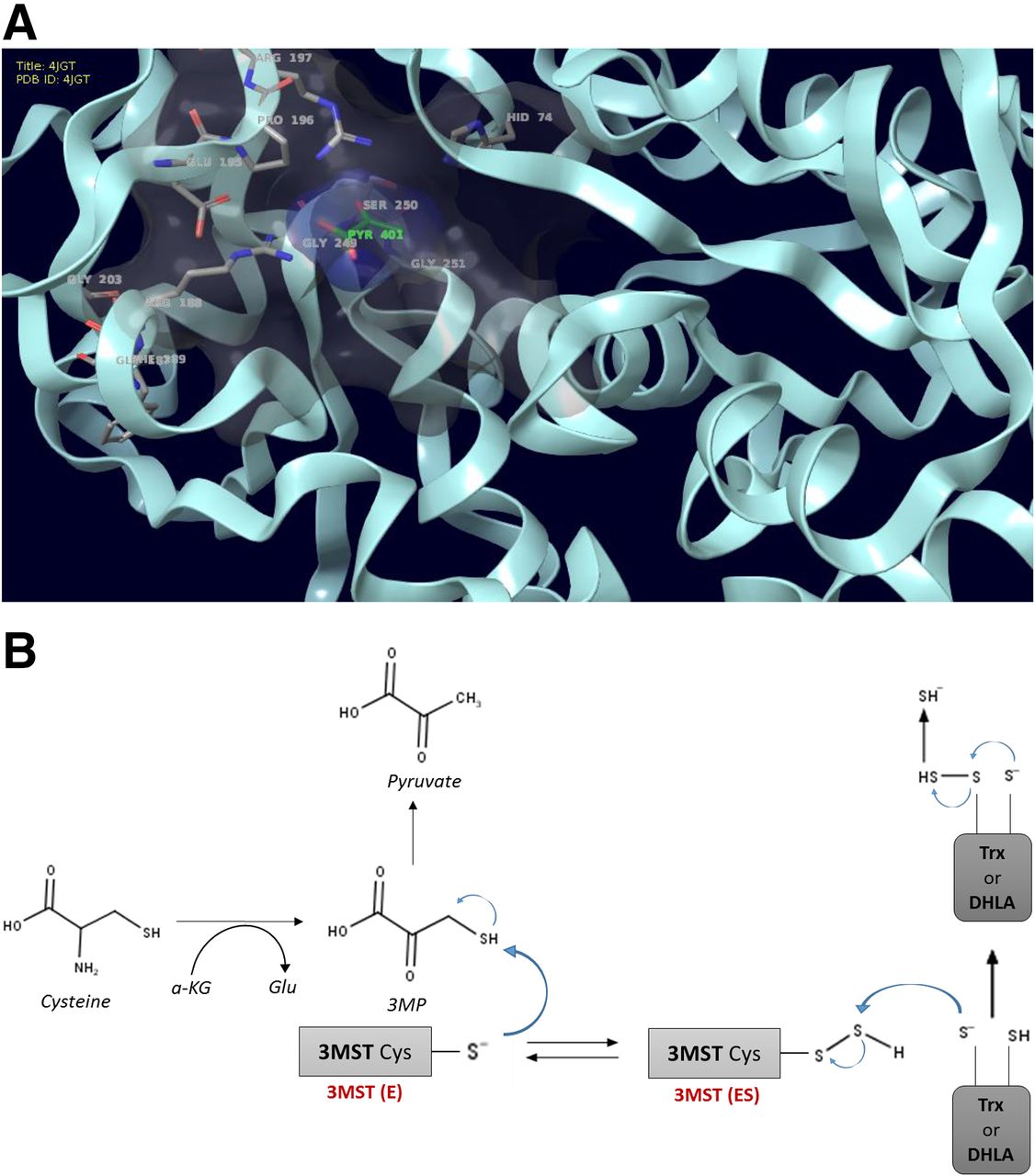
(A) 3-MST is a 32.8-kDa protein comprising an N-terminal catalytically inactive domain and a C-terminal catalytically active domain. The catalytic site, Cys247, is redox-active and is oxidized to form sulfenyl cysteine. The sulfenyl cysteine is then reduced to the active form by thioredoxin (Trx). The catalytic site, Cys247, serves as an intrasubunit redox-sensing switch. (B) The production of H2S by 3-MST in the presence of Trx or DHLA. 3-MST reacts with 3MP to produce H2S via a persulfide intermediate. Trx or DHLA accepts a sulfur atom from a persulfide intermediate that is attacked by another thiol and releases H2S.
Up until very recently, the only compounds with inhibitory activity against 3-MST consisted of a diverse collection of chemicals, including hypotaurine and methanesulfinic acid, and substrate mimics, like pyruvate, phenylpyruvate, oxobutyrate, oxoglutamate, 2- mercaptopropionic acid, and 3-mercaptopropionic acid (Porter and Baskin, 1995; Porter and Baskin, 1996; Wróbel and Jurkowska, 2007). All of these compounds are characterized by low potency (IC50 values in the millimolar range) (Wing and Baskin, 1992; Porter and Baskin, 1995; Porter and Baskin, 1996) and lack of selectivity, making them essentially unsuitable for biologic studies. Moreover, as discussed above, AOAA, through inhibition of CAT, decreases cellular 3-MP production, which indirectly suppresses H2S production by 3-MST.
Hanaoka et al., 2017 in a recent high-throughput screen of 174,118 compounds reported the characterization of several 3-MST inhibitors with micromolar potencies that shared a common aromatic ring-carbonyl-S-pyrimidone structure. One of them, 2-4-hydroxy-6-methylpyrimidin-2-yl-sulfanyl-1-naphthalen-1-yl-ethan-1-one (or “Compound 3”) (Table 2) showed high selectivity for 3-MST over other H2S-producing enzymes and rhodanese. By using cocrystallization studies with two of these inhibitors, as well as theoretical calculations, the authors proposed that the mechanism of inhibition involved the formation of a unique long-range electrostatic interaction between the positively charged carbonyl carbon of the pyrimidone moiety of the inhibitor with the persulfurated cysteine in the active site. The recently identified 3-MST inhibitors will undoubtedly help in shedding light on the biologic roles of 3-MST in health and disease. Given the ubiquitous expression of 3-MST, targeting a specific cell type or tissue in the body may be challenging. As increased 3-MST levels have been observed in glioma, melanoma, and lung cancer (Wróbel et al., 2014; Panza et al., 2015; Szczesny et al., 2016) and in erythrocytes from patients with polycythemia vera (Frendo and Wróbel, 1997), selective 3-MST inhibitors might be of therapeutic value in these conditions.
XXIX. Alternative Means to Decrease Biologic H2S Levels
Similar to the alternative approaches related to H2S donation, there are also alternative approaches to H2S biosynthesis inhibition through the pharmacological modulation of substrate availability (Table 1). Intracellular cysteine pools are being replenished by extracellular circulating l-cysteine, the cellular uptake mainly occurring through the uptake of its dimer (l-cystine, CSSC) via the xCT− CSSC/L-glutamate antiporter (SLC7A11) (Banjac et al., 2008). Inhibition of this transport system may be sufficient to restrict intracellular l-cysteine levels and suppress H2S biosynthesis and inhibit a variety of other l-cysteine-dependent intracellular processes, including glutathione biosynthesis (as shown by Chung et al., 2005), although it is possible that some cells may maintain their intracellular l-cysteine levels through import via additional transporters and/or via upregulation of endogenous l-cysteine synthesis pathways. Another approach, as demonstrated by Cramer et al. (2017), is the reduction of extracellular l-cysteine levels using infusion of recombinant CSE enzyme. Although this approach may increase circulating (extracellular) H2S levels, it is expected to decrease intracellular H2S levels, which is likely to be detrimental for tumor types that rely on H2S (or on other l-cysteine-dependent processes) for their growth and survival. Indeed, extracellular CSE was shown to reduce tumor cell viability in cell culture experiments in vitro; it also caused a rapid and sustained suppression of circulating l-cysteine levels in vivo and suppressed the growth of tumors in a mouse xenograft model (Cramer et al., 2017).
Indirect ways to reduce CBS-dependent H2S production involve approaches that decrease homocysteine levels. Many such interventions have been developed and tested experimentally in the context of the therapy of patients with CBS mutations and associated hyperhomocysteinemia. (In this context, the goal was to find ways to metabolize homocysteine via routes independent of CBS; the intended goal of this approach was to reduce homocysteinemia, but an additional effect of this approaches is that CBS-dependent homocysteine conversion, and, therefore, H2S production is also suppressed). The first such approach aimed to restrict the level of methionine in the diet and supplement with cysteine (Komrower et al., 1966; Perry et al., 1966; Sardharwalla et al., 1968; Gupta et al., 2016). A second approach involved betaine supplementation. Betaine is cosubstrate of the enzyme BHMT, which catalyzes the formation of methionine from homocysteine in the liver. Thus increased betaine was found to lower homocysteine levels by decreasing the homocysteine pool and increasing the methionine pool (Wilcken et al., 1983; Gupta et al., 2016).15 Additional, enzyme-based approaches to suppressing biologic H2S production may relate to the modulation of various enzyme cofactors (e.g., inhibiting SAM biosynthesis or perhaps suppression of SAM binding to CBS); these have not yet been explored experimentally. Naturally, inhibition of the levels of the enzyme (either by downregulating its expression or perhaps by enhancing their proteolytic degradation) may be additional approaches that should be explored in future experiments.
Another way to reduce biologic H2S levels may be H2S scavenging. Although scavengers have many inherent problems (specificity, selectivity, delivery issues, and, unless they are catalytic, the fact they are consumed in the reactions and typically require large concentrations/doses), in theory, this approach may also be of some merit for further exploration. However, the current state-of-the-art of H2S scavengers is in an embryonic stage; although heme-containing proteins (e.g., hemoglobin, myoglobin, neuroglobin) are known to scavenge H2S (Brunyanszki et al., 2015; Bostelaar et al., 2016; Ruetz et al., 2017; Vitvitsky et al., 2017), they also scavenge many other reactive species including nitric oxide.
The “mirror-image” of the other nontraditional approaches listed in section XXIV are not feasible in our opinion. For instance, there are currently no known drugs or mechanisms for on-demand upregulation of various H2S degrading enzymes.
XXX. Conclusions and Future Directions
Substantial progress has been made in the field of H2S donors over the last decade. Multiple classes of H2S donors have been synthesized, with various characteristics (different half-life; different release profiles, including compounds that respond to specific cellular environments such as pH or oxidative stress; and as donors targeted to specific cellular compartments such as the mitochondria).
H2S donors are commonly used as experimental tools to delineate the roles of H2S in various physiological and pathophysiological conditions (although the effects elicited by them may not necessarily reflect the roles and functions of endogenous H2S). As already discussed in detail, using salt-based H2S donors (Na2S, NaHS) from most commercial sources will create a mixture of species (Fig. 2) (including various forms of H2S in solution and polysulfides), the resulting biologic effects result from effects elicited by these various species. Even bubbling of pure H2S through physiological solutions will create some small amount of additional sulfur species (e.g., polysulfides). As these various sulfur-species react with various biologic constituents (oxidants, free radicals, NO, thiols, proteins), additional (secondary and tertiary) species will form to create a mixture of species. Although this may resemble the situation in a biologic system (where, it is also likely that various sulfur-species are present simultaneously), the relative proportions and effects of the species created by H2S donors is probably not identical to the conditions that apply to endogenously produced H2S. The proportion and biologic role of the various secondary and tertiary species are likely to be dependent on the cell type, the experimental condition, the time of exposure, the source and purity of the donor, the concentration of the donor, and many additional factors. These complexities and variabilities are likely to contribute to the diverse biologic effects reported with H2S donors in various experimental studies.
By using slow-release and/or cell-compartment-targeted H2S, donors have substantial advantages over the use of the salt-based H2S donors. Nevertheless, even with the use of these donors, it is unavoidable that in biologic systems multiple secondary and tertiary species (each with its own characteristic effect) are created. It is also becoming increasingly clear that different H2S donors, although they all can induce similar outcomes, e.g., anti-inflammatory and cytoprotective effects at low concentrations or proinflammatory and cytotoxic effects at high concentrations, can stimulate different sets of cellular processes. For instance, rapid-release H2S donors tend to induce more pronounced increases in cellular cGMP levels than slow-acting donors or mitochondrially targeted ones. Moreover, rapid-release H2S donors tend to cooperate with NO-related signaling processes, whereas slow-acting donors (or mitochondrially targeted ones) work largely independently from NO (Chatzianastasiou et al., 2016). Mitochondrially targeted donors, by design, tend to primarily affect mitochondrial processes (e.g., electron transport or mitochondrial DNA repair) and tend to have lesser effects on cytoplasmic signaling processes (e.g., PI3K/Akt phosphorylation) or membrane channels (e.g., KATP channel opening). However, rapid-releasing donors can also have mitochondrial effects, especially in the initial stages of the experiments when they generate a high burst of H2S that will reach the mitochondrial compartment; such a response, in fact, may act as a short-term “chemical hypoxia” or “preconditioning” effect, which, in turn, may induce secondary cellular signaling processes. It should be emphasized that the systematic characterization of the signaling processes activated by various classes of H2S donors remains to be completed; Fig. 26 organizes these processes according to the current (admittedly, fragmented) state-of-the-art.

An overview of the cellular signaling processes elicited by H2S donors. The cellular effects of H2S donors can be different, depending on the rate of the H2S release and the targeted versus nontargeted nature of the donor. For example, mitochondrially targeted H2S donors preferentially activate mitochondrial processes (e.g., protection against mitochondrial oxidative stress, or facilitation of mitochondrial DNA repair processes, or electron donation to the mitochondrial electron transport chain) and have lesser effect on cytoplasmatic signaling pathways. High concentrations of mitochondrial H2S donors may also suppress mitochondrial electron transport by inhibiting mitochondrial Complex IV. When fast-acting H2S donors are applied to cells or animals, the initial high H2S concentration may be sufficient to inhibit mitochondrial Complex IV to induce a short-lasting chemical hypoxia, which, in turn, may stimulate compensatory (preconditioning type) processes. Fast-acting H2S donors tend to be more potent activators of cGMP-dependent processes than slow-release H2S donors. Fast-acting H2S donors also tend to exert their action in cooperation with NO synthase-dependent signaling processes. Please note that the downstream pathways activated by the various H2S donors have not yet been characterized in a systematic manner.
The above considerations should be taken into account when designing mechanistic biologic studies using H2S donors. As a rule of thumb, it is recommended that 1) multiple classes of donors should be used; increases in cellular H2S levels may also be achieved by overexpressing H2S-producing enzyme(s) and/or by treating the cell with substrates of the endogenous H2S-producing enzymes; 2) careful time-course studies and concentration-response studies should be incorporated; 3) the results should be tested in multiple cell types or cell lines; and 4) it should be kept in mind that the compounds used may have additional pharmacological actions (i.e., effects unrelated to H2S donation). Whenever possible, control groups using “spent” donors should be incorporated into the experimental design. Because H2S has important interactions with NO (and, in many systems, acts as an enhancer/amplifier of endogenous NO/cGMP signaling) the contribution of NO to the effects seen with H2S donors can be easily tested (e.g., by pretreating the system with a NOS inhibitor). With the availability of cell-based H2S detecting techniques, the targeting of H2S into various cellular compartments can now be confirmed and visualized (although the determination of absolute concentrations of H2S remains challenging).
As far the clinical development of H2S donors, the situation is challenging, as well. Although many H2S donor compounds have undergone cell-based and animal-based testing, most of the donors discussed have not (yet) progressed beyond the stage of chemical synthesis and in vitro characterization in simple buffers or other cell-free systems. Generally, the concept of therapeutic H2S donation is well justified, because there are many pathophysiological conditions where endogenous H2S levels are suppressed, and donation (i.e., “replacement therapy”) makes pathophysiological and experimental therapeutic sense (Fig. 27, left side). There are also several indications where endogenous H2S levels are not suppressed, and yet H2S donation may be beneficial or warranted, e.g., approaches formulated around the antiviral effects of H2S donation.
