
Abstract
Kidney excretion of numerous organic anionic drugs and endogenous metabolites is carried out by a family of multispecific organic anion transporters (OATs). Two closely related transporters, SLC22A6, initially identified by us as NKT and also known as OAT1, and SLC22A8, also known as OAT3 and ROCT, are thought to mediate the initial steps in the transport of organic anionic drugs between the blood and proximal tubule cells of the kidney. Coding region polymorphisms in these genes are infrequent and pairing of these genes in the genome suggests they may be coordinately regulated. Hence, 5′ regulatory regions of these genes may be important factors in human variation in organic anionic drug handling. We have analyzed novel single nucleotide polymorphisms in the evolutionarily conserved 5′ regulatory regions of the SLC22A6 and SLC22A8 genes (phylogenetic footprints) in an ethnically diverse sample of 96 individuals (192 haploid genomes). Only one polymorphism was found in the SLC22A6 5′ regulatory region. In contrast, seven polymorphisms were found in the SLC22A8 5′ regulatory region, two of which were common to all ethnic groups studied. Computational analysis permitted phase and haplotype reconstruction. Proximity of these non-coding polymorphisms to transcriptional regulatory elements (including potential sex steroid response elements) suggests a potential influence on the level of transcription of the SLC22A6 and/or SLC22A8 genes and will help define their role in variation in human drug, metabolite and toxin excretion. The clustering of OAT genes in the genome raises the possibility that nucleotide polymorphisms in SLC22A6 could also effect SLC22A8 expression, and vice versa.
Similar content being viewed by others


Introduction
The organic anion transporters (OATs) constitute a subfamily within the SLC22 family of solute carriers (Eraly et al. 2003). Two members of this subfamily, SLC22A6 (also known as OAT1), originally identified as novel kidney transporter (NKT; Lopez-Nieto et al. 1996, 1997) and SLC22A8 (also known as OAT3), originally identified as reduced in osterosclerosis (ROCT) (Brady et al. 1999), function as the major basolateral transporters of a wide variety of drugs and toxins excreted via the proximal tubule of the kidney. These drugs include nonsteroidal anti-inflammatory drugs, antibiotics, antivirals (such as adefovir and cidofovir), antihypertensives, diuretics, methotrexate and many other commonly prescribed drugs (Eraly et al. 2004a, b). They are also responsible for the excretion of toxins such as Ochratoxin A and mercurials, and are believed to be essential for the mediation of the renal toxicity of these agents (Sweet 2005). In addition, recent mouse knockout studies suggest that these two genes are sufficient to explain most of the excretion of certain prototypical organic anions like para-aminohippurate (PAH), estrone sulfate and taurocholate (Sweet et al. 2002; Eraly et al. 2005), consistent with the notion that these two genes mediate the rate-limiting step for uptake of organic anion drugs from the blood.
For these reasons, there has been much interest in the possibility that polymorphisms in SLC22A6 and SLC22A8 may be partly responsible for variation in the handling and efficacy of the many commonly used drugs that are transported by these transporters. Likewise, such polymorphisms could conceivably explain susceptibility to the toxicity of drugs and environmental toxins. Recent studies have thus analyzed coding region polymorphisms in these genes (Xu et al. 2005; Bleasby et al. 2005; Fujita et al. 2005). However, unlike other members of this subfamily such as SLC22A11 (also known as OAT4) and SLC22A12 (also known as URAT1 and originally identified as RST in mouse), there appear to be relatively infrequent nonsynonymous coding region human polymorphisms in SLC22A6 and SLC22A8 (Xu et al. 2005). Thus, while some variation in drug/toxin handling might be explained by coding region polymorphisms in these genes, the apparent paucity of such polymorphisms suggests the need to also analyze noncoding region variation in these genes, which might, for example, affect the transcription of SLC22A6 and SLC22A8 and thereby the total levels of functional protein.
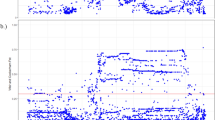
SLC22A6 and SLC22A8 exist as a tandem pair on human chromosome 11 (Fig. 1). Given the complex genomic structure and the as yet undetermined promoter regions of SLC22A6 and SLC22A8, we have utilized the computational technique of phylogenetic footprinting to identify evolutionarily conserved noncoding regions of the SLC22A6 and SLC22A8 genes (Fig. 1); these regions are likely to contain sequences important for regulating gene expression (Kim et al. 2004; Prohaska et al. 2004). We identified three such regions in the SLC22A6 gene and five regions in the SLC22A8 gene. We then sequenced these regions in an ethnically diverse panel of human DNA samples in order to identify single nucleotide polymorphisms (SNPs) likely to affect the expression of the SLC22A6 and SLC22A8 transcripts. The data suggest a number of polymorphisms that could affect SLC22A8 expression and a more limited number that could affect SLC22A6 expression. This approach can be more broadly applied to identify potential regulatory region SNPs for SLC22A6 and SLC22A8, as well as other SLC22 family members. It will be important to consider these polymorphisms, along with those found in coding regions, in clinical studies aimed at understanding variation in drug handling and toxin susceptibility mediated via OATs.

Locations of single nucleotide polymorphisms (SNPs) on phylogenetic footprints (PFs) of human SLC22A6 and SLC22A8 on chromosome 11. a Locations of SLC22A6 and SLC22A8 on human chromosome 11. The genomic distance is relative to the centromere, derived from the University of California Santa Cruz (UCSC) genome database. SLC22A6 and SLC22A8 are separated by a small interval of 8 kb, and the direction of their transcription is indicated by arrows. The proximal locations of the evolutionarily conserved regulatory regions of PFs are indicated relative to the transcriptional start sites. The locations of the PFs are relative to each other, and the distances between them are not proportional. b Conserved regions on the promoter/enhancer SLC22A8 (PFus) are depicted relative to the transcription start site (arrow). Selected putative transcription factor sites are listed according to general localization on the PFus. Positions of the SNPs, by distance to the transcription start site, are marked to indicate the proximity to the transcription factor binding sites
Materials and methods
Phylogenetic footprinting
We have previously defined phylogenetic footprints (PFs) of SLC22A6 and SLC22A8 5′ regulatory regions (Eraly et al. 2003). Briefly, reference SLC22A6 and SLC22A8 regulatory sequences for human, chimpanzee and mouse were acquired from the University of California Santa Cruz (UCSC) Genome Browser (http://www.genome.ucsc.edu/). An 8 kb region of sequence upstream from the first translational start site for the SLC22A6 and SLC22A8 genes was obtained for each species (Fig. 1). PFs were then identified through sequence alignment using pairwise BLAST (http://www.ncbi.nlm.nih.gov/BLAST) and then realigned using ClustalX (http://bips.u-strasbg.fr/fr/Documentation/ClustalX/). Homology between human, chimpanzee, and mouse was confined to three regions on the promoter/enhancer of SLC22A6 (designated PFi1, PFi2, and PFi3) and five regions on the promoter/enhancer of SLC22A8 (designated PFu1 to PFu5). PFi1, PFi2, and PFi3 started at −239, −3,700, and −7,600 base pairs (bp) upstream from the SLC22A6 transcription start site. PFu1, PFu2, PFu3, PFu4, and PFu5 started at −2,055, −147, −2,608, −6,800, and −3,500 bp upstream from the SLC22A8 transcription start site.
SNP detection
Ninety-six DNA samples, 52 of which were from females and 44 from males, representative of a generally healthy population and chosen from 11 ethnic groups, were obtained from the Coriell panel. PCR primers encompassing the PF of human SLC22A6 and SLC22A8 genes were designed using Primer3 (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3.cgi). PF sequences were amplified by PCR from 16 ng DNA in a final volume of 20 μl. Residual primers were removed by treatment with exonuclease 1 and shrimp alkaline phosphatase. Sequencing was performed on an ABI3100 automated sequencer with BigDye terminators (Applied Biosystems, Foster City, CA), and analyzed using the Phred/Phrap/Consed suite of software to arrive at base quality scores (http://www.phrap.org). Polymorphisms and heterozygosity were located using Polyphred and were then also manually confirmed.
Haplotype determination
Genotypes were created for each individual and haplotypes were inferred using PHASE 2.1.1 (http://www.stat.washington.edu/stephens/software.html). PHASE employs an algorithm that partitions each locus into segments and then creates statistically probable haplotypes for each individual; haplotype frequencies are then determined for the population and extrapolated to a population of 10,000 in order to satisfy the requirement of the algorithm. To discern the phylogenetic lineage of each haplotype, a network was created using the software package ARLEQUIN (http://anthro.unige.ch/arlequin/). This network also incorporates the extrapolated haplotypes inferred by PHASE from a larger population size.
Results and discussion
SNP detection
SLC22A6 (OAT1)
An A to G substitution was identified on one of the conserved portions of the SLC22A6 5′ regulatory region (Pfi2), −3,655 bp from the transcription start site of SLC22A6 (Table 1). This SNP was located 8 bp downstream from the consensus Wilm’s tumor gene (WT1) recognition site, a gene critical for the development of a functional kidney. This polymorphism was found in a single individual of Pacific Islander descent (minor allele frequency 0.07 in the Pacific Islander sample, or 0.005 in the total population sample).
SLC22A8 (OAT3)
There were seven SNPs located on the PFs of SLC22A8 (Table 1). Three of these SNPs were found on PFu1. The first SNP (C to G) was found at position −1,901 in an individual of Southeast Asian descent (minor allele frequency 0.05 in the Southeast Asian sample, 0.005 in the total sample). A second SNP (G to C substitution) at position −1,882 was common to all ethnic groups studied (minor allele frequency ranging from 0.10 to 0.64 within specific populations and 0.47 for the total sample). A third SNP (G to A) at −1,851 was also found in all ethnic groups (minor allele frequency within groups ranging from 0.07 to 0.35 and a minor allele frequency of 0.20 in total population). Of note, G1882C and G1851A are located 8 bp upstream and 5 bp downstream, respectively, of the consensus steroid hormone recognition element (SRE).
There were two SNPs located on PFu2 of SLC22A8 (Fig. 1b). The first was an A to G substitution, located 30 bp downstream from the SLC22A8 transcription start site in an individual of Southeast Asian origin (minor allele frequency 0.05, 0.005 overall). The second SNP, a C to A substitution, was found 41 bp downstream in an individual of Japanese descent (Table 1).
Two SNPs were found on PFu3 of SLC22A8 (Fig. 1b). The first was a C to T substitution at position −2,521, found in an individual of the South American Andean gene pool (minor allele frequency of 0.05, 0.005 overall). The second SNP on PFu3 region was an A to G substitution at position −2,520 in an individual of South Saharan African decent (Table 1).
Haplotype determination
There was primarily one SLC22A6 5′ regulatory region haplotype, identical to the chimpanzee sequence for this region. The majority of SLC22A8 5′ regulatory region haplotypes had one of the three-nucleotide sequences at these positions −2,521, −2,520, −1,901, −1,882, −1,851, 30, and 41: CACCGAC (haplotype 1); CACGGAC (haplotype 2); or CACGAAC (haplotype 3) (Fig. 2). The major human SLC22A8 haplotype (haplotype 1) appeared to be derived from the chimpanzee/ancestral haplotype (haplotype 2).

SLC22A8 5′ regulatory region haplotypes. Haplotypes were inferred using PHASE 2.1.1 (http://www.stat.washington.edu/stephens/software.html). Haplotype networks were generated by ARLEQUIN. Each circle represents the corresponding haplotype number. Haplotypes that are directly related and have a relationship of one (a single nucleotide difference) are connected with one solid line. Dashed lines symbolize alternate relationships. The major human SLC22A6 5′ regulatory region haplotype is the same as the chimpanzee/ancestral haplotype (not shown). There are 11 potential human SLC22A8 5′ regulatory region haplotypes; however, over 95% of human SLC22A8 5′ regulatory region haplotypes are either haplotype 1, 2 or 3. The major human SLC22A8 5′ regulatory region haplotype (haplotype 1) appears to have been derived from the chimpanzee/ancestral haplotype (haplotype 2)
These analyses have focused on evolutionarily conserved SLC22A6 and SLC22A8 5′ regulatory regions (phylogenetic footprints designated as Pfis and Pfus, respectively) in order to identify SNPs that could potentially alter the expression of SLC22A6 and SLC22A8 (Fig. 1). With the exception of a G to C transversion at −1,882 (rs11231306) on the SLC22A8 5′ regulatory region, the SNPs identified in this study are novel. Only one polymorphism was found on the SLC22A6 5′ regulatory region, suggesting that transcription of this gene is highly conserved. In contrast, seven polymorphisms were found on the SLC22A8 5′ regulatory region, two of which were present among all ethnic groups studied, suggesting increased nucleotide diversity in the SLC22A8 5′ regulatory region (nucleotide diversity of 0.012) compared to that in the 5′ regulatory region of SLC22A6 (nucleotide diversity of 0.006) (Fig. 1).
The most common SLC22A8 regulatory region haplotype (haplotype 1 from Fig. 2) appears to have been derived from the chimpanzee haplotype by a G to C transversion at −1,882, consistent with either genetic drift or selection pressure for this haplotype in the human population. Five other minor haplotypes (4, 6, 8, 10, and 11) differ from haplotype 1 by other single nucleotide substitutions but retain the C allele at −1,882. As this otherwise conserved position lies adjacent to a SRE, changes at this location could potentially alter SLC22A8 expression in response to steroid signaling.
Analysis of a considerably larger, ethnically diverse, set of human samples will be required to more fully understand less common and population-specific variations in OAT genes. Nevertheless, our data indicate that variation in the noncoding regions of these genes, particularly SLC22A8, may have an impact on human variation in handling of drugs and toxins by the basolateral OATs. Such regulatory polymorphisms are especially important given the low level of nonsynonymous polymorphisms so far observed in coding regions of these genes compared with other members of family such as SLC22A11 and SLC22A12 (Xu et al. 2005). Similar considerations apply to understanding the roles of SNPs in this subfamily on uric acid handling in gout, uric acid nephrolithiasis and human variations in serum uric acid, since OAT1, OAT3, and URAT1 are encoded by three genes proposed to regulate renal urate handling (reviewed in Hediger et al. 2005).
In mice, knockout data strongly suggest that most of the basolateral uptake of prototypical organic anions such as PAH, estrone sulfate and taurocholate, not to mention drugs like loop and thiazide diuretics, can be explained by the combined action of SLC22A6 and SLC22A8 (Sweet et al. 2002; Eraly et al. 2005). It is likely that this also applies to other common organic anion drugs, including NSAIDs, ACE inhibitors, methotrexate, certain antibiotics and certain antivirals (such as adefovir and cidofovir).
Interestingly, in mice with a SLC22A8 functional deletion, SLC22A6 function appears also to be reduced (Sweet et al. 2002). However, the impact of particular noncoding region polymorphisms on the transcription of SLC22A6 and SLC22A8 will need to be evaluated in an appropriate expression system, ideally one utilizing cloned human proximal tubule cultured cells known to express SLC22A6 and SLC22A8 message and protein. From the literature, it is not clear that such a system currently is available. Therefore, as a result of functional redundancy and the possible co-regulation of these closely linked or paired genes, polymorphisms on SLC22A6 or SLC22A8 5′ regulatory and/or coding regions might affect organic anion transport to a much greater degree than polymorphisms in other gene pairs or families.
Together with the continuing analysis of coding region polymorphisms in OATs and other SLC22 family members, the type of approach we describe should eventually provide a useful database of polymorphisms that are potentially of functional importance in either regulating the actual transport (or, conceivably, targeting) process or message/protein levels. This set of polymorphisms can then be analyzed in well-defined clinical populations to determine whether variations correlate with differences in drug handling, susceptibility to toxicity or metabolic abnormalities, as evident in SLC22A12 coding region variants of which affect serum uric acid levels (Enomoto et al. 2002; Anzai et al. 2005).
References
Anzai N, Enomoto A, Endou H (2005) Renal urate handling: clinical relevance of recent advances. Curr Rheumatol Rep 7:227–234
Bleasby K, Hall LA, Perry JL, Mohrenweiser HW, Pritchard JB (2005) Functional consequences of single nucleotide polymorphisms in the human organic anion transporter hOAT1 (SLC22A6). J Pharmacol Exp Ther 314:923–931
Brady KP, Dushkin H, Fornzler D, Koike T, Magner F, Her H, Gullans S, Segre GV, Green RM, Beier DR (1999) A novel putative transporter maps to the osteosclerosis (oc) mutation and is not expressed in the oc mutant mouse. Genomics 56:254–261
Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H (2002) Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417:447–452
Eraly SA, Hamilton BA, Nigam SK (2003) Organic anion and cation transporters occur in pairs of similar and similarly expressed genes. Biochem Biophys Res Commun 300:333–342
Eraly SA, Bush KT, Sampogna RV, Bhatnagar V, Nigam SK (2004a) The molecular pharmacology of organic anion transporters: from DNA to FDA? Mol Pharmacol 65:479–487
Eraly SA, Monte JC, Nigam SK (2004b) Novel slc22 transporter homologs in fly, worm, and human clarify the phylogeny of organic anion and cation transporters. Physiol Genomics 18:12–24
Eraly SA, Vallon V, Vaughn DA, Gangoiti JA, Richter K, Nagle M, Monte JC, Rieg T, Truong DM, Long JM, Barshop BA, Kaler G, Nigam SK (2005) Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knockout mice. J Biol Chem 281:5072–5083
Fujita T, Brown C, Carlson EJ, Taylor T, de la Cruz M, Johns SJ, Stryke D, Kawamoto M, Fujita K, Castro R, Chen C-W, Lin ET, Brett CM, Burchard EG, Ferrin TE, Huang CC, Leabman MK, Giacomini KM (2005) Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1). Pharmacogenet Genomics 15:201–209
Hediger MA, Johnson RJ, Miyazaki H, Endou H (2005) Molecular physiology of urate transport. Physiology 20:125–133
Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV (2004) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 24:5923–5936
Lopez-Nieto CE, You G, Barros EJG, Beier DR, Nigam SK (1996) Molecular cloning and characterization of a novel transport protein with very high expression in the kidney (Abstract). J Am Soc Nephrol 7:1301
Lopez-Nieto CE, You G, Bush KT, Barros EJ, Beier DR, Nigam SK (1997) Molecular cloning and characterization of NKT, a gene product related to the organic cation transporter family almost exclusively expressed in the kidney. J Biol Chem 272:6471–6478
Prohaska SJ, Fried C, Flamm C, Wagner GP, Stadler PF (2004) Surveying phylogenetic footprints in large gene clusters: applications to Hox cluster duplications. Mol Phylogenet Evol 31:581–604
Sweet DH (2005) Organic anion transporter (Slc22a) family members as mediators of toxicity. Toxicol Appl Pharmacol 204:198–215
Sweet DH, Miller DS, Pritchard JB, Fujiwara Y, Beier DR, Nigam SK (2002) Impaired organic anion transport in kidney and choroid plexus of organic anion transporter 3 (Oat3 [Slc22a8]) knockout mice. J Biol Chem 277:26934–26943
Xu G, Bhatnagar V, Wen G, Hamilton BA, Eraly SA, Nigam SK (2005) Analyses of coding region polymorphisms in apical and basolateral human organic anion transporter (OAT) genes [OAT1 (NKT), OAT2, OAT3, OAT4, URAT (RST)]. Kidney Int 68:1491–1499
Acknowledgments
This work was supported by NIH Grant R01AI05769, K08DK064839 and the Department of Veterans Affairs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bhatnagar, V., Xu, G., Hamilton, B.A. et al. Analyses of 5′ regulatory region polymorphisms in human SLC22A6 (OAT1) and SLC22A8 (OAT3). J Hum Genet 51, 575–580 (2006). https://doi.org/10.1007/s10038-006-0398-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0398-1
Keywords
This article is cited by
-
Organic anion transporter 1 is an HDAC4-regulated mediator of nociceptive hypersensitivity in mice
Nature Communications (2022)
-
Pathophysiological and Genetic Basis of Tenofovir-Induced Acute Renal Dysfunction: Strategies and Recent Developments for Better Clinical Outcomes
Current Pharmacology Reports (2022)
-
Uraemic syndrome of chronic kidney disease: altered remote sensing and signalling
Nature Reviews Nephrology (2019)
-
A novel SNP in the 5’ regulatory region of organic anion transporter 1 is associated with chronic kidney disease
Scientific Reports (2018)
-
What do drug transporters really do?
Nature Reviews Drug Discovery (2015)
