




Abstract
LIS1 is mutated in the human neuronal migration defect lissencephaly and along with NDEL1 (formerly NUDEL ) participates in the regulation of cytoplasmic dynein function during neuronal development. Targeted disruption of Ndel1 suggested that NDEL1 could have other molecular targets that regulate microtubule organization for proper neuronal migration. To further understanding the molecular mechanism of LIS1 and lissencephaly, we identified the katanin p60 microtubule-severing protein as an additional molecular target of NDEL1. We demonstrate that phosphorylation of NDEL1 by Cdk5 facilitates interaction between NDEL1 and p60, suggesting that P-NDEL1 regulates the distribution of katanin p60. Abnormal accumulation of p60 in nucleus of Ndel1 null mutants supports an essential role of NDEL1 in p60 regulation. Complete loss of NDEL1 or expression of dominant negative mutants of p60 in migrating neurons results in defective migration and elongation of nuclear-centrosomal distance. Our results suggest that NDEL1 is essential for mitotic cell division and neuronal migration not only via regulation of cytoplasmic dynein function but also by modulation of katanin p60 localization and function.
INTRODUCTION
The microtubule cytoskeleton performs a variety of tasks during the cell-division cycle, providing a scaffold for organelle organization and vesicle movement in interphase, and forming a spindle that segregates duplicated chromosomes in mitosis ( 1 – 3 ). The establishment of cell polarity and cell division requires microtubules and makes use of their intrinsic capacity for rapid structural rearrangement ( 4 – 6 ). Numerous modulators play an essential role in microtubule rearrangement to adapt to various biological phenomena ( 7 , 8 ). For example, microtubule stability is promoted to a large degree by microtubule-associated proteins (MAPs) ( 9 ). Classical MAPs, such as MAP2 and Tau, bind to the surface of the microtubule, bridging several tubulin subunits and possibly neutralizing the repulsive negative charge on the microtubule surface ( 10 ). Microtubule-end-binding MAPs, such as CLIP-170 and EB1, may copolymerize with new tubulin subunits, selectively bind to a specific conformation of the microtubule end and/or serve as attachments for growing or shortening microtubules to kinetochores or cellular membranes through interaction with proteins such as the adenomatous polyposis coli (APC) protein and CLIP-associated proteins (CLASPs) ( 11 – 14 ). Binding of the dynein–dynactin motor complex, along with proteins such as LIS1, to microtubule ends and cortical sites or the kinetochore also appears to stabilize microtubules ( 15 ). In contrast, the microtubule-destabilizing factor katanin functions as a severing factor, generating new ends lacking a GTP cap ( 16 – 19 ).
Dynamic regulations of microtubule organization are also considered to play an important role in brain morphogenesis ( 20 , 21 ). Accumulating evidence indicates that mutations of many factors involved in regulation of microtubule dynamics results in defects characterized by abnormal cortical lamination ( 20 ). Lissencephaly is a human brain malformation characterized by a smooth cerebral surface and a disordered organization of the cortical layers resulting from a defect in neuronal migration ( 22 , 23 ). LIS1 was identified as a gene on chromosome 17p13 responsible for a large percentage of classical lissencephaly ( 24 ). Independently, LIS1 was cloned and identified as a subunit of intracellular platelet-activating factor acetylhydrolase ( 25 ). Insights into the microtubule-regulatory function of LIS1 have come from studies of its homologue, nudF, in the filamentous fungus Aspergillus nidulans ( 26 , 27 ). nudF mutations cause defects in nuclear migration, a microtubule-mediated event ( 28 ). Mutations in nudF and nudA, which encodes the heavy chain of cytoplasmic dynein (CDHC), share a similar nuclear migration phenotype, and double mutations have the same phenotype as each single mutation ( 29 ). This suggests that dynein and nudF act in the same nuclear migration pathway ( 30 , 31 ). CDHC is a minus-end of microtubule-directed large motor protein, which is essential for mitotic cell division, organelle transport and nuclear migration ( 32 , 33 ). Lis1/nudF mutations are thought to result primarily from the loss of proper regulation of CDHC ( 30 ).
The Aspergillus NUDE protein is thought to function as a downstream effector of NUDF in regulating nuclear migration ( 34 ). In mammals, two nudE homologues, Ndel1 and Nde1 , were cloned from yeast two-hybrid screens using LIS1 protein ( 35 – 37 ). Nde1 -disrupted mice were viable and exhibited normal cortical lamination, although the mutant cortex has fewer neurons and very thin superficial cortical layers (II–IV) ( 38 ), suggesting that NDE1 plays an important role in neurogenesis, but not in neuronal migration. In contrast, silencing Ndel1 expression by RNA interference (RNAi) resulted in Golgi apparatus fragmentation and cell death ( 39 , 40 ). In addition, loss of function of Ndel1 , Lis1 or dynein by RNAi in developing neocortex impaired neuronal positioning and causes the uncoupling of the centrosome and nucleus ( 41 ).
To address NDEL1 function in neuronal migration, we generated Ndel1-disrupted mice ( 42 ). Surprisingly, Ndel1 , unlike Nde1 , is an essential gene that is also required for neuronal migration in a pathway that partially overlaps with Lis1 . In addition, Ndel1 and Lis1 are genetically interacting for proper regulation of neuronal migration. Interestingly, Ndel1 -disrupted mice displayed similar but distinctive phenotypes with Lis1 mutants. Ndel1 could have an additional role as a regulator of microtubule organization. NDEL1 and NDE1 bind cytoplasmic dynein, and this raises three non-exclusive possibilities: (1) NDEL1 is the major homologue of NUDE in mammals, (2) NDEL1 is more important for dynein regulation and/or (3) NDEL1 binds to other proteins. To address the last possibility, we searched for other interacting proteins and found that NDEL1 has another target, katanin p60, a protein that regulates the remodeling of microtubule organization ( 16 – 19 ). NDEL1 promotes the accumulation of katanin p60 in a P-NDEL1-dependent manner. This interaction appears to be important for proper mitotic cell division and neuronal migration, suggesting that LIS1 and NDEL1 are involved in the regulation of microtubule transport via CDHC and microtubule organization via p60 katanin.
RESULTS
LIS1 and NDEL1 interact with katanin
We performed yeast two-hybrid analysis using LIS1 or NDEL1 as bait, to identify other proteins that interact with LIS1 or NDEL1 and mediate their action. We identified katanin p60 and p80, a heterodimeric protein complex with microtubule-severing activity localized preferentially at the centrosome ( 16 – 19 ). p60 is a member of AAA family of ATPases, and has microtubule-stimulated ATPase and microtubule-severing activity. p80 is a protein containing WD repeats similar to those in LIS1 that are frequently involved in protein–protein interactions ( 17 ). We determined the binding domains between LIS1, NDEL1, CDHC, p60 and p80 by performing directed yeast two-hybrid experiments (Fig. 1 A). p80 and NDEL1 bound the N-terminal domain of p60. In contrast, p60 and NDEL1 bound the C-terminal domain of p80, which is conserved among p80 homologues (conserved domain) ( 19 ). LIS1 bound p80 within the proline-rich domain, but it did not bind to p60. The D1 module which belongs to the AAA (ATPases associated with various cellular activities) domain of CDHC ( 43 ) bound to the microtubule-binding domain of p60 and to the WD domain of p80, suggesting that each protein is capable of localizing at the centrosome in a CDHC-dependent manner. These results were also completely supported by pull-down assays using recombinant proteins (Fig. 1 B). p60 binds NDEL1, p80 and CDHC D1 domain, whereas p80 binds NDEL1, LIS1, CDHC D1 domain and p60. We further examined protein–protein interaction in vivo by immunoprecipitation (IP) after transfection of expression vectors for each protein in COS7 cells, and these in vitro protein interactions were reproduced in cells (Fig. 1 C). Although direct interaction between p60 and LIS1 was observed neither in yeast two-hybrid nor in pull-down assays, we found that p60 was precipitated by LIS1 in cells (Fig. 1 C), suggesting that this interaction is indirect via a complex containing p80 and/or NDEL1.
Phosphorylation of NDEL1 by cdk5 or cdc2 enhances binding affinity with katanin p60
We determined p60 or p80 binding sites in NDEL1 by yeast two-hybrid (Fig. 2 A). p60 and p80 bind NDEL1 at distinct positions. Interestingly, p60 and 14-3-3ε share the same region of NDEL1 for interaction ( 44 ). To examine whether phosphorylation by CDK5/CDC2 might influence interaction with p60, we performed IP in the presence of roscovitine to suppress CDK5 and CDC2 (Fig. 1 C). The suppression of CDK5 and CDC2 significantly diminished interaction between p60 and NDEL1. In contrast, there was no significant effect on the interaction between p80 and other proteins (Fig. 1 C). We also tested whether mutations introduced into the phosphorylation sites of NDEL1 influence the affinity between NDEL1 and p60. The interaction between NDEL1 and p60 was greatly diminished by alanine substitution of the three sites of serine/threonine phosphorylation by CDK5/CDC2 of NDEL1 (Fig. 2 A). In contrast, those mutations had no effect on binding with p80, because p80 binds at the coiled-coil region of NDEL1. The importance of these phosphorylation sites on interaction with p60 was also supported by cell-free pull-down assays (Fig. 2 B). We further examined the effect of phosphorylation in vivo by IP by cotransfection assays using wild-type and mutant forms of Ndel1 (Fig. 2 C). Mutation of NDEL1 sites to alanine clearly reduced its interaction with p60. In contrast, mutation of NDEL1 sites to glutamic acid slightly enhanced its affinity with p60. These IP results were reproduced by pull-down assay using brain lysate (Supplementary Material, Fig. S1). p60 was efficiently precipitated by GST-NDEL1(E), when compared with GST-NDEL1(WT) or GST-NDEL1(A). We tested whether phosphorylation of NDEL1 facilitates binding with p60 using recombinant CDK5, NDEL1 and p60 by pull-down assays (Fig. 2 D). Phosphorylated NDEL1 precipitated p60 more efficiently than non-phosphorylated NDEL1. We further quantitated protein affinity by Biacore using recombinant proteins (Fig. 2 E). p60 appeared to have very low affinity for LIS that was not concentration-dependent, indicating that p60 does not bind LIS1. In contrast, p60 displayed moderate affinity with NDEL1, and that affinity was significantly enhanced by phosphorylation of NDEL1. p80 had moderate affinity with all these proteins, which was not influenced by phosphorylation of NDEL1.
NDEL1 is phosphorylated during mitosis and regulated katanin p60 distribution
To analyze the phosphorylation of NDEL1 in vivo , we generated monoclonal antibodies that specifically recognize phosphorylated NDEL1 by CDK5/CDC2 (Supplementary Material, Fig. S2). Using this anti-phospho NDEL1 antibody, we characterized the phosphorylation of NDEL1 during the cell cycle and in migrating neurons. NDEL1 was preferentially distributed at the centrosome in interphase as shown in previous reports ( 36 , 37 ), but it was not phosphorylated at this stage (Fig. 3 A). Phosphorylation of NDEL1 commenced in mitotic prophase, which preceded an accumulation of total NDEL1 in metaphase and anaphase. Phosphorylated NDEL1 was restricted to the centrosome although a small amount was diffusely localized in the nucleus, where it remained during the progression of mitosis. Although substantial amounts of total NDEL1 were present at telophase, phosphorylated NDEL1 decreased at this stage, suggesting degradation or active dephosphorylation of phosphorylated NDEL1. Specific phosphorylation of NDEL1 was confirmed by using synchronized HeLa cells (Fig. 4 ). The amount of total NDEL1 protein changed during the cell cycle, with a peak at M-phase ( 40 ) [detected by C-6 antibody ( 37 )]. In contrast, accumulation of phosphorylated NDEL1 was maximal at M-phase, and no obvious signal was detected when cells were released from S-phase arrest. We also investigated the cell-cycle-specific interaction of NDEL1 with p60 or p80 by IP. p80 accumulation was similar with total NDEL1 levels [detected by the C-6 antibody ( 37 )], whereas p60 levels were more similar with levels of phosphorylated NDEL1 (detected by the monoclonal antibody).
The intimate association of P-NDEL1 with katanin suggested two possible mechanisms of action of P-NDEL1: (1) P-NDEL1 may activate the microtubule-severing activity of katanin or (2) P-NDEL1 may regulate the distribution of katanin to the centrosome. To examine the first possibility, we made recombinant proteins of katanin p60 and p80. We incubated microtubules in the presence of various combinations of the recombinant proteins, and measured monomerized tubulin (Supplementary Material, Fig. S3). As p60 by itself had severing activity of microtubules, significant activation by NDEL1 was not observed. Therefore, we examined the subcellular localization and colocalization of NDEL1, P-NDEL1, p60 and p80 in epithelial cell lines. Katanin p60 and p80 were centrosomal during interphase and prophase, whereas P-NDEL1 preferentially colocalized to the centrosome only after the initiation of mitosis during prophase in HeLa cells (Fig. 3 B), suggesting that phosphorylated NDEL1 may be sequestered by p60 at the centrosome at beginning of mitosis.
To analyze the role of NDEL1 and/or LIS1 in the distribution of katanin, we generated mouse embryonic fibroblast (MEF) from Ndel1 or Lis1 compound heterozygotes (cko/-), and made Ndel1 or Lis1 null MEFs by CRE-mediated recombination (Supplementary Material, Fig. S4). RFP-tagged CRE was efficiently expressed in the MEFs (Supplementary Material, Fig. S4). While the signal of NDEL1 or LIS1 was already weak in the MEFs from compound heterozygotes, these proteins were completely undetectable by immunohistochemistry 3 days after CRE expression. Using these Ndel1 or Lis1 null cells, we examined the distribution of katanin p60 and p80. CRE expression did not have any influence on the distribution of katanin p60 or p80 in wild-type MEFs (Fig. 5 A, Supplementary Material, Fig. S5). Surprisingly, katanin p60 abnormally accumulated in the nucleus in the NDEL1 null cells (Fig. 5 A), whereas katanin p80 exhibited relatively normal distribution (Supplementary Material, Fig. S4). Loss of LIS1 only mildly affected the distribution of either katanin p60 or p80 (Fig. 5 A, Supplementary Material, Fig. S5). Mislocalization of p60 by loss of endogenous NDEL1 was efficiently rescued by exogenous expression of wild-type NDEL1-GFP (Fig. 5 B), but the mutated NDEL1-GFP in which phosphorylation sites by CDK5 were replaced by alanine was much less efficient at rescue (Fig. 5 B). Interestingly, NDE1-GFP was either less efficient at rescue (Fig. 5 B). p60 binding site of NDEL1 was not conserved between NDEL1 and NDE1 (Supplementary Material, Fig. S6A), suggesting that NDE1 might have weaker affinity to p60. IP also supported this hypothesis (Supplementary Material, Fig. S6B).
Next, we examined microtubule organization in NDEL1 or LIS1 null MEFs. Loss of either LIS1 or NDEL1 caused profound changes in the organization of microtubules of MEFs. Microtubules exhibited a perinuclear concentration in Lis1 -disrupted MEFs ( 31 ) (Fig. 5 C). In contrast, microtubules in Ndel1 -disrupted MEFs were extended and exhibited an amorphous pattern (Fig. 5 C), suggesting that loss of NDEL1 causes aberrant microtubule organization distinct from the disorganization observed in Lis1 mutants. We further examined microtubule organization of MEFs in which wild-type or mutant p60 was overexpressed. The mutant p60 cannot hydrolyze ATP and acts as a dominant inhibitor of the wild-type enzyme (Fig. 5 D) ( 45 ). Overexpression of wild-type p60 resulted in significant loss of microtubule network (Fig. 5 D). In contrast, inactivation of p60 by dominant negative p60 caused amorphous organization of microtubules, which resembled microtubules in Ndel1 -disrupted MEFs (Fig. 5 D). Interestingly, overexpression or inactivation of p60 also resulted in reduction of NDEL1 localization (Fig. 5 D), suggesting that p60 and NDEL1 distributions at centrosome are inter-dependent.
Katanin p60 is localized in brain and neurons, and is essential for proper neuronal migration
We next addressed the brain localization of these proteins in the embryo and adult. p60 and p80 exhibited a punctate distribution pattern at E15.5, similar to the centrosomal LIS1 and NDEL1 pattern previously described ( 36 , 37 ), suggesting that these proteins are colocalized at the centrosome during neuronal development (Fig. 6 ). p80 changed its distribution from this punctate centrosome pattern to a streak-like axonal pattern in postnatal cortex, similar to LIS1 and NDEL1 ( 36 , 37 ), whereas p60 always exhibited a punctate distribution pattern.
We examined phosphorylation of NDEL1, using the anti-phosphorylated NDEL1 antibody, and colocalization with katanin in migrating neurons, using granule cells isolated from cerebellum and cortical neurons isolated from fetal cortex. Although total NDEL1 distributed preferentially at the centrosome, substantial amounts of NDEL1 were present in the soma and axon (Fig. 7 A). In contrast, phosphorylated NDEL1 was more restricted to the centrosome. Total NDEL1 displayed a similar distribution pattern with p80, whereas phosphorylated NDEL1 exhibited a distribution pattern closer to p60 (Fig. 7 A).
Next, we examined p60 distribution and microtubule organization in NDEL1 or LIS1 null neurons. We isolated granule neurons from the cerebellum of Ndel1 or Lis1 compound heterozygotes (cko/-), and made Ndel1 or Lis1 null neurons by CRE-mediated recombination. p60 exhibited relatively normal localization in LIS1 null neurons, whereas substantial amount of p60 was distributed in the nucleus in NDEL1 null neurons as in the case of MEFs (Fig. 7 B). Microtubules formed a perinuclear cage-like structure, converging into the centrosome and projecting into the leading process from the centrosome ( 21 , 41 , 46 ). This microtubule organization maintains nucleus–centrosome coupling (N–C coupling), which is essential for proper nuclear translocation in migrating neurons. We examined N–C coupling in the Ndel1 -disrupted neurons as an indicator of microtubule organization in migrating neurons. In LIS1 null neurons, the distance between nucleus and centrosome was extended two-fold compared with wild-type neurons, suggesting that N–C coupling was defective in LIS1 null neurons ( 41 , 46 ). Similarly, the distance between nucleus and centrosome was elongated in NDEL1 null neurons. The magnitude of extension in NDEL1 null neurons was slightly greater than that in the LIS1 null neurons (Fig. 8 B). We further examined N–C coupling of neurons in which wild-type or mutant p60 was overexpressed. Wild-type neurons in which wild-type p60 was overexpressed displayed clear elongation the distance between nucleus and centrosome compared to untransfected neurons (Fig. 7 B), presumably due to abnormal fragmentation of microtubules. Similarly, overexpression of mutated p60 also resulted in N–C coupling defects (Fig. 7 B), presumably due to lack of normal severing of microtubules. These data suggest that N–C coupling depends on katanin activity and dynein activity.
To define the mechanistic roles of NDEL1, LIS1 and p60 in mammalian neuronal migration, we used mouse cerebellar granule neurons in an in vitro migration assay in wild-type or mutant neurons, or wild-type neurons expressing wild-type or mutated katanin p60. Migration distances in neurons transfected with RED-CRE alone were positioned indistinguishably from untransfected neurons, suggesting that RED-CRE transfection itself had no effect on migration (Fig. 8 ). LIS1 null granule neurons displayed severe migration defects compared with wild-type neurons characterized by a leftward shift of the bin distribution of migration distance, and the mean distance decreased by ∼60% from the WT level (Fig. 8 ). Similar migration defects were observed in NDEL1 null neurons (Fig. 8 ), but the magnitude of these migration defects was slightly milder than those of LIS1 null granule neurons. We also examined the effect of wild-type and mutant p60 overexpression on neuronal migration in wild-type neurons. Neurons in which wild-type p60 was overexpressed displayed mild migration delay compared to untransfected neurons (Fig. 8 ). Similarly, overexpression of mutated p60 resulted in more severe migration defects (Fig. 8 ). These neuronal migration defects uncovered by reaggregation assays are consistent with N–C coupling defects and suggest that NDEL1-dependent p60 recruitment plays an essential role for appropriate regulation of microtubule dynamics in migrating neurons.
DISCUSSION
Ndel1 is one of the two mouse homologues of Nude from Aspergillus , which was identified as a multicopy suppressor of a mutation in the nudF gene, the Aspergillus homologue of LIS1 ( 34 ). LIS1 and NDEL act in the CDHC pathway, which is involved in numerous functions including microtubule organization, vesicle transport (especially from the endoplasmic reticulum to the Golgi apparatus), chromosome separation and nuclear distribution ( 32 , 33 ). To understand the in vivo role of NDEL1, we generated Ndel1 -disrupted mice ( 42 ). Complete loss of Ndel1 caused embryonic lethality at the peri-implantation stage and a deficiency of cell proliferation in the inner cell mass (ICM). In contrast, Nde1 -disrupted mice were viable and exhibited normal cortical lamination, although the mutant cortex has fewer neurons and very thin superficial cortical layers (II–IV) ( 38 ), suggesting that NDE1 plays an important role in neurogenesis, but not in neuronal migration. We found that reduction of Ndel1 dosage resulted in mild neuronal migration defects and Ndel reduction genetically interacted with reduction of Lis1 dosage ( 42 , 47 ), suggesting that Ndel1 and Lis1 participate in a partially overlapping pathway. Examination of LIS1 and NDEL1 functions on dynein regulation, microtubule organization and neuronal migration in vitro suggested that NDEL1 and LIS1 and act in a common pathway to regulate dynein but each has distinct roles in the regulation of microtubule organization and neuronal migration. These findings suggest that NDEL1 may have additional targets, and we searched for other NDEL1 interacting proteins.
We identified katanin as a novel molecular target of NDEL1. Phosphorylation of NDEL1 by CDK5 and/or CDC2 significantly enhanced their interaction. We also demonstrated that NDEL1 was specifically phosphorylated at the centrosome during mitotic cell division and in the perinuclear area in migrating neurons. Katanin p60 and p80 bound CDHC, and target katanin to centrosome in a P-NDEL1-dependent fashion. Our findings suggest that phosphorylation of NDEL1 by CDK5 and/or CDC2 recruits katanin and facilitates the generation of hexamers of p60 that are required for its function ( 18 ). NDEL1-mediated accumulation of p60 at the centrosome may trigger the dynamic reorganization of the microtubule network for chromosomal segregation and neuronal migration.
In Cre-expressing Ndel1 compound heterozygous MEFs, complete inactivation of NDEL1 resulted in drastic alterations of katanin p60 distribution. These results suggest that the embryonic lethality observed in NDEL1 null mutants might be at least partially attributed to the loss of katanin p60 at the centrosome, and/or its abnormal accumulation in the nucleus. These findings also support the hypothesis that NDEL1 and NDEL1 phosphorylation are essential for the correct accumulation of katanin p60 at centrosome. The abnormal microtubule organization in Ndel1 -disrupted MEFs may be attributed to abnormal dynein function due to lack of normal dynein regulation, abnormal katanin function due to lack of normal distribution of p60 or both. LIS1 may have a more generalized role, perhaps as a regulator of CDHC throughout the cell cycle, whereas NDEL1 may have a more temporally specific role, perhaps to regulate the localization of katanin as well as participate in dynein regulation. Dominant negative experiments reveal a role for p60 in N–C coupling and neuronal migration in a cerebellar cluster reaggregate assays, and suggests that the interaction of p60 katanin and NDEL1 is likely to play a role in neuronal migration. Additionally this N–C coupling defect in NDEL1 null neurons could also be partially attributed to aberrant microtubule regulation due to lack of p60 targeting to the centrosome.
Other recent findings are consistent with a model in which phosphorylation of NDEL1 is critical for its function. Loss of 14-3-3ε results in neuronal migration defects, abnormalities of dynein motor function and reduced steady-state levels of P-NDEL1. P-NDEL1 binds directly to 14-3-3ε, which protects P-NDEL1 from phosphatase attack ( 44 ). 14-3-3ε is preferentially localized to the centrosome ( 48 , 49 ). Thus, the maintenance of P-NDEL1 at the centrosome may be critical to its function. In this article, we have demonstrated that one of these functions is to correctly localize the katanin microtubule-severing complex to the centrosome.
Other studies in vitro and in humans suggest that microtubule-severing activity is important for neuronal function. First, a recent report suggests that axonal growth is sensitive to the levels of katanin p60 ( 50 ). As expression levels of p60 are high during rapid phases of axonal growth, but diminish as axons reach their targets. Similarly, in neuronal cultures, katanin levels are high when axons are allowed to grow but drop when the axons are presented with target cells that cause them to stop growing. Second, mutations in Spastin are the most common cause of the autosomal dominant hereditary spastic paraplegia ( 51 ), which is a retrograde axonopathy. Spastin also a member of the ATPases associated with diverse cellular activities (AAA) protein family, and regulates microtubule stability to modulate synaptic structure and function ( 51 – 53 ). Based on the homology of Spastin's ATPase domain to that found in katanin, Spastin has been hypothesized to have microtubule-severing activity. Our observations also suggest a potential role of the AAA protein family in neuronal migration.
NDEL1 appears to be at a critical nexus that integrates upstream signals to modulate dynein and microtubule-severing activities during neuronal migration, and possibly mitosis. NDEL1 binds to LIS1 and is phosphorylated by CDK5, a kinase whose activation depends upon the binding of the p35 and p39 activators ( 54 – 56 ). Binding of 14-3-3ε protects P-NDEL1 from phosphatase attack ( 44 ). LIS1, CDK5, p35, p39 and 14-3-3ε are all required for normal neuronal migration ( 44 , 47 , 54 – 56 ), as is NDEL1. P-NDEL1 binds to CDHC and katanin, and activates dynein activity as well as localization of microtubule-severing activity of katanin. These two activities are complementary in the movement of cargo around in the cell, as well as the nucleus during neuronal migration. It is well known that a microtube cage surrounds the nucleus ( 21 ). The movement of this cage is guided by centrosomal proteins, including LIS1, DCX, NDEL1, dynein and PAR6α ( 41 , 46 , 57 ). As katanin is localized to the centrosome by P-NDEL1, it is in an appropriate place to mediate the reorganization of the microtubule cage during neuronal migration. The mitotic spindle is also regulated by dynein activity originating in the centrosome ( 58 ), and the localization of katanin at the centrosome during mitosis allows it to be in a location necessary for reorganization of spindle microtubules during cell division.
These findings further suggest that neuronal migration uses common biochemical mechanisms with mitotic cell division, providing a linkage between cell division and corticogenesis. Most of the genes specifically activated during mitosis, such as cyclins, are down-regulated in post-mitotic neurons. Interestingly, we have found genes in a common pathway regulating mitosis such as Lis1 , Ndel1 , p60 and p80 which are highly expressed in migrating and adult neurons. These genes may play an essential role in the regulation of migration and maintenance of neuronal networks. Exploration of the common and/or divergent function of these genes during mitosis and neuronal development could provide unique insights into role of such genes in neurogenesis, neuronal migration and post-mitotic neurons.
MATERIALS AND METHODS
Yeast two-hybrid screening and pull-down assay
Full-length Ndel1 or Lis1 cDNA was conjugated to pLexA (Clontech), and used to screen a fetal mouse brain cDNA yeast two-hybrid library as described ( 37 ). To determine the site of binding of NDEL1, LIS1 and CDHC with p60 or p80, several truncated cDNAs of p60 or p80 were cloned into pLexA . Mutated NDEL1 proteins conjugated to pLexA were made by site-directed mutagenesis (Stratagene). The lacZ activity was measured as described ( 37 ).
For the pull-down assay, we generated GST-tagged full-length recombinant p60 and p80 by Bac-to-Bac baculo system (Invitrogen) using SF-9 or High Five insect cells (Pharmingen). For generation of [S 35 -Met]-labeled recombinant proteins of LIS1, NDEL1, p60, p80 and CDHC, full-length cDNAs of Lis1 , Ndel1 , p60 and p80 and Hind III– Sac I cDNA fragment of CDHC carrying D1 domain (1890–3065) were cloned into p TnT vector and translated by the TNT in vitro translation kit (Promega). Each recombinant protein was mixed and incubated 4°C for 1 h in 100 µl of 10 m m Tris–HCl pH 7.5, 150 m m NaCl, 0.1% NP-40 and protease inhibitor cocktail (Roche), followed by incubation with GST-Sepharose beads (Pharmacia) at 4°C for 1 h. The resulting precipitates were washed five times in incubation buffer and were subjected to SDS-PAGE separation, followed by autoradiography. For IP in cells, we cloned each cDNA into pEGFP (Clontech) or pcDNA3.1/His (Invitrogen), and transfected each of them into COS-7 cells. Mutated NDEL1 constructs in which phosphorylation sites were changed to alanine or glutamic acid were generated by Quick-Change (Stratagene). After transfection, cells were lysed by the lysis buffer [10 m m Tris–HCl pH 7.5, 150 m m NaCl, 0.1% NP-40 and protease inhibitor cocktail (Roche) and were incubated with anti-GFP rabbit polyclonal antibody (Clontech) in 100 µl by the same condition, followed by IP. Immunoprecipitates were subjected to SDS-PAGE analysis, followed by immunoblotting using anti-His mouse monoclonal antibody (Clontech). To examine phosphorylation by CDC2/CDK5, non-synchronized HeLa cells were treated with 25 µ m roscovitine for 18 h.
Synchronization of HeLa cells
HeLa cells were cultured in MEM (Sigma Chemical Co.) supplemented with 10% FCS. For mitosis synchronization, HeLa cells were exposed to 2 m m thymidine for 16 h and then resuspended in fresh medium supplemented with 24 µ m 2′-deoxycytidine and allowed to grow for 9 h. Thymidine (2 m m ) was added again for 16 h, causing cells to accumulate near the G1/S boundary. After release from the double thymidine block, cells were harvested at 2-h intervals.
BIAcore analysis
Real-time biospecific interaction analysis was performed using BIAcore 2000 (Pharmacia Biosensor). For generation of analytes, recombinant LIS1 expressed in the insect cells was used. To avoid endogenous phosphorylation, recombinant NDEL1 was expressed in bacteria using pGEX vector (Amersham). Full-length NDEL1 was not soluble. Thereby, the BclI-BsrDI fragment of Ndel1 (1–298) was cloned and expressed in Escherichia coli. Phosphorylation of recombinant NDEL1 was performed by recombinant CDK5/P35 21 . Each protein was immobilized to CM5 biosensor microchips using N -hydroxy-succinimide and N -ethyl- N ′-dimethylaminopropyl carbodiimide after cleavage of GST-tag by thrombin to avoid GST effect for interaction. For ligands, katanin p60 or p80 was generated by Bac-to-Bac baculo system (Invitrogen) as mentioned earlier. Kinetic binding experiments were performed under conditions that mimic the katanin-severing experiments. The same running buffer was used (50 m m KCl, 20 m m HEPES pH 7.4, 1 m m DTT, 0.1% NP-40) and the instrument was equilibrated at 24°C. Kinetic data were collected by injecting various concentration of p60 or p80 (p60: 125, 250 and 500 n m , p80: 31.25, 62.5 and 125 n m ). Kinetic rate constants were determined by fitting the corrected response data to a simple bimolecular interaction model, A + B = AB . Kinetic constants ka and kd are the association constant and dissociation rate constants, respectively.
Generation of the antibodies against p60, p80 and phosphorylated Ndel1
To make anti-p60 or p80 antibodies, we immunized a KLH-conjugated p60 (343–355 or 478–498 AAs) or p80 (288–297 or 324–366 AAs) oligopeptide to New Zealand white rabbits. The antisera were purified by immunized oligopeptide-coupled HiTrap columns (Amersham). Six-week-old BALB/c mice were immunized with 50 µg of KLH-conjugated oligopeptide (CTRKSAPSS 198 PTLD, CLSLPAT 219 PVGKFT and CENSFPS 231 PKAIPN) in Freund's complete adjuvant (Difco), with a second injection in Freund's incomplete adjuvant (Difco) 14 days later. The splenocytes of the immunized mouse were fused to SP2/0 mouse myeloma cells using a standard polyethylene glycol protocol. The fused cell population was resuspended in hypoxanthine aminotropterin thymidine selection medium (Life) and plated into 96-well flat-bottomed culture plates. Positive clones that specifically recognize phosphorylated NDEL1 were subcloned to obtain clonal hybridomas.
Histological examination and immunohistochemistry
After perfusion with Bouin's or 4% PFA fixative, tissues from wild-type and various mutant mice were subsequently embedded in paraffin and sectioned at 5 µm thickness. After deparaffination, endogenous peroxidase activity was blocked by incubating the sections in 1.5% peroxide in methanol for 20 min. The sections were then boiled in 0.01 mol/l citrate buffer, pH 6.0, for 20 min and cooled slowly. Before staining, the sections were blocked with rodent block (LabVision) for 60 min. Some samples were fixed in 2% paraformaldehyde and 0.2% glutaraldehyde for 10 min at RT and blocked in 1 mg/ml NaBH 4 for 30 min at 4°C. After primary culture from each tissue, cells were fixed in cold methanol (−20°C) and post-fixed by 3% formaldehyde. Incubation with 0.15% Triton X-100 was used for cell permeabilization after fixation. Immunohistochemistry was performed based on the standard procedure.
Primary cell culture, transfection and immunofluorescence
Establishment of cerebellar granule cells or MEF was performed as previously described ( 41 , 46 ). HeLa cells or granule neurons were examined for localization of phosphorylated NDEL1, katanin p60 and p80 by immunohistochemistry using anti-phosphorylated NDEL1 antibody, anti-katanin p60 antibody or anti-katanin p80 antibody. An RFP -tagged Cre expression vector was introduced into these primary culture cells using LIPOFECTAMINE 2000 reagents (Invitrogen). After CRE-mediated inactivation of Lis1 or Ndel1 gene, these cells were subjected to immunohistochemistry using anti-katanin p60 antibody or anti-katanin p80 antibody, or rescue experiments. For rescue experiments, GFP-conjugated wild-type Ndel1 [NDEL1(WT)-GFP], mutated Ndel1 [NDEL1(MT)-GFP] or Nde1 [NDE1-GFP] was simultaneously introduced with CRE expression vector. Microtubule organization was detected by monoclonal anti-β-tubulin antibody (Clone TUB 2.1; Sigma).
Reaggregate neuronal migration assay
Cerebellar granule neurons were dissociated from postnatal 5-day mice ( 46 , 47 ) and transfected with various vectors by using LipoTrust SR transfection reagents (Hokkaido System Science Co., Ltd) according to the manufacturer's instruction with a small modification. Briefly, 2×10 6 cells were transfected with 3 µg DNA vector in 4 µl LipoTrust SR reagents in 100 µl d -MEM media without fetal bovine serum for 20 h, resulting in 30–40% transfection efficiency. Reaggregates were transferred to poly- l -lysine-(Sigma-Aldrich) and laminin-(Sigma-Aldrich) treated slides. After 15 h, the distance between cell bodies and the edge of reaggregates was measured. The details of the experimental procedure are described ( 41 , 46 ). To generate a dominant inhibitor of mouse p60, we introduced a single amino acid substitution of the P-loop lysine for alanine (K255A) ( 45 ), and fused this mutant p60 with RFP driven by CMV promoter. An RFP-tagged Cre expression vector, a wild-type p60 vector and a mutated p60 vector were transfected as described above.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We thank Yoshihiko Funae, Hiroshi Iwao, Toshio Yamauch, Yoshitaka Nagai and Robert L. Nussbaum for generous support and encouragement. We are grateful to Yuzuru Yamauchi, Shino Okumura, Michiyo Ishida, Katsumi Hagiwara, Nobuko Tominaga, Masami Suzuki and Masashi Harada for mouse breeding and technical support. We thank Shin-Ichi Hisanaga for providing a recombinant protein of cdk5/p35 and Francis J. McNally for providing mutated kinesin. This work was supported by an NIH grant (NS41030) to A.W.-B., a Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Science, Sports and Culture of Japan to A.Y., Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan to K.T.-O. and a Grant-in-Aid for Scientific Research (B) from the Ministry of Education, Science, Sports and Culture of Japan to S.H. This work was also supported by Nissan Science Foundation to K.T.-O. and Sankyo Foundation of Life Science and Japan Brain Foundation to S.H.
Conflict of Interest statement . None declared.
Present address: Department of Anatomy, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan.
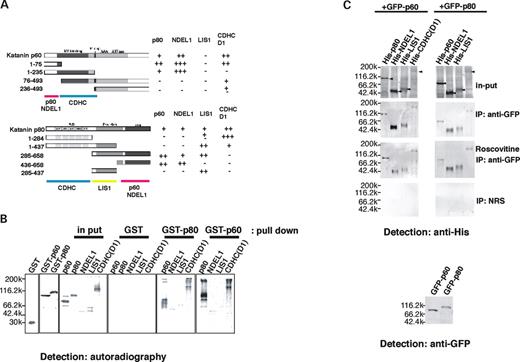
Figure 1. Protein interaction of LIS1, NDEL1, CDHC, katanin p60 and p80. ( A ) Determination of binding sites between katanin p60 and p80 with LIS1, NDEL1 and CDHC by yeast two-hybrid analysis. Katanin p60 and p80 fragments used for binding are shown on the left-hand side. Lac Z activity is shown on the right-hand side. Summary of binding domains of each combination is shown at the bottom of each panel. ( B ) Examination of protein interaction by pull-down assay. S 35 -methionine labeled recombinant proteins made by TNT in vitro translation system were incubated with GST-tagged katanin p60 or p80 made by baculo system, and pulled down by GST column. Input lane indicates 1/20th the amount of proteins that were subjected to pull-down assay. Recombinant proteins used for pull down are indicated at the top. Molecular size is indicated on the left-hand side. The same protein interactions observed in yeast were reproduced. ( C ) Examination of protein interaction in vivo by IP. Expression vectors for GFP-tagged katanin p60 or p80 were transfected into COS7 cells with expression vectors for His-tagged katanin p60 , p80 , Lis1, Ndel1 or CDHC . IP was performed using anti-GFP antibody, followed by detection with anti-His antibody. To inhibit CDK5/CDC2 activity, roscovitine was administrated to culture media. Molecular size was indicated on the left-hand side. NRS: normal rabbit serum. Input lanes indicate 1/20th amount of cell lysate used for IP.
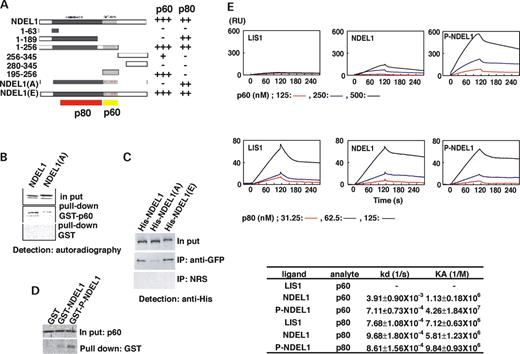
Figure 2. Phosphorylation of NDEL1 enhances binding affinity with katanin p60. ( A ) The effect of phosphorylation of NDEL1 on interaction with katanin p60 and p80 was examined. A yeast two-hybrid assay was performed. Three phosphorylation sites of yeast two-hybrid construct were changed to alanine or glutamic acid. Mutations to alanine (NDEL1(A)) specifically abrogated interaction between NDEL1 and katanin p60, whereas mutations to glutamic acid (NDEL1(E)) did not. Summary of binding domains of each combination is shown at the bottom of each panel. ( B ) A pull-down assay using recombinant proteins expressed in insect cells was carried out. Recombinant proteins used for pull down were indicated at the right-hand side. Input lane indicates 1/20th amount of proteins that were subjected to pull-down assay. Mutation to alanine (NDEL1(A)) significantly reduced the binding between NDEL1 and katanin p60. ( C ) IP was performed using wild-type NDEL1 or mutated NDEL1 in which phosphorylation sites were changed to alanine (His-NDEL1(A)) or glutamic acid (His-NDEL1(E)). Wild-type NDEL1 and His-NDEL1(E) were efficiently precipitated with GFP-p60. In contrast, the efficiency of IP was dramatically reduced by His-NDEL1(A). ( D ) Pull-down assay was performed using in vitro phosphorylated NDEL1. To test whether phosphorylation of NDEL1 by CDK5 facilitates binding with p60, in vitro phosphorylated NDEL1 was used for pull down, and compared with non-phosphorylated NDEL1. Phosphorylation of NDEL1 significantly facilitated pull down of p60. Input lane indicates 1/20th amount of p60 which was subjected to pull-down assay. ( E ) Quantitation of the protein interaction using BIAcore. Characterization of the binding of recombinant protein of LIS1, NDEL1 and phosphorylated NDEL1 (P-NDEL1) with the recombinant p60 or p80 using surface plasmon resonance detection. LIS1, NDEL1 and P-NDEL1 were immobilized on the sensor chip surface. The traces are representative of three different experiments. The specific binding signal shown was obtained by subtracting the signal measured in the absence of immobilized recombinant proteins from the signal in the presence of the fusion protein. The relative change in refractive index expressed as a change in resonance angle (RU) as binding of p60 or p80 to an immobilized LIS1, NDEL1 and P-NDEL1 on a sensor surface in the BIAcore (BIAcore AB) apparatus was monitored over time. p80 was a relatively insoluble protein. Therefore, the concentration used was one-fourth of p60. Kinetic analysis is summarized in the bottom table. Consistent with yeast two-hybrid and pull-down assays, p60 binds NDEL1, and p80 binds both LIS1 and NDEL1. Note that NDEL1 phosphorylation by CDK5 significantly enhanced the affinity with p60, whereas such enhancement was not observed with p80.
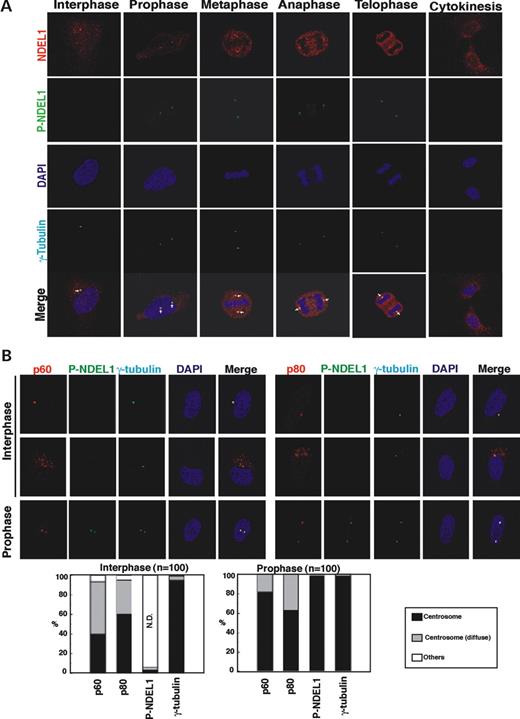
Figure 3. Analysis of NDEL1 phosphorylation by the monoclonal antibody against phosphorylated NDEL1 and localization of katanin. ( A ) Immunocytochemistry revealed that NDEL1 was phosphorylated at M-phase in HeLa cells. Arrows indicate centrosome at interphase and spindle poles at M-phase. ( B ) NDEL1 phosphorylation and colocalization of P-NDEL1, katanin p60 and p80 during mitosis in HeLa cells. Accumulation of phosphorylated NDEL1 and p60 in prophase spindle poles. Upper panel: immunofluorescence staining with antibodies against P-NDEL1, p60 and p80 indicates colocalization of P-NDEL1, p60 and p80 in HeLa cells. Bottom panel: statistical analysis of distribution of P-NDEL1, p60 and p80 in HeLa cells demonstrates accumulation of P-NDEL1 and p60 in prophase spindle poles. n is the number of HeLa cells measured for each examination. Staining patterns of HeLa cells were classified into three categories as follows: centrosome, tightly stained at the centrosome as a spot or spots; centrosome (diffuse), diffusely stained in centrosome and surrounding centrosomal areas; others, other staining patterns than centrosome and centrosome (diffuse); N.D., not detected.
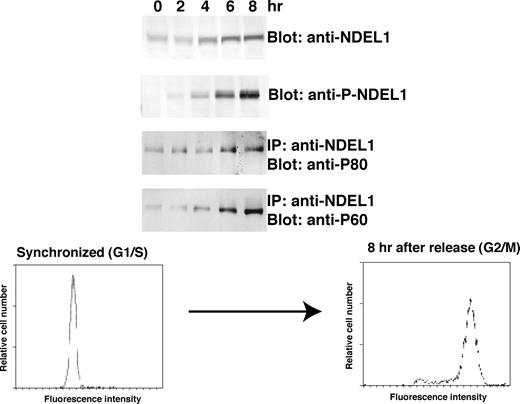
Figure 4. Synchronized HeLa cell culture. HeLa cells synchronized in G1/S with double thymidine block were lysed after release of block at the given time. FACS pattern of synchronized HeLa was displayed at the bottom. NDEL1 and phosphorylated NDEL1 were detected by the C-6 antibody and the anti-phosphorylated NDEL1 monoclonal antibody, respectively. NDEL1 and P-NDEL1 were immunoprecipitated with the C-6 antibody. Coprecipitated p60 or p80 was detected by western blot. Note p60 displayed a similar pattern as phosphorylated NDEL1, whereas the pattern of p80 was similar to the pattern of total NDEL1.
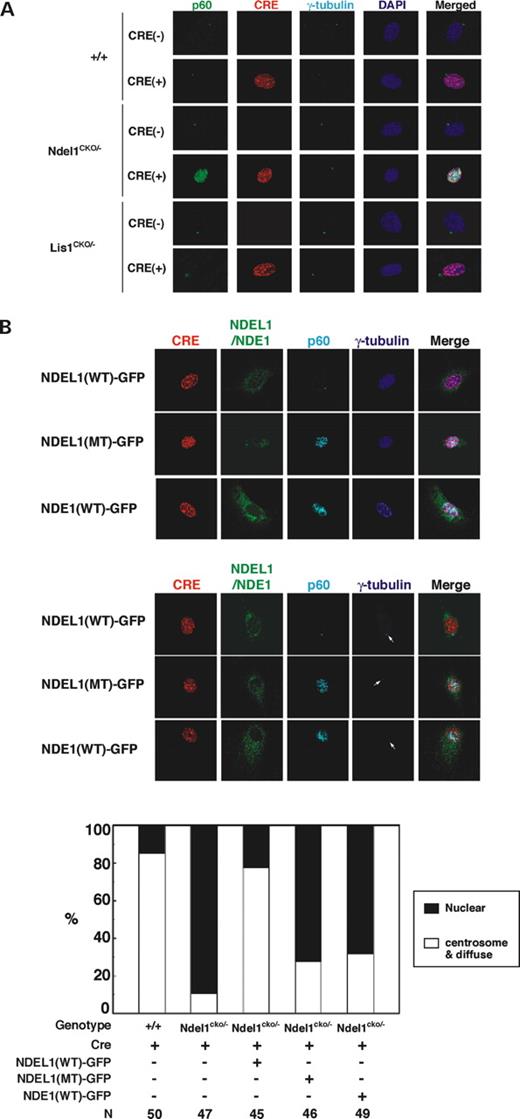
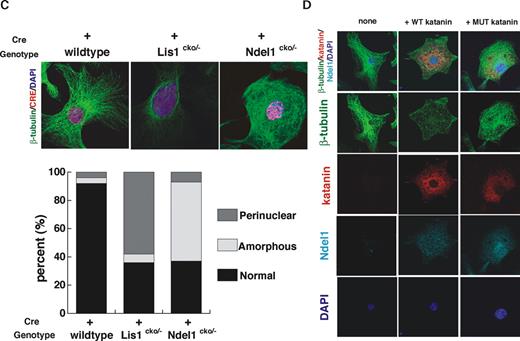
Figure 5. Abnormal distribution of katanin p60 in NDEL1 null cells. ( A ) RFP-tagged Cre expression plasmid which was driven by elongation factor promoter was introduced into MEFs derived from wild-type embryo, Ndel1 compound heterozygous embryo and Lis1 compound heterozygous embryo. Three days after transfection, distribution of katanin p60 was examined. Katanin p60 exhibited abnormal nuclear distribution in Ndel1 null cells. ( B ) Transfection of wild-type NDEL1-GFP, but neither triple mutant NDEL1-GFP nor NDE1-GFP rescued the mislocalization of katanin p60 in Ndel1 null cells. Upper panel: immunofluorescence staining with anti-katanin p60 in wild-type NDEL1-GFP(NDEL1(WT)-GFP), triple mutant NDEL1-GFP(NDEL1(MT)-GFP) or NDE1-GFP(NDE1-GFP) transfected Ndel1 null cells. RFP-tagged Cre and NDEL1(WT)-GFP, NDEL1(MT)-GFP or NDE1-GFP were cotransfected into Ndel1 compound heterozygous MEF cells. Three days after transfection, distribution of katanin p60 was examined. Bottom panel: statistical analysis of distribution of katanin p60 in various Ndel1 -deficient cells. White arrows indicate centrosome. n is the number of MEFs measured for each examination. Staining patterns of MEFs were classified into two categories: nuclear, predominantly stained in nucleus; centrosome and diffuse, tightly stained at centrosome or diffusely stained in centrosome and its surrounding areas. ( C ) Aberrant microtubule organization in LIS1 or NDEL1 null MEFs. Loss of LIS1 causes an enrichment of microtubules near the nucleus, whereas loss of NDEL1 results in extended amorphous microtubules. The frequency of aberrations is summarized at the bottom. n is the number of MEFs measured for each examination. ( D ) Overexpression of wild-type or dominant negative katanin p60, and their effects on microtubule organization and NDEL1 distribution. Overexpression of wild-type p60 resulted in significant reduction of β-tubulin signal due to fragmentation of microtubules (left). In contrast, inhibition of p60 by dominant negative p60 resulted in an extended amorphous pattern of microtubules, which resembled the disruption of microtubule organization seen in the Ndel1 -disrupted MEFs (right). Centrosomal localization of NDEL1 became reduced in both cases, suggesting that functionally NDEL1 and p60 are tightly related.
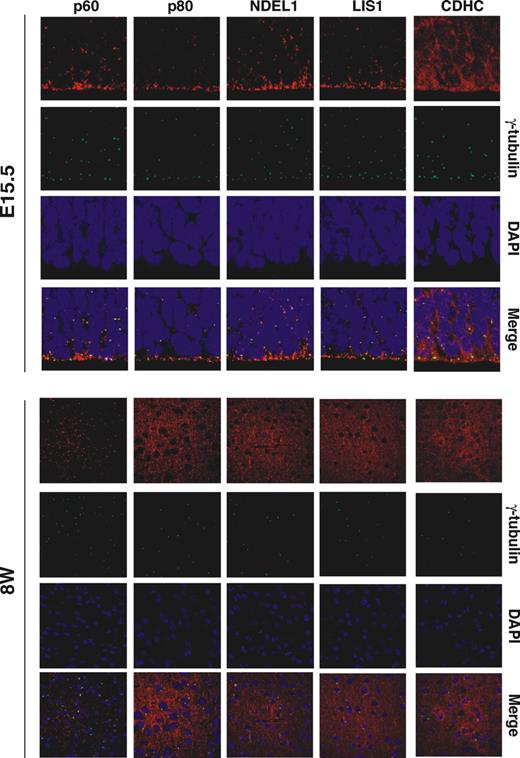
Figure 6. Katanin p60 and p80 localization in the developing brain. Distribution of p60, p80, LIS1, NDEL1 and CDHC in brains was examined. Each protein displayed a punctate distribution pattern in the developing brain (E15.5). In contrast, katanin p80, NDEL1, LIS1 and CDHC changed to streak-like axonal distribution pattern in the adult brains (8W), suggesting that they were localized to the axon. Katanin p60 displayed a punctate distribution pattern in the embryo and the adult.
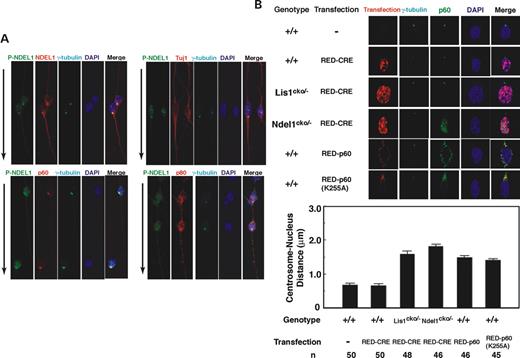
Figure 7. Subcellular localization of katanin p60 and p80 in migrating neurons. ( A ) P-NDEL1 predominantly localized at the centrosome in migrating granular neurons from cerebellum. Double immunostaining with anti-P-NDEL1 mAb and anti-NDEL1 polyclonal Ab (upper left), anti-P-NDEL1 mAb and Tuj1 class III β-tubulin (upper right), anti-P-NDEL1 mAb and anti-katanin p60 polyclonal Ab (bottom left) and anti-P-NDEL1 mAb and anti-Katanin p80 polyclonal Ab (bottom right). Arrows indicate the direction of migration. ( B ) Aberrant distribution of p60 and N–C coupling defect in granule neurons. Localization of p60 in Lis1 null neurons was relatively normal, whereas p60 exhibited abnormal nuclear distribution in Ndel1 null neurons. Wild-type neurons expressing RFP-CRE display normal N–C coupling compared to untransfected neurons. Lis1 -deficient neurons displayed N–C coupling defect characterized by the extended distance between a nucleus and centrosome. N–C coupling defect was also observed in Ndel1 null neurons, and neurons in which wild-type katanin or mutated katanin was overexpressed. n is the number of neurons measured for each examination.
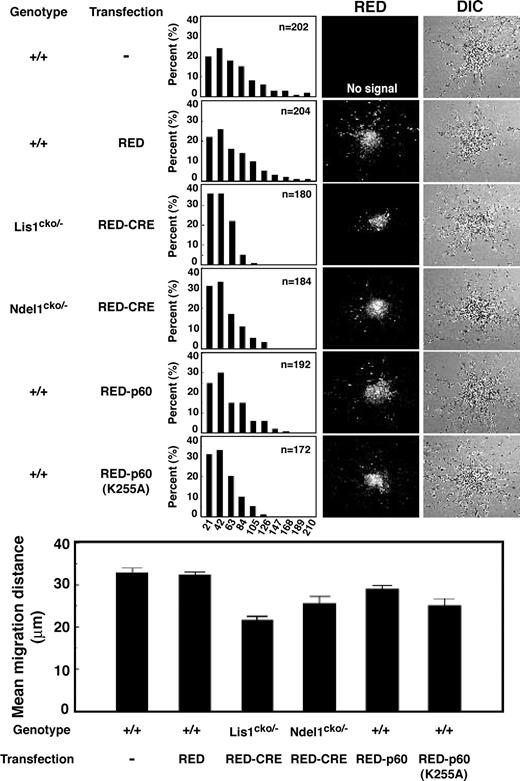
Figure 8. Migration defect associated with aberrant distribution of p60 or mutated p60. Migration assay using cerebellar granule neurons. The migration distance of each neuron in 15 h is binned. Wild-type neurons expressing RFP-CRE display normal migration distance compared to untransfected neurons, whereas Lis1 or Ndel1 null neurons display a shift in the distribution of bins toward the right. Neurons expressing wild-type p60 show mild migration delay. In contrast, neurons expressing mutated p60 exhibit more severe migration defect. Mean migration distances are summarized at the bottom. n is the number of neurons measured for each examination.
References
Blagden, S.P. and Glover, D.M. (
Karcher, R.L., Deacon, S.W. and Gelfand, V.I. (
McIntosh, J.R., Grishchuk, E.L. and West, R.R. (
Jaspersen, S.L. and Winey, M. (
Pearson, C.G. and Bloom, K. (
Rieder, C.L. and Salmon, E.D. (
Hirokawa, N. (
Teng, J., Takei, Y., Harada, A., Nakata, T., Chen, J. and Hirokawa, N. (
Harada, A., Oguchi, K., Okabe, S., Kuno, J., Terada, S., Ohshima, T., Sato-Yoshitake, R., Takei, Y., Noda, T. and Hirokawa, N. (
Mathur, J., Mathur, N., Kernebeck, B., Srinivas, B.P. and Hulskamp, M. (
Mimori-Kiyosue, Y., Grigoriev, I., Lansbergen, G., Sasaki, H., Matsui, C., Severin, F., Galjart, N., Grosveld, F., Vorobjev, I., Tsukita, S. and Akhmanova, A. (
Rickard, J.E. and Kreis, T.E. (
Watanabe, T., Wang, S., Noritake, J., Sato, K., Fukata, M., Takefuji, M. Nakagawa, M., Izumi, N., Akiyama, T. and Kaibuchi, K. (
Tai, C.Y., Dujardin, D.L., Faulkner, N.E. and Vallee, R.B. (
Hartman, J.J., Mahr, J., McNally, K., Okawa, K., Iwamatsu, A., Thomas, S., Cheesman, S., Heuser, J., Vale, R.D. and McNally, F.J. (
Hartman, J.J. and Vale, R.D. (
McNally, F.J. and Vale, R.D. (
McNally, K.P., Bazirgan, O.A. and McNally, F.J. (
Gupta, A., Tsai, L.H. and Wynshaw-Boris, A. (
Rivas, R.J. and Hatten, M.E. (
Dobyns, W.B. (
Dobyns, W.B., Reiner, O., Carrozzo, R. and Ledbetter, D.H. (
Reiner, O., Carrozzo, R., Shen, Y., Wehnert, M., Faustinella, F., Dobyns, W.B., Caskey, T. and Ledbetter, D.H. (
Hattori, M., Adachi, H., Tsujimoto, M., Arai, H. and Inoue, K. (
Morris, R.N., Efimov, V.P. and Xiang, X. (
Morris, R.N. (
Xiang, X., Osmani, A.H., Osmani, S.A., Xin, M. and Morris, N.R. (
Willins, D.A., Liu, B., Xiang, X. and Morris, N.R. (
Faulkner, N.E., Dujardin, D.L., Tai, C.Y., Vaughan, K.T., O'Connell, C.B., Wang, Y. and Vallee, R.B. (
Smith, D.S., Niethammer, M., Ayala, R., Zhou, Y., Gambello, M.J., Wynshaw-Boris, A. and Tsai, L-H. (
Vallee, R.B., Williams, J.C., Varma, D. and Barnhart, L.E. (
Efimov, V.P. and Morris, N.R. (
Feng, Y., Olson, E.C., Stukenberg, P.T., Flanagan, L.A., Kirschner, M.W. and Walsh, C.A. (
Niethammer, M., Smith, D.S., Ayala, R., Peng, J., Ko, J., Lee, M., Morabito, M. and Tsai, L. (
Sasaki, S., Shionoya, A., Ishida, M., Gambello, M.J., Yingling, J., Wynshaw-Boris, A. and Hirotsune, S. (
Feng, Y. and Walsh, C.A. (
Liang, Y., Yu, W., Li, Y., Yang, Z., Yan, X., Huang, Q. and Zhu, X. (
Yan, X., Li, F., Liang, Y., Shen, Y., Zhao, X., Huang, Q. and Zhu, X. (
Shu, T., Ayala, R., Nguyen, M.D., Xie, Z., Gleeson, J.G. and Tsai, L.H. (
Sasaki, S., Mori, D., Toyo-Oka, K., Chen, A., Garrett-Beal, L., Muramatsu, M., Miyagawa, S., Hiraiwa, N., Yoshiki, A., Wynshaw-Boris, A. and Hirotsune, S. (
Mocz, G. and Gibbons, I.R. (
Toyo-Oka, K., Shionoya, A., Gambello, M.J., Cardoso, C., Leventer, R., Ward, H.L., Ayala, R., Tsai, L.H., Dobyns, W., Ledbetter, D., Hirotsune, S. and Wynshaw-Boris, A. (
Buster, D., McNally, K. and McNally, F.J. (
Tanaka, T., Serneo, F.F. Higgins, C., Gambello, M.J., Wynshaw-Boris, A. and Gleeson, J.G. (
Hirotsune, S., Fleck, M.W., Gambello, M.J., Bix, G.J., Chen, A., Clark, G.D., Ledbetter, D.H., McBain, C.J. and Wynshaw-Boris, A. (
Andersen, J.S., Wilkinson, C.J., Mayor, T., Mortensen, P., Nigg, E.A. and Mann, M. (
Pietromonaco, S.F., Seluja, G.A., Aitken, A. and Elias, L. (
Karabay, A., Yu, W., Solowska, J.M., Baird, D.H. and Baas, P.W. (
Errico, A., Ballabio, A. and Rugarli, E.I. (
Sherwood, N.T., Sun, Q., Xue, M., Zhang, B. and Zinn, K. (
Trotta, N., Orso, G., Rossetto, M.G., Daga, A. and Broadie, K. (
Chae, T., Kwon, Y.T., Bronson, R., Dikkes, P., Li, E. and Tsai, L.H. (
Ko, J., Humbert, S., Bronson, R.T., Takahashi, S., Kulkarni, A.B., Li, E. and Tsai, L.H. (
Ohshima, T., Ward, J.M., Huh, C.G., Longenecker, G., Veeranna, Pant, H.C., Brady, R.O., Martin, L.J. and Kulkarni, A.B. (
Solecki, D.J., Model, L., Gaetz, J., Kapoor, T.M. and Hatten, M.E. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}