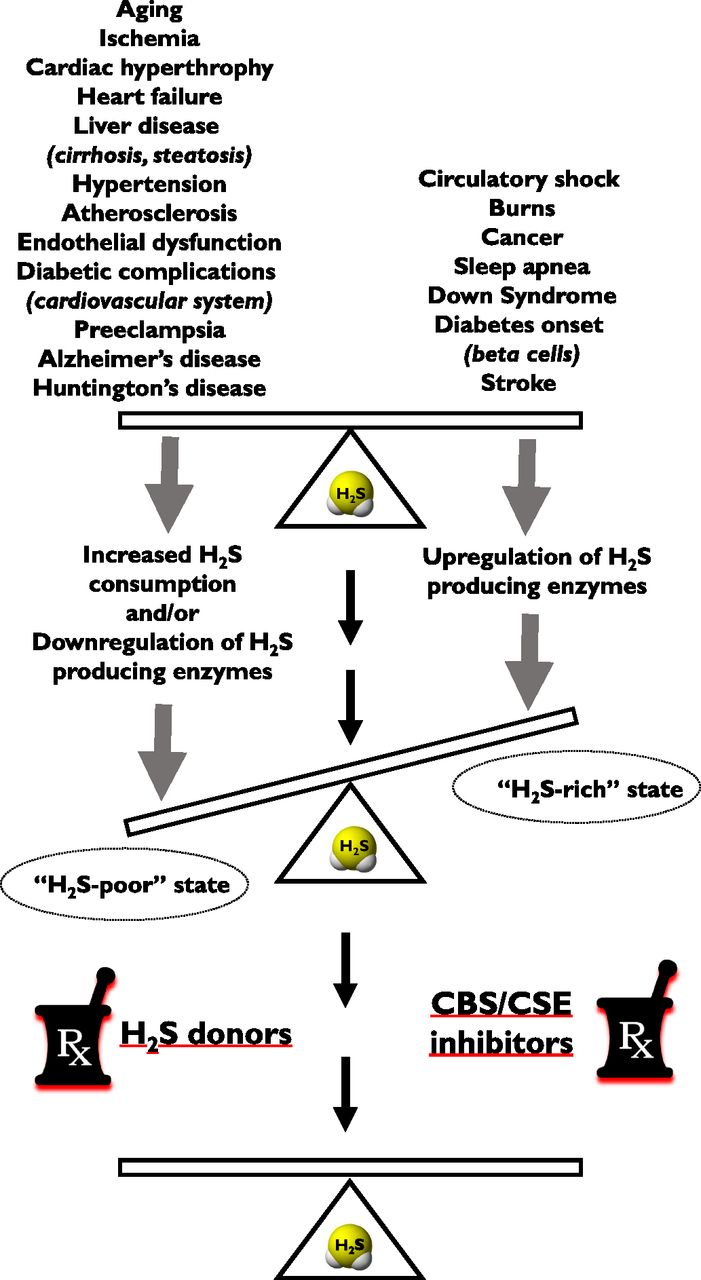
Therapeutic effects of H2S donors and H2S biosynthesis inhibitors: a simplified overview. Some pathophysiological states are associated with H2S deficiency; this can be corrected by H2S donors (a form of replacement therapy) (left side). Other pathophysiological states are associated with H2S overproduction; this can be corrected by H2S biosynthesis inhibitors (right side). The scheme represents an oversimplification for a number of reasons. For example, the same pathophysiological condition can manifest itself with both H2S overproduction and H2S deficiency. In diabetes, the pancreatic beta cell destruction is linked to H2S overproduction; diabetes can also elevate H2S levels in the liver, with pathophysiological consequences. At the same time, the cardiovascular consequences of diabetes include vascular H2S deficiency, which contributes to vascular complications. In addition, in some diseases (e.g., cancer or burn injury), both systemic H2S biosynthesis inhibition and H2S donation can exert beneficial effects through different sets of biologic actions.
Several conceptual and practical challenges can be identified with respect to H2S donation. 1) The very foundation of this approach, i.e., that one delivers an endogenous molecule (“hormone replacement”: supplementation of a “known entity” to the human body) is an attractive feature. At the same time, H2S replacement is not likely to correct the underlying cause of the H2S deficiency: the biologic reason(s) why H2S levels are decreased will remain. This means that H2S therapy will probably need to continue in the long term, perhaps life-long. This will require chronic safety studies (including carcinogenicity and teratogenicity) for clinical development and registration. Due to the bell-shaped pharmacological character of H2S, boosting the levels of H2S beyond the desired tissue concentrations (which are, in fact, hard to quantify using current methods) will cause adverse effects, including, at the end of the spectrum, suppression of mitochondrial respiration/inhibition aerobic ATP generation, as well as potential adverse effects on the genetic material. Given the toxicological profile of H2S, it is conceivable, if not likely, that chronic administration of H2S donors at high doses will cause adverse effects, possibly including genotoxic effects and carcinogenic and teratogenic effects. These effects, on their own, may not mean the “kiss of death” for the development candidate but may narrow the choice of therapeutic indications. 2) The rate of H2S release is a key issue. Although it is clear that the salt-based H2S “donors” produce H2S too rapidly and, at the other end of the spectrum, GYY4137 may have a H2S release profile that may be too slow for many of the potential indications, the exact “optimal” H2S release rate from a donor remains to be established (and it is probably dependent on the indication and the route of the donor’s administration). It should also be emphasized that the rates of H2S release from the different types of donors have only been measured in buffered solutions, so far. H2S production under these in vitro conditions might be drastically different than the rate at which these donors liberate H2S in biologic systems. 3) The fact that the currently available H2S donors are not sufficiently targeted to the site of the actual H2S deficiency (i.e., to specific cells or tissues) means that some of the cells and tissues may “see” too much H2S, whereas those cells that lack H2S may or may not experience a complete degree of restoration. Some of the most ingenious medicinal chemistry approaches, e.g., pH-dependent H2S releasers or compounds where H2S release is triggered by oxidants, may partially mitigate some of these deficiencies, although, curiously, these more “rational” approaches have the least amount of published in vivo data. Oral use of pH-triggered H2S donors will probably require special formulations to avoid premature “dumping” of H2S in the acidic environment of the stomach. Targeted H2S delivery is probably most needed where H2S delivery is aimed at cancer cells; with nontargeted donors, it is likely that the doses of the donors that yield sufficiently high concentrations of H2S within the tumor tissue will also deliver high H2S fluxes to nontumorous tissues (possibly inducing adverse effects). 4) The fact that H2S donor molecules, after delivering their “load,” will also produce “leftover” molecules, represents another challenge. These remainder molecules, ideally should be benign and should be cleared from the body without accumulation or overt intrinsic toxicity. In this respect, natural compounds (e.g., garlic-derived polysulfides) or compounds that are closely associated with small molecules that are likely to be handled by the body’s metabolic systems (e.g., amino acid-like small molecules) may be preferred. 4) The very processes that are needed for some of the H2S donors to produce H2S intracellularly use thiols and other biomolecules; this may affect the balance of the body’s thiol and antioxidant pools, possibly resulting in adverse effects, especially in the long term. Although the above list of “issues” seems substantial, many of the same potential issues apply to NO donors. Yet there are many classes of successful, relatively safe, and clinically widely used NO donors (e.g., glycerin trinitrate); although, to be fair, the intensive research in the field of NO over the last three decades, although yielding many excellent NO donor experimental “tools,” has not produced the kind of new clinical approvals of novel NO donors that we had hoped for.
The “combined donors” (new compounds that link various H2S donating groups to clinically used drugs) represents a distinct field of research. The main pharmacological character of the combined donor compounds is determined by the properties of the clinically used “parent” compound: the H2S donation adds an additional “feature” (such as improved gastrointestinal safety). Some of these combined donor compounds are already in clinical development and it is hoped that this work will eventually result in drug approvals. Nevertheless, it must be mentioned that the combination approaches, although, in principle, they sound elegant, fairly straightforward, and anticipated to be safe, can have their own unexpected complicating issues. This is exemplified by ATB-346, where the H2S donor group unexpectedly also had an effect on the core pharmacological action (COX inhibition) of the compound (see section XXIII). It should also be mentioned that, although the concept of combined donors sounds promising, a similar concept was previously tried with NO. Combined NO-NSAID compounds, often championed by the same groups of investigators who now work on H2S-NSAID technologies, have progressed all the way into Phase III clinical trials, but, regrettably, have not gained regulatory approvals. It is hoped that the lessons and experiences learned from the NO-NSAID projects will help with the design and execution of the H2S-NSAID clinical programs.
Similar to the field of H2S donors, substantial progress has also been made in the field of H2S biosynthesis inhibitors over the last decade. Multiple classes of H2S biosynthesis inhibitors have been identified, either by screening or by rational design. The biggest deficiency in the field (the lack of pharmacological inhibitors of 3-MST) has now also been rectified, as novel 3-MST inhibitors, with considerable potency (both on isolated enzyme and in cell-based system) and specificity have recently been described.
H2S biosynthesis inhibitors are commonly used as experimental tools to delineate the roles of H2S in various physiological and pathophysiological conditions. Ideally, one wishes to have pharmacological inhibitors with high specificity (i.e., an ideal inhibitor should inhibit the desired H2S-producing enzyme target, should not inhibit H2S production from the other H2S-producing enzymes, and should definitely not affect other enzymes unrelated to H2S homeostasis). Moreover, it should exhibit high potency in cell-based systems in vitro and in animal studies in vivo. As already discussed in detail, many of the CSE and CBS inhibitors exert their effects through actions on the PLP prosthetic group of these enzymes. This means that they often (but not always) inhibit both CSE and CBS, and they also often inhibit other (most commonly, PLP-dependent) enzymes. However, this does not mean that CBS or CSE inhibitors inhibit all (or most) PLP-dependent enzymes; neither does this mean that most PLP-dependent enzyme inhibitors are also CSE or CBS inhibitors. For instance, the CBS/CSE inhibitor AOAA inhibits several PLP-dependent enzymes (e.g., GOT and GABA-T), whereas benserazide (a well-known inhibitor of the PLP-dependent enzyme DOPA decarboxylase) was recently identified as a fairly potent inhibitor of CBS.
Currently, the recommended choice of CSE inhibitor is PAG (its L-isoform, not the racemic form, which is also commercially available and is sometimes used in publications); this compound has negligible inhibitory effects on the other two H2S producing enzymes (although it is likely to inhibit several other known enzymes and probably some others of which we are not yet aware). Moreover, PAG is not a very potent inhibitor; in cell-based studies, millimolar concentrations are needed. A recent inhibitor (Compound 2a) emerges as a potential next-generation compound, although the body of biologic data with this compound is currently rather limited. AOAA remains the recommended choice of CBS inhibitor, although this compound also inhibits CSE, as well as several other PLP-dependent enzymes (including CAT, which, in turn, will decrease H2S production by 3-MST). It is much less potent in cell-based assays than what would be expected from its enzyme-based potency. Cell-based potency can somewhat be improved by using lipophilic (and therefore cell-permeable) AOAA prodrugs. Recent work has identified several classes of additional CBS inhibitors (some of which have some degree of selectivity for CBS over CSE); however, most of these compounds have not yet been characterized in sufficient detail (especially in cell-based systems or in vivo). As discussed earlier and in Druzhyna et al. (2016), newly identified compounds (e.g., the compound NSC67078), although potent and somewhat selective for CBS, have additional pharmacological targets, rendering their practical utility questionable.
All of the above issues related to H2S biosynthesis inhibitors should be taken into account when planning mechanistic biologic studies. As a rule of thumb, it is recommended that 1) multiple inhibitory approaches should be used and pharmacological inhibitors should be supplemented with studies where transient or permanent silencing H2S-producing enzyme(s) is achieved; mice lacking CSE, CBS, or 3-MST are also available; 2) careful concentration-response studies should be incorporated and concentrations/doses higher than what is needed to achieve full inhibition of H2S biosynthesis should be avoided; 3) the effects of the inhibitors should be evaluated in multiple cell types; and 4) it should be kept in mind that the compounds used may have additional pharmacological actions (i.e., effects unrelated to inhibition of H2S biosynthesis). Finally, 5) attempts should be made to reverse the effects of the inhibitors by either using excess substrate of the enzyme or by the application of H2S donors (functional antagonism). With the availability of cell-based H2S detecting techniques, inhibition of H2S biosynthesis can be confirmed and visualized in cell-based studies. In vivo, the inhibitory effect of the H2S biosynthesis inhibitors can be confirmed by measuring circulating H2S levels. As discussed here and elsewhere, different H2S-detecting methods give different absolute plasma H2S values (with the methylene blue method producing the highest/unrealistic values); nevertheless, directionally, the effects of the inhibitors can be confirmed by any of the available detection methods. We recommend using the monobromobimane-based method, although this method is not perfect either, because it is relatively cumbersome and labor and equipment intensive and may also not be fully selective for free H2S (as it may “pick up” signals from reactive species other than the ones present in the “free” circulating H2S “pool”).
As far as the clinical development of H2S biosynthesis inhibitors, the situation is much less advanced than it is with H2S donors. Generally, the concept of therapeutic H2S inhibition is well justified, because there are many pathophysiological conditions in which endogenous H2S levels are elevated and inhibition of its biosynthesis makes experimental therapeutic sense (Fig. 27, right side). Although many H2S inhibitor compounds have been used in cell-based and animal-based experiments, none of them have advanced into clinical trials, at least not as H2S biosynthesis inhibitors. The exception is the curious case of AOAA, which has been in clinical trials in the 70s and 80s as a GABA-T inhibitor, for the experimental therapy of neurologic diseases (e.g., Huntington’s disease and tinnitus) (as reviewed in Hellmich et al., 2015). A recent line of work raised the possibility that modified versions of AOAA (exemplified by the AOAA methyl ester compound YD0171) may become clinical development candidates (e.g., for the experimental therapy of various forms of cancer that are associated with the overproduction of H2S within the cancer cell) (Chao et al., 2016). Although, for the last few decades, neither PLP-dependent inhibitors nor irreversible enzyme inhibitors (and AOAA happens to be both) were generally considered as prime pharmaceutical development candidates, the thinking has changed in recent years; there are, in fact, several clinically used drugs that target PLP-dependent enzymes (Amadasi et al., 2007) and there are many approved, clinically used drugs that are covalent modifiers/irreversible enzyme inhibitors (Robertson, 2005). In addition, there is some newly found interest of the pharmaceutical industry in considering irreversible enzyme inhibitors for formal clinical development (Singh et al., 2011).
One of the fundamental questions with H2S biosynthesis inhibitors is their safety and their potential side-effect profile. Some of these issues are mechanism-based (e.g., inhibition of CBS in the liver is expected to suppress the biologic elimination of homocysteine, and the resulting homocysteinemia may be viewed as a side effect and a cardiovascular risk factor) and others are potentially related to off-target effects of the inhibitors (e.g., on other PLP-dependent enzymes). The potential side effects related to CBS inhibitors were discussed recently (Hellmich et al., 2015); as with any small molecule, the real answers can only be given after conducting formal safety studies in several animal species followed by human clinical trials. Depending on the safety profile of the H2S biosynthesis inhibitors, the potential development indications may need to be narrowed. However, in our view, a CBS/CSE inhibitor for cancer therapy is acceptable even if it causes some degree of homocysteinemia.
Just as H2S replacement therapy will not correct the fundamental underlying cause of the H2S deficiency, H2S biosynthesis inhibitors will probably not rectify the underlying cause of the disease (and therefore may need to be given chronically or possibly in an intermittent manner and possibly in combination with other drugs targeting different mechanisms and pathways of the underlying disease).
There are currently no known clinical development efforts based around CSE inhibitors or 3-MST inhibitors. In the majority of published studies, CSE deficiency sensitizes to disease development, rather than protect from it. However, there are several potential disease indications based on studies using CSE knockout mice, in which CSE inhibition may be therapeutically justified, including acute liver injury (Shirozu et al., 2014), pancreatitis (Ang et al., 2013), sleep apnea (Peng et al., 2017), certain forms of sepsis (Ahmad et al., 2016a), and burn injury (Ahmad et al., 2017). Whether 3-MST inhibitors may have potential therapeutic applications is currently not known and remains to be determined. Some of the future research and development directions with H2S biosynthesis inhibitors may also include targeted compounds (e.g., compounds that specifically target tumor cells that overexpress H2S-producing enzymes).
The complexity of H2S biology is well illustrated by the fact that sometimes in the same pathophysiological conditions both H2S biosynthesis inhibitors and H2S donors can exert therapeutic effects. This is well illustrated through the example of the experimental therapy of cancer (overviewed in Szabo, 2016), where inhibition of H2S production by the cancer cells takes away some of the supporting roles (bioenergetics, proliferative signaling, angiogenesis) of H2S (produced due to the upregulation of H2S-producing enzymes within the cancer cells), whereas delivery of additional H2S to the tumor cell drives the cells into apoptosis due to the high/cytotoxic levels of H2S (Fig. 28).
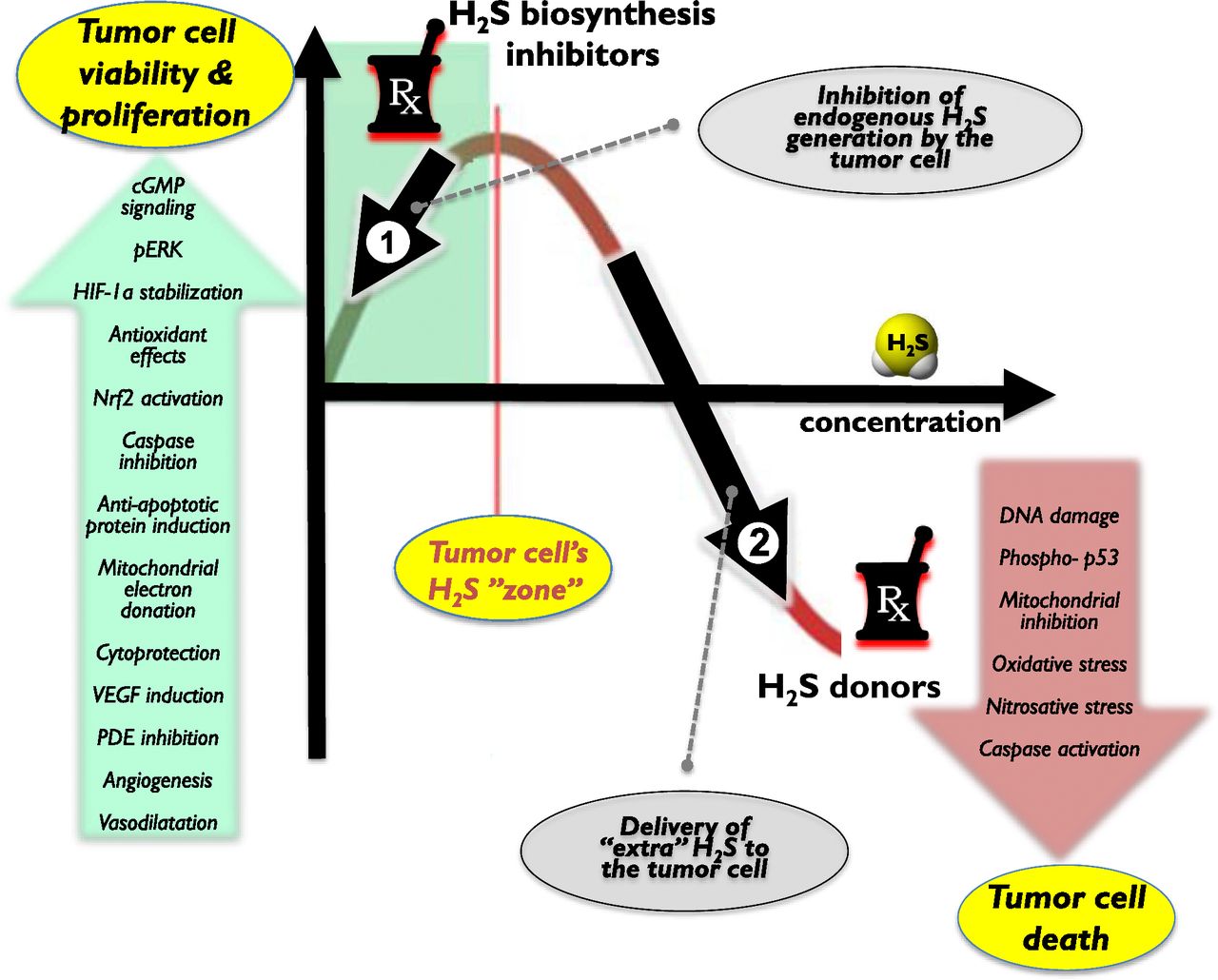
Mechanisms underlying the therapeutic effects of H2S biosynthesis inhibitor (left side) and H2S donors (right side) in cancer. Because of the bell-shaped pharmacological profile of H2S, both H2S biosynthesis inhibition and H2S donation can exert therapeutic effects. Low concentrations of H2S that are produced endogenously by CBS, CSE, and/or 3-MST can support tumor growth and tumor angiogenesis through a variety of pathways shown in the green arrow. Pharmacological inhibition of these responses (depicted by arrow #1) can be of therapeutic benefit, either on its own, or to sensitize the tumor cell to standard anticancer therapies. On the other hand, high concentrations of H2S can be cytostatic or cytotoxic through a variety of pathways shown in the red arrow. Thus therapeutic administration of H2S (depicted by arrow #2), which induces high concentrations of H2S in the tumor cell can be used to induce anticancer effects and/or to potentiate anticancer chemo-or radiotherapy.
Taken together, the field of H2S donors and H2S biosynthesis inhibitors has substantially advanced over the last decade. It is hoped that the information presented in the current article will be useful to help with the use of H2S donors and H2S biosynthesis inhibitors for basic experimental studies. It may also serve as directional and conceptual support for translational efforts in this challenging, unconventional, unusual, but fascinating field of biology.
Acknowledgments
The helpful comments of Dr. John Wallace and Dr. Matt Whiteman and the editorial help of Dr. Anita Marton are appreciated.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Szabo, Papapetropoulos.
Footnotes
The research of C.S. in the field of H2S is supported by the US National Institutes of Health National Cancer Institute [Grant R01CA175803], National Institute of General Medical Sciences [Grant R01GM107846], and the US Cancer Prevention Research Institute of Texas (CPRIT, DP150074). The research of A.P. in the field of H2S is supported by an Excellence in Research IKY/Siemens grant.
↵1Please also note that the measurements of plasma H2S levels remain a heavily debated issue, and the absolute levels reported in the literature are very much dependent on the method used (Furne et al., 2008; Whitfield et al., 2008; Olson, 2009; Wintner et al., 2010; Olson et al., 2014; Papapetropoulos et al., 2015).
↵2This effect of H2S has been known for many decades, and was, for a long time, viewed as the primary pharmacological effect of H2S in the mitochondria, especially in the context of environmental toxicology.
↵3This delivery method, in fact, may parallel the delivery of NO for pulmonary hypertension—a Food and Drug Administration-approved therapeutic for the treatment of the pulmonary hypertension of the newborn—the so-called “blue baby syndrome.”
↵4It should be mentioned that the same report has also unveiled a severe, potentially lethal interaction between H2S inhalation and volatile anesthetics (Li et al., 2012); although the underlying mechanisms remain to be further explored, this effect certainly needs to be kept in mind for any potential future translation of H2S gas-based therapeutic approaches.
↵5Certain cell types, for example intestinal epithelial cells, due to their biological function to limit the systemic absorption of H2S produced by bacteria of the intestinal microbiota, have high H2S-consuming capacity (Abou-Hamdan et al, 2015; Beaumont et al., 2016).
↵6Note that polysulfide formation is not an exclusive feature of fast-releasing H2S donors. Polysulfides can also be formed in biological matrices after exposure to slow-releasing H2S donors [a class of H2S releasing compounds, reviewed in Kimura (2015)], as part of a set of complex biological reactions (Longen et al., 2016).
↵7Even the way the H2S solution is added to the cell culture could make a difference, e.g., the ratio of the stock solution added and the volume of the culture medium, whether the solution is slowly pipetted to the top of the solution or “shot” to the bottom onto the cells, whether the cell culture is shaken or stirred after the administration of the solution, etc.
↵8This may also provide a potential explanation of the counterintuitive observation that, in some instances, inhibition of endogenously produced H2S can attenuate the effects of exogenously added H2S.
↵9Interestingly, short periods of boiling significantly increase the H2S-releasing capacity of garlic extracts, whereas longer boiling periods decrease it (Tocmo et al., 2017). Although the mechanisms have not been clarified, it may be related to the interconversion of various sulfur species.
↵10It should be mentioned that with some commercially available sources of GYY4137, some purity issues remain. For instance, it is often unclear how much residual solvent (e.g. dichloromethane complex xCHCl2) remains in the preparations.
↵11Shorter (e.g. “overnight”) protocols are insufficient, because they will not eliminate all of the compound’s H2S releasing capacity.
↵12Typically, measurements of cyclic nucleotide levels include phosphodiesterase (PDE) inhibitors (e.g., IBMX) as part of the assay. Since the effect of H2S on cGMP levels is due to inhibition of cGMP phosphodiesterase inhibition, PDE inhibitors like IBMX will mask the effect of H2S. Therefore, to study H2S-related effects, the assay conditions must be modified to omit “external” PDE inhibitors.
↵13Although not discussed in the H2S literature, one should mention that benzamide, on its own, has distinct pharmacological effects as an inhibitor of poly(ADP-ribose) polymerase (PARP); PARP inhibition, on its own, is known to exert cytoprotective and anti-inflammatory effects, as reviewed in Virág and Szabó (2002) and Jagtap and Szabó (2005). Whether actions on PARP may contribute to the effects of H2S-donating compounds needs to be explored.
↵14It should be noted that copper, in addition to inhibiting CBS activity, also reacts directly with H2S. In fact, copper chemistry has been used in some assays to detect H2S. Copper, therefore, can be viewed as a combined CBS inhibitor and H2S “trap.” In biological contexts, the binding of H2S to copper plays a key role in the H2S-mediated inhibition of Complex IV, and H2S-copper reactions are responsible for the sensitive detection of H2S by the olfactory nerves. Whether sulfur-copper coordination plays a role in pathophysiological conditions that are associated with free copper intra- or extracellularly remains to be determined.
↵15Although both methionine restriction and betaine supplementation are expected to suppress CBS-dependent H2S production in vivo (Hine and Mitchell, 2015), the effect of these approaches on CBS-dependent H2S production and overall changes in circulating H2S levels remain to be directly confirmed.
Abbreviations
- ADT
- 5-(4-methoxyphenyl)-3H-1,2-dithiole-3-thione
- ADT-OH
- 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione
- Akt
- protein kinase B
- AOAA
- aminooxyacetic acid
- AVG
- aminoethoxyvinylglycine
- BCA
- β-cyano-l-alanine
- BNP
- brain natriuretic peptide
- CAT
- cysteine aminotransferasem
- CBS
- cystathionine-β-synthase
- COS
- carbonyl sulfide
- COX
- cyclooxygenase
- CSE
- cystathionine-γ-lyase (also CGL or CTH)
- DADS
- diallyl disulfide
- DATS
- diallyl trisulfide
- eNOS
- endothelial isoform of nitric oxide synthase
- GABA-T
- 4-aminobutyrate aminotransferase
- GOT
- aspartate transaminase
- GSH
- glutathione
- IL
- interleukin
- KATP channel
- ATP-sensitive potassium channel
- LPS
- endotoxin (bacterial lipopolysaccharide)
- 3-MP
- 3-mercaptopyruvate
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MSN
- mesoporous silica nanoparticle
- 3-MST
- 3-mercaptopyruvate sulfurtransferase
- NAC
- N-acetylcysteine
- NO
- nitric oxide
- NOS
- nitric oxide synthase
- Nrf2
- nuclear factor erythroid 2 (NFE2)-related factor 2
- NSAID
- non-steroidal anti-inflammatory drug
- NTA
- N-thiocarboxyanhydride
- PAG
- propargylglycine
- PARP
- poly(ADP-ribose) polymerase
- PDE
- phosphodiesterase
- PEG
- polyethylene glycol
- PGE2
- prostaglandin E2
- PLP
- pyridoxal 5′-phosphate
- ROS
- reactive oxygen species
- SAC
- S-allylcysteine
- SAM
- S-adenosylmethionine
- SATO
- S-aroylthiooxime
- SPRC
- S-propargyl-l-cysteine
- STS
- sodium thiosulfate
- TNFα
- tumor necrosis factor α
- TPP
- triphenylphosphonium
- TTM
- ammonium tetrathiomolybdate
- TUM1
- tRNA thiouridin modification protein 1
- VCAM
- vascular cell adhesion molecule
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
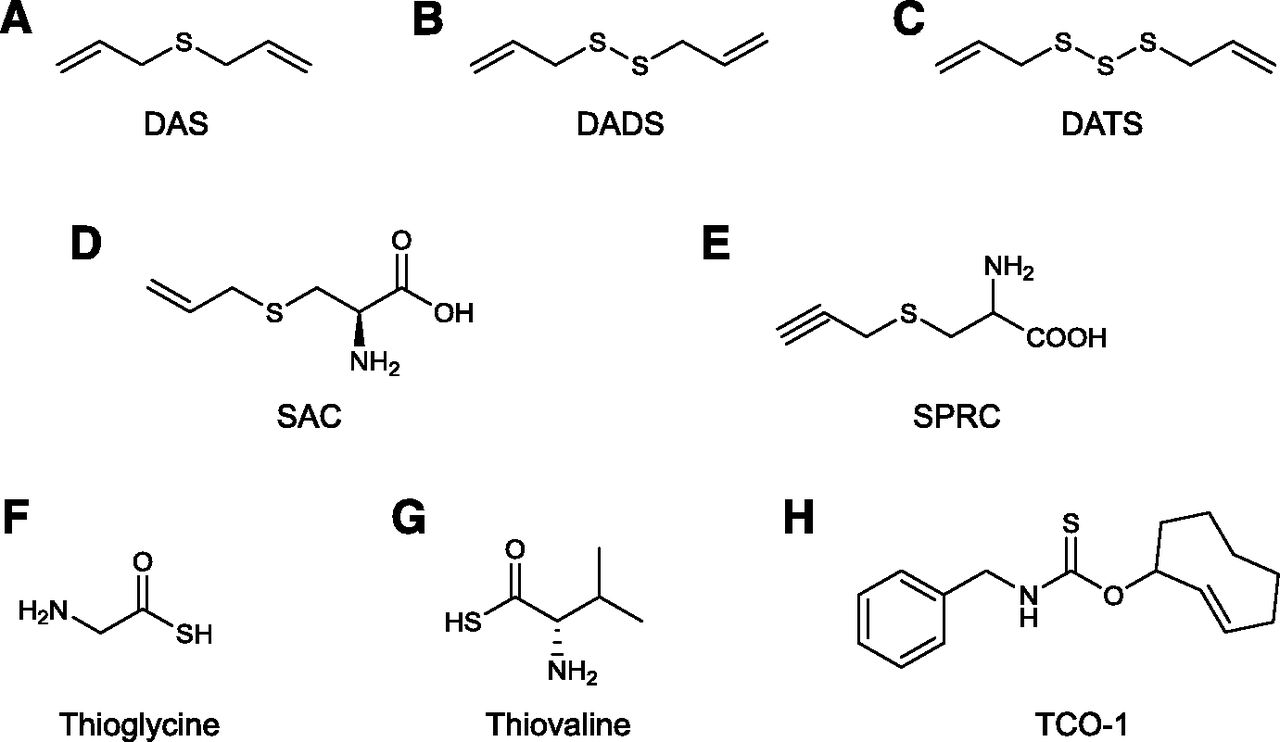{kind=link}
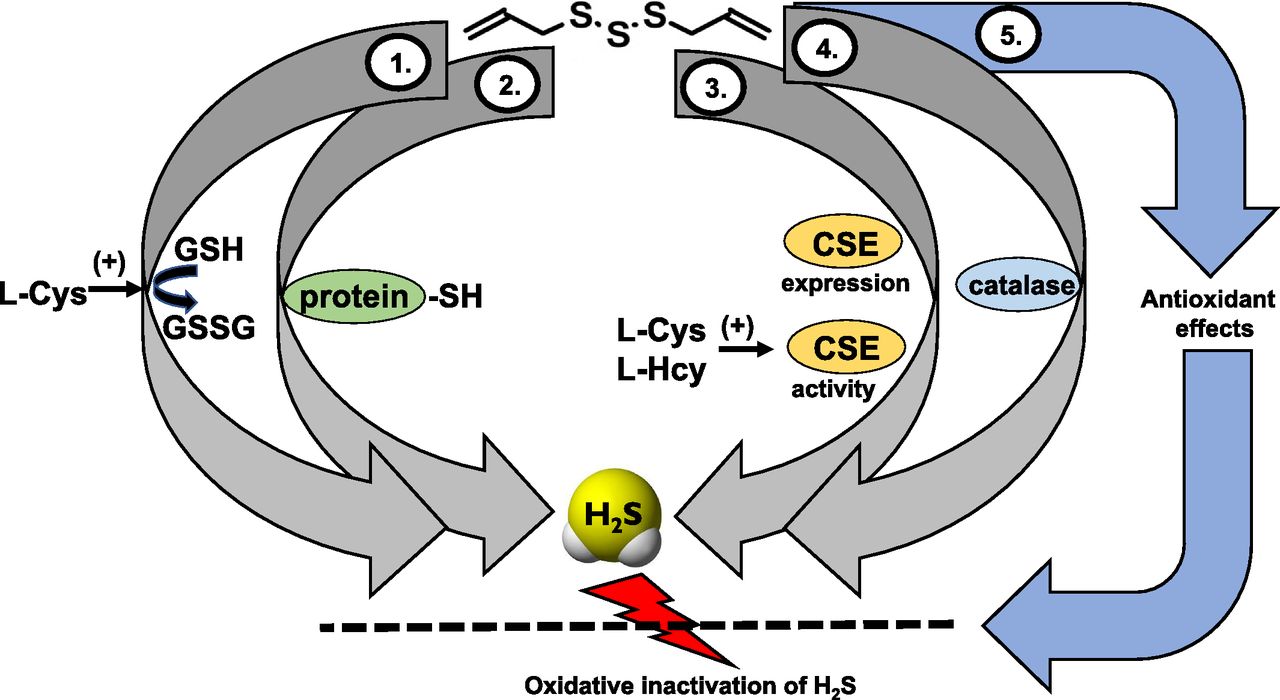{kind=link}
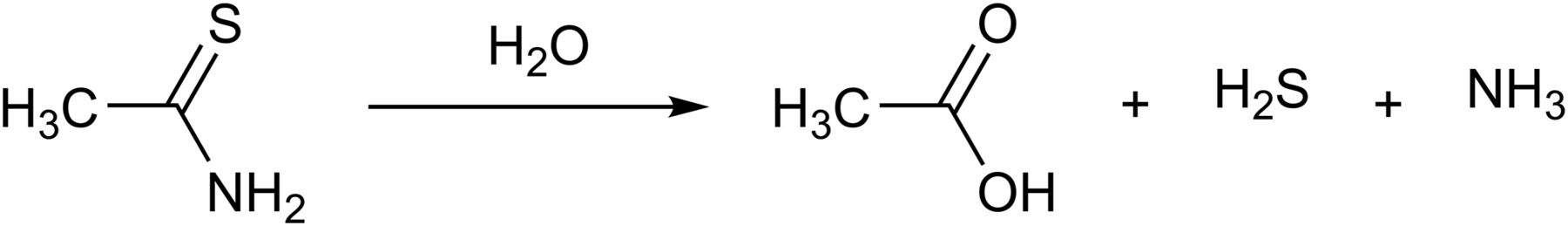{kind=link}
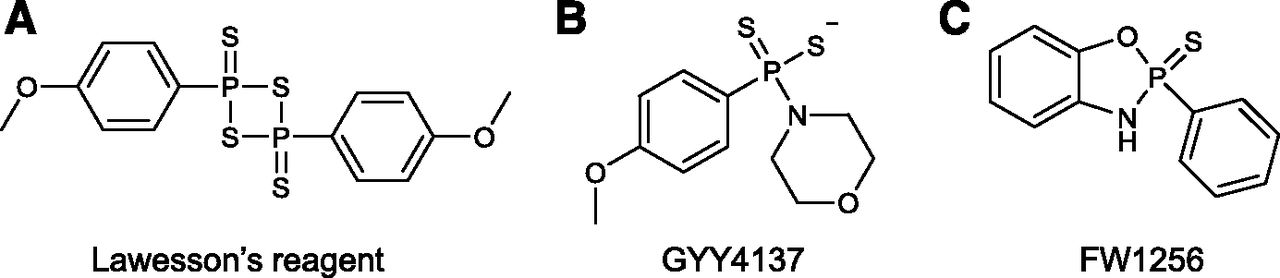{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
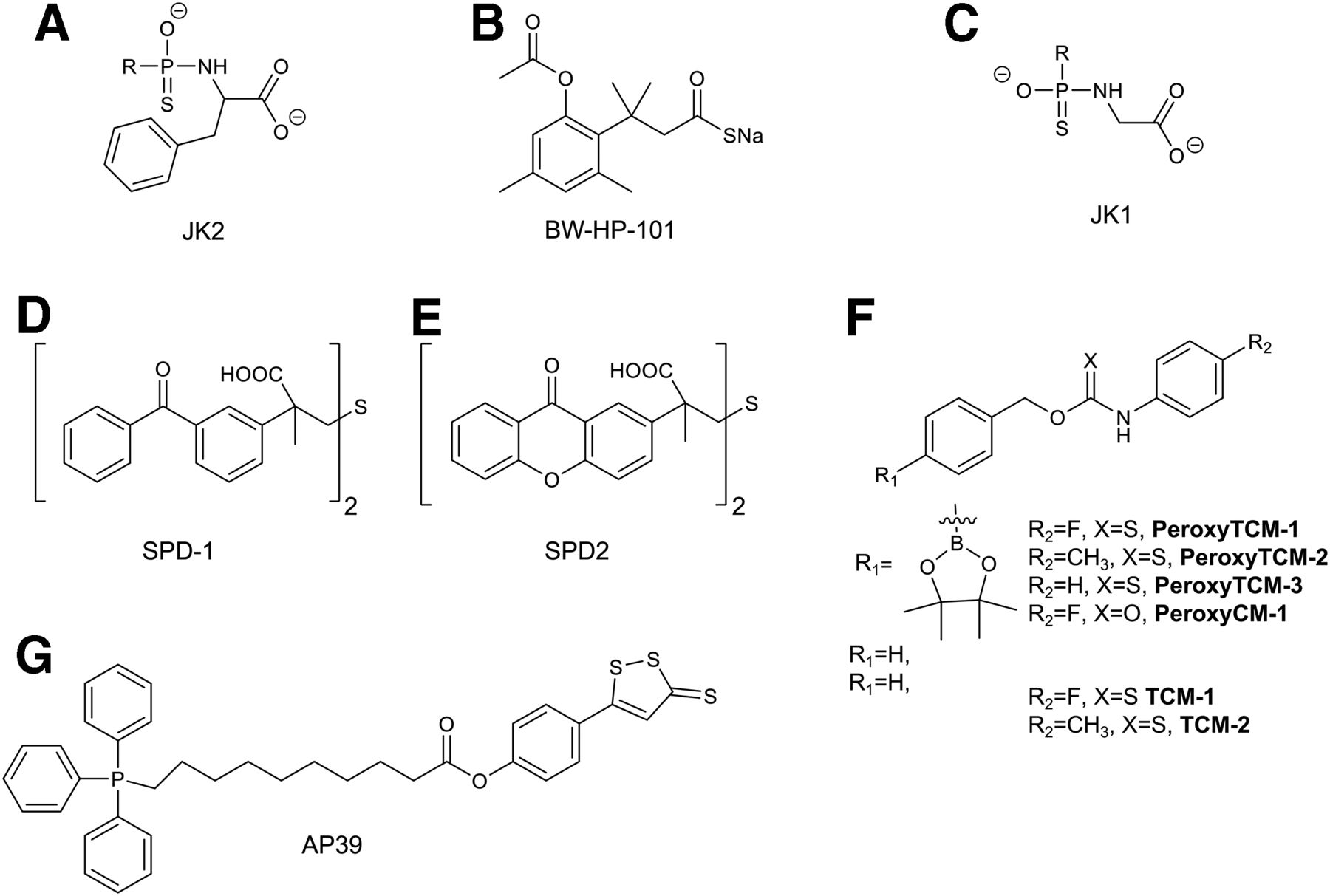{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
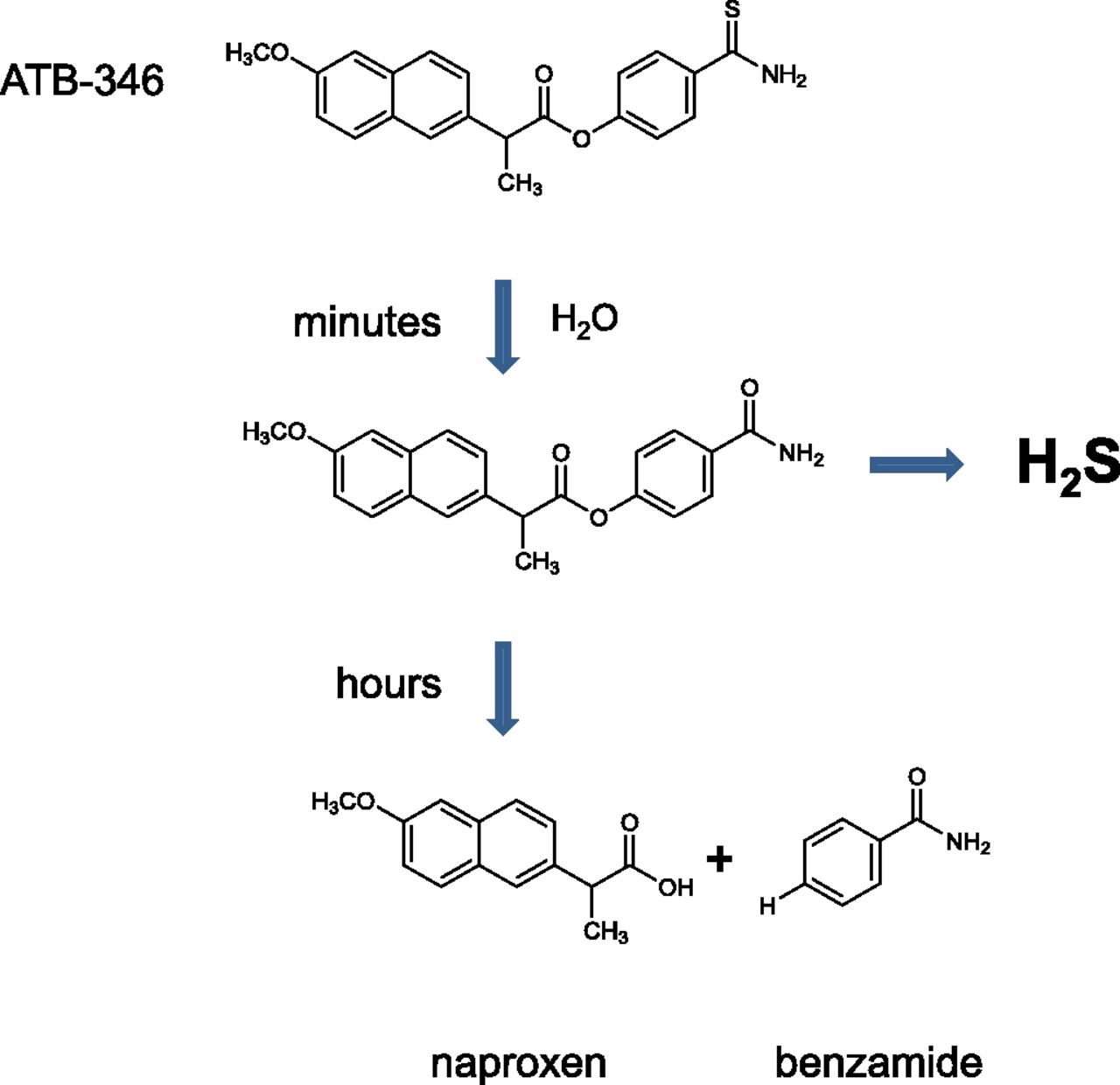{kind=link}
{kind=link}
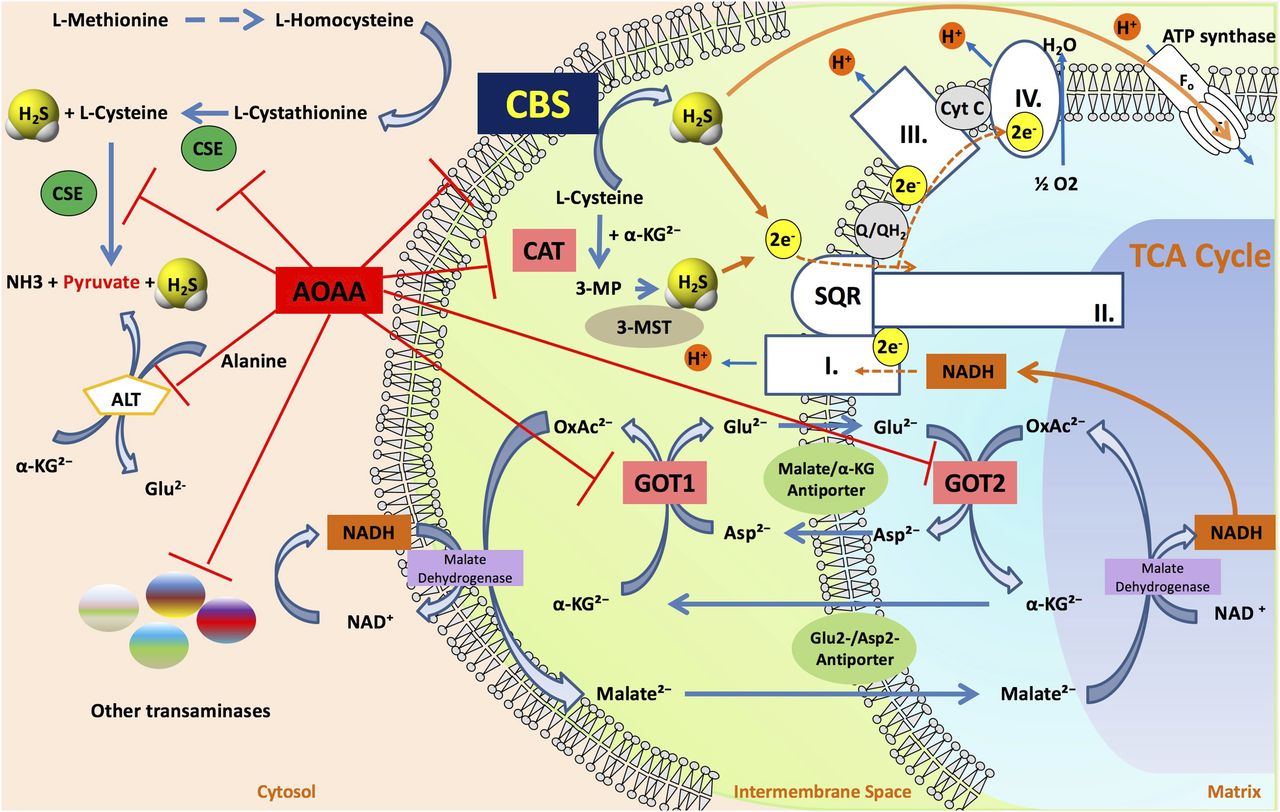{kind=link}
{kind=link}
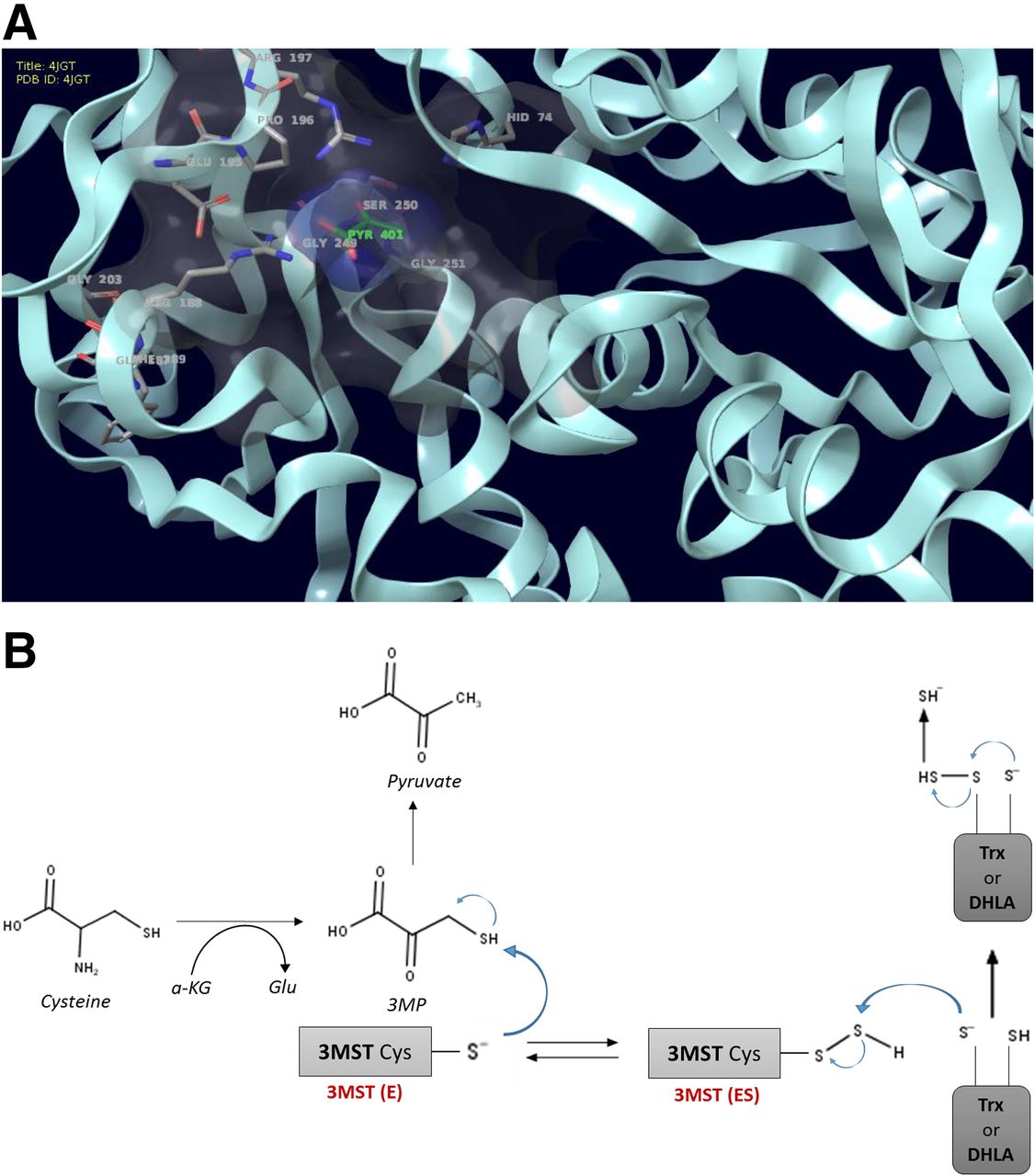{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. The History of H2S as an Environmental Toxin
- III. H2S, as an Endogenous Biologic Mediator: Physiologic Roles
- IV. “H2S-Rich” and “H2S-Poor” Pathophysiological Conditions
- V. The Modes of H2S’s Biologic Actions
- VI. H2S Delivery via Inhalation of H2S Gas
- VII. Sulfide Salts (“Rapid-Release H2S Donors”)
- VIII. Sodium Polythionate (SG-1002)
- IX. IK-1001, a Pharmaceutically Acceptable, Parenteral Injectable Formulation of H2S
- X. Natural H2S Donors
- XI. S-Propargyl-Cysteine
- XII. “Old School” Spontaneous H2S Generators: Thioacetamide and Lawesson’s Reagent
- XIII. GYY4137 and Other Phosphinodiothioate Derivatives
- XIV. Carbonyl Sulfide and its Prodrugs
- XV. Nonregulated, Nontargeted, Miscellaneous H2S Donors
- XVI. pH-Controlled H2S Donors
- XVII. Redox-Activated H2S Donors
- XVIII. Photoactivated H2S Donors
- XIX. Esterase-Activated H2S Donors
- XX. Mitochondrially Targeted H2S Donors
- XXI. NO/H2S Hybrid Donors
- XXII. H2S-Donating Polymers and Special Pharmaceutical Formulations
- XXIII. Combined (or Hybrid) Molecules: H2S-Donating Derivatives of Clinically Used Drugs
- XXIV. Alternative Means to Increase Biologic H2S Levels
- XXV. A Brief Overview of Endogenous H2S Sources
- XXVI. Pharmacological Inhibitors of Cystathionine-γ-lyase
- XXVII. Pharmacological Inhibitors of Cystathionine-β-synthase
- XXVIII. Pharmacological Inhibitors of 3-Mercaptopyruvate Sulfurtransferase
- XXIX. Alternative Means to Decrease Biologic H2S Levels
- XXX. Conclusions and Future Directions
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters