
Key Points
-
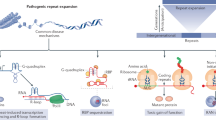
Progress in understanding the molecular pathogenesis of repeat expansion diseases has proceeded at a tremendous pace. Here, we review recent developments in the field, including themes and mechanistic pathways that are unexpectedly shared among different repeat expansion diseases.
-
One of the most striking developments has been the discovery that the pathogenesis of some repeat expansion diseases is mediated by toxic RNA species. New insights into the pathogenesis of myotonic dystrophy type 1 and type 2 have revealed that expanded repeats in RNA sequester and deplete the activity of RNA-binding proteins, which leads to widespread defects in splicing. Evidence is discussed suggesting that this mechanism is involved in the pathogenesis of other repeat diseases.
-
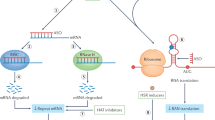
Autophagy is a catabolic process in which cell constituents, such as organelles and proteins, are delivered to the lysosomal compartment for degradation. It has recently become evident that autophagy has an important role in some repeat expansion diseases. As autophagy is amenable to pharmacologic manipulation, this has created optimism about the possibility of targeting autophagy for therapeutic benefit. But is autophagy activated or impaired in repeat expansion disease? And should the aim be to activate autophagy or to suppress it? Answers to these questions have evolved as the role of autophagy in disease has been illuminated.
-
Post-translational modifications profoundly influence the toxicity of polyglutamine disease proteins. The characterization of individual post-translational modifications — including phosphorylation, acetylation and sumoylation — has revealed unanticipated insights into polyglutamine disease pathogenesis.
-
The unexpected finding of polyglutamine inclusions in spinocerebellar ataxia type 8 (SCA8) mice and patients with SCA8, a disease caused by the pathological expansion of a CTG trinucleotide repeat, led to the discovery of bidirectional transcription at the SCA8 locus. Therefore it seems that SCA8 pathogenesis might involve two mechanisms: toxic gain-of-function poly-CUG RNA encoded by the sense strand and expression of polyglutamine-expanded peptide encoded by the antisense strand. Could this mechanism apply to other repeat expansion diseases?
-
Microsatellites have been implicated in regulating chromatin organization and utilization. Emerging evidence suggests that pathological repeat expansions may impair this epigenetic regulation.
Abstract
Repeat expansion mutations cause at least 22 inherited neurological diseases. The complexity of repeat disease genetics and pathobiology has revealed unexpected shared themes and mechanistic pathways among the diseases, such as RNA toxicity. Also, investigation of the polyglutamine diseases has identified post-translational modification as a key step in the pathogenic cascade and has shown that the autophagy pathway has an important role in the degradation of misfolded proteins — two themes that are likely to be relevant to the entire neurodegeneration field. Insights from repeat disease research are catalysing new lines of study that should not only elucidate molecular mechanisms of disease but also highlight opportunities for therapeutic intervention for these currently untreatable disorders.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
La Spada, A. R., Wilson, E. M., Lubahn, D. B., Harding, A. E. & Fischbeck, K. H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79 (1991).
Verkerk, A. J. et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991).
Ashley, C. T. et al. Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nature Genet. 4, 244–251 (1993).
Bell, M. V. et al. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell 64, 861–866 (1991).
Heitz, D. et al. Isolation of sequences that span the fragile X and identification of a fragile X-related CpG island. Science 251, 1236–1239 (1991).
Aslanidis, C. et al. Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature 355, 548–551 (1992).
Brook, J. D. et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808 (1992).
Harley, H. G. et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 355, 545–546 (1992).
Jansen, G. et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nature Genet. 13, 316–324 (1996).
Harris, S., Moncrieff, C. & Johnson, K. Myotonic dystrophy: will the real gene please step forward! Hum. Mol. Genet. 5, 1417–1423 (1996).
Klesert, T. R. et al. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nature Genet. 25, 105–109 (2000).
Sarkar, P. S. et al. Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nature Genet. 25, 110–114 (2000).
Ranum, L. P., Rasmussen, P. F., Benzow, K. A., Koob, M. D. & Day, J. W. Genetic mapping of a second myotonic dystrophy locus. Nature Genet. 19, 196–198 (1998).
Ricker, K. et al. Proximal myotonic myopathy. Clinical features of a multisystem disorder similar to myotonic dystrophy. Arch. Neurol. 52, 25–31 (1995).
Day, J. W. et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 60, 657–664 (2003).
Liquori, C. L. et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867 (2001).
Timchenko, L. T. Myotonic dystrophy: the role of RNA CUG triplet repeats. Am. J. Hum. Genet. 64, 360–364 (1999).
Mankodi, A. et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1773 (2000). This paper shows that expression of a CUG repeat expansion in a non-repeat disease RNA is sufficient to produce a myotonic dystrophy-like phenotype in mice. This was an important step in validating the RNA gain-of-function toxicity model.
Philips, A. V., Timchenko, L. T. & Cooper, T. A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280, 737–741 (1998). This study showed a role for altered splicing in the pathogenesis of myotonic dystrophy and also offered an explanation for how the DM1 gene defect could affect a variety of different cell types and tissues.
Kanadia, R. N. et al. A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980 (2003). This work implicated muscleblind in the splicing pathology caused by the CUG repeat expansions in myotonic dystrophy and provided evidence for the genesis and nature of the splicing alterations in this disease.
Miller, J. W. et al. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448 (2000).
Artero, R. et al. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol. 195, 131–143 (1998).
Begemann, G. et al. muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development 124, 4321–4331 (1997).
Du, H. et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nature Struct. Mol. Biol.24 Jan 2010 (doi:10.1038/nsmb.1720).
Hagerman, R. J. et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57, 127–130 (2001).
Jacquemont, S. et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 72, 869–878 (2003).
Greco, C. M. et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 125, 1760–1771 (2002).
Jin, P. et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 39, 739–747 (2003).
Willemsen, R. et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum. Mol. Genet. 12, 949–959 (2003).
Jin, P. et al. Pur-α binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55, 556–564 (2007).
Sofola, O. A. et al. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55, 565–571 (2007).
Katsuno, M. et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–854 (2002).
Klement, I. A. et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95, 41–53 (1998).
McLeod, C. J., O'Keefe, L. V. & Richards, R. I. The pathogenic agent in Drosophila models of 'polyglutamine' diseases. Hum. Mol. Genet. 14, 1041–1048 (2005).
Li, L. B., Yu, Z., Teng, X. & Bonini, N. M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453, 1107–1111 (2008).
Kundu, M. & Thompson, C. B. Autophagy: basic principles and relevance to disease. Annu. Rev. Pathol. 3, 427–455 (2008).
Sapp, E. et al. Huntingtin localization in brains of normal and Huntington's disease patients. Ann. Neurol. 42, 604–612 (1997).
Kegel, K. B. et al. Huntingtin expression stimulates endosomal–lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 20, 7268–7278 (2000).
Petersen, A. et al. Expanded CAG repeats in exon 1 of the Huntington's disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum. Mol. Genet. 10, 1243–1254 (2001).
Nagata, E., Sawa, A., Ross, C. A. & Snyder, S. H. Autophagosome-like vacuole formation in Huntington's disease lymphoblasts. Neuroreport 15, 1325–1328 (2004).
Vig, P. J., Shao, Q., Subramony, S. H., Lopez, M. E. & Safaya, E. Bergmann glial S100B activates myo-inositol monophosphatase 1 and co-localizes to Purkinje cell vacuoles in SCA1 transgenic mice. Cerebellum 8, 231–244 (2009).
Zander, C. et al. Similarities between spinocerebellar ataxia type 7 (SCA7) cell models and human brain: proteins recruited in inclusions and activation of caspase-3. Hum. Mol. Genet. 10, 2569–2579 (2001).
Montie, H. L. et al. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 18, 1937–1950 (2009).
Yuan, J., Lipinski, M. & Degterev, A. Diversity in the mechanisms of neuronal cell death. Neuron 40, 401–413 (2003).
Yue, Z. et al. A novel protein complex linking the δ2 glutamate receptor and autophagy. Neuron 35, 921–933 (2002).
Nedelsky, N. B., Todd, P. K. & Taylor, J. P. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta 1782, 691–699 (2008).
Taylor, J. P. et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 12, 749–757 (2003).
Ravikumar, B., Duden, R. & Rubinsztein, D. C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 (2002).
Qin, Z. H. et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12, 3231–3244 (2003).
Berger, Z. et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 15, 433–442 (2006).
Pandey, U. B. et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 (2007).
Ravikumar, B. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature Genet. 36, 585–595 (2004). This study showed that pharmacological induction of autophagy ameliorated neurodegeneration in animal models of HD.
Pandey, U. B., Batlevi, Y., Baehrecke, E. H. & Taylor, J. P. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 3, 643–645 (2007).
Sarkar, S. et al. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum. Mol. Genet. 17, 170–178 (2008).
Tanaka, M. et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nature Med. 10, 148–154 (2004).
Davies, J. E., Sarkar, S. & Rubinsztein, D. C. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 15, 23–31 (2006).
Sarkar, S., Davies, J. E., Huang, Z., Tunnacliffe, A. & Rubinsztein, D. C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J. Biol. Chem. 282, 5641–5652 (2007).
Kiffin, R., Bandyopadhyay, U. & Cuervo, A. M. Oxidative stress and autophagy. Antioxid. Redox Signal. 8, 152–162 (2006).
Lum, J. J. et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248 (2005).
McCray, B. A. & Taylor, J. P. The role of autophagy in age-related neurodegeneration. Neurosignals 16, 75–84 (2008).
Komatsu, M. et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 (2006). This study, and the related study by Hara et al . (reference 62), showed the importance of basal, quality-control autophagy in the CNS. Impairment of basal autophagy was found to cause neurodegeneration with accumulation of ubiquitin-positive inclusions.
Hara, T. et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006).
Komatsu, M. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 (2005).
Komatsu, M. et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl Acad. Sci. USA 104, 14489–14494 (2007).
Hollenbeck, P. J. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J. Cell Biol. 121, 305–315 (1993).
Hara, T. et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006).
Kamaya, H., Hayes, J. J. Jr & Ueda, I. Dissociation constants of local anesthetics and their temperature dependence. Anesth. Analg. 62, 1025–1030 (1983).
Nixon, R. A., Yang, D. S. & Lee, J. H. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590–599 (2008).
Atwal, R. S. et al. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 16, 2600–2615 (2007).
Li, X. et al. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol. Cell. Biol. 29, 6106–6116 (2009).
Li, X. et al. Disruption of Rab11 activity in a knock-in mouse model of Huntington's disease. Neurobiol. Dis. 36, 374–383 (2009).
La Spada, A. R. & Taylor, J. P. Polyglutamines placed into context. Neuron 38, 681–684 (2003).
Emamian, E. S. et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38, 375–387 (2003).
Johnson, L. N. & Lewis, R. J. Structural basis for control by phosphorylation. Chem. Rev. 101, 2209–2242 (2001).
Chen, H. K. et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113, 457–468 (2003).
Lim, J. et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452, 713–718 (2008). This study showed that polyglutamine expansion alters the ratio of ataxin 1 between two alternative protein complexes. This discovery has important implications: it suggests that polyglutamine disease pathogenesis might involve subtle alteration of native protein function rather than an entirely novel gain of function.
Humbert, S. et al. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves huntingtin phosphorylation by Akt. Dev. Cell 2, 831–837 (2002).
Rangone, H. et al. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur. J. Neurosci. 19, 273–279 (2004).
Warby, S. C. et al. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum. Mol. Genet. 14, 1569–1577 (2005).
Pardo, R. et al. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J. Neurosci. 26, 1635–1645 (2006).
Difiglia, M. et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat-brain neurons. Neuron 14, 1075–1081 (1995).
Engelender, S. et al. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 6, 2205–2212 (1997).
Kalchman, M. A. et al. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nature Genet. 16, 44–53 (1997).
Li, S. H., Gutekunst, C. A., Hersch, S. M. & Li, X. J. Interaction of huntingtin-associated protein with dynactin P150Glued. J. Neurosci. 18, 1261–1269 (1998).
Li, X. J. et al. A huntingtin-associated protein enriched in brain with implications for pathology. Nature 378, 398–402 (1995).
Gauthier, L. R. et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138 (2004).
Altar, C. A. et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 389, 856–860 (1997).
Baquet, Z. C., Gorski, J. A. & Jones, K. R. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J. Neurosci. 24, 4250–4258 (2004).
Zala, D. et al. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum. Mol. Genet. 17, 3837–3846 (2008).
Colin, E. et al. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 27, 2124–2134 (2008).
Warby, S. C. et al. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol. Cell. Neurosci. 40, 121–127 (2009).
Aiken, C. T. et al. Phosphorylation of threonine-3: implications for huntingtin aggregation and neurotoxicity. J. Biol. Chem. 284, 29427–29436 (2009).
Schilling, B. et al. Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J. Biol. Chem. 281, 23686–23697 (2006).
Gu, X. et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64, 824–840 (2009).
Thompson, L. M. et al. IKK phosphorylates huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187, 1083–1099 (2009).
Fei, E. et al. Phosphorylation of ataxin-3 by glycogen synthase kinase 3β at serine 256 regulates the aggregation of ataxin-3. Biochem. Biophys. Res. Commun. 357, 487–492 (2007).
LaFevre-Bernt, M. A. & Ellerby, L. M. Kennedy's disease. Phosphorylation of the polyglutamine-expanded form of androgen receptor regulates its cleavage by caspase-3 and enhances cell death. J. Biol. Chem. 278, 34918–34924 (2003).
Glozak, M. A., Sengupta, N., Zhang, X. & Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23 (2005).
Mookerjee, S. et al. Posttranslational modification of ataxin-7 at lysine 257 prevents autophagy-mediated turnover of an N-terminal caspase-7 cleavage fragment. J. Neurosci. 29, 15134–15144 (2009).
Jeong, J. W. et al. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 111, 709–720 (2002).
Jeong, H. et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137, 60–72 (2009).
Steffan, J. S. et al. SUMO modification of huntingtin and Huntington's disease pathology. Science 304, 100–104 (2004).
Subramaniam, S., Sixt, K. M., Barrow, R. & Snyder, S. H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324, 1327–1330 (2009).
Day, J. W., Schut, L. J., Moseley, M. L., Durand, A. C. & Ranum, L. P. Spinocerebellar ataxia type 8: clinical features in a large family. Neurology 55, 649–657 (2000).
Koob, M. D. et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nature Genet. 21, 379–384 (1999).
Moseley, M. L. et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nature Genet. 38, 758–769 (2006). This paper showed that expansion of the SCA8 triplet repeat is sufficient to produce neurological disease, and demonstrated that transcription of a non-coding RNA on one strand and transcription of a coding CAG RNA on the opposite strand, which gives rise to a polyglutamine protein, both occur at the SCA8 disease locus.
Mutsuddi, M., Marshall, C. M., Benzow, K. A., Koob, M. D. & Rebay, I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr. Biol. 14, 302–308 (2004).
Daughters, R. S. et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 5, e1000600 (2009).
Margolis, R. L. et al. A disorder similar to Huntington's disease is associated with a novel CAG repeat expansion. Ann. Neurol. 50, 373–380 (2001).
Takeshima, H., Komazaki, S., Nishi, M., Iino, M. & Kangawa, K. Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22 (2000).
Rudnicki, D. D. et al. Huntington's disease-like 2 is associated with CUG repeat-containing RNA foci. Ann. Neurol. 61, 272–282 (2007).
Nishi, M. et al. Motor discoordination in mutant mice lacking junctophilin type 3. Biochem. Biophys. Res. Commun. 292, 318–324 (2002).
Wang, Y. H., Gellibolian, R., Shimizu, M., Wells, R. D. & Griffith, J. Long CCG triplet repeat blocks exclude nucleosomes: a possible mechanism for the nature of fragile sites in chromosomes. J. Mol. Biol. 263, 511–516 (1996).
Ohlsson, R., Renkawitz, R. & Lobanenkov, V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 17, 520–527 (2001).
Filippova, G. N. et al. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nature Genet. 28, 335–343 (2001). This study indicated that altered CTCF binding at the DM1 locus could be involved in DM1 pathology. It also implicated CTCF in a trinucleotide repeat disease for the first time, and suggested that CTCF-binding sites are commonly associated with repeat tracts that are susceptible to disease-causing expansion.
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Kim, T. H. et al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128, 1231–1245 (2007).
Nguyen, P. et al. CTCFL/BORIS is a methylation-independent DNA-binding protein that preferentially binds to the paternal H19 differentially methylated region. Cancer Res. 68, 5546–5551 (2008).
Cho, D. H. et al. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 20, 483–489 (2005).
Chao, W., Huynh, K. D., Spencer, R. J., Davidow, L. S. & Lee, J. T. CTCF, a candidate trans-acting factor for X-inactivation choice. Science 295, 345–347 (2002).
De Biase, I., Chutake, Y. K., Rindler, P. M. & Bidichandani, S. I. Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS ONE 4, e7914 (2009).
Pearson, C. E., Nichol Edamura, K. & Cleary, J. D. Repeat instability: mechanisms of dynamic mutations. Nature Rev. Genet. 6, 729–742 (2005).
Libby, R. T. et al. CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet. 4, e1000257 (2008).
Core, L. J., Waterfall, J. J. & Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848 (2008).
He, Y., Vogelstein, B., Velculescu, V. E., Papadopoulos, N. & Kinzler, K. W. The antisense transcriptomes of human cells. Science 322, 1855–1857 (2008).
Preker, P. et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854 (2008).
Seila, A. C. et al. Divergent transcription from active promoters. Science 322, 1849–1851 (2008).
Ladd, P. D. et al. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 16, 3174–3187 (2007).
Seong, I. S. et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 19, 573–583 (2009).
Kim, M. O. et al. Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation. J. Neurosci. 28, 3947–3957 (2008).
Mulders, S. A. et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl Acad. Sci. USA 106, 13915–13920 (2009).
Wheeler, T. M. et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339 (2009).
Nedelsky, N. B., Todd, P. K. & Taylor, J. P. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta 1782, 691–699 (2008).
Mukherjee, S., Thomas, M., Dadgar, N., Lieberman, A. P. & Iniguez-Lluhi, J. A. Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. J. Biol. Chem. 284, 21296–21306 (2009).
Thomas, M. et al. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J. Biol. Chem. 279, 8389–8395 (2004).
Palazzolo, I. et al. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet. 16, 1593–1603 (2007).
Terashima, T., Kawai, H., Fujitani, M., Maeda, K. & Yasuda, H. SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. Neuroreport 13, 2359–2364 (2002).
Shen, L. et al. Research on screening and identification of proteins interacting with ataxin-3. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 22, 242–247 (2005).
Acknowledgements
The authors' repeat disease research is supported by the US National Institutes of Health (grants R01 NS041648, R01 GM059356 and R01 EY014061 to A.R.L.S., and grants R01 NS053825 and R01 AG031587 to J.P.T.) and by grants from the Muscular Dystrophy Association to A.R.L.S. and J.P.T.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
DATABASES
OMIM
fragile X mental retardation syndrome
spinal and bulbar muscular atrophy
FURTHER INFORMATION
Glossary
- Post-translational modification
-
A covalent chemical modification of a protein that takes place after translation.
- Haploinsufficiency
-
A condition in a diploid organism in which a single functional copy of a gene results in a phenotype, such as a disease.
- Contiguous gene syndrome
-
A multi-symptom disorder caused by the deletion of a large sequence of DNA that encodes several genes.
- Phenocopy
-
A phenotype that is closely similar to a phenotype determined by a different gene.
- Anticipation
-
The tendency of certain diseases to have an earlier age of onset and increasing severity in successive generations.
- Myotonia
-
The failure of muscle to relax immediately after voluntary contraction has stopped.
- Myopathy
-
A disease of the muscle.
- Premutation
-
An unstable mutation that has no phenotypic effect but that is highly likely to mutate to a full mutation during transmission through the germ line, as is seen with some expanding trinucleotide repeats.
- Inclusions
-
Accumulations of proteins and other materials that are visualized as discrete entities at the light microscope level, often after the application of special stains or antibodies.
- Endocytosis
-
The process whereby cells engulf extracellular material through invagination of the plasma membrane to create an endocytic vesicle.
- Neurotrophic factor
-
A small protein that promotes the growth and/or survival of neurons.
- Nucleosome
-
The basic unit of chromatin. A nucleosome contains approximately 146 bp of DNA wrapped around a histone octamer.
- Heterochromatin
-
Parts of chromosomes with an unusual degree of contraction and that consequently have different staining properties from euchromatin at nuclear divisions. Largely composed of repetitive DNA, heterochromatin forms dark bands after Giemsa staining.
Rights and permissions
About this article
Cite this article
La Spada, A., Taylor, J. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11, 247–258 (2010). https://doi.org/10.1038/nrg2748
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrg2748
This article is cited by
-
RNA structure promotes liquid-to-solid phase transition of short RNAs in neuronal dysfunction
Communications Biology (2024)
-
LUSTR: a new customizable tool for calling genome-wide germline and somatic short tandem repeat variants
BMC Genomics (2024)
-
Senataxin helicase, the causal gene defect in ALS4, is a significant modifier of C9orf72 ALS G4C2 and arginine-containing dipeptide repeat toxicity
Acta Neuropathologica Communications (2023)
-
Gelation of cytoplasmic expanded CAG RNA repeats suppresses global protein synthesis
Nature Chemical Biology (2023)
-
The relationship between common mutations in CFTR, AR genes, Y chromosome microdeletions and karyotyping abnormalities with very severe oligozoospermia in Iranian men
Genes & Genomics (2023)
