


Abstract
Antimicrobial resistance presents us with a potential global crisis as it undermines the abilities of conventional antibiotics to combat pathogenic microbes. The history of antimicrobial agents is replete with examples of scaffolds containing halogens. In this review, we discuss the impacts of halogen atoms in various antibiotic types and antimicrobial scaffolds and their modes of action, structure-activity relationships, and the contributions of halogen atoms in antimicrobial activity and drug resistance. Other halogenated molecules, including carbohydrates, peptides, lipids, and polymeric complexes, are also reviewed, and the effects of halogenated scaffolds on pharmacokinetics, pharmacodynamics, and factors affecting antimicrobial and antivirulence activities are presented. Furthermore, the potential of halogenation to circumvent antimicrobial resistance and rejuvenate impotent antibiotics is addressed. This review provides an overview of the significance of halogenation, the abilities of halogens to interact in biomolecular settings and enhance pharmacological properties, and their potential therapeutic usages in preventing a postantibiotic era.
Significance Statement Antimicrobial resistance and the increasing impotence of antibiotics are critical threats to global health. The roles and importance of halogen atoms in antimicrobial drug scaffolds have been established, but comparatively little is known of their pharmacological impacts on drug resistance and antivirulence activities. This review is the first to extensively evaluate the roles of halogen atoms in various antibiotic classes and pharmacological scaffolds and to provide an overview of their ability to overcome antimicrobial resistance.
I. Introduction
Antimicrobial resistance (AMR) is a phenomenon whereby pathogenic microbes like bacteria, fungi, viruses, and parasites can adapt and grow in the presence of antimicrobial compounds to which they were once susceptible (Founou et al., 2017). AMR leads to prolonged illness and hospital admissions, the use of more expensive second-line drugs, and treatment failures. It also adversely affects health economies; for example, in 2019, estimates of additional annual burdens were nine billion euros in Europe and 20 billion dollars in the United States (Dadgostar, 2019). Microbes have several drug resistance mechanisms in their arsenals, such as modifying drug targets, limiting drug uptake, enhancing drug efflux, or deactivating drugs that are deployed based on antibiotics administered (Reygaert, 2018). Furthermore, acquired resistance mechanisms based on drug-modifying β-lactamases and carbapenemases, multidrug efflux pumps, drug transport proteins, and conformation-modification are components of pathogen resistomes and further enhance drug recalcitrance (Davies and Davies, 2010).
Pathogen virulence factors also confer antimicrobial resistance. Biofilm formation is a significant virulence factor that provides physical protection from antibiotics and facilitates horizontal gene transfer of resistance genes among microbial communities (Molin and Tolker-Nielsen, 2003). Regulation systems like the two-component systems and quorum sensing also modulate resistance and virulence when pathogens are exposed to external stresses (Haque et al., 2018). Other virulence factors like toxin production, adhesion, iron metabolism, immune response evasion, and bacterial secretory systems further heighten antimicrobial resistance (Beceiro et al., 2013). Thus, acquired resistance mechanisms and virulence factors ensure the survival of pathogens in various ecological niches and nosocomial settings. Countermeasures and alternate therapeutic strategies are warranted to combat increasingly resistant strains and maintain beneficial commensal microbes, which are increasingly resistant to common antibiotics.
Halogens are important in several classes of antibiotics and antimicrobial scaffolds and are present in ∼25% of licensed drugs and ∼40% of actively tested lead compounds (Xu et al., 2014) (Fig. 1). Notably, pharmaceuticals referred to as “blockbuster drugs” are usually halogenated (Suárez-Castro et al., 2018). Furthermore, of the 50 compounds approved by the Food and Drug Administration (FDA) in 2021, 14 contained halogens (Benedetto Tiz et al., 2022). Halogen bond formation is one of the primary drivers underlying the inclusion of halogens in antimicrobial drugs. Halogenated compounds are capable of forming multiple covalent interactions with ligands and can act as both electrophiles and nucleophiles (Turunen and Erdélyi, 2020). Furthermore, the introduction of a carbon-halogen (C-X) bond variously influences biological activities, such as the thermal and oxidative stabilities of enzymes, ligand binding abilities, and intracellular delivery (Fang et al., 2019; Bhutani et al., 2021). They can also use their bulk to have agonistic or antagonistic activities on bioactive targets (Zhou et al., 2018b; Liu et al., 2020). Thus, adsorption, distribution, metabolism, and excretion (ADME) parameters of compounds like drug binding affinity, membrane permeabilization, and lipophilicity can be altered by halogen bonding (Fig. 2) (Hernandes et al., 2010). Interestingly, halogenated antimicrobial agents contribute significantly to the natural defense systems of several plants, marine algae, fungi, aquatic animals, and halobionts, which supports the notion that halogenation plays an important role in antimicrobial activity (Fig. 2) (Mardirossian et al., 2021).

Properties of halogens (A), proportions of drugs containing halogens approved by the FDA between 1988 and 2006 (Hernandes et al., 2010) (B), and numbers of small-molecule drugs containing fluorine, sulfur, phosphorus, or boron approved by the FDA from 2015 to June 2020 (Bhutani et al., 2021) (C). Ar, atomic radius; e-, electronegativity value.

The sources, applications, and advantages of halogenated antimicrobial agents.
Previous reviews have addressed diverse synthetic and naturally occurring halogenated compounds, their modes of action, and therapeutic potentials. The roles and importance of halogens, especially fluorine and chlorine in medicinal chemistry and FDA-approved drugs (Böhm et al., 2004; Gillis et al., 2015; Swallow, 2015; Fang et al., 2019; Mardirossian et al., 2021), and their contribution to the functionalities of halogen-containing compounds (Wilcken et al., 2013; Cavallo et al., 2016) have been previously reviewed. However, this is the first comprehensive review of the antimicrobial and antivirulence effects of halogenated antibiotics and pharmacological scaffolds activity on resistant microbes (Fig. 2). The structural features, structure-activity relationships (SARs), and the mechanism of actions (MOAs) of halogenated pharmacological scaffolds are provided. In addition, halogenated polymeric materials, proteins, carbohydrates, and lipids exhibiting antimicrobial activity against resistant microbes and factors affecting the bioactivities, pharmacokinetic and pharmacodynamic properties of halogenated antimicrobials and their relationships with gut microbiota are reviewed. We also provide an overview of the rejuvenation of impotent antibiotics by halogenation and of the resistance evasion conferred by halogenated scaffolds and offer perspectives regarding the clinical use of halogenated antimicrobials.
II. Halogenated Antibiotic Classes
Antibiotics are a cornerstone of chemotherapy against bacteria and fungi. The discovery of penicillin by Alexander Fleming in the 20th century revolutionized general medicine and led to the discovery of several other classes of antibiotics. However, antibiotic abuse resulted in the development of microbe drug resistance, and today, novel antibiotics are in constant demand to combat increasing trends of microbial resistance (Pulingam et al., 2022). Starting with the discovery of chloramphenicol in 1947, halogens have now become prominent components of several classes of antibiotics and antibiotic scaffolds. Furthermore, several other halogenated metabolites have been isolated from various sources like microorganisms, algae, and several plants and animal sources (Hutchings et al., 2019). Antibiotic classes are usually classified by molecular structure, mode of action, or activity spectrums. Here, we discuss and classify halogenated antibiotic classes, in which the halogen is responsible for the antimicrobial activity or its enhancement against drug-resistant microbes based on their modes of action.
A. Cell Wall Synthesis Inhibitors (Penicillins, Cephalosporins, Glycopeptides, and Lipopeptides)
Inhibitors of cell membranes and cell wall synthesis are among the most effective antibiotics (Bhattacharjee, 2016). Inhibition of the cell envelope at various stages of cell development provides one of the most efficient antibacterial strategies as these agents lodge themselves in cell membranes, compromise membrane integrity, and cause cell swelling and lysis (Sarkar et al., 2017). Major antibiotic classes like penicillins, lipopeptides, cephalosporins, and glycopeptides inhibit bacteria in this manner.
Penicillin was the first class of antibiotics discovered in penicillium molds and possesses broad-spectrum antibactericidal activity. Halogenated isoxazolyl penicillin derivatives like cloxacillin, dicloxacillin, and flucloxacillin are members of this class of antibiotics (Fig. 3, A–C) (Yasuda and Shimada, 1971). Chloro or dichloro substitutions of phenyl-, naphthyl-, or quinolyl-penicillins are potent inhibitors of Gram-negative Escherichia coli and Klebsiella aerogenes (Cole et al., 1972).

Cloxacillin (A), dicloxacillin (B), flucloxacillin (C), SAR of cloxacillin (D), cefiderocol (E), cefaclor (F), loracarbef (G), cefazaflur (H), cefazedone (I), flomoxef (J), SARs of cefiderocol (K), and effects of halogenation on penicillins and cephalosporins (L).
Penicillins inhibit the crosslinking of peptidoglycans during cell wall biosynthesis by inhibiting the activities of associated penicillin-binding proteins, thereby disrupting cell structural integrity (Yocum et al., 1980). Penicillins contain the β-lactam ring, which binds to the penicillin-binding protein and disrupts peptidoglycan synthesis (Fig. 3D) (Hou and Poole, 1971). However, this ring is susceptible to degradation by β-lactamases produced by bacteria. Oxacillin derivatives like cloxacillin, dicloxacillin, and flucloxacillin are impervious to β-lactamase activity because their large chains prevent enzyme binding. Chlorine substitution in these antibiotics aid penicillin-binding protein binding and augments the antimicrobial effects of these antibiotics (Fig. 3L) (Neu, 1986). Although a clear role of halogens in preventing β-lactamase binding is not mentioned, halogen substitution being the only structural difference in the above-mentioned antibiotics might play a larger role in the reduced binding affinity with the enzyme.
Cephalosporins are also β-lactam antibiotics and inhibit the peptidoglycan synthesis required for cell wall formation. Several halogenated cephalosporins like cefazaflur, cefazedone, loracarbef, cefaclor, flomoxef, and cefiderocol (Fig. 3, E–J) are present across generations, although halogen components are not part of their core structures. The cephalosporin cephem nucleus contains a β-lactam ring, which is essential for antibacterial activity. Other groups attached to the cephem nucleus, like the aminothiazole, carboxylic acid, or dimethyl oxime groups, enhance binding to penicillin-binding proteins (Fig. 3K) (Aoki et al., 2018). Halogens are present in different parts of the substructure of cephalosporins, such as in the cephem nucleus in cefaclor or the chlorocatechol group in cefiderocol, which makes the role of the halogen atom difficult to determine (Aoki et al., 2018). The locations of halogen atoms in these structures may dictate the effects of these antibiotics, for example strengthening binding to penicillin-binding proteins in cefaclor or augmenting the chelation of iron in cefiderocol (Fig. 3, K and L) (Sato and Yamawaki, 2019). As cephalosporins have a β-lactam ring in their substructure that is subject to enzymatic degradation, strategic halogenation of the cephalosporins has the potential to attenuate resistance as observed in penicillins.
Glycopeptide antibiotics include glycosylated monocyclic or polycyclic nonribosomal peptides with effects similar to those of β-lactam antibiotics. They inhibit cell wall peptidoglycan synthesis by binding to the d-Ala-d-Ala termini of pentapeptide-ending precursors localized at the outer surfaces of cytoplasmic membranes, which blocks the reticulation of peptidoglycan by inhibiting associated transglycosylases and transpeptidases (Kang and Park, 2015; Van et al., 2017). Halogenated glycopeptides include first-generation teicoplanin (Fig. 4A) and vancomycin (Fig. 4B) and the second-generation semisynthetics dalbavancin and oritavancin (Allen, 2010; Binda et al., 2014). Pentapeptide binding is further enhanced by dimerization, and the presence of a halogen or a sugar moiety is believed to be responsible for the stronger interaction conferred by homo-dimerization as was reported for vancomycin (Fig. 4, B and E) and oritavancin (Kang and Park, 2015). The addition of lipid groups to the side chains of the core structures of glycopeptides enhances binding to the lipid bi-layer and thus increases permeability (Fig. 4B) (Kang and Park, 2015). Although halogen atoms are secondary to target binding, they play significant roles by enhancing bioactivity as was demonstrated by the dehalogenation and halogenation of balhimycin, which contains a chlorine atom. Interestingly, fluorobalhimycin, bromobalhimycin, and balhimycin were reported to be up to 8 times as potent as dechlorobalhimycin at combating microbial infections validating a crucial role in glycopeptides (Ashford and Bew, 2012). Vancomycin-resistant Enterococci species were found to alter the d-Ala-d-Ala termini to d-Ala-d-Ser or d-Ala-d-Lac, thereby preventing vancomycin binding, but these resistant species were sensitive to teicoplanin, which also has halogen substitutions on its sugar moiety but at different positions in comparison with vancomycin (Yushchuk et al., 2020). Therefore, halogenation of key binding sites of the sugar moiety can increase binding to the termini of the resistant strains and restore efficacy of the glycopeptides.

Teicoplanin (A), SAR of vancomycin (B), taromycin A (C), SAR of taromycin B (D), and effects of halogenation on glycopeptides (E). The DXDG motif refers to the conserved peptide motif of Asp-X-Asp-Gly in the lipopeptide backbone.
Lipopeptides like glycopeptides, has peptides in their core bound to lipid entities giving them their name. The action mechanisms of lipopeptides differ from those of glycopeptides. Lipopeptides insert themselves into cell membranes in a phosphatidylglycerol-dependent manner, aggregate, and produce holes that leak ions (Reynolds et al., 2018), causing rapid depolarization, membrane potential loss, and, ultimately, cell death. This mechanism was observed in the daptomycin series of lipopeptide antibiotics. The taromycin series, which was derived from the marine bacterium Saccharomonospora sp., is the only halogenated antibiotic series in the lipopeptide antibiotic class. Taromycin A and B displayed potent antimicrobial activity against methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecium (Fig. 4, C and D) (Reynolds et al., 2018). Daptomycin has tryptophan and kynurenine groups in its peptide structure, and these are essential for membrane binding. The additional chlorine substitution in the taromycin lipopeptide series takes place at key tryptophan and kynurenine groups (Fig. 4D), suggesting that chlorine might be responsible for their antimicrobial activity, although the impacts of these substitutions on efficacy have yet to be determined (Reynolds et al., 2018). SAR studies of the halogenated tryptophan and kynurenine groups would give better insights into the role of halogens and their incorporation into other lipopeptide-based antibiotics. Halogen substitution might have aided in improved activity against the methicillin-resistant S. aureus and vancomycin-resistant E. faecium. Lipopeptide-resistant strains are not a development yet, and halogenation of the lipopeptides, especially the tryptophan and kynurenine groups, could afford minimal resistance and enhanced binding to the bacterial membrane.
B. Cell Proliferation Inhibitors (Sulfonamides, Trimethoprims, and Quinolones)
Cell division is an attractive antibacterial target as the bacterial cell divisome is essential and conserved in many infectious pathogens. Also, access to associated protein targets of the cell divisome is made easier by their external locations (Lock and Harry, 2008). Common strategies for suppressing bacterial cell proliferation include inhibiting DNA synthesis or proteins, such as those of the Fts family associated with cell division, and targeting metabolic processes (Lock and Harry, 2008). Different antibiotics like sulfonamides, trimethoprim, and quinolones inhibit cell proliferation by inhibiting DNA synthesis by disrupting different stages of the DNA biosynthetic pathway.
Sulfonamides, more commonly known as sulfa drugs, are sulfonamide-derivatized anilines. Sulfonamide antibiotics disrupt enzymatic reactions involving para-aminobenzoic acid (PABA) by competitively inhibiting the enzyme dihydropteroate synthase (Kumar Verma et al., 2020). Bacteria require PABA to produce folic acid, which is required for purine and pyrimidine synthesis. The para-amino group attached to the aromatic benzene ring and the sulfanilamide group are essential for the activities of these antibiotics as they mimic the binding site of dihydropteroate synthase, which is responsible for the synthesis of PABA (Fig. 5D) (Kumar Verma et al., 2020). The native scaffold of sulfonamides is devoid of halogens, but a series of di-halogenated sulfonamides were found to be potent inhibitors of the three β-class carbonic anhydrases in drug-resistant Mycobacterium tuberculosis (Fig. 5, A–C) (Maresca et al., 2013). Typically, sulfonamides are ineffective against mycobacteria, in part because of their low membrane permeabilities. Halogenation might aid permeability and increase the antimicrobial activity of the sulfonamide pharmacophore. Furthermore, substitution patterns on the aromatic ring affect activity; for example, halogen substitution at ortho positions enhances antimicrobial activity (Fig. 5, D and S). Also, these halogenated derivatives have potential use against other multidrug-resistant (MDR) pathovars like S. aureus and Streptococcus pneumoniae (Aspatwar et al., 2019). Since halogenated sulfonamides were active against several MDR pathogens, these derivatives might be effective at bypassing MDR-associated efflux pumps and decreasing membrane permeability commonly associated with MDR pathovars.

4-Amino-3-bromo-5-chlorobenzenesulfonamide (A), 4-amino-3-chloro-5-iodobenzenesulfonamide (B), 4-amino-3,5-dibromobenzenesulfonamide (C), SAR of dihalogenated sulfonamide (D), chlorotrimethoprim (E), bromotrimethoprim (F), iodotrimethoprim (G), SAR of halogenated trimethoprim derivatives (H), sitafloxacin (I), rufloxacin (J), levofloxacin (K), 8-chloroquinolone (L), ciprofloxacin (M), flumequine (N), moxifloxacin (O), norfloxacin (P), sparfloxacin (Q), SAR of ciprofloxacin (R), and effects of halogenation on sulfonamides, trimethoprim, and quinolones (S).
Trimethoprim is another antibiotic that inhibits DNA synthesis by inhibiting the conversion of dihydrofolic acid to tetrahydrofolic acid, which acts as a precursor in the thymidine synthesis pathway (an important requirement for bacterial DNA synthesis). The amine group present in the fourth position of trimethoprim is essential for antimicrobial activity as it enables the competitive inhibition of the dihydrofolate reductase enzyme by mimicking the carbonyl oxygen of the 5,6-dihydrofolate substrate (Blaney et al., 1984; Watson et al., 2007). Trimethoprim has no halogen atoms in its core structure, but halogenated derivatives (Fig. 5, E–G) were successfully synthesized (Nilchan et al., 2018). Halogenated trimethoprim derivatives synthesized by substituting one of its methoxy groups with a halogen resulted in 5 times more activity against the panel of microbes tested (Fig. 5H) (Kompis and Wick, 1977). Furthermore, docking analysis revealed that this increased potency was the result of bond formation between the Ser49 amino acid residue of dihydrofolate reductase and the halogen atom, which supported the notion that the inclusion of a halogen provides additional enzyme binding affinity and increases antimicrobial activity (Fig. 5S) (Nilchan et al., 2018). This can be an effective way to minimize trimethoprim resistance as halogenated trimethoprim derivatives can facilitate and enhance binding affinity for modified dihydrofolate reductase in resistant strains.
Quinolones employ a different strategy to inhibit cell division. Fluoroquinolones constitute one of the largest halogenated antimicrobial classes and are commonly used clinically (Fig. 5, I–Q). Fluoroquinolones inhibit the ligase activity of type II topoisomerase, DNA gyrase, and topoisomerase IV, which are responsible for negative supercoiling in the replication fork and the decatenation of DNA after replication. The inhibition of these enzymes leads to single- or double-stranded breaks in the DNA and eventually to cell death (Fàbrega et al., 2009). Binding to DNA gyrase is achieved by the two carbonyl groups present at the third and fourth positions of the bicyclic core of fluoroquinolones (Fig. 5R). The fluorine atom, which is generally located at the sixth position of the bicyclic core, enhances antimicrobial activity by 10-fold for gyrase inhibition and increases antimicrobial activity by 100-fold. In this case, the halogen plays a subsidiary role by enhancing antimicrobial activity (Fig. 5R) (Domagala, 1994).
Despite the substantial potency of fluoroquinolones, microbes were able to develop resistance by reducing membrane permeability, efflux of compounds, or modifying target configurations, though this increased resistance can be counteracted using adjuvant efflux pump inhibitors or strategies that improve permeability. Interestingly, halogenation of efflux pump inhibitors also increased the efficacy of halogenated chalcone adjuvants of fluoroquinolones against NorA and MepA multidrug efflux pumps in S. aureus (Freitas et al., 2021) and halogenated pyridylpiperazine-based allosteric inhibitors of resistance nodulation cell division (RND)-type multidrug efflux pump in E. coli (Plé et al., 2022). In addition, a novel bactericidal 8-chloroquinolone was synthesized that was 30 and 128 times more potent than trovafloxacin against two clinical isolates of methicillin-resistant S. aureus (MRSA) (Fig. 5L) (Kuramoto et al., 2003). Mutation in DNA gyrase also imparts quinolone resistance by substitution of Leu or Trp for Ser-83 and Asn or Tyr for Asp-87 in the GyrA protein, leading to an electron-deficient helix microenvironment. But halogenation of the quinolone at the C8 position reduces the probability of the development of quinolone resistance (Lu et al., 1999) and can aid in higher efficacy and activity of several quinolone antibiotics against resistant strains.
C. Protein Synthesis Inhibitors (Aminoglycosides, Tetracyclines, Macrolides, Lincosamides, and Oxazolidinones)
Protein synthesis inhibitors act at different translation stages to inhibit the synthesis of proteins. These inhibitors are highly specific for prokaryotic 70S ribosomes. Translation in prokaryotes involves the assembly of ribosomes, binding to mRNA, and tRNA binding to A, P, and E sites to further translate to polypeptide sequences. Common antibiotics like aminoglycosides, tetracyclines, macrolides, lincosamides, and oxazolidinones disrupt this pathway at different translation stages.
Aminoglycosides are a Gram-negative combating class of antibiotics that contain an amino-modified glycoside, which acts by penetrating the bacterial cell wall and disturbing peptide elongation at the 30S subunit of the ribosome, which, in turn, leads to improper mRNA translation and truncated protein biosynthesis or amino acid alterations. Aminoglycoside-to-ribosome binding is mediated by the aminoglycoside neamine core, which binds specifically to the decoding site (site A) of ribosomes (Carter et al., 2000; François et al., 2005). The OH and amine groups in ring 1 of aminoglycosides are required for stacking interactions with ribosomes (Fig. 6B) (Hobbie et al., 2005, 2006). Antibiotic resistance is due to aminoglycoside structural modifications by catalytic enzymes like aminoglycoside phosphotransferases, nucleotidyltransferases, and acetyltransferases (Azucena and Mobashery, 2001; François et al., 2005).

SAR of aminoglycosides as represented by 5-deoxy-5-fluoro-kanamycin B (A), 3′-deoxy-3′-fluoro-kanamycin A (B), demeclocycline (C), eravacycline (D), and SAR of chlortetracycline (E) and effects of halogenation on aminoglycosides and tetracyclines (F).
The aminoglycoside class of antibiotics does not contain any halogen, but fluorination produced derivatives that successfully recovered the potencies of parent aminoglycosides against resistant bacteria (Fig. 6, A and B). Fluorinated derivatives of many mainstream aminoglycosides like kanamycin A, tobramycin, amikacin, dibekacin, gentamycin, paromomycin, and neomycin have significant antimicrobial activities against drug-resistant and non–drug-resistant microbes (Fig. 6F) (Tsuchiya et al., 1985; Mingeot-Leclercq et al., 1999). Fluorine substitution alters glycosidic bonds and reduces the binding affinities of fluorinated aminoglycoside analogs with modifying enzymes rather than binding affinity with the ribosome site (Fig. 6B) (Shitara et al., 1992). The fluorinated derivatives maintained their similar intrinsic activity against both sensitive and resistant strains (Mingeot-Leclercq et al., 1999).
Tetracyclines also target the 30S subunit of ribosomes and inhibit the formation of the mRNA-ribosome complex by blocking the aminoacyl tRNA binding site (Chukwudi, 2016). Tetracycline possesses four hydrocarbon rings with functional substitutions of alkyl, halogen, hydroxyl, amine, and others on the upper and lower periphery of the tetracycline rings (Fig. 6, C–E). Modifications on the lower periphery of the rings diminished bioactivity, whereas modifications of the upper periphery enhanced activity as exemplified by substitutions of highly electronegative groups, including halogens on the C7 and C9 positions of the D ring (Fig. 6E) (Chopra and Roberts, 2001).
Tetracyclines do not have halogen atoms in their native structures, but several important tetracyclines like chlortetracycline, demeclocycline, meclocycline, and eravacycline and their associated semisynthetic derivatives contain halogen atoms (Fig. 6, C–E) (Lamberth and Dinges, 2016). A bromine-substituted tetracycline 7-bromotetracycline was synthesized, though the rate of halogenation was significantly less than that of its chlorine analog (Doerschuk et al., 1959). Halogen substitution on the D ring improved antibacterial activity but at the cost of stability. Furthermore, halogen substitution is not essential for bioactivity and might only enhance binding to the ribosomal unit (Fig. 6F) (Dong et al., 2012). Common tetracycline resistance mechanisms involve efflux pumps and the ribosomal protection protein, which inhibits tetracycline binding (Hobson et al., 2021). Although halogenation has not been used to counter these mechanisms, we hypothesize strategic halogenation on the upper periphery of the tetracycline pharmacophore might thwart the binding of resistance enzymes and the efficacy of efflux pumps and prevent recalcitrance.
In contrast to tetracyclines and aminoglycosides, macrolides target the 50S subunit of ribosomes. Macrolides block the approach to the exit tunnel of elongated peptides, which leads to the premature release of the peptidyl-tRNA complex (Gaynor and Mankin, 2003). Macrolides are a class of natural polyketide antibiotics that typically contain a large macrocyclic 15-16 membered lactone ring with one or more attached deoxy sugars. The C5 desosamine sugar moiety and the three hydroxy groups present on the lactone ring are essential for antimicrobial activity as they form polar contacts and hydrogen bonds with the residues of the 50S ribosomal subunit (Mazzei et al., 1993).
Macrolides have no halogen atom in their core structure, but several halogenated ketolide derivatives have been synthesized, like flurithromycin and solithromycin, which are the fluorinated members of this class of antibiotics (Fig. 7, A and B) (Nguyen and Chung, 2005). Macrolides also include brominated phorboxazoles isolated from the marine sponge Phorbas sp. Furthermore, halogenated hydrophosphoryl derivatives of pimaricin and mycophetin were reported to be much more potent against Candida sp. than their nonhalogenated counterparts (Belakhov and Shenin, 2007; Belakhov et al., 2008). The addition of fluorine at position 2 or 8 of the macrolide scaffold enhances binding to the ribosomal subunit and improves pharmacokinetic properties (Fig. 7, B and L). In this case, halogen substitution enhanced macrolide antimicrobial activity (Chellat et al., 2016). To a large extent, macrolide resistance involves efflux pumps, as is the case for quinolones (Schroeder and Stephens, 2016), and adjuvant halogenated efflux pump inhibitors offer an excellent strategy for diminishing macrolide resistance. However, no study has yet reported on the efficacies of halogenated macrolide derivatives against multidrug-resistant strains. Halogenation of the macrolide pharmacophore might be an effective strategy to subside AMR and increase antimicrobial efficacy.

Solithromycin (A), SAR of flurithromycin (B), lincomycin (C), pirlimycin (D), SAR of clindamycin (E), linezolid (F), posizolid (G), SAR of sutezolid (H), tedizolid (I), radezolid (J), and contezolid (K) and effects of halogenation on macrolides and lincosamides (L).
Lincosamides compete with macrolides as peptidyl transferase inhibitors and bind to the peptidyl transferase center of the 23S portion of the 50S subunit of bacterial ribosomes. Binding takes place via the mycarose sugar moiety, which has structures that overlap with peptidyl transferase, and leads to the premature dissociation of peptidyl-tRNA containing two, three, or four amino acid residues and ultimately inhibits peptidyl transferase activity by steric hindrance as is observed for lincomycin (Fig. 7C) (Schlünzen et al., 2001).
There are two lincosamide halogenated antibiotics, namely clindamycin and pirlimycin (Fig. 7, D and E) (Spížek and Řezanka, 2017). Chlorine atom substitution on the 7-carbon importantly enhanced the activity of clindamycin (Fig. 7E), and E. coli was 20 times more susceptible to clindamycin than lincomycin, elucidating halogen-promoted enhancement (Douthwaite, 1992). Lincosmide and macrolides are competing antibiotics and elicit similar patterns of antibiotic resistance involving efflux pumps and target-site modifications. A novel ketolide derivative was developed by linking macrolide and quinolone units (macrolones) to combat resistance against macrolides and lincosamines. The halogenated derivatives of these macrolones were the most potent derivatives of the pharmacophore examined and potently reduced the viabilities of several multidrug-resistant strains of E. coli and S. pneumoniae (Fajdetić et al., 2010; Čipčić et al., 2016). Also, halogenated analogs of 4’’-O-(ω-quinolylamino-alkylamino) propionyl derivatives of macrolides potently inhibited macrolide-resistant Streptococcus sp., Staphylococcus sp., and Haemophilus influenzae (Fajdetić et al., 2010). This demonstrates the ability of halogen derivatives in overcoming resistance mechanisms like efflux pumps and ribosomal modifications associated with the tested resistant strains.
Like macrolides and lincosamides, oxazolidinones target the 50S subunit of ribosomes, but they act by disrupting the formation of the initiation complex and the translocation of peptidyl-tRNA from the A to the P site of the 50S ribosomal subunit to ultimately inhibit protein synthesis (Bozdogan and Appelbaum, 2004). Most of the antibiotics in this class have halogens, especially fluorine, in their native structures. Linezolid, posizolid, sutezolid, tedizolid, radezolid, and contezolid are examples of fluorinated oxazolidinones (Fig. 7, F–K) (Diekema and Jones, 2001). The n-aryl group in the 2-oxazolidone nucleus is important for antimicrobial activity (Fig. 7H). Also, the fluorine atom of the fluorophenyl core (a fluorinated phenyl ring) is essential for antimicrobial activity as the fluorine atom is perfectly situated to form bonds with the 50S ribosomal subunit (Fig. 7H) (Phetsang et al., 2014; Kumar et al., 2015). Mutations of the 23S rRNA of the 50S ribosomal subunit diminish oxazolidinone affinity and antibiotic resistance. Several synthesized halogenated oxazolidinone derivatives, such as derivatives with a halogenated pyrrolidone moiety at the α- or β-position, have exhibited antimicrobial activity against drug-resistant MRSA, vancomycin-resistant S. aureus, and several other Gram-negative bacteria (Bhattarai et al., 2012). No significant changes were observed in the minimum inhibitory concentrations (MICs) of these derivatives against drug-sensitive and drug-resistant strains except S. aureus strains, which had a twofold increase in MICs against resistant strains. Oxazolidinones containing a halostilbene pharmacophore substitution demonstrated significant potency against Gram-positive bacteria and increased efficacy against drug-resistant pathogens (Sciotti et al., 2002), and fluorine-containing benzoxazinyl-oxazolidinones have been used to treat multidrug-resistant tuberculosis (Zhao et al., 2017). The use of halogenation to combat oxazolidinone resistance is largely unexplored. However, strategic halogenation to facilitate binding to a modified resistant 50S ribosome unit may be a judicious strategy to combat resistance.
III. Halogenated Antimicrobial Scaffolds
Natural antimicrobials isolated from different sources, including plants, animals, and prokaryotic and eukaryotic organisms, and their derivatives account for a third of the drugs approved by the FDA (Quave, 2016). Their applications are often limited by narrow bioactivity spectrums, low toxicities and yields, enzymatic degradation, and microbial resistance. Halogen incorporation represents an innovative means of enhancing or restoring the pharmacological efficacies of simple and complex natural and synthetic products (Molchanova et al., 2020; Hurtová et al., 2022). As a result, studies now focus on the control of emerging and reemerging drug-resistant pathogens using halogenated products. Here, we discuss the roles of these derivatives based on chemical groups and/or scaffold robustness. This classification includes halogenated polyphenolics, essential oils, alkaloids, benzopyrones, phenazines, azoles, repurposed drugs, and other bioactive compounds. Each section provides details of efficacies against resistant pathogens, the roles of halogens, their cytotoxicities, and their mechanisms.
A. Polyphenolic Compounds
Phenolics contain at least a benzene ring and a hydroxyl group attached directly to a phenyl group. Natural phenolics are secondary metabolites produced via phenylpropanoid pathways and are found in fruits, vegetables, and other food plants (Laura et al., 2019; Hamad, 2021). These compounds include phenolic acids, tannins, lignans, flavonoids, stilbenes, catechols, magnolols, hydroquinones, lignans, and chalcones and are used as food supplements and intermediate or active ingredients in pharmaceutical products (Hamad, 2021). Furthermore, their halogenated derivatives (Fig. 8, A–X) display a variety of pharmacological properties, including antimicrobial activities.

Structures of halogenated polyphenols and essential oils. Catechols (A), magnolols (B–F), resveratrols (G–I), flavonoids (J–Q), and cinnamaldehydes (R–X) and effects of halogenation on catechols, resveratrols, and flavonoids (Y). Full chemical names are listed in Supplemental Fig. 1.
1. Catechols
Catechol is a water-soluble compound found in plants and marine mussels and is used in pesticides and bioadhesives and as a starting material in the perfume and pharmaceutical industries (Ahn, 2017; Razaviamri et al., 2021). Halogenated catechol, 6-chlorodopamine methacrylamide loaded into hydrogel remarkably inhibited Gram-positive and -negative drug-resistant bacteria. It showed >8-log colony-forming unit reduction against MRSA, vancomycin-resistant enterococci (VRE), multidrug-resistant Acinetobacter baumannii, Pseudomonas aeruginosa, and carbapenem-resistant K. pneumoniae as compared with its nonhalogenated analog (Liu et al., 2021). In addition, the biofilm-forming capacities of P. aeruginosa and MRSA, which aid in antibiotic resistance, were inhibited by the chlorinated catechol. The halogenated catechol, being electrically neutral, oxidizes to produce negatively charged semiquinone capable of preventing biofilm formation. The incorporation of chlorine in the catechol moiety increased cell membrane rupture and, eventually, cell death (Fig. 8Y). This mechanism was independent of H2O2 production since the incorporation of the Cl- group reduced rates of catechol oxidation (Liu et al., 2021). Notably, catechol itself enhanced adherence for contact killing of bacteria, whereas chlorine presence may have enhanced its permeability and bioavailability.
In addition, chlorinated catechol inhibited bacterial fatty acid synthesis via the formation of enoyl-acyl carrier enzyme complex (DMA-Cl/FabI/NAD+) to disrupt fatty acyl carbon chain elongation, and, importantly, it exhibited little toxicity against 3T3-E1 fibroblasts (Liu et al., 2021). Chlorocatechol (Fig. 8A) present at the C3 position was identified as a core moiety in a drug (cefiderocol) recently approved for the treatment of complicated urinary tract infections (Fig. 3E). This moiety is responsible for siderophore-iron binding activities, improved potency, and stability against drug-degrading enzymes like the β-lactamases (Sato and Yamawaki, 2019; Nishimura et al., 2022). Overall, chlorocatechol could inhibit resistant bacteria by chelating iron, interfering with fatty acid synthesis and membrane disruption.
2. Magnolols
Magnolol is a natural biphenolic with two para-allyl groups isolated from the bark of Magnoliae officinalis. This scaffold has established pharmacological activities, including antimicrobial, antioxidative, antianxiety, anti-inflammatory, and antiproliferative activities (Khadke et al., 2019). Halogenation of magnolol enhanced its antimicrobial activity. More specifically, dibromo-magnolol (Fig. 8B) had a low MIC (1 to 2 µg/mL), which was 8–64 times more superior to magnolol, ciprofloxacin, and erythromycin (MIC: 8–64 µg/mL) against MRSA and VRE. Similarly, dichloro and diiodo magnolols (Fig. 8, C and D) at 4–8 µg/mL were 4–16 times more effective than magnolol (32 µg/mL) and erythromycin (32–64 µg/mL) against MRSA and VRE (Jada et al., 2012). Dihalogenation, especially with bromine, on the aromatic rings of biaryl groups (Fig. 8F) increased potency against resistant pathogens, whereas the mono I- and Cl- substitution showed lesser activity (Jada et al., 2012; Li et al., 2021b). Parent magnolol inhibits MRSA by binding with FtsZ protein, which is involved in cell division (Liu et al., 2014). However, the mechanisms of its derivatives against MRSA and VRE were not investigated. A similar derivative 5,5′-diallyl-3,3′-dibromo-[1,1'-biphenyl]-2,2'-diol (Fig. 8E) inhibited the growth and virulence of B. cinera by disrupting mitochondrial functions, membrane permeability, and hyphal integrity (Li et al., 2021b). Bacterial structures present similar targets, with the exception of hyphal formation, which suggests that halogenated magnolol might target the cell division process (FtsZ protein), cell membrane, and/or adenosine triphosphate (ATP) generation in bacteria. We recommend a detailed investigation of mechanisms and toxicities and how halogens modulate uptake by the pathogen.
3. Resveratrols
Resveratrol is a phytoalexin found in red wine, grapes, soybeans, mulberries, cranberries, and Japanese knotweed, which possesses numerous activities against microbes and other biological conditions (Zhang et al., 2021; Kim et al., 2022). Halogenated resveratrols are considered active compounds that might overcome antimicrobial resistance (Li et al., 2012; Di Fermo et al., 2020). For instance, 2-chloro-resveratrol (Fig. 8G) and 2-bromo-resveratrol (Fig. 8H) at MIC (3.9 μg/mL) inhibited C. albicans and were ∼30-fold and threefold more potent than resveratrol (125 μg/mL) and fluconazole (12.5 μg/mL), respectively (Li et al., 2012). Similarly, 4-[(E)-2-(4-chlorophenyl)ethenyl]phenol (Fig. 8I) inhibited resistant strains of Helicobacter pylori 11F/11 and ATCC 43629 at 3.1 and 12.5 μg/mL and were 64 and 16 times more potent than the parent compound (200 μg/mL) (Di Fermo et al., 2020). Parent resveratrol was reported to show less activity against Gram-negative bacteria due to the efflux pump system, which reduced interaction with cytoplasmic or periplasmic targets in the bacteria (Vestergaard and Ingmer, 2019; Mattio et al., 2020). Interestingly, halogenated resveratrol (Fig. 8I) displayed the capacity to suppress the efflux system and restore antimicrobial activity as evident by the synergism with levofloxacin to control two drug-resistant H. pylori at sub-MIC concentrations (Di Fermo et al., 2020).
Virulence factors are promising targets for the control of resistant pathogens. Halogenated resveratrol at sub-MICs suppressed H. pylori biofilm formation and motility, which are essential for the colonization of gastric mucosa (Di Fermo et al., 2020). Halogenated resveratrols exhibited diverse antimicrobial activities, which were attributed to the introduction of Cl- or Br- atoms on the aromatic A ring. Furthermore, this introduction improved lipophilicity, facilitated diffusion into the bacterial membrane, and subsequently triggered the production of peroxyl radicals (LOO*) (Fig. 8Y). Similarly, the halogenated resveratrols were reported to interfere with ATP generation and adhesion to confer antivirulence effects (Li et al., 2012; Di Fermo et al., 2020). Chlorinated resveratrol (Fig. 8I) caused cell elongation, indicating interference with cell division, and protected Galleria mellonella against H. pylori infection (Di Fermo et al., 2020). In addition, chlorinated resveratrol was nontoxic to G. mellonella larvae (Di Fermo et al., 2020) and was reported to be nonhemolytic to human red blood cells, which could be influenced by the size or numbers of halogen present (Li et al., 2012).
Previously, -OH substituents located at the 4- and 4′- carbon positions of trans-resveratrol were reported to be required for its antiproliferative activities (Savio et al., 2009). Other defects associated with resveratrol are poor bioavailability, metabolite production, and poor stability resulting from excessive metabolism in the intestine and liver (Walle, 2011; Francioso et al., 2014). However, halogenated resveratrol was reported to show a cardioprotective effect with a higher bioavailability and inhibited sirtuins associated with neurologic or metabolic disorders better than the native scaffold, indicating halogen’s tendency to alleviate pharmacological issues (Bourgault et al., 2011; Nguyen et al., 2013). Hence, we believe that halogenation of resveratrol could favor the antibacterial and antivirulence effects, potentiate activities as reported against H. pylori strains, and alleviate issues associated with their pharmacology. However, further study is required to elucidate their mechanisms and to what extent halogenation could shield resveratrol from the problems of excessive metabolism.
4. Flavonoids
Flavonoids are ubiquitously found in plants as phytoalexins, detoxifying agents, and signal molecules (Rana and Gulliya, 2019). They play protective roles in plants, and the same capacity is being harnessed for the treatment of human infections (Biharee et al., 2020). Halogenated flavonoid derivatives such as BrCl-flav (Fig. 8J) were targeted against resistant E. faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter spp. pathogens. Gram-positive MRSA and S. pneumonia were highly susceptible to BrCl-flav (Fig. 8J) at very low MICs (0.24–0.48 µg/mL), which were superior to gentamycin (3.9–62.5 µg/mL) and chloramphenicol (3.9–31.2 µg/mL), respectively (Moldovan et al., 2022). Conversely, it was less effective against Gram-negative pathogens such as E. cloacae, K. pneumonia, S. enterica, and P. aeruginosa except for E. faecium (7.8 µg/mL) (Moldovan et al., 2022). The presence of an outer membrane in Gram-negative bacteria confers extra protection, and hence, the low activities observed suggest an inability to traverse bacterial membranes. Interestingly, when used in combination with fluconazole, BrCl-flav showed a synergetic effect against fluconazole-resistant C. albicans by reducing fluconazole MIC by 128-fold (Babii et al., 2021).
Halogenated flavonoids such as BrCl-flav (Fig. 8J) and ClCl-flav (Fig. 8K) exhibited antimicrobial activities against bacteria by causing the leakage of bacterial intracellular materials, increasing cell permeability to biocidal agents, and damaging cell morphology (Fig. 8Y) (Babii et al., 2018; Moldovan et al., 2022). Also, compound 8J inhibited Candida spp. by irreversibly damaging cellular morphology, and preventing hyphal transition (Babii et al., 2021). The replacement of hydrogen with halogen at the C4 position of compound 8K in addition to the 1,3-dithiolium ring increased bactericidal activities (Babii et al., 2018). Additionally, upon co-incubation with the breakpoint concentrations of respective antibiotics, flavonolignan 8-bromo-2,3-dehydrosilybin AB (Fig. 8L) reversed the colistin-resistant phenotype in P. aeruginosa to a sensitive strain. A similar effect was reported for 6,8,21- tribromosilybin A (Fig. 8M) and 6,8,21-tribromosilybin B (Fig. 8N) at 40 µM against the gentamicin-resistant S. aureus phenotype (Hurtová et al., 2022). The bromination of flavonolignan (Fig. 8, L–N) was reported to significantly enhance the activity and reversed resistant phenotypes probably by inhibition of the efflux pump and bacterial communication as well as improving selectivity index (Hurtová et al., 2022).
Furthermore, interference with bacterial communication represents a viable strategy to regulate bacterial virulence. Halogenated flavonolignan (Fig. 8, L–N) inhibited the autoinducer-1 (AI-1) and autoinducer-2 (AI-2) in Vibrio campbellii and controlled the adhesion of S. aureus and P. aeruginosa to the surface, subsequently preventing biofilm formation (Hurtová et al., 2022). Similarly, BrCl-flav caused loss of hyphal formation and impeded the biofilm formation of C. albicans by 80% at 15.0 µg/mL. It also prevented and disrupted biofilms of A. baumannii by 95% and 58% at 62.5 and 3.9 µg/mL, respectively (Babii et al., 2021; Moldovan et al., 2022). In addition, ClCl-flav reduced the biofilm formation of S. aureus, E. coli, and K. pneumonia by ∼75% at 0.97–7.81 μg/mL (Babii et al., 2018). The forgoing also suggests the capacity of the halogenated flavonoids to combat resistance by targeting the virulence factors.
Notably, halogenated flavonoids such as ClCl-flav (Fig. 8K) were nontoxic to Vero or mammalian HeLa cells at low concentrations (2 μg/mL) but mildly toxic at higher concentrations (4–32 μg/mL) (Babii et al., 2018). BrCl-flav (Fig. 8J) was slightly cytotoxic, with an IC50 value of ∼80 µg/mL. Notably, it enhanced cell viability at doses ranging from 5 to 25 µg/mL, probably due to cellular metabolization. Taken together, halogenated flavonoids could serve as adjuvants with known antibiotics or target the virulence factors to reverse or control resistance in pathogens with low selective pressure.
B. Essential Oil Compounds
Essential oils (EOs) are highly concentrated hydrophobic liquids derived from a variety of plants and classified based on their chemical and physical properties. The pharmacological effects of EOs have been extensively examined and include antimicrobial, antioxidant, anti-inflammatory, and immunomodulatory activities (Masyita et al., 2022). Structurally, the components of EOs can be classified as terpenes, terpenoids, phenylpropanoids, and others (Pandey et al., 2017). Chemical halogenation is a prominent strategy used to enhance the uptakes and activities of EOs and other natural products. Here, we describe the bioactivities of halogenated essential oil compounds such as thymol and cinnamaldehyde.
1. Thymols
Thymol is a bioactive compound found in thyme oil (Thymus vulgaris) and the Origanum genus (Marchese et al., 2016). Thymol is an isomer of carvacrol, which is used to control zoonotic and food pathogens. Synthetic thymols have been reported to have antiseptic effects in toothpaste and are used to flavor cough syrups as well as chewing gum (Kaur et al., 2014; Kim et al., 2022). Chlorothymol (Fig. 8O) at 32 µg/mL had 16-fold greater inhibitory effects against MRSA than carvacrol (512 µg/mL), whereas thymol was without activity (Kim et al., 2022). However, chlorothymol inhibited the sensitive S. aureus strain at 12.5 µg/mL which is ∼3 times lower than what was required for MRSA control (Kaur et al., 2014). Chlorothymol was more active against sensitive than resistant strains. Interestingly, the combination of chlorothymol with antibiotic oxacillin further improved activity against MRSA (MIC: 8 μg/mL) suggesting its capacity to act optimally and overcome resistance via synergism (Kim et al., 2022). In addition, C. albicans was better inhibited by chlorothymol than thymol at 100 μg/mL and in a manner comparable to amphotericin B (Kaur et al., 2014).
As part of strategies to control resistance, halogenated thymols (Fig. 8O) exhibited antivirulence potential. The sub-MIC concentrations prevented MRSA biofilm formation, disrupted mature MRSA biofilms, restricted motility, and decreased staphyloxanthin production involved in bacterial evasions of oxidative stress and host immune system (Xue et al., 2019; Kim et al., 2022). Furthermore, the SAR study revealed that the presence of chlorine at the C4 position of thymol was crucial to antimicrobial activity, compared with the bromo- and dibromo- derivatives which lost activity (Fig. 8, P and Q), suggesting a small-sized halogen is required for activity (Kaur et al., 2014; Kim et al., 2022). We surmise that halogens like chlorine enhanced the lipophilicity and membrane localization of thymol to improve membrane penetration. This is corroborated by reports that the hydrophobic nature and enhanced affinity for the membrane lipid bilayer of chlorothymol were responsible for MRSA inhibition via membrane integrity disruption (Kowalczyk et al., 2020; Kim et al., 2022).
2. Trans-Cinnamaldehydes
Trans-cinnamaldehyde (CNMA) largely accounts for the antimicrobial activities of Cinnamomum spp. and was approved by FDA for application in the food industry as a flavoring agent (Shreaz et al., 2016; Friedman, 2017). The halogenation of CNMA was reported to offer a potential means of countering antibiotic-resistant pathogens. 2,4-Dichloro- and fluoroCNMA (Fig. 8, R and S) inhibited penicillin-resistant S. pyogenes and S. aureus (16–64 µg/mL) and were better than cinnamic acid and ciprofloxacin (>128 µg/mL) against resistant S. pyogenes (Li et al., 2015). Comparatively, the derivatives (Fig. 8, R and S), as expected, required lesser doses (4–16 µg/mL) to achieve inhibition against sensitive S. aureus. However, sensitive S. pyogenes was more tolerant (≥128 µg/mL) and required twice the dose for activity than the resistant strain (Li et al., 2015). This may suggest a specific resistance mechanism that is not available in the sensitive strain as the target of the derivatives. Also, p-brominated CNMA (Fig. 8T) with an MIC of 0.51 mM against S. aureus and E. coli, respectively, was approximately four- to fivefold more effective than the parent CNMA (1.93–2.32 mM) (Doyle et al., 2019). Also, 4-bromophenyl–substituted (Fig. 8U) and 2,4-dichlorinated CNMA (Fig. 8R) were effective against MRSA and A. baumannii (64 µg/mL), respectively (Chai et al., 2022), but ineffective against extensively drug-resistant A. baumannii. However, they became effective when coincubated with an efflux pump inhibitor, indicating that drug efflux contributed to the intrinsic resistance displayed. Of note, the outer membrane of A. baumannii was reported to be about 100 times more impervious than those of other Gram-negative bacteria, especially E. coli, due to the absence of nonspecific porins (Zgurskaya et al., 2015; Chai et al., 2022). Therefore, further halogen modulation of the CNMA scaffold is required to enhance efficacy against multidrug-resistant A. baumannii. Mechanistically, halogenated CNMAs (Fig. 8, R, S, and U) similarly inhibited the bioactivities of S. aureus and A. baumannii strains by suppressing cell proliferation. Specifically, they caused cell elongation and inhibited FtsZ protein polymerization as well as GTPase activity, which led to abnormal cell division and subsequent cell death (Li et al., 2015; Chai et al., 2022).
Halogenated CNMAs also target bacterial virulence factors. For instance, 4-bromoCNMA, 4-chloroCNMA, 4-fluoroCNMA, and dichloroCNMA (Fig. 8, T and V–X) decreased protease activities, biofilm formation, cell surface hydrophobicities, motilities, fimbria production, quorum sensing, and pigment production in C. albicans and Vibrio spp., and these effects were corroborated by downregulations of virulence- and biofilm-related genes (Brackman et al., 2011; Faleye et al., 2021; Khadke et al., 2022).
The preclinical safety evaluations showed that the halogenated derivatives 8R, 8U, and 8W were devoid of hemolytic activity in human blood and nontoxic as determined by their effects on the growth and metabolism of HepG2 mammalian liver cells and the survival of the Caenorhabditiselegans model (Chai et al., 2022). Depending on concentrations, 4-bromoCNMA (Fig. 8T) had a none to slightly toxic effect on the survival of G. mellonella larvae. Notably, the dichlorophenyl- and bromophenyl CNMA (Fig. 8, R and U) lacked an off-target effect on human tubulin polymerization (Chai et al., 2022). Given the nontoxicity observed in preliminary in vivo and in vitro toxicity assays of halogenated CNMAs, we recommend further toxicity tests in advanced models, especially animals for possible future applications like the parent CNMA in food applications.
C. Alkaloids
Alkaloids play an essential role in the natural defense systems of organisms and human medicine and constitute ∼20% of known secondary metabolites in plants and microbes. In plants, alkaloids protect against predators and regulate growth. Therapeutically, alkaloids are used as anesthetics and cardioprotective and anti-inflammatory agents. Well known clinical alkaloids include morphine, strychnine, quinine, ephedrine, and nicotine (Heinrich et al., 2021). Here, we discuss the bioactivities, toxicities, and pharmacokinetic properties of alkaloids functionalized with halogens (Fig. 9, A–Z).

Structures of halogenated alkaloids. Indoles (A–P), streptochlorins (Q and R), quinones and quinolines (S–Z), and 4-oxoquinolizines (A1–A4) and effects of halogenation on indoles, streptochlorins, and quinones (A5). Full chemical names are listed in Supplemental Fig. 2.
1. Indoles
More than 85 species of Gram-negative and Gram-positive bacteria possess the tryptophanase (tnaA) gene and produce indole as a signaling molecule directing certain bacterial functions (Lee and Lee, 2010; Kumar et al., 2021). Indole-based compounds such as vincristine, reserpine, and amedalin are used clinically as anticancer, antihypertensive, and antidepressant agents, respectively (Kumar et al., 2020a). Halogenated indole derivatives have been used as antimicrobial controls against antibiotic-resistant pathogens (Qin et al., 2020; Raorane et al., 2020). CZ74 (Fig. 9A) at 2–4 μg/mL was reported to exhibit highly potent activity against ampicillin-resistant S. aureus, MRSA, and VRE. The activity was attributed to the presence of the trifluoromethyl group (CF3)(Yuan et al., 2019). Furthermore, 5-iodoindole (Fig. 9B) inhibited the resistant strains of A. baumanii at a MIC of 50 µg/mL, which was superior to gentamicin and colistin but comparable to ciprofloxacin at a dose of 200 µg/mL (Raorane et al., 2020). Insertion of iodine at the C5 position of indole resulted in more effective antimicrobial activity against A. baumannii than other positions by enhancing selective transportation via the membrane (Raorane et al., 2020). In addition, potent brominated metabolites from the Red Sea sponge Callyspongia siphonella, viz. 5‐bromotrisindoline (Fig. 9C), and 6‐bromotrisindoline (Fig. 9D) inhibited MRSA at concentrations of 8 and 4 μM, respectively (Sayed et al., 2020). Some halogenated indoles (Fig. 9, A, C, and D) exhibited limited activities against Gram-negative bacteria, probably because being neutral, they were unable to establish electrostatic interactions (Yuan et al., 2019; Sayed et al., 2020), which led to the inability to transverse the outer bacterial membrane. Thus, the addition of positively charged groups, such as primary amines, was proposed to expand their activity spectra (Huigens et al., 2013).
Halogenated indoles can also target some virulence factors as reported for 5-iodoindole, 4-fluoroindole, and 7-chloroindole (Fig. 9, B, E, and F), which eradicated MRSA and E. coli persister cells by multiple folds as compared with the parent indole (Lee et al., 2016). Interestingly, these halogenated indoles inhibited staphyloxanthin production by S. aureus strains at sub-MIC concentrations as well as the motility and pellicle formation of A. baumannii at 25 and 50 µg/mL, respectively (Lee et al., 2016; Raorane et al., 2020). In addition, 4-chloroindole, 5-chloroindole, and 5-chloro 2-methyl indole (Fig. 9, J–L) inhibited biofilm formation and restricted motility, curli, fimbriae, protease production, and the cell surface hydrophobicity of V. parahaemolyticus and uropathogenic E. coli (Sathiyamoorthi et al., 2021; Boya et al., 2022).
Various mechanisms underlie the antimicrobial activities of halogenated indoles on resistant pathogens. Specifically, CZ74 (Fig. 9A) was reported to interfere with FtsZ polymerization and inhibited GTPase activity, resulting in the inhibition of bacterial cell division (Yuan et al., 2019). Also, 5-iodoindole (Fig. 9B) increased reactive oxygen species generation, which caused the leakage of cytoplasmic contents leading to oxidative stress-mediated A. baumannii cell membrane disruption (Raorane et al., 2020). However, 9B did not affect the cell shapes or membranes of E. coli or S. aureus (Lee et al., 2016). We believe this might be related to the dosages used and the pathogen intrinsic factors. Other halogenated indoles (Fig. 9, G and H) effectively inhibited enzymes related to protein synthesis and mycobacterial wall synthesis in E. coli and Mycobacterium, respectively (Sapnakumari et al., 2017; Rathod et al., 2018). Also, 5‐bromotrisindoline (Fig. 9C) acted as a dual enzyme inhibitor of DNA gyrase and pyruvate kinase, which are involved in DNA replication, staphylococcal biofilm formation, resistance, pyruvate, and ATP generation (Vasu et al., 2015; Thomsen and Liu, 2018; Sayed et al., 2020). Conversely, 6‐ bromotrisindoline (Fig. 9D) was found to be a potent gyrase‐B inhibitor, indicating that the location of bromine contributed to selectivity and potency (Sayed et al., 2020). Importantly, fluorinated indole (Fig. 9I) potently inhibited S. aureus (SA-1199B), known to overexpress norA, a drug efflux protein responsible for efflux activity and viability (Lepri et al., 2016).
Halogenated indoles demonstrated non- to mild toxicity in various in vivo, in vitro, and in silico studies. An ADME study of chloroindoles (Fig. 9, J–L) revealed they are noncarcinogenic to mice, have minimal acute fish toxicities, and do not violate Lipinski’s rule of five (Boya et al., 2022). Halogenated trisindoles (Fig. 9, C and D) exhibited excellent drug‐like attributes, good oral absorption, reasonable toxicities, and high bioavailabilities (Sayed et al., 2020). Similarly, 5-chloroindole (Fig. 9K) was nontoxic to the MN9D mouse dopaminergic cell line (Kholodar et al., 2021). Fluorinated indoles (Fig. 9, E, M, and N) were reported to be nontoxic to C. elegans at 100 mg/L. However, at high doses (ca. 100 × MIC), 7-fluoroindole was mildly toxic (Raorane et al., 2022). It is worth noting that halogenation levels may alter the pharmacokinetics of the indole scaffold (Fig. 9A5). For example, a fluorine atom at the C2 position of a fluorinated indole (Fig. 9O) improved the pKa and directly affected pharmacokinetics more so than its difluorinated indole congener (Fig. 9P) (Manallack, 2007; Lepri et al., 2016). Contrary to the notion that halogens enhance metabolic stability, the presence of fluorine in an indole derivative (Fig. 9O) resulted in rapid oxidation, molecular instability, and the production of two metabolites, including M-90 (N-dealkylation) and M-68 (N-dealkylation) (Lepri et al., 2016). Given the diverse, versatile activities displayed by halogen-containing indoles against various resistant pathogens and their virulence factors, we speculate that these compounds offer promising solutions to bacterial resistance and that, to a greater extent, the antimicrobial and pharmacological properties of halogenated indoles are dependent on the positions, numbers, and types of halogens present and the bacterial cell type. However, further in vivo assessments are required to identify suitable drug candidates.
2. Streptochlorins
Streptochlorin (4-chloro-5-(1H-indol-3-yl)oxazole) is a small alkaloid of indole origin found in marine bacteria like Streptomyces sp. The streptochlorin scaffold is privileged, and it is found in natural compounds like labradorin, pimprinine, and almazole. The scaffold exhibits a variety of biological activities but with narrow-spectrum antimicrobial activity (Zhou et al., 2018a; Song et al., 2021). However, halogen modification of the scaffold was used to increase potency (Fig. 9, Q, R, and A5). For instance, 4-bromo-5-(1H-indol-3-yl)oxazole (Fig. 9Q) and ethyl 2-(3-4-bromooxazol-5-yl)-1H-indol-1-yl)-2-fluoroacetate (Fig. 9R) potently inhibited some fungal pathogens (Jia et al., 2018). Notably, the replacement of Cl- with Br- at the C4-position of the oxazole ring improved the activities of these derivatives (Jia et al., 2018). In addition, streptochlorin could bind strongly with Thermus thermophiles Leucyl-tRNA synthetase to inhibit protein synthesis like Tavaborole (a small-molecule medication for fungal infections) (Gao et al., 2021). Modified streptochlorin (Fig. 9, Q and R) were not investigated for their effects on drug-likeness; however, parent streptochlorin was shown to exhibit high hepatic clearance using mouse liver microsomes and liver S9 fractions. It also showed low oral bioavailability, reduced plasma concentrations, short half-life, high systemic plasma clearance, and metabolization by glucuronidation or monooxygenation of the indole structure (Zhou et al., 2018a). Hence, further structural tuning is required to avert metabolic stability problems and improve other pharmacokinetic profiles or to develop compatible topical applications.
3. Quinone and Quinolines
Quinones are aromatic cores containing two oxygen atoms that form two carbonyl bonds and are found in plants, fungi, and bacteria (Niaz et al., 2020). They are metabolites formed by the oxidation of hydroquinone and have found applications in approved clinical drugs and those in the developmental pipeline. Quinones have considerable pharmacological potential, and their biological activities are due to their involvement in redox cycling processes (Campos-Xolalpa et al., 2021; Sahoo et al., 2022). Quinones have been modified to enhance their efficacies and alter their physiochemical features (Fig. 9, S–Z). Fluorinated carbazoloquinones (Fig. 9, S and T) inhibited MRSA at MIC 25–50 µg/mL, and the effect of the compound (9S) was comparable to linezolid and vancomycin (28–30 µg/mL) (Chakraborty et al., 2017). Interestingly, hydrophilic fluorine at C6 on one benzene ring and the incorporation of the hydrophobic methyl group at the C2 or C3 of the other benzene in carbozoloquinones were crucial for anti-MRSA activity (Chakraborty et al., 2017).
In addition, 7-bromohydroxyquinoline and 5,7-dichlorohyroxyquinoline (Fig. 9, U and V) exhibited similar inhibitory activities against clinically important Gram-negative pathogens but weaker activities against Gram-positive bacterial and fungal isolates (Cherdtrakulkiat et al., 2016). The presence of Cl- or Br- at the C5 and/or C7 positions of these compounds enhanced lipophilicity and improved their effectiveness against Gram-negative bacteria. However, this was not the case for Gram-positive pathogens as unsubstituted and low lipophilic 8-hydroquinoline adequately penetrated the bacterial membrane (Cherdtrakulkiat et al., 2016). This observation challenges the status quo, and it suggests the possibility of improved affinity conferred by halogenation against a specific target protein in Gram-negative species. Also, bromoquinol (Fig. 9X) at 1 µM demonstrated 95% killing against the invasive fungus Aspergillus fumigatus by rapidly inducing oxidative stress and apoptosis (Ben Yaakov et al., 2017). Bromoquinol functions optimally at low metal concentrations, as is observed in infected hosts, but becomes chelated and inactive in the presence of iron, copper, or zinc. Consistently, the incorporation of bromine residues at C5 and C6 accounted for its efficacy to parent 8-hydroxyquinoline (Ben Yaakov et al., 2017). In addition, to improving lipophilicity, halogenation might enhance the iron-chelating effect of the 8-hydroxyquinoline moiety to display antibacterial activity (Fig. 9A5). This is corroborated by the activity of clioquinol, a hydroxyquinoline drug that contains chlorine and iodine and was reported to exert various antimicrobial activities by chelating metals, including Cu, Zn, and Fe (Das et al., 2019).
Preclinical evaluations have shown that halogenated quinoline (Fig. 9W) is not toxic to HeLa cells at 100 µg/mL (Desai et al., 2017). Bromoquinol (Fig. 9X) protected the larvae of G. mellonella against aspergillosis as much as amphotericin B but was ineffective against invasive pulmonary aspergillosis in a murine model at 8 mg/kg (Ben Yaakov et al., 2017). The disparity observed between in vivo and in vitro efficacies may have been due to physiologic responses, drug properties, or drug-pathogen interactions. According to transcriptional profiling, A. fumigatus in response to bromoquinol repressed energy-dependent cellular activities such as protein synthesis and activated genes responsible for efflux, detoxification, and reactive radical formation (Ben Yaakov et al., 2017). Although we are aware that fungi and bacteria are continuously evolving due to exposure to diverse environmental cues or stressors, further investigation might provide better insight. Furthermore, halogenated aminoquinones (Fig. 9, Y and Z) were found to be noncarcinogenic and nonmutagenic, like the antibiotics ciprofloxacin, vancomycin, and cefoxitin, and were predicted to have high gastrointestinal absorption profiles. Additionally, no toxic/hemolytic effect was reported on healthy human blood, and the presence of a sugar moiety on the halogenated scaffold contributed to the nontoxic properties and non-CYP3A4 inhibition (Dias et al., 2018). Based on the forgoing, these halogenated derivatives were less toxic than the parent quinone, suggesting the possible role of halogenation in alleviating side effects. Conversely, certain halobenzoquinones were reported to demonstrate genotoxic, carcinogenic, and cytotoxic effects, but confirmation from in vivo studies is scarce (Matos et al., 2015). Despite the suggested aforementioned toxicological effects, quinones demonstrated cytoprotective potentials via anti-inflammatory activities, detoxification enzyme induction, and redox status modification (Bolton and Dunlap, 2017). Due to these differential toxicity profiles of the various halogenated quinone analogs, more detailed elucidation of in vivo toxicity is recommended.
4. 4-Oxoquinolizines
The 4H-4-oxoquinolizine scaffold represents the bioisostere of quinolones, and its derivatives were synthesized as alternatives to combat the limitations of fluoroquinolones, which include low intrinsic activity toward Gram-positive bacteria and the increased quinolone-resistant pathogens (Ma et al., 1999). The halogenated 4-oxoquinolizine displayed better antibacterial activity than fluoroquinolones. For example, CC-195776, CC-195767, and CC-195820 (Fig. 9, A1–A3) exhibited excellent antibacterial activities against drug-resistant A. baumannii at very low MICs of 0.09–0.19 µg/mL and were superior to ciprofloxacin, ofloxacin, and levofloxacin (8–64 µg/mL) (Na et al., 2017). Also, GC-072 (Fig. 9A4), which is currently undergoing clinical trials, exhibited better antibacterial activities against drug-resistant Burkholderia pseudomallei strains (MIC: ≤0.004–0.25 µg/mL) than ciprofloxacin (0.25–16 µg/mL) (Shearer et al., 2019). Another prodrug TNP-2092 containing 4H-4-quinolizine pharmacophore, and rifamycin inhibited resistant H. pylori strains (0.008–16 μg/mL) and was superior to clarithromycin and levofloxacin (0.016–128 μg/mL) (Wang et al., 2018).
Previously, mutations that occured at the quinolone resistance-determining region of GYrA and ParC subunits were attributed to fluoroquinolone resistance (Madurga et al., 2008). Halogenated 4-oxoquinolizines displayed the capacity to bind these mutated enzymes to exert antimicrobial activities against quinolone-resistant bacteria. For instance, CC-195776, CC-195767, and CC-195820 exhibited notable inhibitory effects on resistant A. baumannii despite amino acid mutations in the quinolone resistance-determining region of GyrA and ParC (Na et al., 2017). Similarly, GC-072 overcame trimethoprim and ceftazidime resistance caused by folA and PenA β-lactamase point mutations, respectively, with activity better than ciprofloxacin (Shearer et al., 2019). They also overcame antibiotic resistance by inhibiting efflux activities as evident in the resistance of GC-072 to efflux export mediated by AmrAB-OprA and BpeAB-OprB in B. pseudomallei. However, it may be susceptible to BpEF-OprC pump activity but with a significantly lower degree of efflux than ciprofloxacin and doxycycline.
Regarding the SAR, GC-072 containing two fluorine atoms and an amine group with conformation different from the piperidine ring in ciprofloxacin potentially contributed to the enhanced activities against quinolone-resistant bacterial strains (Shearer et al., 2019). Previously, the substituent in the C-8 position of the oxoquinolizine scaffold was reported to influence target affinity, drug access to enzymes, DNA binding, and overall activity (Peterson, 2001). The addition of free halogen at this location or in combination with the methyl group enhanced activity against certain anaerobes and fluoroquinolone-resistant bacteria (Ledoussal et al., 1999).
Furthermore, halogenated 4-oxoquinolizines, like fluoroquinolones, target bacterial DNA gyrase and the topoisomerases IV but display better selectivity and also possess a separate binding mode from the fluoroquinolones (Shearer et al., 2019). Halogenated derivatives, including CC-195776, CC-195767, and CC-195820, showed a high degree of nontoxicity. They exhibited strong selective toxicity toward A. baumannii and were nontoxic to human HeLa and U937 cells at lower doses (25 µg/mL) but became toxic at 50 µg/mL, which is 100 times higher than their MICs (0.09, 0.19, and 0.19 µg/mL, respectively) (Na et al., 2017). Overall, halogenated 4H-4-oxoquinolizines have shown the capacity to outcompete fluoroquinolones in overcoming drug resistance. It is our opinion that increased interest in halogenation of the C8 position will further improve tolerability, specificity, and pharmacokinetics and increase the activity spectrum.
D. Benzopyrone and Phenazines
Benzopyrones are common phytochemicals and have produced promising results in clinical applications, especially when used with other therapies. Coumarins and some bioflavonoids are common agents that have been used to treat asthma, lymphedema, infections, and neoplasms (Matos et al., 2015; Hu and Piller, 2017).
1. Coumarins
The coumarins are bicyclic, heterocyclic benzopyrans that contain an aromatic ring fused to a six-membered lactone ring (Rehuman et al., 2021). Both natural and synthetic coumarins are commonly used in the perfume and cosmetics industries (Khan et al., 2019). The charge-transport properties and electron-rich structure of coumarins confer several pharmacological and biological properties (Matos et al., 2015). Novel halogenated coumarins, such as bromoacetyl derivatives (Fig. 10, A–C), inhibited a panel of clinically important bacteria, including Bacillus coagulans, B. cereus, Micrococcus luteus, S. faecalis, E. coli, and S. aureus, in the MIC range of 0.75–1.5 mg/mL. The addition of bromine to the acetyl group - 3-(2-bromoacetyl) and 3-(2, 2-dibromoacetyl) at the C3 position of the coumarin nucleus favored antimicrobial activities (Kasumbwe et al., 2014). Also, dihalogenation at C6 and C8 of the chromene ring (Fig. 10, D and E) enhanced potency against fungal (A. fumigatus, A. flavus, and Rhizopus spp.) and bacterial pathogens (E. coli and B. cereus) as compared with mono substitution at C6 (Khan et al., 2019). Notably, the para-bromination increased lipophilicity and stability of the coumarin structure due to less electronegativity (Fig. 10A1). However, substituting fluorine at the -ortho position led to destabilization (Završnik et al., 2011). It appears that the efficacy of halogenated coumarin depends on halogen number, position, and electronegativity.

Structures of halogenated benzopyrones, phenazines, and azoles. Coumarins (A–F), phenazines (G–P), and azoles (Q–Z) and effects of halogenation on coumarins, phenazines, and azoles (A1). Full chemical names are provided in Supplemental Fig. 3.
Regarding their MOAs, halogenated coumarins exhibited a binding affinity for glucosamine-6-phosphate (GlcN-6-P) synthase and lanosterol 14α-demethylase (CYP51), which are responsible for viability in bacteria and ergosterol biosynthesis and viability in fungi, respectively. Furthermore, this affinity was greater for bacterial GlcN-6-P than fungal lanosterol, indicating better efficacy against bacteria (Khan et al., 2019). The halogenated coumarin-chalcone CC2 (Fig. 10F) was able to cross the blood-brain barrier and was nontoxic to Vero cells at a concentration 100 times higher than the effective concentration used for biological evaluations. In addition, it halted H2O2-induced cellular damage by scavenging reactive oxygen species (Rehuman et al., 2021). Typically, coumarin and some coumarin derivatives were reported to undergo phase I metabolism and subsequently become hepatotoxic (Hu and Piller, 2017). However, the effects of coumarin halogenation on these contraindications have not been determined; hence, further investigation is recommended.
2. Phenazines
Phenazines are found in Streptomyces and Pseudomonads as secondary metabolites and confer survival advantages (Huigens et al., 2022). Two phenazines, pyocyanin and phenazine-1-carboxylic acid, produced by P. aeruginosa, serve as electron acceptors for intracellular redox homeostasis (Schiessl et al., 2019). Phenazines have several pharmacological properties, ranging from anti-inflammatory, neuroprotective, antimicrobial, antimycobacterial, and antibiofilm to anticancer activities. The halogenated congeners were reported to possess better antimicrobial activities against resistant pathogens (Huigens et al., 2022). Halogenated phenazines (Fig. 10, G–L) demonstrated excellent inhibitory activities against MRSA, methicillin-resistant S. epidermis (MRSE), and VRE, at MICs of 0.04–1.17 μM, and were ∼3- to 23-fold superior to the parent phenazine (1.17–4.69 μM) and antibiotics—vancomycin, linezolid, and daptomycin (0.39–100 μM) (Yang et al., 2017; Liu et al., 2022). The presence of Cl- or -Br- at the C6 or C8 positions of chlorinated or tri-brominated phenazines (Fig. 10, G, H, and M) enhanced membrane permeability and metalloprotein targeting (Yang et al., 2017).
Halogenated phenazines (Fig. 10, M–O) also exhibited ∼fourfold antitubercular potential (6.25 μM) against slow-growing Mycobacterium spp. compared with the parent (25 μM) (Garrison et al., 2016; Yang et al., 2017). Two Cl- atoms at the C7- and C8-positions of phenazine enhanced antimycobacterial activity, but their replacement with two -Br atoms led to a loss of activity, suggesting that halogen size critically affected bioactivity (Fig. 10, N and O) (Garrison et al., 2016). Conversely, these derivatives displayed limited activity against Gram-negative A. baumannii and no activity against E. coli (Liu et al., 2022). This is the opposite as observed for halogenated quinones, which showed better activities against Gram-negative bacteria. We opine that the difference was due to cellular uptake capacity and possibly the compound structural properties. For instance, the importance of the pair of nitrogen atoms on the phenazine scaffold was evident by a multiple-fold reduction in activity when one of the nitrogen atoms of 2,4-dibromo-7,8-difluorophenazin-1-ol (Fig. 10P1) was replaced with C-H to obtain halogenated acridine - 1,3-dibromo-6,7-difluoroacridin-4-ol (Fig. 10P2) (Liu et al., 2022).
Regarding antivirulence potentials, halogenated phenazines (Fig. 10, I, J, and L) eradicated MRSA, MRSE, and VRE biofilms (0.30–37.5 μM) and their persister cells many times more effectively than the membrane lysing quaternary ammonium cation-10 (2.3–93.8 μM) (Garrison et al., 2016; Liu et al., 2022). Also, the control antibiotics (vancomycin, daptomycin, and linezolid) failed to eradicate biofilms at a concentration of >2000 μM (Yang et al., 2017). The replacement of bromine with iodide in 10L resulted in a significant increase in biofilm-eradicating activity (Garrison et al., 2016). These reports demonstrate that the halogenation of phenazine is a promising source of antibiofilm agents capable of effectively controlling chronic and persistent bacterial infections.
Halogenated phenazines employ several mechanisms against resistant pathogens and their virulence factors. In addition to bacterial membrane crossing and metalloprotein targeting, they enhance phenolic acidity, which may be essential for metal chelation (Fig. 10A1). Furthermore, 2,4-dibromo-1-hydroxyphenazine inhibited RNA and protein biosynthesis but not DNA synthesis in MRSA, obviously suggesting RNA as its target (Yang et al., 2017). Also, the biofilm-forming ability of MRSA was inhibited by a non–membrane disrupting mechanism, specifically iron starvation, which was corroborated by the excellent iron-binding properties of the fluorinated halogenated phenazines and multiple-fold upregulations of the iron uptake genes sbnC and isdB (Liu et al., 2022). Unlike the iron-specific antibiofilm mechanism, other halogenated phenazines may also employ other metal (II)-dependent mechanisms (Garrison et al., 2016; Liu et al., 2022). Of note, the presence of the phenazine efflux pump gene hprS, responsible for survival against P. aeruginosa in the host environment, was reported in S. aureus (Fu et al., 2021), suggesting the possibility of evolutionary resistance to the basic phenazine scaffold. However, the resistance of S. aureus to 2,4-dibromo-8-chlorophenazin-1-ol (10M) was difficult to develop, probably due to the enhanced potency and reduced selective pressure conferred by the halogen (Fu et al., 2021), which implies another merit of halogenation.
Different preclinical evaluations have revealed that halogenated phenazines are not hemolytic to mammalian red blood cells (Yang et al., 2017; Liu et al., 2022), and reportedly, they were mild to nontoxic against HeLa cells at IC50 > 100 μM, with excellent selectivity indices (>1000- to 2000-fold) toward MRSA and MRSE cells (Garrison et al., 2016; Yang et al., 2017; Liu et al., 2022). Generally, pathogens develop resistance to drugs with little or no selectivity. Therefore, high selectivity indices, multitarget inhibition, improved activity spectrum, and low tendency for resistance development exhibited by different halogenated phenazines highlight their potential as drugs that target resistant pathogens (Fig. 10A1).
E. Azoles
The azoles are a class of heterocyclic compounds with a five-membered ring carrying an atom of nitrogen, sulfur, or oxygen. Based on the heteroatoms present, azoles include imidazole, oxadiazole, triazole, tetrazole, isoxazole, thiazole isoxazole, pyrazole, 1,2,3-triazole, thiadiazole, and others (Emami et al., 2023). Importantly, azoles are often used in the design of antifungal compounds, including ketoconazole, fluconazole, and voriconazole. In addition to their antifungal efficacies, azoles also possess anti-inflammatory, anticancer, immunosuppressant, and antidiabetic effects (Ghani, 2020). They are known to target the cytochrome P450–dependent enzymes required for sterol biosynthesis and the formation of antifungal byproducts (Vardanyan and Hruby, 2016). However, fungal pathogens have developed countermeasures, such as the upregulation of lanosterol 14α-demethylase encoding (CYP51) and efflux pump genes (Cowen et al., 2014). To address these developments and minimize unfavorable pharmacokinetics, a series of azole derivatives, including halogenated azoles, have been developed (Fig. 10, Q–Z). Previously, the importance of fluorine in benzoxazole compounds was reviewed (Al-Harthy et al., 2020), but we focused herein on the influence of different halogens in the azole scaffold with respect to resistance inhibition, mechanisms, and pharmacokinetics.
Halogenated benzothiazole-urea derivatives (Fig. 10, Q and R) inhibited MRSA at 0.78 μmol/L, which was 8- to 128-fold better than the antibiotics cefoxitin and linezolid (6–100 μmol/L) but comparable to ofloxacin (1.56 μmol/L). Relative to the sensitive S. aureus (0.39 μmol/L), twice the MIC (0.78 μmol/L) of 10QR was required to control MRSA (Zha et al., 2022). Most importantly, the passage assay revealed that MRSA could not gain resistance against 10Q, which supports its potential to combat drug resistance. Improved lipophilicity and solubility probably mediated by the presence of para-CF3 and 3,4-difluoro on the phenylurea moiety as shown in Fig. 10, S and A1 were responsible for the activity (Zha et al., 2022). Furthermore, halogen containing 1,2,3-trizole-derived naphthalimides inhibited E. coli at 0.5 μg/mL, which was 1000-, 32-, and fourfold more potent than its precursor, chloromycin, and norfloxacin, respectively. The introduction of -Cl on the C3 and C4 of the benzyl group of compounds enhanced activity against E. coli (Lv et al., 2014). Similarly, halogenated benzoxazoles (Fig. 10, T and U) more strongly inhibited the clinical pathogens E. coli, S. aureus A. terrus, and Penicillium brocae than benzoxazole (Rodrigues et al., 2022). These improved activities were attributed to the presence of -Br or -Cl and the aminopyrazole group (Rodrigues et al., 2022).
Also, halogenated azoles are highly effective at controlling virulence factors. Halogenated benzothiazole-urea (Fig. 10, Q and R) exhibited better biofilm-eradicating effects against MRSA at 4 to 5 μM than the antibiotics ofloxacin (7 to 8 μM) and erythromycin (>200 μM) (Zha et al., 2022). In addition, other derivatives (Fig. 10, X and Y) inhibited biofilm formation by S. epidermidis and had no hemolytic effect on human erythrocytes at 50 µM (Zhao et al., 2014). Further modification of fluconazole with halogens resulted in broad-spectrum activity against the in vitro and systemic viabilities and biofilm formation of fluconazole-resistant C. albicans isolates at an MIC of 0.25–2 µg/mL compared with fluconazole (≥64 µg/mL) (Shafiei et al., 2021).
Regarding the MOA, halogenated azoles exhibited various antimicrobial activities by targeting the bacterial membrane and DNA replication. For instance, compound 10R killed MRSA by damaging its cell membrane and bound favorably to the active site of S. aureus DNA gyrase to disrupt replication (Zha et al., 2022). In addition, 10T-U were predicted to inhibit the active site of S. aureus UDP-N-acetylenolpyruvylglucosamine reductase (MurB), which participates in peptidoglycan biosynthesis (Rodrigues et al., 2022). Similarly, the halogenated azoles (Fig. 10, V and W) targeted sterol 14-α demethylase to exhibit anticandidal activity and DNA gyrase to inhibit E. coli (Kumar et al., 2022). Also, 10Z is effectively intercalated into DNA to block replication in E. coli and could be transported by human serum albumin in the blood plasma via electrostatic interactions to enhance the drug distribution (Lv et al., 2014). Other halogenated derivatives (Fig. 10, X and Y) interfered with the enzymatic activity of YycG (histidine kinase), which is essential for the viability and cell wall synthesis of Staphylococcus spp. (Zhao et al., 2014).
Preclinical examinations revealed that halogenated analogs (Fig. 10, Q and R) exhibited low cytotoxicity at 25 µM in HepG-2 cells, with a >75% survival rate. Furthermore, 10R was well tolerated in a mouse model and did not induce central hepatic hilum necrosis or notable steatosis at a concentration of 200 mg/kg (Zha et al., 2022). Functionalized fluconazole was nontoxic to mammalian cells like the human dermal fibroblast, epidermoid (A-431), and human pancreatic cancer (PANC-1) but could produce a mild effect at 200× the MIC (100 µg/mL). It was predicted with no carcinogenic tendencies, medium-risk hERG inhibition, and bioavailability similar to the antifungal drugs fluconazole and ravuconazole (Shafiei et al., 2021). These diverse biological activities, coupled with reasonable toxicities, showed that halogenated azoles provide a promising means of combating resistance either alone or in combination with natural or conventional antibiotics.
F. Repurposed Halogenated Drug Scaffolds
The processes involved in drug discovery are expensive and time consuming, and these restraints have limited the numbers of approved drugs amid infection concerns. Drug repurposing, which entails the discovery of new medicinal applications or clinical indications for existing clinically important drugs, could play an essential role in reducing these constraints and facilitating the discovery of drugs. Here, we review various halogen modifications made to approved drugs. The scaffolds include, but are not limited to, isoniazid, aspirin, haloperidol, phenothiazines, and salicylanilides (Fig. 11, A–T).

Structures of repurposed scaffolds. Isoniazids (A and B), azo-aspirins (C and D), pyrrolopyrimidines (E–G), phenothiazines (H and I), and salicylanilides (J–T) and effects of halogenation on isoniazides, phenothiazines, and salicylanilides (U). Full chemical names are listed in Supplemental Fig. 4.
1. Isoniazids
Isoniazid (also known as isonicotinic acid hydrazide) is used to treat tuberculosis. Due to the nature of the isoniazid structure, only the hydrazine moiety can be altered. Isoniazid mainly targets the mycobacterial cell wall; however, since toxicity to hepatic cells and drug resistance development have been reported, they are usually used in combination with other antituberculosis drugs (Gegia et al., 2017). To address these shortcomings, the aniline moiety was halogenated (Pflégr et al., 2021). For instance, the 4-triflouromethoxy- and 4-iodophenyl isoniazid (Fig. 11, A and B) displayed inhibitory effects on multidrug-resistant M. tuberculosis strains (8–64 μM), whereas isoniazid was inactive. As expected, the sensitive M. tuberculosis strain was more susceptible to the derivatives and isoniazid at lower MICs of 0.03–0.25 μM and 0.5 μM, respectively (Pflégr et al., 2021). Mutation of inhA genes encoding enoyl-acyl carrier protein reductase involved in mycolic acid synthesis was reported to cause isoniazid resistance in M. tuberculosis (Palomino and Martin, 2014). Hence, the inhibition of MDR M. tuberculosis by halogenated compounds (Fig. 11, A and B) compared with the inactivity of isoniazid suggests the capacity to counter mutated enoyl-acyl carrier protein enzyme (InhA) and overcome resistance via improved permeability and drug uptake. Their application as adjuvants with antibiotics is recommended to further potentiate inhibitory activity. Interestingly, the enhancement of lipophilicity and mycobacterial cell wall penetration as improved by the presence of trifluoromethoxy (-OCF3) and iodine (I) groups at the C4 of the aniline ring of 11A-B supported the effect (Fig. 11U) (Pflégr et al., 2021).
Furthermore, unlike the hepatotoxic effect reported for isoniazid (Gegia et al., 2017), halogenated isoniazids, 11A-B showed no cytotoxic or cytostatic activity in HepG2 cells (a human hepatocellular cell line) at >50 μM, in addition to high selectivity index toward Mycobacterium spp. (Pflégr et al., 2021). Also, INH-c (brominated isonicotinoylhydrazone derivative) had no to mild toxicity and good biocompatibility with fibroblasts at 1.75 mg/mL, and its LD50 revealed that it is 10 times less toxic than the parent isoniazid (Fig. 11U). It also showed the lowest signs of hepatic parenchyma alteration and vascular congestion compared with isoniazid (Dragostin et al., 2019), thus indicating halogenation as a promising alternative for alleviating hepatic toxicity typical of antimycobacterial drugs.
2. Azo Aspirins
This drug is otherwise known as acetylsalicylic acid and is a representative nonsteroidal anti-inflammatory drug. Its precursor aspirin i.e. salicylic acid is present in willow tree bark. Aspirin is weakly acidic and has been used to treat fever and pain and as an anti-inflammatory drug but possesses weak antibacterial activity (Nordin et al., 2017). Halogen-containing derivatives were synthesized to enhance their antibacterial activity and interactions with targets. Halogenated azo aspirin (Fig. 11C) displayed considerable antibacterial activity better than aspirin against E. coli and S. aureus (Ngaini and Mortadza, 2019). Iodine substitution in addition to hydroxyl and carboxylic groups increased lipophilicity and subsequent easy cell membrane penetration to enhance the antibacterial activity (Ngaini and Mortadza, 2019). Compound 11C was predicted to bind effectively with phosphatidylinositol-specific phospholipase C of S. aureus and phospholipid-binding protein MIaC of E. coli. A halogen bond interaction was observed with the active site of E. coli MIaC through iodine binding with –C=O and–NH of Gln48 (Ngaini and Mortadza, 2019). Although other aspirin derivatives with weak antibacterial activities have been reported (Nordin et al., 2017), this review shows that halogenation is suitable to potentiate the antimicrobial effects of azo aspirin.
3. Haloperidols
Haloperidol is a butylbenzoic antipsychotic agent cleared by the liver via glucuronidation and CYP3A4-mediated oxidation. Haloperidol is tightly bound in human plasma and extensively metabolized by the liver (Osacka et al., 2022). Haloperidol showed promising antifungal properties against fluconazole-resistant C. albicans. Specifically, fluorinated derivative 11D at 32 μg/mL inhibited the invasive fungi better than fluconazole (>64 μg/mL). Fluorine substitution on the C4 position of the terminal benzene ring was reported to be crucial for antifungal activity and may have improved the metabolic stability, toxicity, and good intrinsic clearance rate compared with haloperidol (Ji et al., 2020). Haloperidol derivative 11D (4-fluorophenyl haloperidol) exhibited mechanisms targeting the growth and virulence factors of pathogens. It also reduced the production of biofilms, melanin, urease, and capsular polysaccharides, which are responsible for cell wall integrity, host immunity resistance, host survival and morbidity, and the pathogenicity of Cryptococcus neoformans (Revankar and Sutton, 2010; O’Meara and Alspaugh, 2012; Ji et al., 2020).
Coadministration of compound 11D with fluconazole further reduced MIC to 4 μg/mL and minimized resistance development by inhibiting efflux pump (MDR1) gene expression and interfering with ergosterol biosynthesis in fluconazole-resistant C. albicans (Ji et al., 2020). Furthermore, bromoperidol derivatives in combination with posaconazole and voriconazole synergistically inhibited azole-resistant C. albicans, which otherwise would have been resistant to either drug (Holbrook et al., 2017). These observations suggest that the application of haloperidol and its halogenated derivatives as adjuvants may reverse antifungal drug resistance by targeting a transporter/efflux pump (MDR1) as corroborated by other studies (Iwaki et al., 2006; Iatta et al., 2017). Haloperidol is associated with severe extrapyramidal side effects because of its strong affinity for the dopamine D2 receptor (Sikazwe et al., 2003). However, 11D exhibited less affinity for this receptor, which suggests a low risk of haloperidol-related side effects (Ji et al., 2020).
4. Pyrrolopyrimidines
The pyrrolopyrimidine scaffold has proven to be versatile in the medicinal chemistry field and has been used in many FDA-approved medications (Tian et al., 2019). The pyrrolopyrimidines are structurally similar to purines and can bind to proteins and antimetabolites during the metabolism of nucleic acids (Adel et al., 2018). Compounds containing this scaffold have antineoplastic, antibiotic, and antiviral effects and are known to bind to similar targets in bacteria but have narrow activity profiles (Zanello and Corsini, 2017; Olsen et al., 2022). Different halogenated pyrimidines were synthesized to target antimicrobial resistance. The halogenated pyrrolopyrimidines 11E and 11F inhibited S. aureus at 8 μg/mL, which was eightfold better than parent pyrrolopyrimidine (64 µg/L) but lacked activity against Gram-negative E. coli (Olsen et al., 2022). This may have been due to the outer membrane–mediated protection of Gram-negative E. coli. Interestingly, the combinatory effects of 11E or 11F with the antimicrobial peptide betatide drastically lowered MICs against S. aureus (1 to 2 mg/L), suggesting their potential use as adjuvants (Olsen et al., 2022). The presence of bromine or iodine at the C4 of the benzylamine group and a -OH group at the -meta or -para position on the 6-aryl unit (Fig. 11G) were essential for increased potency toward S. aureus. The bromophenyl derivative 11E moderately inhibited thymidylate monophosphate kinase, which is responsible for essential nucleoside synthesis and a possible target in S. aureus. However, 11E showed very low activity against human kinases, suggesting the possibility of low in vivo activity (Olsen et al., 2022). Since pyrimidine is an essential component of nucleic acid, we envisage that halogenated pyrrolopyrimidines would have better in vivo compatibilities and thus be leveraged for application in drug development.
5. Phenothiazines
Phenothiazines are tricyclic compounds and represent a large group of antipsychotic medications. Some phenothiazine derivatives, including thioridazine, were reported to inhibit extensively drug-resistant M. tuberculosis, increased MRSA susceptibility to β-lactam drugs, and controlled P. aeruginosa, K. pneumonia, and A. baumannii persister cells (Thorsing et al., 2013; Mohiuddin et al., 2022). However, the difficulty of separating their central effects and other pharmacological activities had limited their applications (Nizi et al., 2020). The versatility of the phenothiazine scaffold makes it an important lead structure, and derivatives with specific biological targets were discovered after halogenation. Compounds 11H-I exhibited excellent antimycobacterial activities better than standard thioridazine or chlorpromazine against M. tuberculosis. Importantly, 11H and 11I inhibited nonreplicating M. tuberculosis under hypoxic conditions, suggesting their potential use to prevent persistent infection (Nizi et al., 2020). The poly halogenation (3,7-dibromo and 1,3,7,9-tetrachloro-) of the phenothiazines enhanced biological activity. Additionally, phenothiazine chlorpromazine inhibited MDR-resistant S. enterica serovar Typhimurium and synergized with norfloxacin and ethidium bromide (Fig. 11U). It was reported to repress the expression of the efflux pump gene acrB in a manner that incapacitated the efflux pump to significantly export the drug, hence increasing the accumulation of chlorpromazine within the Salmonella cells (Bailey et al., 2008).
Phenothiazine derivatives exhibited different mechanisms for their antimicrobial actions. For example, 3,7-dibromo- (11H) and 1,3,7,9-tetrachloro- phenothiazines (11I) disrupted oxygen consumption and NADH oxidation, which are central to the survival of M. tuberculosis. It was inferred that poly-halogenated derivatives targeted type II NADH dehydrogenase (NDH-2) to exert their antitubercular activities. Of note, NDH-2 transfers NADH-derived electrons into the mycobacterial respiratory chain and thereby plays an essential role in ATP generation through oxidative phosphorylation in mycobacteria (Nizi et al., 2020). These outcomes suggest that halogenation may have conferred the scaffold with novel potential and mechanisms other than blocking D2 dopaminergic receptors, muscarinic M1 receptors, and histamine H1 receptors.
Compounds 11H and 11I were metabolically stable and showed synergism with rifampin but may be hepatotoxic only at high concentrations. Typically, phenothiazines are not used as anti-infective agents because of their effects on the central nervous system (Dougherty and Marraffa, 2014). However, halogenated phenothiazines showed lower affinities for central nervous system receptors and could probably alleviate side effects (Fig. 11U) (Nizi et al., 2020). Also, parent phenothiazine seems to produce photoallergic and phototoxic effects, which are usually triggered by exposure to UV radiation and result in eczematous eruption at the exposed sites (Kowalska et al., 2021). However, reports regarding the potential of halogen modulation to curb this anomaly are scarce; hence, further investigation is recommended.
6. Salicylanilides
The salicylanilide scaffold contains the amide of salicylic acid and aniline, which acts by restricting the two-component regulatory systems in bacteria, limiting nutrient uptake and structural damage to microbial cells (Paraskevopoulos et al., 2017). Although the phenolic group on the scaffold seems to confer antimicrobial activity, it is sometimes responsible for irritation and uncoupling activity. Hence, modification is required to temporarily block these activities and improve membrane penetration, bioavailability, and bioactivity (Ienașcu et al., 2022). Halogenated salicylanilide derivatives were reported as anthelmintic agents, disinfectants, and antimicrobial agents against resistant pathogens. Niclosamide (Fig. 11J), as an FDA-approved anthelmintic drug and oxyclozanide (Fig. 11K), effectively inhibited daptomycin-, linezolid-, methicillin-, and vancomycin-resistant S. aureus by disrupting the cell envelope (Rajamuthiah et al., 2015). Other derivatives (11L-M) effectively controlled multidrug and extensively drug-resistant Mycobacterium (0.5 μM) better than standard isoniazid (16 μM). The monohalogenation at C4 of the salicylic ring (Fig. 11, L and M) and the introduction of -CF3 at C4 of the aniline ring improved antimycobacterial activities (Paraskevopoulos et al., 2017).
Furthermore, a panel of MRSA and VRSA were inhibited at very low MICs (0.03–0.50 μg/mL) by trifluoromethylphenyl and 4-chlorophenyl analogs (Fig. 11, N–P). Particularly, 4’-bromo-3′-trifluoromethylphenyl congener (11N) at 0.03–0.06 μg/mL concentration was 32- to 1024-fold better than methicillin (32–64 μg/mL) and vancomycin (1 to 2 μg/mL) against MDR S. aureus and VRSA (Lal et al., 2021). Relative to the sensitive S. aureus (0.25–0.50 μg/mL), 11N was more effective, implying the capacity to break resistance and exert strong antibacterial activity. As revealed by the SAR study (Fig. 11Q), the activity of compound 11N-P against MDR strains was potentiated by the presence of 3′-trifluoromethyl and 4’-bromo on the salicylanilide moiety (Lal et al., 2021). Also, halogenated salicylanilides (11R-S) effectively controlled different strains of colistin-resistant K. pneumonia and A. baumannii by 128- to 2048-fold at 5 μM (Nemeth et al., 2020). The replacement of the 3,5-bistrifluoromethyl groups with dibromo substituents and the addition of fluorine enhanced the activity of 11R and 11S, respectively, against the colistin resistance (Nemeth et al., 2020).
However, niclosamide (11J) and oxyclozanide (11K) lacked activity against Gram-negative pathogens like K. pneumoniae, A. baumannii, P. aeruginosa, and E. aerogenes (Rajamuthiah et al., 2015). It seems that the presence of bulky halogens (Br, CF3) on the salicylanilide scaffold (Fig. 11, R and S) could facilitate membrane penetration and uptake as compared with -Cl on niclosamide and oxyclozanide (Fig. 11, J and K). Perhaps, the bromine and trifluoromethyl groups were able to create better dipole moment and interact with polar amino acids in the constriction zone of the outer membrane proteins of Gram-negative bacteria. Typically, the biological activities of salicylanilides are influenced by their hydrophobicities (Ienașcu et al., 2022), which suggests that different halogen additions might have modulated this property.
Halogenated salicylanilides could also potentiate activity against microbial virulence factors capable of increasing drug resistance (Fig. 11U). For instance, compound 11N disrupted preformed S. aureus biofilms better than vancomycin and levofloxacin at the same doses (Lal et al., 2021). Similarly, niclosamide (Fig. 11J) at 0.5–5 μM attenuated the filamentation, hyphae, and biofilm formation of sensitive and azole-resistant C. albicans (Garcia et al., 2018). Niclosamide downregulated the virulence genes related to the cell wall, filamentation, ergosterol, sphingolipids, and phosphoglycerides in Candida spp.; inactivated a nucleotide release factor Mge1; and disrupted mitochondrial membrane potential, thereby affecting mitochondrial functionality (Garcia et al., 2018). The excellent antivirulence activity by 11J was attributed to the stability of the C-Cl bond, which enabled effective binding to virulence targets. Notably, niclosamide (Fig. 11J) is currently undergoing a phase 2 clinical trial for the treatment of S. aureus infections (Butler and Paterson, 2020).
Toxicity profiling of various halogenated salicylanilides demonstrated they are safe for clinical applications (Fig. 11U). Niclosamide and oxyclozanide protected host cells against fungal invasion, significantly reduced damage to HT-29 enterocytes, attenuated damage to intestinal epithelial cells, and were nontoxic to sheep erythrocytes (Rajamuthiah et al., 2015; Garcia et al., 2018). Also, niclosamide rescued C. elegans from MRSA infection and prolonged worm survival but was toxic to cancer lines (HepG2 and HEK293). The anticancer effect of niclosamide and its ability to interfere with signaling pathways could explain this observation. (Ye et al., 2014; Rajamuthiah et al., 2015). Furthermore, analog 11R had no measurable toxicity against epithelial 4T1 cells up to 200 μM, whereas others (Fig. 11, N and O) showed better specificity toward bacterial cells than host cells and improved selectivity indices (Nemeth et al., 2020; Lal et al., 2021). Typically, halogenated salicylanilides share some pharmacological features, such as prolonged elimination half-life, high plasma binding, and limited metabolism (Swan, 1999). Information as to whether halogens influence these parameters is not available. However, insight was provided by the contribution of halogens to toxicity minimization, as was reported for the trifluoromethylphenyl derivative (11T), whereby the introduction of an additional halogen alleviated cytotoxicity (Paraskevopoulos et al., 2017). It does suggest that halogenation might potentiate the activity of salicylanilides against resistant pathogens, minimize toxicity, and provide a safe scaffold for in vivo administration.
G. Other Halogenated Bioactive Compounds
1. β-Nitrostyrene and Nitrovinylfurans
β-Nitrostyrene is an aromatic compound and nitroalkene isolated from plants and bacteria. 2-Nitro-4-((E)-2-nitrovinyl) phenol was isolated from Sonneratia acids Linn. F and an arctic sea ice bacterium, Salegentibacter sp. The bioactivities and ease of synthesis of β-nitrostyrene generated renewed interest in their derivatives (Shafi et al., 2016; Tsai et al., 2017). Also, nitrovinylfuran has biological activities that have been harnessed in clinical and industrial settings. Dermofural ointment is used to treat nail/human skin infections, and Furvinol is used in veterinary medicine (Allas et al., 2016). Brominated nitrovinylfuran could exert broad-spectrum antibacterial activity against sensitive and resistant bacteria (Fig. 12U). For example, Furvina (12A) and its conversion product bromonitromethane (12B) exhibited the same MICs of 4 and 2 µg/mL against both sensitive and resistant S. aureus, respectively, which was comparable to fosfomycin (4 µg/mL) but superior to cnicin (>32 µg/mL) (Scholz et al., 2013). The broad potency observed was attributed to the presence of a bromonitromethyl group in the scaffold (Scholz et al., 2013).

Structures of other halogenated bioactive compounds. β-Nitrostyrene and nitrovinylfuran (A–G), aminothiopines (H–L), isophthalonitrile (M), and β-amino amides and alboflavusin (N–T) and effects of halogenation on β-nitrostyrene, aminothiopenes, and β-amino amides and alboflavusins (U). Full chemical names are listed in the Supplemental Fig. 5.
Similarly, halogenated nitrostyrene (Fig. 12, C–E) exhibited antibacterial and antifungal activities against E. coli, B. subtilis, S. aureus, E. faecalis, and C. albicans (Lo et al., 2012; Cornell et al., 2014). Bromine addition to β-nitrostyrenes (Fig. 12C) at the C4 position of the aromatic ring and the β-position of the alkene side chain of nitropropenyl arenes resulted in improved activities (Cornell et al., 2014). Inhibition of E. coli by the fluorinated analogs (12D-E) was due to the high electronegativity and hydrophilic properties induced by fluorine, which alters the ability to penetrate Gram-negative bacteria cell walls (Lo et al., 2012). A comparison of the inactivity of the fluorinated derivative 12F to the potency of a similar compound (12D) against E. coli demonstrated the importance of the scaffold to which halogens are attached. Specifically, direct attachment of fluorine to β-methyl-β-nitrostyrene favored efficacy more than methylenedioxy attachment (Lo et al., 2012; Cornell et al., 2014). This corroborates a report that the ability of fluorine to alter physicochemical properties like lipophilicity (logP) depends on the scaffold and proximal functionality (Al-Harthy et al., 2020). Regarding antivirulence potentials, Furvina inhibited the 3-oxo-C12-homoserine lactone-dependent quorum-sensing system of P. aeruginosa and reduced biofilm formation and the production of quorum sensing–dependent virulence factors (Borges et al., 2017).
MOA studies revealed that the nitrovinylfuran derivatives (12A-B) irreversibly inhibited MurA (an enzyme involved in peptidoglycan biosynthesis) and E. coli MetAP. They also exerted pleiotropic effects on numerous proteins by interfering with cysteine residues, like the standard cysteine residue inhibitor Fosfomycin. Notably, like antibiotics, Furviva targeted actively growing E. coli cells rather than nongrowing cells (Allas et al., 2016). Additionally, it disrupted translation initiation by interfering with the P-site of the 30S subunit (Fabbretti et al., 2012; Oliveira et al., 2021).
Furvina (Fig. 12A), however, displayed in vitro instability as it transformed rapidly into different intermediates, attributing its antimicrobial effects to the combinatory effects of its immediate reactivity and those of its reaction products (Allas et al., 2016). The phenomenon was improved in acidic conditions but may impair target binding or cellular uptake (Scholz et al., 2013). Furvina and its conversion products showed long-term toxic effects against mammalian cell lines and were rather suggested for use in the design of cysteine residue inhibitors in protein targets as indicated by the irreversible inhibition of MurA cysteines and bacterial methionine aminopeptidase (Scholz et al., 2013; Allas et al., 2016). In addition, 4-chloro-β-nitrostyrene (12G) was reported to exhibit vasorelaxant effects courtesy of the electrophilic capacity of the substituted chloro-nitrostyrene (Sousa‐Brito et al., 2021). Overall, the halogenation of β-nitrostyrene and nitrovinylfuran improved the antimicrobial and antivirulence properties, but further studies on their pharmacokinetics are recommended.
2. 2-Aminothiopenes
2-Aminothiophenes are a group of heterocyclic compounds with five rings and exhibit diverse biological functions, including antifungal, antiviral, antitubercular, and anticancer effects and allosteric agents. The aminothiophene scaffold can be found in approved drugs such as Zyprexa and drug candidates (Bozorov et al., 2017). Despite the versatility of this scaffold, low water solubility remains a challenge. As a result, new derivatives with lower octanol-water partition coefficients, improved water solubilities, antimicrobial activities, and pharmacokinetic profiles were synthesized (Luna et al., 2021). The halogenated 2-aminothiophene 12H at 1 μg/mL inhibited all dermatophyte fungal strains, including Trichophyton tonsurans and T. rubrum, and other congeners (Fig. 12, I and J) inhibited them in a manner comparable to fluconazole (Luna et al., 2021). The position and size of the halogen substituent at the benzylidene moiety determined antifungal activity, and the presence of fluorine, chlorine, or bromine at the C4 position increased derivative potencies (Fig. 12U). Halogenation type had a differential influence on the antimicrobial activities of the 2-aminothiopenes. The presence of bromine (Fig. 12J) caused sensitivity in tested fungal strains, whereas the dichloro- substitution (Fig. 12I) at the -ortho position increased the spectrum of activity (Luna et al., 2021). Furthermore, two halogenated 2-aminothiopene derivatives (Fig. 12, K and L) reversed ciprofloxacin and erythromycin resistance in S. aureus by acting as NorA and MrsA efflux pump inhibitors independent of growth inhibition. The presence of the 2-aminotrifluoroacetyl group was responsible for the increased potentiation of ciprofloxacin and erythromycin to overcome S. aureus resistance (Fig. 12U) (da Cruz et al., 2020).
Preclinical toxicity evaluations of halogenated aminothiophenes (Fig. 12, I and J) showed that they were not toxic to three nontumor cell lines (VERO, MRC-05, and 3T3) at 1–100 μM or to Tetrahymena pyriformis, a protozoan employed to assess in vivo toxicity. Similarly, the 2-aminotrifluoroacetyl derivative (Fig. 12K) was nontoxic to the murine macrophage cells, whereas (Fig. 12L) showed potential toxic effects, and the cost-benefit of its application should be considered (da Cruz et al., 2020). In addition, ADME analysis revealed that halogenated aminothiophenes (Fig. 12, H and J) had excellent oral bioavailabilities and complied with Lipinski's rule of five and Veber's parameters. In addition, they were negative for hERG inhibition, had high gastrointestinal absorptions, were nonhepatotoxic, and had no skin sensitization effects but had limited brain-blood barrier permeabilities and low skin permeabilities (Luna et al., 2021). This could, however, impair distribution and topical application, respectively. We consider halogenated aminothiophenes promising oral drug candidates that could act as antibiotic adjuvants for therapy against drug-resistant infection. However, further structural modulation and study are required to improve pharmacokinetic properties and unveil the antimicrobial MOAs.
3. Isophthalonitrile
Isophthalonitrile derivatives remain one of the most essential organic cyanides and halogenated isosteres, which are usually referred to as virtual analog halogens. These derivatives have antiviral, anti-inflammatory, and insecticidal activities, and their polyhalogenated derivatives, especially polyfluoro-isophthalonitrile derivatives, also possess anticancer, anti-inflammatory, and insecticidal activities and other applications of pharmaceutical interest (Senapati et al., 2017). Halogenated isophthalonitrile derivatives showed excellent antimicrobial activities against clinically important pathogens, including S. aureus, B. cereus, and C. albicans. Specifically, these derivatives (Fig. 12M) inhibited the pathogens at 0.4–0.7 µg/mL, which compared favorably with norfloxacin and fluconazole in the range of 0.5–3 µg/mL (Huang et al., 2013). It was reported that fluorine substitutions at the C2 and C4 positions and benzyl amino substitution at the C6 position contributed to antibacterial and antifungal activities. The group speculated that the cell wall/membrane was probably targeted by halogenated isophthalonitrile for antimicrobial activity (Huang et al., 2013). The potency shown by halogenated isophthalonitriles further confirms the indispensable role of halogens in medicinal scaffolds. However, available data on fluorinated and chlorinated isophthalonitriles is limited, and thus further studies are required to fill gaps in addition to investigating the effects of other halogens like bromine and iodine.
β-Amino Amides and Alboflavusins
β-Amino acids are essential monomers present in natural products, including Taxol and peptidomimetics (Paulsen et al., 2016), and the synthesis of peptidomimetics like α,α -disubstituted β-amino amides, which mimic the activities of larger antimicrobial peptides against bacterial cell membranes, has received great attention. Structurally, β-amino acids contain two cationic moieties and a pair of bulky lipophilic groups, that are resistant to α-chymotrypsin degradation, and maintain their stability in aqueous solutions, even at pH 7.4 (Paulsen et al., 2019). β-Amino acids were reported to possess excellent antibacterial and antibiofilm activity against S. aureus through membrane disruption (Ausbacher et al., 2014). Despite these features, they are susceptible to phase I oxidation, and, importantly, their electron-rich aromatic sidechains are easily oxidized. Halogenation of β-amino amides was employed as a tool to improve activity and control metabolic instability (Fig. 12U) (Paulsen et al., 2019). Halogenated disubstituted β-amino amides exhibited antibacterial activities against a panel of bacterial strains, including 30 MDR clinical isolates (Paulsen et al., 2019). In particular, halogenated diamine and diguanidine derivatives (12N-Q) containing 3,5-Br-Ph and 3,5-CF3-Ph substituents respectively inhibited multidrug-resistant S. aureus, E. faecium, and K. pneumoniae at low concentrations (MIC: 4–8 µg/mL) but not P. aeruginosa (MIC: 32 µg/mL) (Paulsen et al., 2019), which may be related to the intrinsic factors of the pathogens. Also, halogenated di-guanidine derivatives (12P-Q) exhibited similar activity against S. aureus, E. faecium, E. coli, P. aeruginosa, K. pneumoniae, and colistin-resistant A. baumannii (MIC: 2–16 µg/mL). Of note, diguanylated analog 3,5-Br-Ph (Fig. 12P) demonstrated effects comparable to oxytetracycline (the positive control) against reference strains (Paulsen et al., 2019). Notably, SAR analysis, as shown in Fig. 12Q, revealed that halogenated lipophilic groups (3,5-Br-Ph and 3,5-CF3-Ph) contributed to the antibacterial effects and low toxicities of the di-guanidine derivatives (Paulsen et al., 2019).
Alboflavusin is a naturally occurring, novel, cyclic hexapeptide antibiotic containing a chlorine atom. It has been isolated from marine S. alboflavus and shown to possess promising antitumor and antibacterial activities and apoptosis induction (Crowe et al., 2021; Li et al., 2021a). Brominated alboflavusins 1 and 2 (Fig. 12, S and T) isolated from S. alboflavus sp. 313 exhibited antibacterial activities against various MRSA strains. Particularly, alboflavusin 2 (Fig. 12T) at an MIC of 3.1–6.2 µM was 4–8 times superior to the parent alboflavusin (12.5–25.0 µM) against four MRSA strains (Li et al., 2021a). Compared with the sensitive strain (6.2 µM), the halogenated congener (12T) showed a comparable to superior effect (3.1–6.2 µM), indicating the potential to overcome resistance and exert equipotent activity. According to the SAR studies, the presence of bromine improved their anti-MRSA activities (Li et al., 2021a).
Regarding toxicity, the halogenated β-amino amides and alboflavusins showed none to mild toxicity and improved metabolic stability. β-Amino amide derivative (Fig. 12P) had no major in vitro toxicity on human lung fibroblasts (MRC-5) or human hepatocyte carcinoma cells (HepG2), and the compounds (Fig. 12, P and Q) were nonhemolytic to red blood cells at an EC50 of >200 µg/mL in addition to having high selectivity indices. Compared with the parent compound, the lipophilic sidechains 3,5-Br-Ph of the diguanylated analog (Fig. 12O) showed resistance to phase I oxidation, suggesting high in vivo metabolic stability (Paulsen et al., 2019). Taken together, the halogenation of β-amino amides and alboflavusins conferred improved antimicrobial efficacy and protected against metabolic degradation. However, target molecules and mechanisms for the various antibacterial effects displayed need further investigation.
IV. Halogenated Polymeric Materials
Biopolymers such as chitosan are nontoxic, biocompatible, bioactive, and biodegradable (Zhang et al., 2018b). Also, the antibacterial polymers, especially the cationic ones, including poly(ethyleneimine), poly(l-lysine), and poly(2-dimethyl(aminoethyl) methacrylate, were reported as promising alternatives to commercial antibiotics (Kyzioł et al., 2020; Qiu et al., 2020). Despite the aforementioned, the majority of biopolymers do not possess inherent antimicrobial activity and therefore require chemical modification (Ganie et al., 2021). This section reviews various halogen-functionalized polymers and the roles played by halogens in their antimicrobial activities (Fig. 13).

Structures of halogenated polymeric materials. Halogenated cellulose grafted hyperbranched polyethyleneimine (A), N,N,N-trimethyl chitosan trifluoroacetate (B), N,N,N-trimethyl chitosan trichloroacetate (C), N,N,N-trimethyl chitosan dichloroacetate (D) and N,N,N-trimethyl chitosan chloroacetate (E), chitosan bearing 2-fluoro-phenylimino)-acetaldehyde (F), 2-chloro-phenylimino)-acetaldehyde (G), 2-bromo-phenylimino)-acetaldehyde (H), fluorinated fabric polymer (I), and polyurethane-AHM-chlorine (J).
The cellulose-grafted, hyperbranched, chlorine-containing polymer shown in Fig. 13A inhibited E. coli, Proteus microbilis, P. aeruginosa, P. vulgaris, E. aerogenes, and S. typhimurium, and this was attributed to the hydrophobicity caused by chlorine substitution (Demircan and Zhang, 2017). Similarly, the halogenated chitosan analogs (Fig. 13, B–E) displayed excellent antifungal activities against Fusarium oxysporum and Phomopsis asparagi, which was attributed to the electron withdrawing of halogen groups and resulting increases in membrane permeability and hydrophobicity. Furthermore, observed increases in antimicrobial activity were proportional to degrees of substitution and halogen electronegativity (Zhang et al., 2018a). In addition, the halogenated Schiff bases of chitosan (Fig. 13, F and H) inhibited Botrytis cinerea by 96.7%, 96.0%, and 95.8%, respectively, compared with 23.8% inhibition by chitosan. Regarding the mechanism, halogen atoms established strong electrostatic interactions with negatively charged fungal cells, causing the leakage of intracellular materials and death (Wei et al., 2021). Moreover, fabric antiadhesive polymer modified with fluorine (Fig. 13I) reduced the presence of S. aureus and E. coli on fabric surfaces due to increased penetration into the lipid domains of bacterial membranes (Lin et al., 2018).
N-halamines constitute another category of halogen-containing polymers. They exist largely as imides, amines, or amides and covalently interact with halogens to produce oxidative bactericidal halogen ions (Demir et al., 2017). The synthesis, application, and antibacterial roles of N-halamines have been reviewed (Dong et al., 2017; Wang et al., 2020). Since new N-halamines have been described, we provide an update and describe the specific roles played by halogens. N-halamine conjugate based on polydopamine was formulated, and the chlorinated polydopamine film produced reduced the viability of E. coli by ∼99.5% after 3 hour of treatment and its surface adhesion by 45% (Nazi et al., 2020). Similarly, a polyurethane functionalized with an N-halamine precursor [2-amino-5-(2-hydroxyethyl)-6-methyl pyrimidine-4-one] (AHM) produced a conjugated PU-AHM-Cl (Fig. 13J) film that showed marked antibacterial activity against S. aureus and E. coli with a 100% reduction in viability after 5 minutes of contact, whereas the parent polyurethane had no effect (Peng et al., 2021). Also, cotton fabric doped with N-halamine–based dopamine/polyethyleneimine (PDA/PEI-Cotton-Cl) containing 0.182% of active chlorine reduced the adhesiveness of S. aureus and E. coli to fabrics by 5.0 and 7.2 Log, respectively (Wan et al., 2022). The MOAs of N-halamine–based polymers are similar and involve the chlorination of N-H bonds to form N-Cl bonds, and the bonds undergo hydrolysis to produce oxidative chloride ions, which disrupt bacterial membranes (Wan et al., 2022).
Antimicrobial polymers are less susceptible to bacterial resistance, and their antibacterial activities are due to cell membrane disruption (Qiu et al., 2020). In the current review, halogenation either restored or further enhanced the penetration of polymers into the cell barriers to display notable antimicrobial activities. A deeper understanding of the effects of halogenated polymers on resistant pathogens is required before they can be considered viable alternatives to traditional antibiotics.
V. Halogenated Biomolecules
Halogen sources are not common in ecosystems, and halogen bonds were not recognized in biological systems during the early part of the 20th century. However, due to the discovery that halogens bond to peptides, lipids, carbohydrates, and other biomolecules (Ho, 2015), halogens are now considered important elements in biological systems.
A. Peptides
Antimicrobial peptides (AMPs) are components of the innate immune defense responses of many organisms. Their net positive charges enable them to bind negatively charged bacterial components and disrupt their activities. The structural makeup of AMPs, like the amino acid substitutions, critically determines their functionalities (Ageitos et al., 2017). For example, proline-rich AMPs penetrate bacterial cytoplasm rather than destroy membranes (Mattiuzzo et al., 2007), and glycine-rich AMPs induce phagocyte-mediated bactericidal mechanisms that differ from the conventional MOAs of AMPs (Huan et al., 2020). Notably, amino acid halogenation enhances antimicrobial potency; for example, the replacement of phenylalanine with a halogenated analog profoundly increased the antibiofilm and antibacterial effect of jelleine-1 (Jia et al., 2019). Halogenation can also be used to specifically target microbes as demonstrated by o-fluorine substitution in phenylalanine residues, which promoted the antimicrobial effect of temporin L on E. coli but had no effect on S. aureus or P. aeruginosa (Setty et al., 2017).
Halogenation of amino acids alters the structural and physiochemical properties of peptide scaffolds influencing peptide membrane permeabilization and catabolic stability (Parisini et al., 2011). Fluorination impacts the proteolytic stabilities of peptides and bulky fluorinated residues, such as hexafluoroleucine or trifluoroisoleucine, and fluorinated amino acids shield polypeptides from proteolytic enzymes (Berger et al., 2017; Huhmann and Koksch, 2018). Halogenated amino acids may be natural or synthetic. Most natural antimicrobial amino acids are bound by sp2 carbons as the sp2 C-X bond is less likely to react with biological nucleophiles like water and amino and hydroxyl groups. For this reason, most of the halogenated amino acids in nature are aromatic, like tyrosine, histidine, and tryptophan (Table 1, A–N) (Peng et al., 2005). Halogenation of the majority of the amino acids occurs post-translationally, and thus effects are only observed in peptide form (Bittner et al., 2007). Although aromaticity favors halogenation, nonessential and aliphatic amino acids with antimicrobial activities have also been isolated from natural sources (Table 1, O–X). Halogenated amino acids are also well represented among nonribosomal peptides and are usually synthesized by one or more specialized nonribosomal peptide synthases (Moravej et al., 2018). A deeper understanding of many halogenated amino acids in nonribosomal peptides and their peptidomimetic characteristics were provided in a previous review (Mardirossian et al., 2021).
Halogenated aromatic, nonessential, and aliphatic amino acids and their sources
Halogenated antimicrobial peptides can be classified into two categories, namely ribosomally synthesized naturally halogenated AMPs and synthetic halogenated AMPs. Halogenation of ribosomally synthesized peptides occurs post-translationally. In most cases, bromine substitution in tryptophan occurs at position 6 of the tryptophan indole ring, as observed in styelin D, hedistin, strongylocin, and centrocins, and in hagfish AMPs isolated from marine sources (Taylor et al., 2000; Uzzell et al., 2003; Tasiemski et al., 2007; Li et al., 2008, 2010; Maffioli et al., 2014; Cruz et al., 2015). The role of bromine substitution on AMP activity has not been determined, though bromine substitution was reported to protect peptides from proteases (Uzzell et al., 2003).
Synthetic halogenated AMPs have been produced to improve antimicrobial potency and modify pharmacokinetics. Fluorination of maganin and melittin enhanced antimicrobial activity and proteolytic resistance. Moreover, halogenated analogs of jelleine-1 displayed an eightfold increase in in vitro activity, and halogenation of tripticin and its amidated analog tritrp1 also increased antimicrobial activities (Lawyer et al., 1996; Gottler et al., 2008; Jia et al., 2019; Arias et al., 2020). Peptidomimetics are synthetic peptide analogs resistant to proteolytic degradation. Several halogenated peptidomimetic libraries have been synthesized, such as halogen battacin derivatives with fluorinated aromatic L-serine substitutions, which increased broad-spectrum activity against several clinically relevant bacteria like P. aeruginosa and Campylobacter jejuni, and a 36-peptoid oligomer library, in which halogenated N-substituted glycine exhibited a multifold increase in antimicrobial activity against S. aureus, E. coli, and P. aeruginosa (Krenk et al., 2015; Molchanova et al., 2020; Glossop et al., 2021).
The antimicrobial activity of halogenated peptides depends on several factors, such as the type and aromaticity of the amino acid substituted, the ability to form halogen bonds with biological systems, and the ease of peptide self-assembly (Molchanova et al., 2020). Protein and peptide halogenation have been used to synthesize defensive compounds in organisms of various ecosystems (Pizzi et al., 2020). A similar approach to the synthesis of AMPs might be fruitful, though the applicability of halogenation in synthetic biology remains largely unexplored due to a lack of knowledge of the impacts of steric and electronic factors. Nonetheless, leveraging halogenation in synthetic biology for the synthesis of peptides would appear judicious as halogenation is a simple modification that induces marked differences in peptide supramolecular behavior. The limited number of reports on the activities of halogenated AMPs against drug-resistant pathogens warrants future studies on their use as alternative therapeutic strategy to overcome drug resistance.
B. Carbohydrates
Halogenated carbohydrates were mentioned earlier in the section on major classes of aminoglycoside, macrolide, and glycopeptide antibiotics (Halogenated Antimicrobial Classes). Here, we discuss other halogenated carbohydrates and polysaccharides in detail. A carbohydrate-based cellulose nanopaper impregnated with chitosan and halogenated, demonstrated good antibacterial properties against S. aureus and E. coli due to the conversion of chitosan amino groups to bactericidal halamines (Fig. 14A) (Du et al., 2021). Furthermore, halogenated methyl β-D-galactopyranoside derivatives were significantly more potent than their nonhalogenated counterparts against B. subtilis and E. coli and showed potential against SARS-CoV-2 (Fig. 14B) (Amin et al., 2022). In another study, yeast and aerobic microbe invasion and accumulation on bamboo shoots were prevented using a coating of chitosan and chlorine dioxide, which significantly extended the postharvest life of fresh bamboo (Yang et al., 2015). Halogenated monosaccharides derived by chlorobenzoylation of methyl α-D-mannopyranoside were reported to be antimicrobial against B. subtlis, S. aureus, E. coli, and A. niger (Yasmin et al., 2021). Others reported that chlorinated sugar-based acyclo C-nucleosides of 1, 2, 4-triazolo[4, 3-a]quinoxaline derivatives were antimicrobial against S. aureus, P. aeruginosa, E. coli, and C. albicans (Fig. 14C) (Ayoup et al., 2016) and that O-acrylamidomethyl-N-[(2-hydroxy-3-trimethylammonium) propyl] chitosan chloride (HTCC) derivatives were bactericidal against S. aureus and E. coli within 20 minutes of contact (Fig. 14D) (Lim and Hudson, 2004). Novel halogenated carbohydrate-based phosphoramidate derivatives were reported to be antibiotic adjuvants. In particular, a fluorinated glucose derivative demonstrated a remarkable 32-fold decrease in the MIC of ampicillin against methicillin-resistant S. aureus (Fig. 14, E and F) (Subratti et al., 2021). The MOAs of these halogenated carbohydrates are substantially unknown, but we believe that the appropriate positioning of halogen groups enables these derivatives to bind to or penetrate cell membranes and attack subcellular targets.

A cellulose nano paper-chitosan derivative (A), methyl β-D-galactopyranoside ester derivatives (B), derivatives of the acyclo C-nucleosides of 1, 2, 4-triazolo [4, 3-a] quinoxaline (C), O-acrylamidomethyl-HTCC derivative (D), and phosphoramidate adjuvants (E and F).
C. Lipids
Fatty acids (FAs) encompass a large part of the lipid class. Various natural halogenated antimicrobial FAs have been discovered, and others have been synthesized. Several natural C18 and C16 FAs isolated from Xestospongia sp. exhibited antimicrobial activity against S. aureus (Hirsh et al., 1987) (Fig. 15A), and a novel series of natural brominated FAs isolated from the sponge Petrosia volcano were antifungal against the pathogenic fungus Mortierella ramannianus (Fig. 15B) (Dembitsky and Srebnik, 2002). Furthermore, natural lactylates of chlorinated FAs (chlorosphaerolactylates A–D) isolated from the cyanobacterium Sphaerospermopsis sp. were inhibitory against S. aureus and C. parapsiloris (Gutiérrez-del-Río et al., 2020) (Fig. 15C), and halogenated FAs isolated from Asparagopsis taxiformis were antimycobacterial against M. pheli (El-Baroty et al., 2007).

Halogenated FAs isolated from Xestospongia sp. (A), natural brominated FAs isolated from the sponge P. volkano (B), natural lactylates of chlorinated FAs, chlorosphaerolactylates (C), and bromine or chlorine substituted vinyl halogenated FAs (D).
Halogenated FAs have also been synthesized to enhance antimicrobial activity. Chlorine or bromine-substituted vinyl halogenated FAs were antimicrobial against methicillin-resistant S. aureus (Fig. 15D) (Sanabria-Rios et al., 2022), and brominated derivatives of vinyl FAs were antileishmanial and inhibited the topoisomerase IB enzyme of Leishmania sp. due to halogen bond formation (Carballeira et al., 2019). In addition, fluorinated polyhydroxyalkanoate-derived FAs at high concentrations had bacteriostatic and mild bactericidal effects against a panel of bacteria (Snoch et al., 2019).
Halogenated FAs incorporated into complex lipopeptide structures were antimicrobial against pathogens. Several chlorinated FAs were isolated from lipopeptides present in marine sponges, for example (S)-4,4,4-trichloro-3-methylbutanoic acid from Lamellodysidea herbacea and (E)-6,6,6-trichloro-3-methoxy-5-methylhex-2-enoic acid and herbacic acid [(E)-6,6,6-trichloro-5-methylhex-2-enoic acid, 62)] from Dysidea (Dembitsky, 2017). (S)-7,7-Dichloro-3-hydroxy-2,2-dimethyloctanoic acid is incorporated into many lipopeptides, predominantly in cyanobacteria like Lyngby sp. and Moorea sp. (Dembitsky, 2022). Interestingly, Gram-positive bacteria are sensitive to FAs, whereas Gram-negative bacteria are usually resistant (Knapp and Melly, 1986). We observed a similar pattern for halogenated FAs; that is, they were predominantly active against Gram-positive bacteria. We suppose halogens enhance FA activity by forming halogen bonds in binding sites, which contrasts with the usual practice in medicinal chemistry wherein halogenation is used to accentuate permeability and warrants extensive studies on the effects of halogen and parent scaffold in permeability enhancement.
The type of halogen substituted may also influence microbial membrane permeability. Fluorine substitution is primarily used to enhance permeability, whereas most halogenated FAs of marine origin identified are brominated. Also, the effect of FA length on antimicrobial potency or cell permeability, the extent of halogen substitution, and the effects of halogen-containing FAs on drug-resistant microbes remain subjects for study. Recently, several studies reported that FAs exhibit antibiofilm and antivirulence activities at low concentrations (Kumar et al., 2020b; Kim et al., 2021; Lee et al., 2021, 2017), which suggests that halogenated FAs could be used to target drug-resistant biofilms and their pathogenesis.
VI. Effects of Halogenation and Factors Affecting the Antimicrobial Activities
A. Halogen Bonding, Pharmacokinetics, and Pharmacodynamics
Halogens are capable of halogen bonding, which makes them useful in medicinal chemistry and drug design (Cavallo et al., 2016). Halogen bonds are generally formed between halogen ligands and Lewis bases present in binding sites (Fig. 16). Halogens are capable of covalent interactions both as electrophiles and nucleophiles (Turunen and Erdélyi, 2020). These interactions were observed based on the electron density of the σ-hole region, in which the electrons of heavy elements like halogens have anisotropic distributions, and three lone pairs of electrons form a cylindrical cloud of higher electron density around the atom that generates a positive electrostatic potential, creating the σ-hole region (Fig. 16) (Zhu et al., 2019). This phenomenon is used to increase drug affinity in biological environments.

Formation of a halogen bond between the less-electron-dense σ-hole region and a Lewis base facilitated by formation of R-X bond. Formation of the covalent bond between the carbon and halogen leads to the depopulation of the valence pz orbital and creates an electropositive crown, whereas the pxy orbitals maintain their electrons to balance the negative charge of the halogen (Mendez et al., 2017).
Halogenation can enhance enzyme activity by destabilizing staggered conformations and stabilizing gauche conformations, thereby reducing activation energy barriers and improving catalysis (Szell et al., 2019). This behavior is useful in enzyme and peptide engineering as the substitution of hydrogen bonds in peptides with a halogen bond increases binding (Parisini et al., 2011). Halogen substitution also increases thermal stability and enzymatic activity at higher temperatures as observed after chlorotyrosine substitution in T4 lysozyme (Carlsson et al., 2018). Conversely, halogenation can be used to design ligands that inhibit enzymes, as was reported for E. coli gyrase, which was inhibited by compounds with a halogen at the p-position, and for the 5-hydroxytryptamine 2B receptor, which was inhibited by a halogenated doxepin ligand (Zhou et al., 2018b; Kolarič et al., 2021).
Halogenated ligands' interactions especially with proteins are highly dependent on the ligand scaffold (Fig. 17). A study was conducted in which different substitutions of iodine, bromine, chlorine, and trifluoromethyl groups on the chlorobenzene scaffold were evaluated. 3-Iodo-1-pyrrole had greater activity than iodobenzene, signifying the importance of the scaffold itself over substituted atoms for antimicrobial activity (El Kerdawy et al., 2012; Wilcken et al., 2013).

Factors affecting the antimicrobial activities of halogenated scaffolds.
Target site conformations also play critical roles in halogen interactions (Fig. 17) as observed by the inhibition of plasminogen activator by 4-bromobenzylamine, inactive 4-iodobenzylamine, and CF3-containing bisphenol AF, which acted as an agonist and antagonist against ERα and Erβ estrogen receptors, respectively (Jiang et al., 2018; Liu et al., 2020). Proteins also contain halogen bonding “hot spots,” which preferentially bind to ligand halogen atoms and increase binding affinity as demonstrated by the 5-hydroxytryptamine receptor, which possesses four halogen binding spots: that is, two each in extracellular and transmembrane regions (Kurczab et al., 2018).
Other stereochemical and bond formation properties of halogen atoms, such as distance between the halogen and the target Lewis base, σ-hole-angle, and spatial orientation of halogen atoms with respect to the Lewis base, also critically determine the strengths of halogen bond interactions (Fig. 17) (Wilcken et al., 2013).
Understanding pharmacokinetic and pharmacodynamic properties is important when manipulating scaffold behavior in medicinal chemistry. Inserting halogen atoms is a common practice in many hits-to-lead or lead-to-drug conversions. The incorporation of halogen atoms improves key pharmacokinetic properties like membrane, blood-brain barrier, and central nervous system permeability (Nunes et al., 2021). This increase in permeability is believed to be due to the presence of halogen bond donor sites in membrane lipid bilayers and the resulting enhancement of anion transportation by the “ion hopping” pathway. The use of specific transporters that act as halogen-bond acceptors is another strategy for delivering halogen-containing molecules to cells (Govindaraj et al., 2019). The influence of halogen bonding on the permeabilities of biological membrane systems was confirmed by interactions between halobenzene derivatives and phosphate or oxygen acceptors in the phospholipid bilayer (Nunes et al., 2021). Further exploration of halogen bonding in membrane systems is required to make further inroads into the pharmacokinetic manipulation of lead compounds or drug scaffolds. Data on parameters such as drug clearance and toxicity are also necessary when deciding on the fates of potential drug candidates.
The toxicity profiles of scaffolds largely depend on the scaffolds themselves, and thus singling out atoms on scaffolds for toxic effects is probably ill advised. For example, several brominated metabolites from natural sources are nontoxic, whereas brominated haloesters are toxic (Khan and Roy, 2021). However, high-throughput in silico scaffold screening enables the safety implications of certain functional groups to be assessed. A thorough point-by-point survey of potential drug candidates during lead-to-drug conversion was performed on halogenated structures, and only two candidates were rejected based on toxicity. This statistic cannot be applied to other functional groups, like nitro and sulfur groups, which are metabolized to harmful byproducts (Hernandes et al., 2010).
Although the carcinogenic and mutagenic profiles of halogenated antibiotics have been extensively tested, those of newer halogenated pharmacological scaffolds remain largely unexplored. Several organochlorines and halogenated hydrocarbons pose serious carcinogenic threats (Fiore et al., 2019; Muslem et al., 2020). Halocarbons, especially aliphatic haloalkanes, are capable of interacting with DNA, accumulating, and inducing mutagenicity (Della-Flora et al., 2023). However, over 3800 halogenated compounds of nonanthropogenic origin found in various ecosystems have no harmful effects (Gul et al., 2020). Thus, we suggest widespread testing be conducted to determine the carcinogenicities, toxicities, and mutagenicities of halogenated antimicrobial compounds using modern in silico toxicity-predicting tools.
Xenobiotic metabolism is another important pharmacologic parameter. The strength of carbon-halogen bonds raises questions regarding the clearance of organohalogens by the cytochrome P450 system (Hernandes et al., 2010; Wang et al., 2019). Biotransformation of halogenated drugs can produce harmful byproducts in some cases, such as the hepato- and nephrotoxicity induced by the glutathione-dependent bioactivation of a small subset of compounds (Anders, 2008). It has been reported that halocarbons can accumulate in rat adipose tissue, making it difficult for excretion (Kania-Korwel and Lehmler, 2016). However, halogenation of the substrates with aromatic derivatives of fluoro, chloro, and trifluoromethyl groups increased microsomal clearing (Sun et al., 2011). Also, strategic halogenation on the scaffold can increase binding affinity with the cytochrome P450 enzymes as observed in coumarin derivatives substituted with heavy halogens like chlorine, bromine and iodine which showed enhanced binding affinity with CYP2B enzymes (Liu et al., 2016). Therefore, the role of halogen in drug clearance is ambiguous, and an elimination study with diverse halogenated compounds is recommended to ascertain the role of halogen atoms in drug clearance.
The topics discussed in this section, viz. halogen bonding, scaffold dependence, target conformation, halogen bond distance, and angle, importantly determine the pharmacological characteristics of halogenated compounds. Although the upsides of halogen bond formation are far reaching, careful consideration should be given to the toxicological profiles of halogenated compounds to weed out potential offenders. In-depth analysis at the molecular level is required to further probe the effects of halogenated compounds, especially in contexts of toxicity and pharmacodynamics.
B. Factors Affecting the Antimicrobial Activities
An understanding of the properties likely to modulate the efficacies of halogenated agents is critical during drug development. Some studies have attributed the limited number of bromine- or iodine-containing drugs approved by the FDA to size, among other factors (Wilcken et al., 2013). Adequate awareness of these factors is essential for improving antimicrobial performance or combating antimicrobial resistance as this would provide researchers with proper rationales or guidance regarding the design, synthesis, and applications of halogenated drugs. Based on the reviewed literature, several critical factors, such as halogen size, substitution patterns, pathogen natures, hydrophobicity, and other physicochemical characteristics, determined antimicrobial activities and are herein elaborated.
1. Halogen Size and Substitution Pattern
Halogen sizes increase from fluorine to iodine as do C-X bond lengths (Fig. 1A), and halogen size and substitution patterns of halogens in the modified haloalkanes and haloarenes may determine antimicrobial potencies (Fig. 17). For instance, the introduction of chlorine or fluorine specifically at the C5 positions of 8-hydroxyquinoline and indole scaffolds strictly favored activities against certain Gram-negative and fungal pathogens (Cherdtrakulkiat et al., 2016; Raorane et al., 2022). Also, the antifungal activity spectrum of fluconazole was increased markedly by further fluorine substitution, whereas chlorine or bromine substitution reduced activity. The group noted that the small size and electronegativity were crucial to the activity (Shafiei et al., 2021). Of note, small halogens such as fluorine and chlorine may also enhance synergism between halogenated compounds and antibiotics or other antimicrobial agents. This is supported by a report that combinations of fluorinated bromperidol with posaconazole, an antifungal agent, showed the highest synergy against fungal pathogens. The small-sized fluorine atom was deemed better for optimal interaction with the fungal cells (Holbrook et al., 2017). Typically, size is essential for permeability and absorption by the cells as smaller molecules diffuse more rapidly and easily across membranes than bulky analogs due to factors related to surface area and volume. Interestingly, this may partially explain the reason why fluorinated and chlorinated drugs have been prominently approved by regulatory bodies. On the other hand, heavy halogens such as bromine and iodine have also been reported to enhance the activities of tricyclic flavonoid derivatives (Bahrin et al., 2016). Similarly, short peptoids (6–8mers) containing bromine or iodine exhibited better activity against resistant S. aureus and S. epidermidis (Molchanova et al., 2020). This relationship between high molecular weight halogen atoms and high bioactivity may be explained by their propensity for halogen bond formation, which promotes ligand binding affinities and confers stability (Xu et al., 2014).
Furthermore, the substitution pattern of halogens on the parent scaffold is also crucial to bioactivity. For example, fluorination at the -ortho position on the aminobenzene ring was solely responsible for the potency of a quinone derivative against C. albicans as modification other than that decreased activity (Wellington et al., 2019). Similarly, the presence of iodine atoms specifically at the C5-position on the antibacterial activity of halogenated indole against A. baumannii decreased from C5, C6, and C7 to the C4 position (Raorane et al., 2020). It is envisaged that the interaction between the halogens and the parent scaffold at these specific locations might have produced sufficient effect to enhance the target binding affinity of the compounds. Therefore, the understanding of these variations could contribute immensely to the drug development and improvement process.
2. Hydrophobicity
Hydrophobic interactions and the conformation of the ligand to the protein determine antimicrobial activities (Yin et al., 2022). These interactions are made possible by halogen bonding, which enhances inhibitory activity by modulating the potentials of halogenated agents to bind to cells, cross membrane barriers and even transported them within cells and organs (Priimagi et al., 2013). Fluorine grafting into chitosan increased hydrophobicity, lowered surface energy, and prevented bacterial surface adherence (Lin et al., 2018). Hydrophobicity may also be influenced by the size and electronegativity of halogens. The presence of bromine or iodine increased the hydrophobicity of peptoids more so than fluorine and notably corresponded to antimicrobial activities (Molchanova et al., 2020). However, hydrophobicity has a threshold beyond which no antimicrobial effect is observed. Iodinated peptoids lost their antimicrobial activities against Gram-positive bacteria when optimal hydrophobicity was attained by an increase in the number of halogen and residue lengths (Molchanova et al., 2020). This loss of activity may have resulted from poor solubility and low bioavailability typical of highly hydrophobic drugs. Prodrugs, emulsion preparation methods, drug complexation, polymeric micelles, drug nanocrystals, and other techniques have been reported to control these anomalies (Vimalson, 2016).
Furthermore, the distribution of halogens on the parent moiety could also affect the hydrophobicity of halogenated agents. For example, two bromine atoms on the phenyl rings of halogenated peptide compared with its presence on all the rings resulted in a hydrophobicity sufficient to exert a 32-fold antibacterial effect against resistant P. aeruginosa (Molchanova et al., 2020). Also, Cl and Br substituents were required for the hydrophobic binding and inhibitory activity of halofuginone on eukaryotic prolyl-tRNA synthetase (ProRS), whereas the presence of the highly electronegative trifluoromethyl group exhibited poor enzyme inhibitory activity (Guo et al., 2020). It thus appears that the space of the hydrophobic pocket at target sites determines which halogen elicits the suitable hydrophobic interaction for antimicrobial activity.
3. Physicochemical/Biophysical Properties
Biophysical/physiochemical properties, such as steric effects, electrophilicity, charge distribution, electronic (σ), and lipophilic attributes, are necessary for binding to target biomolecules and can also influence the target site affinities (El Hage et al., 2011; Fang et al., 2019). Firstly, steric effects may give rise to electronic conditions that facilitate interactions or impair target protein functionality or biological activity (Fang et al., 2019). For example, -para-substituted bromocinnamaldehyde was 10-fold more active against E. coli and S. aureus than the ortho derivative. The ortho location of Br- and its steric bulk were believed to hinder nucleophilic interaction at the electrophilic β-carbon as steric interference increased (Doyle et al., 2019). Similarly, the inactivity of halogenated pyrimidines against S. aureus growth and biofilm formation was attributed to steric hindrance and low electron-withdrawing potentials that impaired electron distribution on the linked ring system (Provenzani et al., 2021).
Furthermore, electrophilicity, which is the affinity of a molecule for electrons in its environment, might also determine antimicrobial potency. It was reported that the more electrophilic p-bromocinnamaldehyde was more potent than its parent compound against S. aureus and E. coli. Notably, the presence of Br- and Cl- substituents caused moderate electrophilic activation, which was independent of para-, ortho-, or meta- position on the phenyl ring, indicating that the type of halogen rather than the position caused the activation and improved activity (Doyle et al., 2019). Halogens can also influence the electrophilicity of neighboring atoms as was reported for melphalan flufenamide, an FDA-approved drug, in which the presence of chlorine increased the electrophilicity of directly attached carbon atoms to make endogenous nucleophilic interactions with components of nucleotide bases (Benedetto Tiz et al., 2022).
Charge distribution on the halogenated compounds is another important biophysical property (Fig. 17). This is dependent on molecular electrostatic potentials that determine relationships between binding activities and electron distributions, which can influence antimicrobial properties (Shafiei et al., 2021). Paulsen et al. (2019) reported that an increase in net positive charge of +2 on halogenated diamines (3,5-Br-Ph, 3,5-CF3-Ph, and 4-F-1-Nal) enhanced their aqueous solubilities and antimicrobial activities against E. coli and P. aeruginosa. Conversely, the charge distribution could also be responsible for a loss of antimicrobial activity as was observed for some halogenated fluconazole derivatives. Specifically, the moderate electron densities in a negative potential region having fluorine would be expected to bind more strongly with a positive target region than those containing Cl or Br atoms. This led to reduced antifungal activity against yeast and filamentous fungi compared with fluorine (Shafiei et al., 2021). The higher affinity of fluorine toward the targets and membrane in comparison with other halogens might be explained by the high electronegativity, which could create a more positive σ-region. Generally, a negative charge on a halogen has little effect on ligand-to-receptor binding energy. However, an increase in the electrostatic potential of a halogenated compound would enhance solvation and electrostatic contributions and thus potentiate ligand-to-receptor binding (Ibrahim et al., 2018). Therefore, the electron distribution charge on the halogenated moiety plays a key role in bioactivity.
Lipophilicity is a measure of the ability of a compound to interact with or dissolve in lipophilic surfaces or substances. Lipophilicity determines antimicrobial effectiveness by influencing the permeability and subsequent bioavailability of halogenated agents in tissues and lipid membranes (Fang et al., 2019). For example, modifications of isoniazid with 4-CF3 or 4-OCF3 at C4 on the aniline ring enhanced lipophilicity and thus facilitated the passive diffusion of compounds into a mycobacterial envelope inaccessible to isoniazid (Pflégr et al., 2021). Furthermore, chlorinated resveratrol penetrated cell membranes and inhibited H. pylori due to its increased lipophilicity (Di Fermo et al., 2020). Additionally, higher electronic and lipophilic values of 3,4-dichloro-cinnamaldehyde increased anti–quorum-sensing activities against Vibrio spp. (Brackman et al., 2011). Usually, lipophilicity increases with the number of halogens on aromatic rings, and when in excess, it leads to improper antioxidant localizations in membranes and reduces radical scavenging potentials, which may be detrimental as evidenced by the loss of antivirulence activities by 2, 4, 6-tribromo-3, 5, 4′-trihydroxystilbene (Li et al., 2012). Excessive lipophilicity could also be detrimental to drug absorption and expose drugs more to degradation by liver enzymes (e.g., cytochrome P450), reduce effective drug delivery at target sites due to reduced solubility, and increase toxic effects (Böhm et al., 2004; Danishuddin and Khan, 2016), hence the need to control lipophilicity to ensure adequate metabolic stability and optimum efficacy.
4. Bacterial Cell Type
Although halogens are known to enhance the membrane permeabilities of halogenated compounds, their activities may be subjected to cellular uptake (Fig. 17). During this review, we observed that the majority of new and existing compounds functionalized with halogens had a greater affinity for Gram-positive bacteria and fungi than Gram-negative bacteria (Doyle et al., 2019; Sanabria-Rios et al., 2022). In addition, despite exhibiting excellent drug-like properties, including blood-brain penetration and high gastrointestinal absorption, Br- and Cl-substituted cyclohexanol benzo[b]thiophene derivatives lacked activity against Gram-negative bacteria at high doses (Masih et al., 2021). However, this was not observed for the majority of halogenated cationic polymers, peptides, or quinones (Paulsen et al., 2019; Molchanova et al., 2020; Nazi et al., 2020; Peng et al., 2021). The presence of peptidoglycan fortified by the outer membrane of Gram-negative bacteria may have been a cause of this differential behavior (Doyle et al., 2019). Furthermore, parent structure properties, testing conditions, pathogen physiologic state, medium pH, halogenation pattern, and enzymatic and thermal stabilities may have also contributed, but unfortunately, these factors were not fully addressed in the studies reviewed. Consequently, it is difficult to infer that halogenated compounds are selective toward certain microbes, and thus we recommend that these properties be given adequate consideration during the design and the antimicrobial studies of halogenated drug candidates.
Also, the efficacies of halogenated agents depend on the state of the pathogen. When pathogens form biofilms, drug permeability is reduced, and virulence and antibiotic resistance are increased. Interestingly, this review revealed that halogenated agents potently control biofilm formation and virulence of several resistant bacteria and fungi. We suppose that increased affinity, permeability, and bioavailability induced by halogenation facilitated the penetration of biofilm matrices and subsequent control.
C. Halogenated Agents and Gut Microbiota
Halogen-containing scaffolds have applications in the food, pharmaceutical, agrochemical, and personal care product sectors as primary or adjuvant agents (Dikeocha et al., 2022). The gut microbiome represents all microbial groups inhabiting the human gastrointestinal tract and is often referred to as a metabolic organ with respect to maintaining the host's overall health (Li et al., 2016). The gut microbiome plays an essential role in metabolism and affects the pharmacology of drug candidates (Dhurjad et al., 2022). Thus, understanding gut microbe-drug metabolism can improve drug development processes. Herein, we discuss the possible impacts of gut microbiota on the administration of halogenated antimicrobial agents.
Gut microbiota contains several drug-metabolizing enzymes, including lyases and oxidoreductases (Koppel et al., 2017), and their interactions with halogenated antimicrobials could produce toxic intermediates, inactivate drugs, and cause crossresistance. For example, gut microbiota converted 5-fluoro-2-deoxyuridine into 5-fluorouridine-5′-monophosphate, which increased toxicity and became fatal to the host (Ke et al., 2020). Gemcitabine, a fluorinated nucleoside, was also inactivated by cytidine deaminase of duodenal γ proteobacteria and resulted in resistance and impaired efficacy (Li et al., 2021c). Also, genes related to resistance to several antibiotics were reported to be abundant in gut microbes exposed to chlorinated water (Nadimpalli et al., 2022). This suggests that the interactions between halogenated and gut microbiota could trigger resistance or produce detrimental or beneficial metabolites.
The interplay could also result in dysbiosis, impaired gut microbial diversity, and metabolome changes. Chlorpyrifos favored the growth of Bacteroides sp. but reduced the growths of Lactobacillus sp. and Bifidobacterium sp. Exposure of mice to 2,4-dichlorophenoxyacetic acid was also reported to increase gut bacteria counts (Ribado et al., 2017; Tu et al., 2019; Nadimpalli et al., 2022). These variations in the gut microbiome are closely associated with the host's response to drug therapy (Li et al., 2016). In addition, gut exposure to halogenated agents could result in translocation whereby gut commensals are introduced to other sites and trigger unwarranted immune responses. Furthermore, halogenated antimicrobials can directly modulate metabolic processes such as pyruvate metabolism, the tricarboxylic acid cycle, protein export, and carbon fixation by gut microbiota (Tian et al., 2020; Sha et al., 2022), which could impair the metabolic functions of gut microbiota and consequently impact host health.
Gut bacteria can also modulate drug metabolism through host functions and are capable of altering drug absorption rates by modulating gut environmental conditions (Enright et al., 2016). In addition, adhesins expressed by most gut bacteria interact with and bind drugs, which could reduce bacterial adhesion to host cells and alter drug absorption and plasma levels (Dhurjad et al., 2022). Although gut microbes are not in direct contact with the liver, they are capable of modulating the expressions of liver-related genes and thus modulating drug metabolism (Selwyn et al., 2016; Dhurjad et al., 2022). Another crucial effect is the competition for active sites whereby host, drug, and microbial metabolites compete for active sites of the host metabolic enzymes and thus influence their efficacies (Dhurjad et al., 2022). Generally, antibiotics are less effective in differentiating between commensals and pathogenic microbes, and the same is being envisaged as a limitation for these halogenated compounds. Also, certain commensals that acquire antibiotic resistance genes were reported to shield pathogens from the impacts of antibiotics, indicating further complication in the management of infection (Gjonbalaj et al., 2020).
There are also reports that attest to the beneficial or indifferent interaction between gut microbiota and halogenated compounds. For instance, the gut microbiota of Bangladeshi children was not perturbed after consumption of chlorinated water but rather increased the beneficial bacteria, which favored gut health (Nadimpalli et al., 2022). Similarly, the exposure to household triclosan in toothpaste and soaps did not cause dysbiosis (Ribado et al., 2017), and at physiologic concentrations, triclosan did not show notable effects on endocrine and metabolic markers in human oral and gut microbiota (Poole et al., 2016). Taken together, the interplay between gut microbes and halogenated antimicrobials generates several effects. Thus, their understanding should be a guide to improve the design and production of halogenated antimicrobials.
VII. Impacts of Halogenation on Antibiotic Resistance Mechanisms
As halogenation increases the abilities of antimicrobials to penetrate membranes and enhances ADME capabilities, it could also rejuvenate antimicrobials that are ineffective against drug-resistant antimicrobial strains to restore antibiotic susceptibility. Halogenated compounds are capable of overcoming several types of resistance mechanisms, including active drug efflux, drug target modification, limiting drug uptake, or drug deactivation, as elaborated below (Fig. 18).

Effects of halogenated compounds on various modes of antimicrobial resistance.
Efflux pumps, which are mainly responsible for drug extrusion from bacterial cells before reaching the intracellular targets, are the most significant cause of antibiotic resistance and have become important therapeutic targets (Nishino et al., 2021). Halogenated agents circumvented antibiotic resistance by inhibiting arrays of efflux pump transporters. For instance, the overexpressing RND pumps, namely AcrAB-TolC, MexAB-OprM, and MexXY-OprM, were inhibited by 3,4-dibromopyrrole-2,5-dione to reverse the multidrug resistance of E. coli to ciprofloxacin, levofloxacin, kanamycin, erythromycin, oxacillin, piperacillin, tetracycline, and chloramphenicol (Whalen et al., 2015). Similarly, ethyl 4-bromopyrrole-2-carboxylate used in combination with antibiotics reduced the overexpressing RND transporters to restore susceptibility to MDR E. coli and P. aeruginosa (Tambat et al., 2022).
Another efflux family controlled by halogenated agents is the proton-dependent pump. Trifluoromethyl-ketone enhanced the activities of ampicillin, tetracycline, and erythromycin antibiotics and improved sensitivity to resistant wild-type E. coli AG100 and E. coli K12 LE140 strain with a tetracycline-resistant plasmid by proton pump inhibition (Spengler et al., 2003; Wolfart et al., 2006). 3-(2-Benzoxazolyl)-1,1,1-trifluoro-2-propanone also inhibited the E. coli proton pump to enhance promethazine activity (Wolfart et al., 2006). Notably, the trifluoro group is a common feature among these compounds and may suggest an important role in proton pump inhibition, but this requires further investigation. Furthermore, halogenated 1,3,5-triphenyl-2-pyrazolines inhibited p-glycoprotein efflux pumps in M. tuberculosis and other bacterial and fungal strains and thus mitigated resistance (Sivakumar et al., 2010).
Halogenated agents are capable of suppressing efflux pump–related genes to limit efflux activities. Chlorpromazine was reported to synergize with norfloxacin to control MDR S. enterica by repressing the expression of the efflux pump gene (acrB), limiting export potential, and enhancing chlorpromazine uptake within the bacterial cell. Also, haloperidol derivative combined with fluconazole overcame fluconazole resistance in C. albicans by downregulating the efflux pump gene (MDR1) (Ji et al., 2020). Herein, the synergistic effect of halogenated agents with antibiotics is notable in efflux pump inhibition. Therefore, the use of halogenated agents as adjuvants with antibiotics wherein halogenated agents remove resistance barriers to allow antibiotics to exert their therapeutic effects could be an effective strategy to treat resistant pathogens. Minimization of efflux pump activities by halogenated compounds has the potential to rejuvenate antibiotic activity against MDR strains via reducing drug expulsion and increasing the accumulation of drugs sufficient to reach intracellular targets and could also act as additives in inhibiting the pathogens themselves.
However, halogenation processes could also impair the efflux pump inhibitory potentials of halogenated agents. It was reported that orfA-mediated halogenation in Streptomyces facilitated the binding affinity of halogenated albofungin toward transglycosylase but limited the antimicrobial potencies against K. pneumoniae and P. aeruginosa by intrinsically altering the biological and physicochemical properties. This affected molecular recognition, hence enhancing the orfL-dependent expulsion of chloro- and bromoalbofungin. OrfL is a proton-dependent oligopeptide transporter belonging to the major facilitator superfamily (Wang et al., 2022). It does suggest that adequate modulation of physicochemical properties is essential when designing potential halogenated efflux pump inhibitors.
Membrane impermeability leading to decreased uptake is a relatively uncommon resistance mechanism. Although research on this aspect is limited, 3,4-dibromopyrrole-2,5-dione and ethyl 4-bromopyrrole-2-carboxylate boosted the uptake and accumulation of Hoechst 33342 (fluorescent efflux pump substrate) in attempts to reverse the MDR in E. coli and P. aeruginosa (Whalen et al., 2015; Tambat et al., 2022). Additionally, halogenated novel bacterial topoisomerase inhibitors against several resistant strains of Staphylococcus sp. and E. coli achieved satisfactory results by adjusting lipophilicity/hydrophobicity ratios (Kolarič et al., 2021). Halogenated agents may act as membrane permeabilizers capable of improving the permeability of the outer membrane and increasing intracellular drug levels to overcome antibiotic resistance.
Halogenation could also potentiate the inhibition of drug-deactivating enzymes responsible for the breakdown of antimicrobials like β-lactamase, providing another viable strategy to combat AMR. Enzyme-inhibition combinatory therapies that included halogenated β-lactamase CMY-10 inhibitors were found to be effectively therapeutic against multidrug-resistant Enterobacteriaceae (Parvaiz et al., 2021). Halogen-substituted triazolethioacetamides displayed potential as metallo-β-lactamase inhibitors (Zhang et al., 2019). Halogen ions also served as potent inhibitors of OXA-enzymes, which are extended-spectrum β-lactamases and can increase the efficacies of antibiotics (Stojanoski et al., 2015).
Halogenation controlled drug resistance mediated by drug target modification or mutation by improving potency and molecular recognition. Fluoroquinolone resistance caused by the modification of gyrase-topoisomerase was circumvented using sparfloxacin (a C-8 halogen-substituted fluoroquinolone) against the gyrase-resistant mutant of M. smegmatis and the gyrase-topoisomerase IV double mutant of S. aureus as an alternative to C-8 hydrogen-substituted ciprofloxacin (Lu et al., 2001). Similarly, quinolone-35 and sitafloxacin exhibited potent activity against quinolone-resistant staphylococci and ciprofloxacin-resistant gyrase (gyrA CipR) mutants of P. aeruginosa, respectively, when substituted with fluorine at the eighth position (Ito et al., 1995; Kitamura et al., 1995). A modification at the 2-position of halogenated quinolines exhibited antibacterial and biofilm eradication activities against MDR strains of Staphylococcus sp. and Enterococcus sp. (Basak et al., 2016).
Several halogenated compounds and antibiotics were active against resistant pathogens as reviewed in Halogenated Antibiotic Classes and Halogenated Antimicrobial Scaffolds. But the novelty of the scaffold itself should be considered when reviewing activity against resistant strains as resistance is subject to only specific antibiotics, resulting in similar MIC values between sensitive and resistant strains. Although the practice of finding novel compounds for chemotherapy against resistant strains is a good initiative, a deeper understanding of the resistance mechanisms would encourage the rejuvenation of older antibiotics, which can be economical and efficient, and also differentiates potential approaches for sensitive and resistant strains. The potential of rejuvenating older antibiotics is possible in the case of resistant strains employing efflux pumps, membrane permeability, and enzyme degradation type of resistance mechanisms as drug target modification is an irreversible occurence. Taken together, halogenated compounds could minimize various AMR mechanisms and restore antibiotic susceptibility via the inhibition of efflux pumps, drug-deactivating enzyme inhibition, enhancement of drug uptake, and resistance to target modifications in MDR strains. A continuous understanding of resistance mechanisms and the effects of halogenated compounds on them would provide better insight into how to best leverage halogen atoms against resistant microbes.
VIII. Current Practices and Prospects
Microorganisms have demonstrated resistance to many approved drugs and evolved multidrug-resistant strains in addition to several drug contraindications. Herein, we have elaborated on the importance of halogenation for the discovery of drugs targeting antimicrobial resistance. The halogenation of various antimicrobial scaffolds has conferred several advantages, including broadening the activity spectrum against resistant pathogens, inducing reductions in virulence factors, such as biofilm formation and toxin production, and substantially increasing antimicrobial scaffold adjuvant activities. Halogenation also improved metabolic stabilities by inhibiting phase I oxidation and alleviating the side effects/toxicities of parent scaffolds. Pharmacokinetic properties such as absorption, distribution, bioavailability, and clearance rate were also significantly improved by halogenation (Fig. 19). Additionally, halogenated derivatives displayed potential for expanding the clinical usages of existing drugs, blocking scaffold defects, serving as efflux pump inhibitors to reverse drug resistance, and restoring the potency of erstwhile ineffective drugs (Nizi et al., 2020; Ienașcu et al., 2022).

Advantages of scaffold halogenation.
Halogenated antimicrobials use diverse mechanisms (Fig. 20). For instance, halogenated derivatives of indole, magnolol, flavonoids, and CNMA seem to possess more diverse mechanisms than their parent scaffolds, probably because of their different physicochemical properties and improved target affinity mediated by halogenation. Also, halogens form salts by directly reacting with metals, and this seems to have conferred metal chelation as an antimicrobial mechanism on halogenated catechols, phenazines, and hydroxyquinolines. Taken together, halogenated derivatives display a wide range of antimicrobial mechanisms. Furthermore, factors such as degree of halogenation, halogen size, pathogen type, halogen position, and the natures of substituents coexisting with halogens determined the bioactivity and pharmacological properties of halogenated antimicrobials. Nonetheless, certain essential aspects of halogenated antimicrobial drug research are underexplored.

Mechanisms underlying the antimicrobial activities of halogenated antibiotics and antimicrobial scaffolds. ADP, adenosine diphosphate; P, phosphate; QS, quorum sensing; and ROS, reactive oxygen species.
Certain halogenated moieties have found application in the development of antibiotics. Chlorocatechol (8A) is a component of cefiderocol, which was recently approved by the FDA, where it conferred novel MOA, such as siderophore-iron binding and stability against enzymatic action (Sato and Yamawaki, 2019). Also, the halogenated hydroxyquinoline derivative 9V has a structure similar to the antibiotic clioquinol, which was approved for antimicrobial treatment (Das et al., 2019). Although these applications further support the importance of halogenated antimicrobial groups, the subject has been underexploited and could be further substantiated to potentiate, modulate, or replace existing antibiotics.
Research groups have synthesized new prodrugs by linking or fusing halogenated scaffolds to known antibiotics to overcome inactivity (Fig. 21). This molecular hybridization approach involves combining molecules to produce more potent drugs that benefit from synergism (Ivasiv et al., 2019; Zha et al., 2022). For instance, halogenated phenazine-erythromycin and cephalosporin-halogenated phenazine prodrugs (Fig. 21, A and B) were reported to enhance the translational release of phenazines. This design conferred on the prodrug's dual MOA as ribosome inhibitor and iron-starving agents (Xiao et al., 2020; Yang et al., 2021). Similarly, fluconazole was hybridized with a halogenated phenyl urea group to overcome resistant C. albicans, and in a similar study, halogenated thiazolo[4,5-day]pyrimidine was employed (Fig. 21, C and D) (Shafiei et al., 2021; Sharapova et al., 2022). The hybridization concept confers affinities for multiple targets and is expected to rejuvenate potencies and stabilities, and to minimize the side effects of existing medications. In addition, it reduces the risk of drug resistance due to multitargeting ability and provides an avenue for developing drugs at lower risks and high cost effectiveness. However, the selection of suitable combinations and linkers to ensure substrate processing and active compound release remains a major challenge (Ivasiv et al., 2019; Zha et al., 2022).

Structures of approved drugs conjugated with halogenated compounds to combat resistant pathogens. Erythromycin-halogenated phenazine (A), cephalosporin-halogenated phenazine prodrug (B), fluconazole-halogenated urea group (C), and fluconazole- halogenated thiazolo[4,5-day] pyrimidines prodrugs (D).
Furthermore, it is essential that the effects of the interplay between halogenated drug candidates and the host be determined with respect to toxicity and other pharmacological effects. Available reviews have sparingly addressed in vitro and in vivo assessments of toxicities. This is a fundamental requirement of preclinical drug trials and should be adequately integrated into future studies to ensure smooth transitions from bench to bedside. Similarly, information on administration routes, excretion pathways, metabolic soft spots, physiologic clearance, drug interference, metabolite formation, and liabilities should be considered.
The use of heavy halogens such as bromine and iodine in drug repurposing or modification may be promising for antivirulence activities. Microbes produce several virulence factors to cope with stressful conditions and, as a result, become more pathogenic (Ji et al., 2020). Antimicrobial agents targeting them in ways that do not affect growth are unlikely to result in resistance development and are viewed as attractive means of combating drug-resistant pathogens. This review shows heavy halogens are present in many scaffolds such as BrCl-flavonoid (8J), 5-iodoindole (9B), and halogenated phenazines (10L), and these agents effectively inhibited biofilm formation and eliminated persister cells by multiple folds (Garrison et al., 2016; Babii et al., 2018; Raorane et al., 2020). Interestingly, these compounds contain only Br or I atoms with or without a smaller halogen like chlorine, which suggests that larger halogens should be included in studies undertaken to reduce drug resistance and control virulence. The utilization of Br and I in drug production has been limited by their reactivities, tendencies to produce metabolites, cost, and individual bias (Herrera-Rodriguez et al., 2011; Wilcken et al., 2013; Jeschke, 2022). Tuning reaction conditions or late introduction of Br or I might resolve reactivity issues (Wilcken et al., 2013; Tan et al., 2017), and also, we envisage that halogen blending might alleviate the limitations assigned to Br and I application. Of note, the phenomenon of halogen bonding and how it tunes antimicrobial activity has gained attention (Edis et al., 2019). However, its relationship with virulence is unknown and worth investigating.
Halogens have the potential to contribute significantly to alternative therapies designed to combat drug resistance. Antiseptics play a critical role in fighting antimicrobial resistance, especially in hospital settings (Maillard et al., 2021). Halogens are used to produce many antiseptics and disinfectants like povidone-iodine, triclosan, dequalium chloride, and cetylpyridium chloride (Wade et al., 2021; Virji et al., 2022), and interestingly, resistance to common iodophor antiseptics like povidone-iodine and cadexomer iodine is minimal despite their widespread use. Their MOAs revolve around the release of free halide ions, which induce lipid membrane peroxidation and DNA damage and inhibit protein synthesis (Shah et al., 2021). However, the precise reason for their prolonged efficacies has not been determined, though it is clear that free halide ion-induced oxidation is something microbes find difficult to counteract.
Cationic polymers are associated with minimal AMR, as indicated in Halogenated Polymeric Materials, which raises expectations regarding oxidative halogen species released by polymers like N-halamines and antiseptics. Constant long-term release of antimicrobial ions is also an attractive AMR strategy to ensure complete microbe killing and prevent recurrent infection. Furthermore, reactive halogen species play a vital role in immune-based defense, and interestingly, it was reported that ozone-depleting halogen cyclic reactions amplified-reactive halogen species enhanced phagocytosis mediated pathogen killing by 100,000-fold (Lu, 2020).
Preventative vaccine therapies significantly aid the fight against microbial recalcitrance (Micoli et al., 2021). Halogens can enhance vaccine efficacies as was observed for a fluorinated cocaine vaccine, a fluorinated carbohydrate cancer vaccine, and a brominated human respiratory syncytial virus vaccine, which was 6.3-fold more efficient than the nonbrominated vaccine (Yang et al., 2011; Cai et al., 2013; Xue et al., 2023). Halogenation of vaccines is incipient, especially for antimicrobial therapy, and may constitute a vanguard approach for combating antimicrobial resistance.
In the same vein, halogen compounds can also act as immunomodulators to trigger immune responses as evidenced by the use of halogenated aryl thiosemicarbazones against leishmaniasis, halogenated thymoquinone analogs against Plasmodium falciparum, and the Th1-immunostimulatory activity of halogenated α-galactosylceramide analogs against parasites (Hossain et al., 2016; da Silva et al., 2017; Johnson-Ajinwo et al., 2018). Although the concept of immunomodulation using halogenated compounds to achieve antimicrobial activity is a relatively unexplored topic compared with parasitism, we feel that its exploration might provide a means of combating AMR.
Halogenated compounds can also augment viral phage therapy by promoting microbe accumulation and lysis. Halobenzenes bind within the internal nonpolar cavity of the Leu99Ala bacteriophage T4 lysozyme mutant, and halogen bonding provides better stabilization than hydrogen bonding (Erdélyi, 2017). Although the implications of halogenated T4 lysozyme analog on the antimicrobial activity are unreported, they may have significant impacts on future phage-based therapeutics, proteins, and engineering. As discussed in Halogen Bonding, Pharmacokinetics, and Pharmacodynamics, replacing hydrogens with halogens in peptides reduces activation energies and increases enzyme binding affinities, and these phenomena could be used to synthesize specific endolysins or phages against a diverse range of resistant or novel pathogens. Similarly, as discussed in Peptides, using halogenation as a tool in peptide engineering has great prospects with improved antimicrobial activity and peptide’s biocompatible nature in comparison with traditional antibiotics.
Many properties, conditions, and actions of halogens may be responsible for the enhanced antimicrobial potencies of halogenated antimicrobial agents, but regardless of the number of halogenated agents available, studies are limited. Interestingly, several machine learning and artificial intelligence–based software packages, such as DeepChem, DeltaVina, Chemputer, Open Drug Discovery Toolkit, AMPlify SCScore, and DeepNeuralNet-QSAR (Peña‐Guerrero et al., 2021; Matsuzaka and Yashiro, 2022; Staszak et al., 2022), are now used for lead optimization and predicting these properties. Application of these platforms reduces the time required for lead optimization and may further advance the halogenated antimicrobial research field by providing more insights into the roles of halogens.
IX. Conclusions
Antimicrobial resistance has multidimensional impacts on global mortality, morbidity, and national economies and could lead to fatalities by simple infections (Ikhimiukor et al., 2022). In addition to judicious surveillance of antibiotic administration and compliance with current health policies, new strategies based on novel therapeutics are required. In this review, we evaluated the benefits of halogen atoms and their impacts as compared with parent antimicrobial scaffolds. In addition, we explored the structure-activity relationships, modes of action, toxicities, and applications of a wide range of halogenated scaffolds, including antibiotics, antiseptics, plant products, polymers, and biomolecules, and the roles of halogen atoms.
The halogenation of scaffolds provides various advantages; for example, it can generate novel antimicrobial mechanisms and targets and has the potential to enhance activity, minimize toxicity, and overcome drug resistance (Fig. 19). Furthermore, the concept of scaffold or molecular hybridization has the potential to improve affinities for multiple targets. However, an extensive survey of antimicrobial agents revealed that the majority of halogenated antimicrobials synthesized were not tested for toxicity, which we consider an essential part of drug discovery. In addition, drug repurposing is gaining attraction because of the slow rate of novel drug approval (Zhan et al., 2022). In our opinion, halogenated compounds are excellent drug-repurposing candidates given their broad-spectrum capabilities and affinity to various biological targets.
Rejuvenation of existing antimicrobials deemed ineffective against resistant pathogens is an increasing trend (Zhan et al., 2022). Halogenation offers a practical means of rejuvenating impotent antimicrobials as discussed in Impacts of Halogenation on Antibiotic Resistance Mechanisms. A deeper understanding of the effect of halogenation on retired antimicrobials against specific resistant strains is required. We also identified a need for the standardized testing of compounds against a more diverse panel of microorganisms and resistant strains. In our opinion, halogenation and the incorporation of halogen atoms in pharmacological scaffolds are feasible strategies for combating antimicrobial resistance because halogenation is simple, economically viable, and readily accessible.
Acknowledgments
The authors would like to thank John Roberts for English proofreading and anonymous referees’ insightful comments.
Data Availability
The authors declare that all the data supporting the findings of this study are contained within the paper and its Supplemental Material.
Authorship Contributions
Participated in research design: Faleye, Boya, J.-H. Lee, J. Lee.
Wrote or contributed to the writing of the manuscript: Faleye, Boya, J.-H. Lee, Choi, J. Lee.
Footnotes
- Received March 7, 2023.
- Revision received August 7, 2023.
- Accepted September 29, 2023.
This study was supported by grants from the Basic Science Research Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1I1A3A04037486, 2020R1A6A1A03044512) and the NRF funded by the Korean government (MSIT) (2021R1A2C1008368).
The authors have no conflict of interest to declare.
↵1O.S.F. and B.R.B. contributed equally to this work as first authors.
↵
 This article has supplemental material available at pharmrev.aspetjournals.org.
This article has supplemental material available at pharmrev.aspetjournals.org.

Abbreviations
- ADME
- adsorption, distribution, metabolism, and elimination
- AMP
- antimicrobial peptide
- AMR
- antimicrobial resistance
- ATP
- adenosine triphosphate
- CNMA
- cinnamaldehyde
- C-X
- carbon-halogen
- EO
- essential oil
- FA
- fatty acid
- FDA
- Food and Drug Administration
- MDR
- multidrug resistant
- MIC
- minimum inhibitory concentration
- MOA
- mechanism of action
- MRSA
- methicillin-resistant S. aureus
- MRSE
- methicillin-resistant S. epidermis
- PABA
- para-aminobenzoic acid
- RND
- resistance nodulation cell division
- SAR
- structure activity relationship
- VRE
- vancomycin-resistant enterococci.
- Copyright © 2023 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
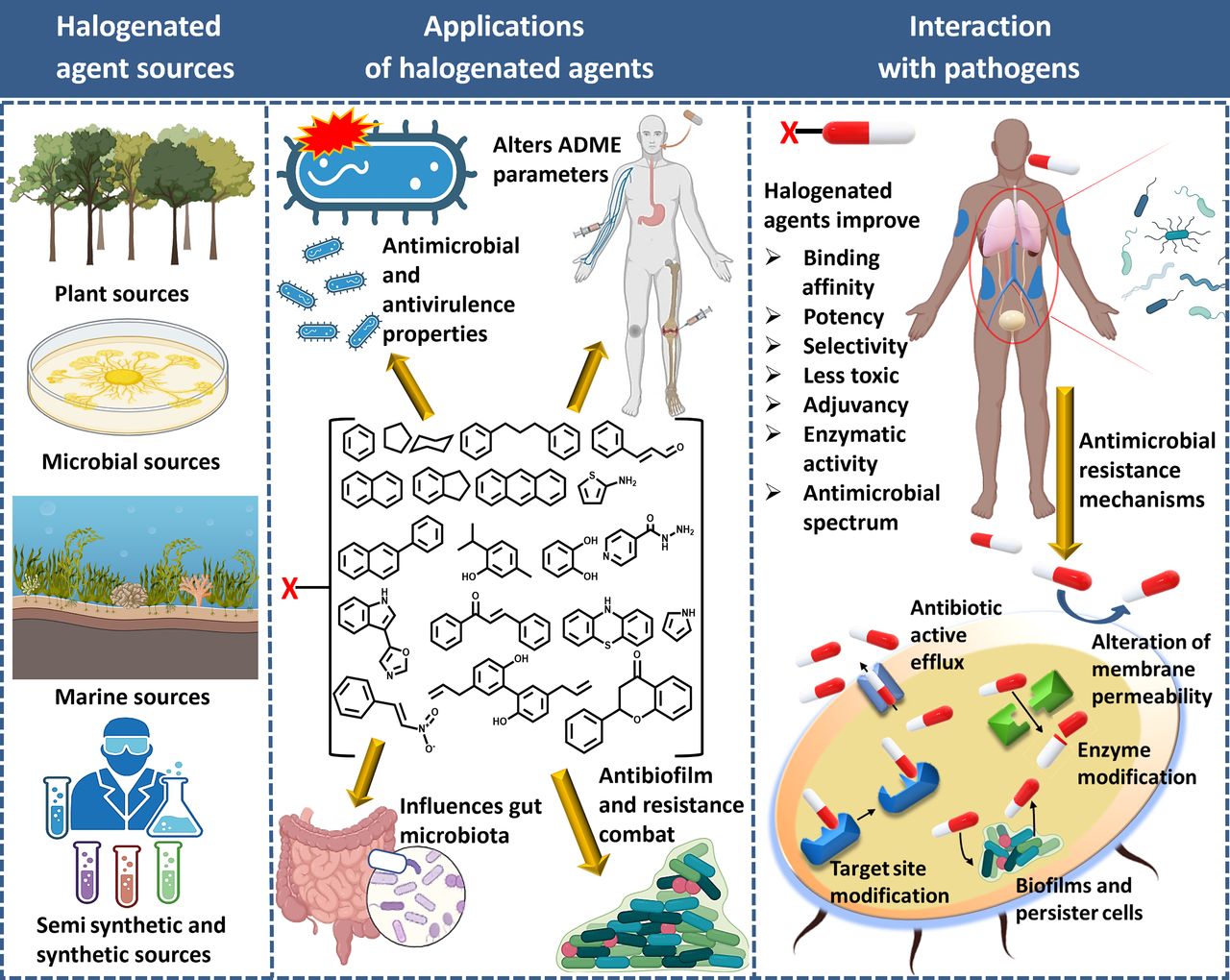{kind=link}
{kind=link}
{kind=link}
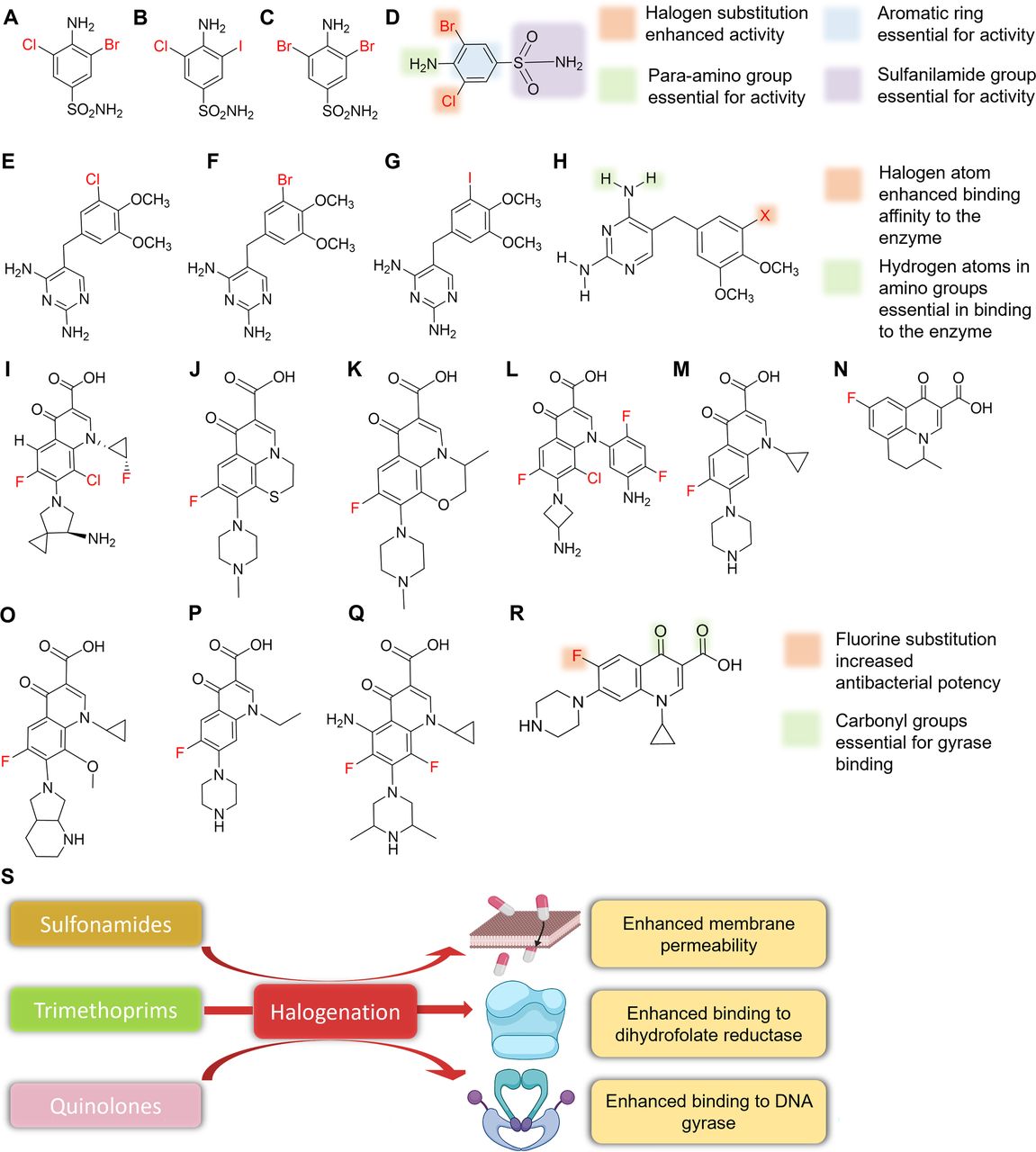{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
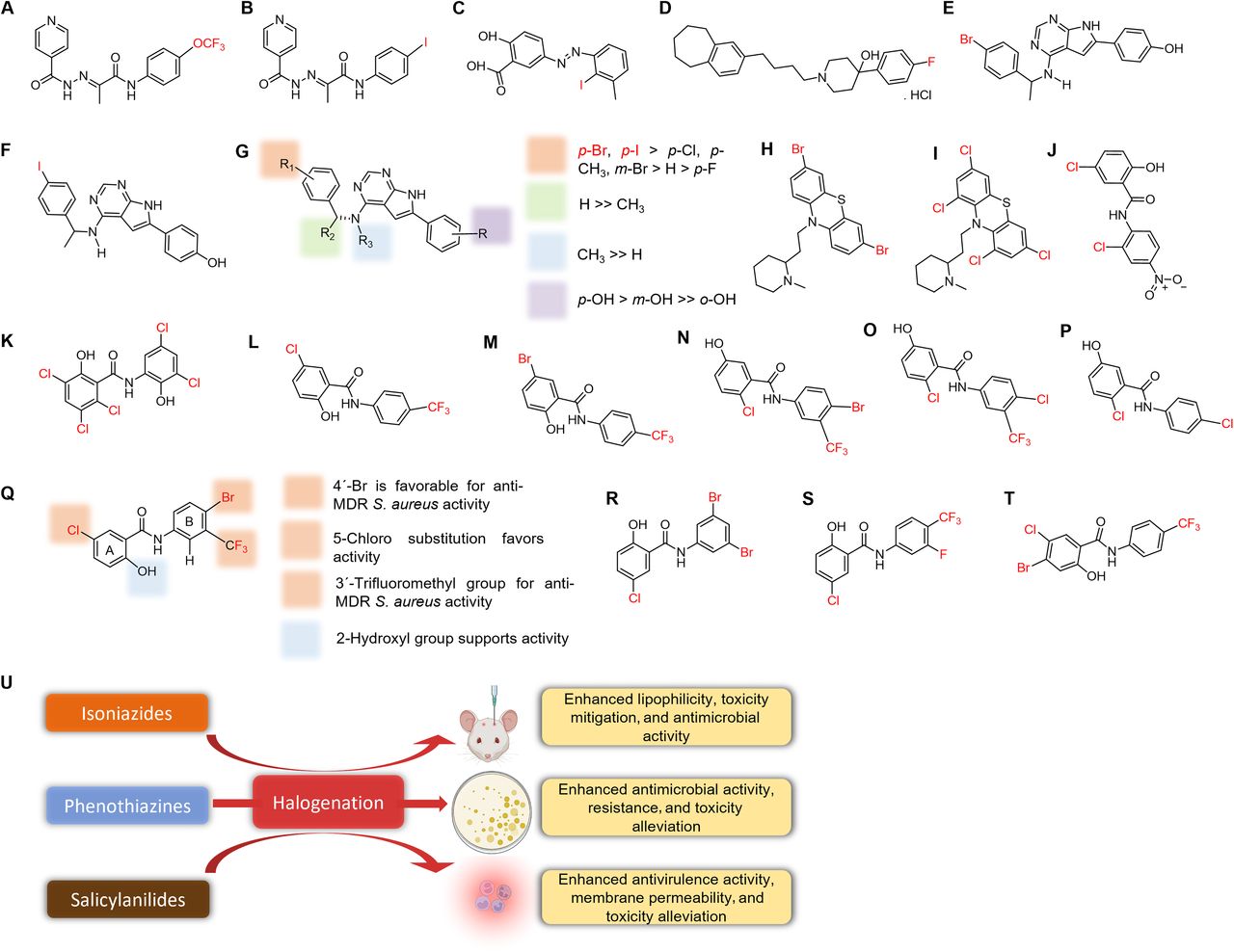{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
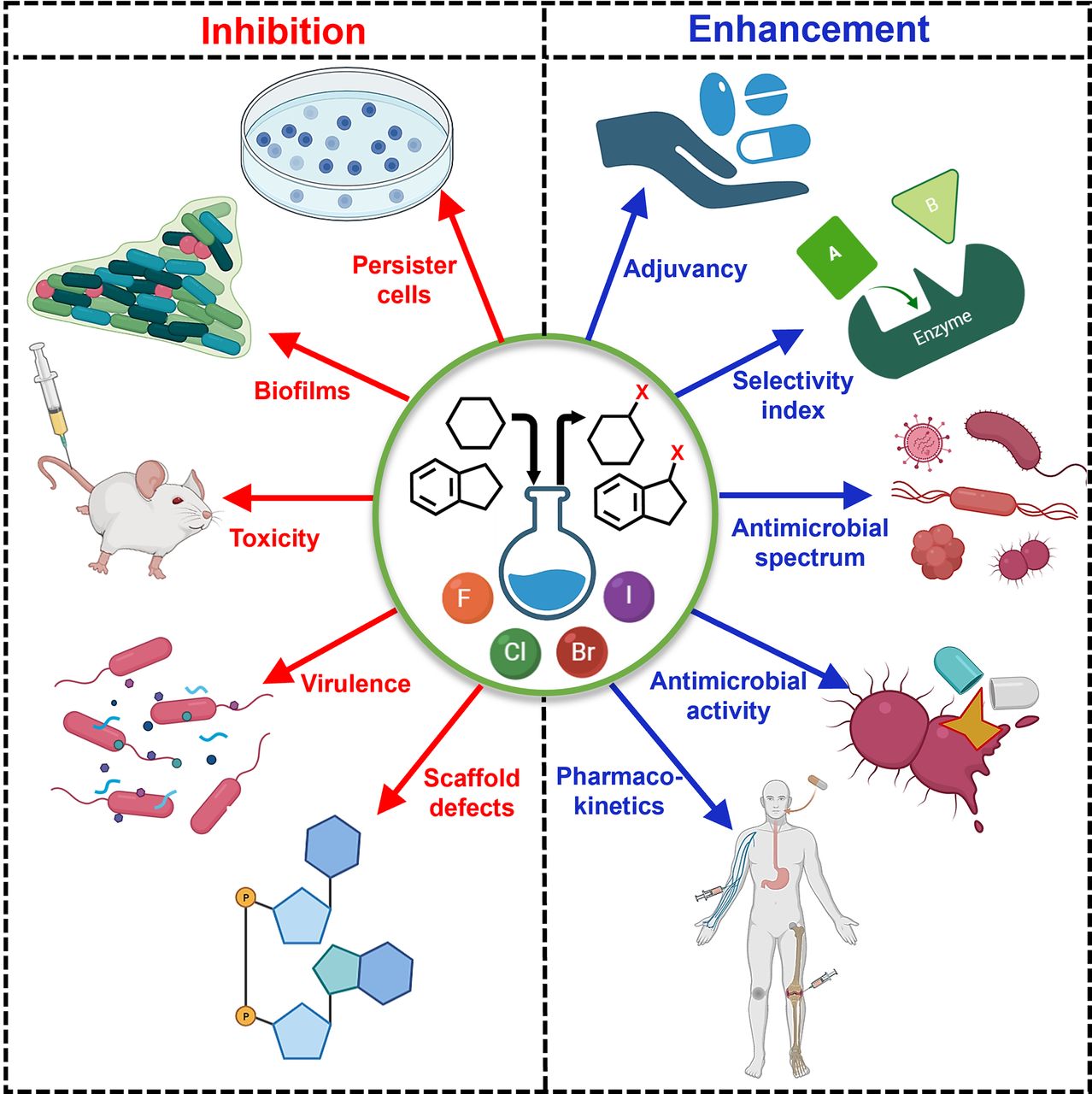{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Halogenated Antibiotic Classes
- III. Halogenated Antimicrobial Scaffolds
- IV. Halogenated Polymeric Materials
- V. Halogenated Biomolecules
- VI. Effects of Halogenation and Factors Affecting the Antimicrobial Activities
- VII. Impacts of Halogenation on Antibiotic Resistance Mechanisms
- VIII. Current Practices and Prospects
- IX. Conclusions
- Acknowledgments
- Data Availability
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI