Aryl Hydrocarbon Receptor Repressor and TiPARP (ARTD14) Use Similar, but also Distinct Mechanisms to Repress Aryl Hydrocarbon Receptor Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
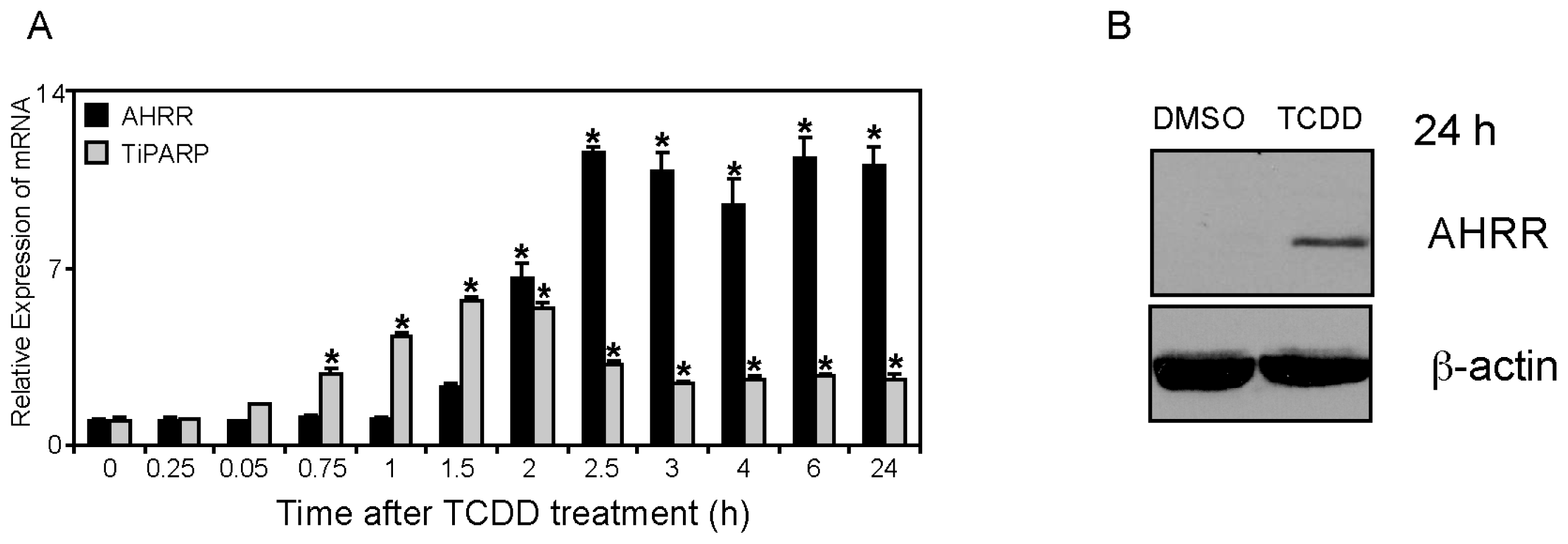
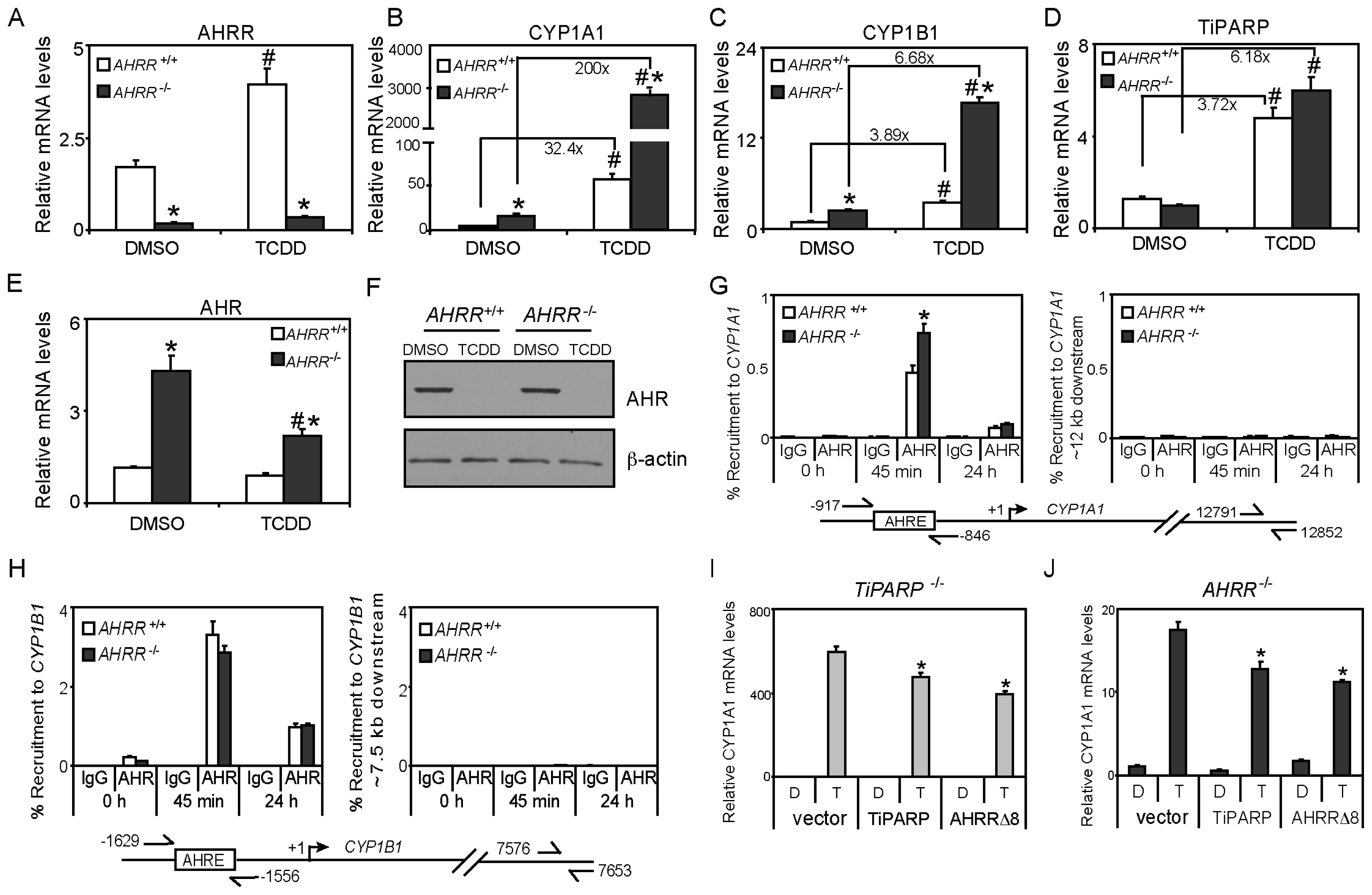
2.1. Differential TCDD (2,3,7,8-Tetrachlorodibenzo-p-dioxin)-dependent Regulation of TiPARP and AHRR (Aryl Hydrocarbon Repressor) Expression
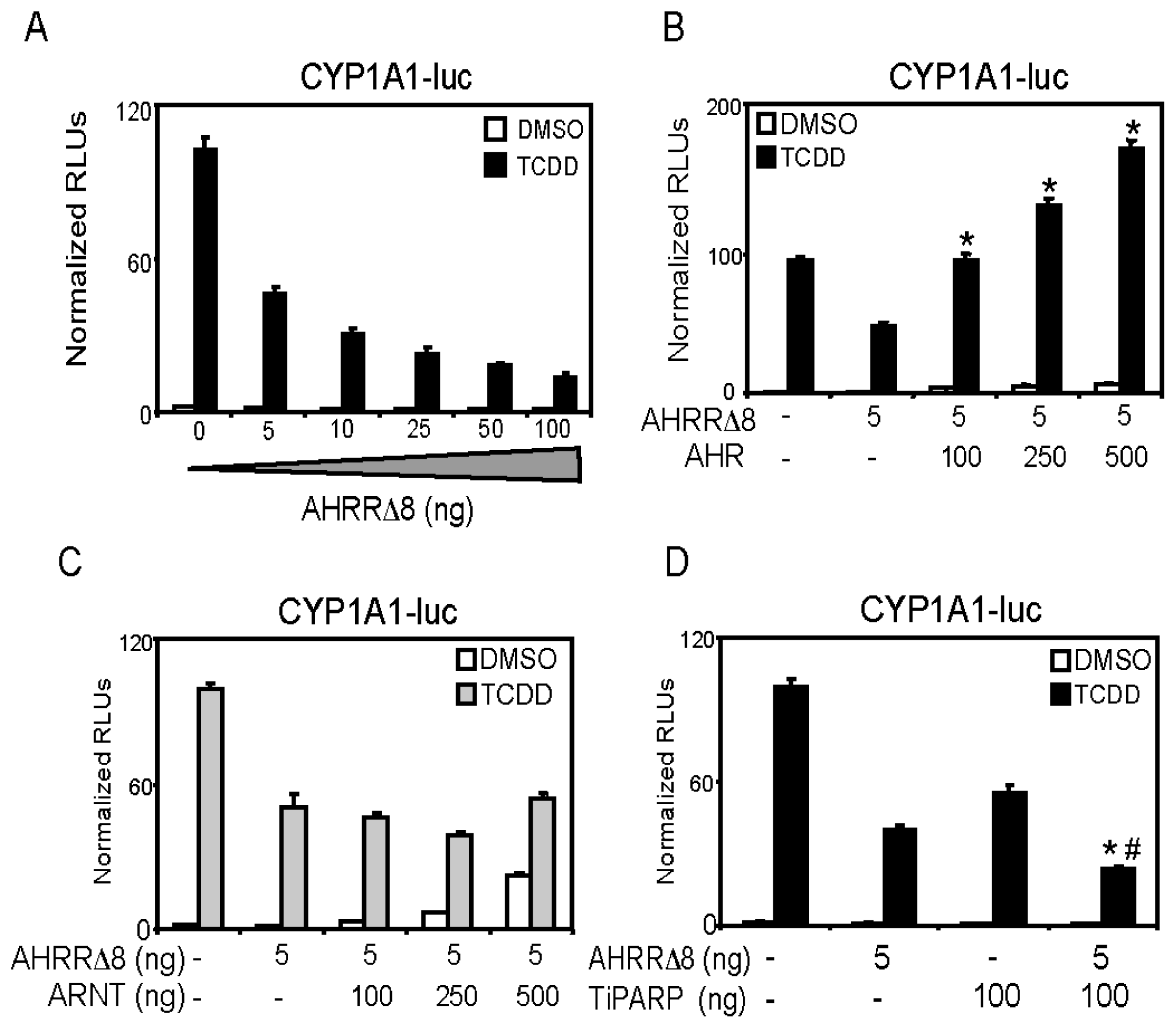
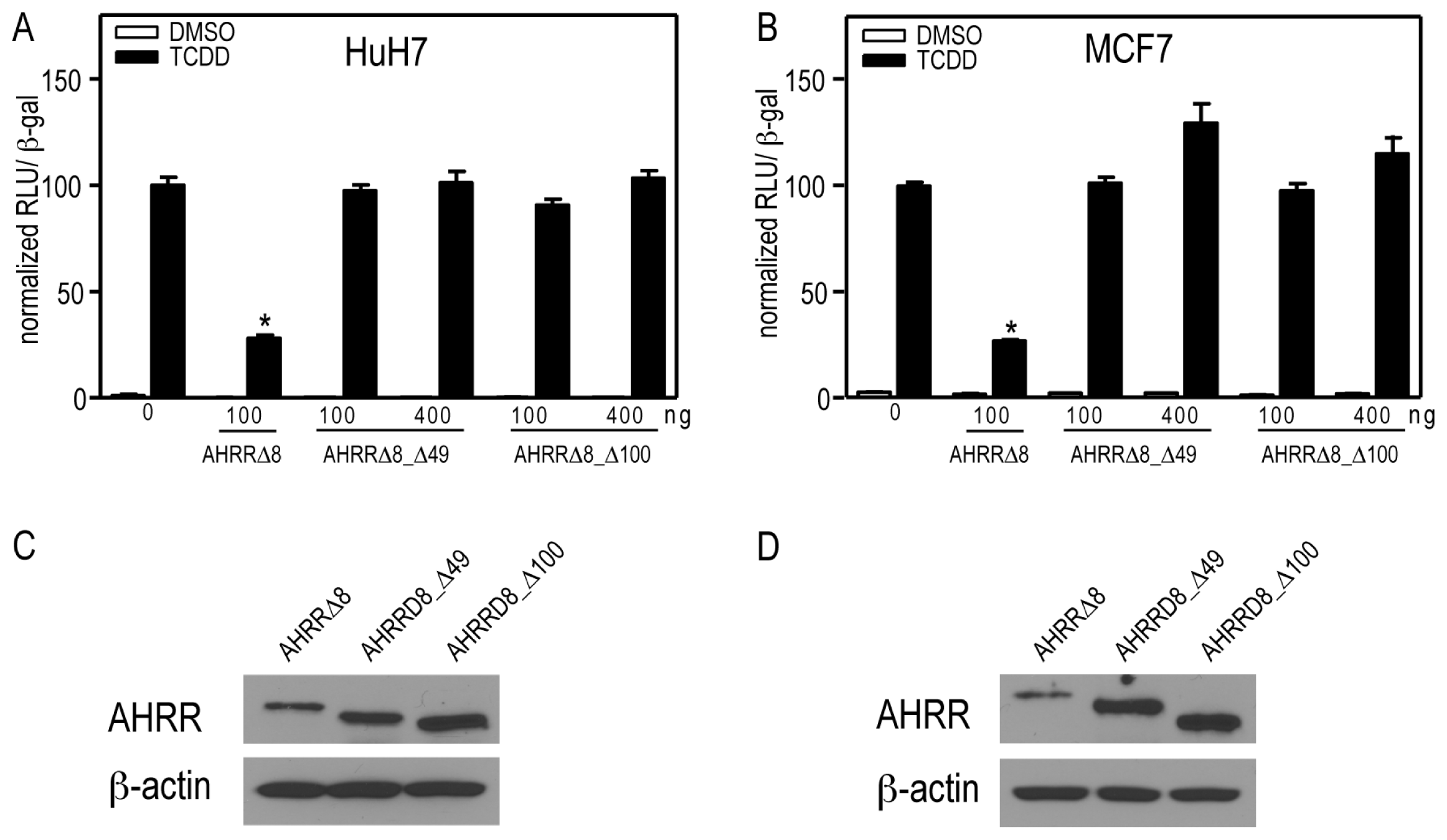
2.2. AHR (Aryl Hydrocarbon Receptor) but not ARNT (Aryl Hydrocarbon Receptor Nuclear Translocator) Overexpression Rescues AHRR-Mediated Repression of CYP1A1 Reporter Gene Activity
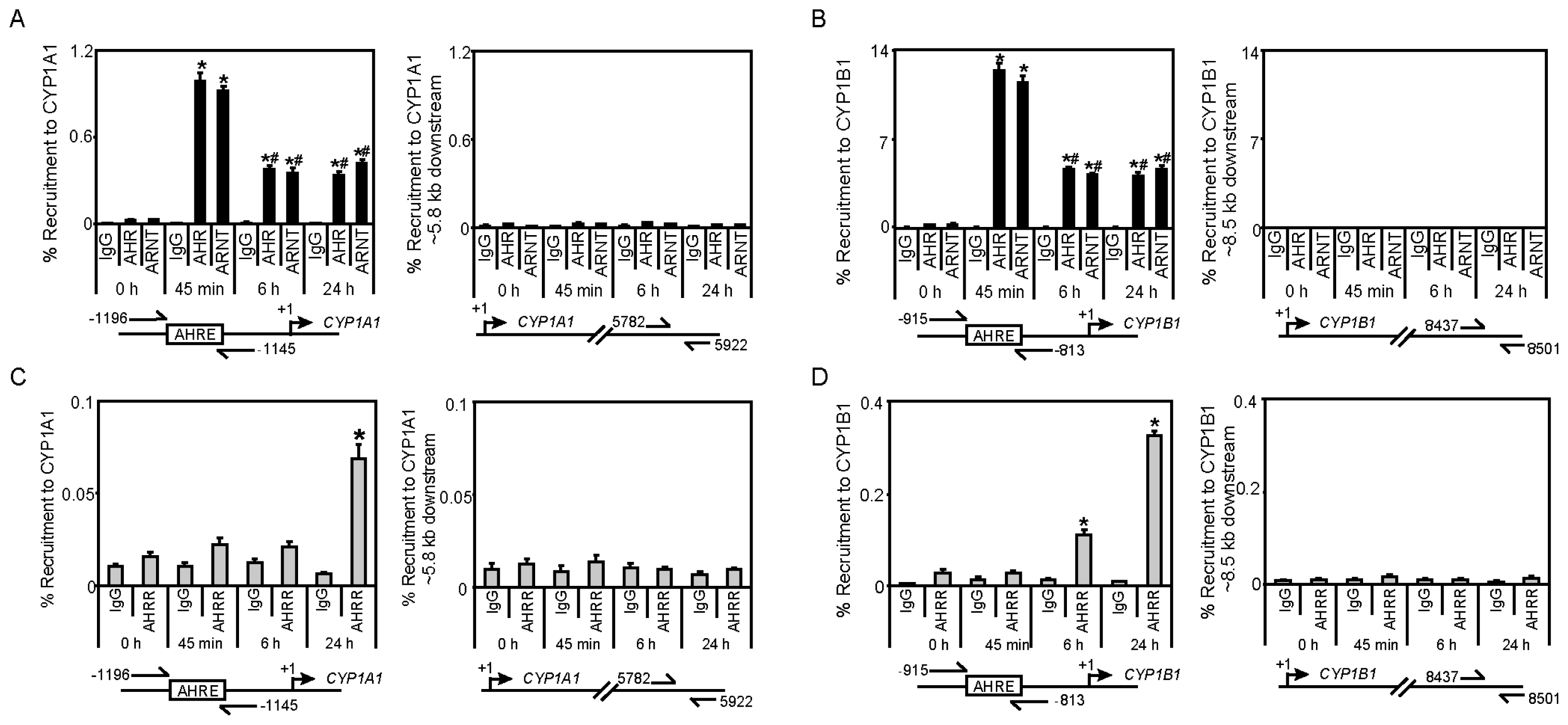
2.3. TCDD-Induced AHRR Recruitment to AHR-Target Genes
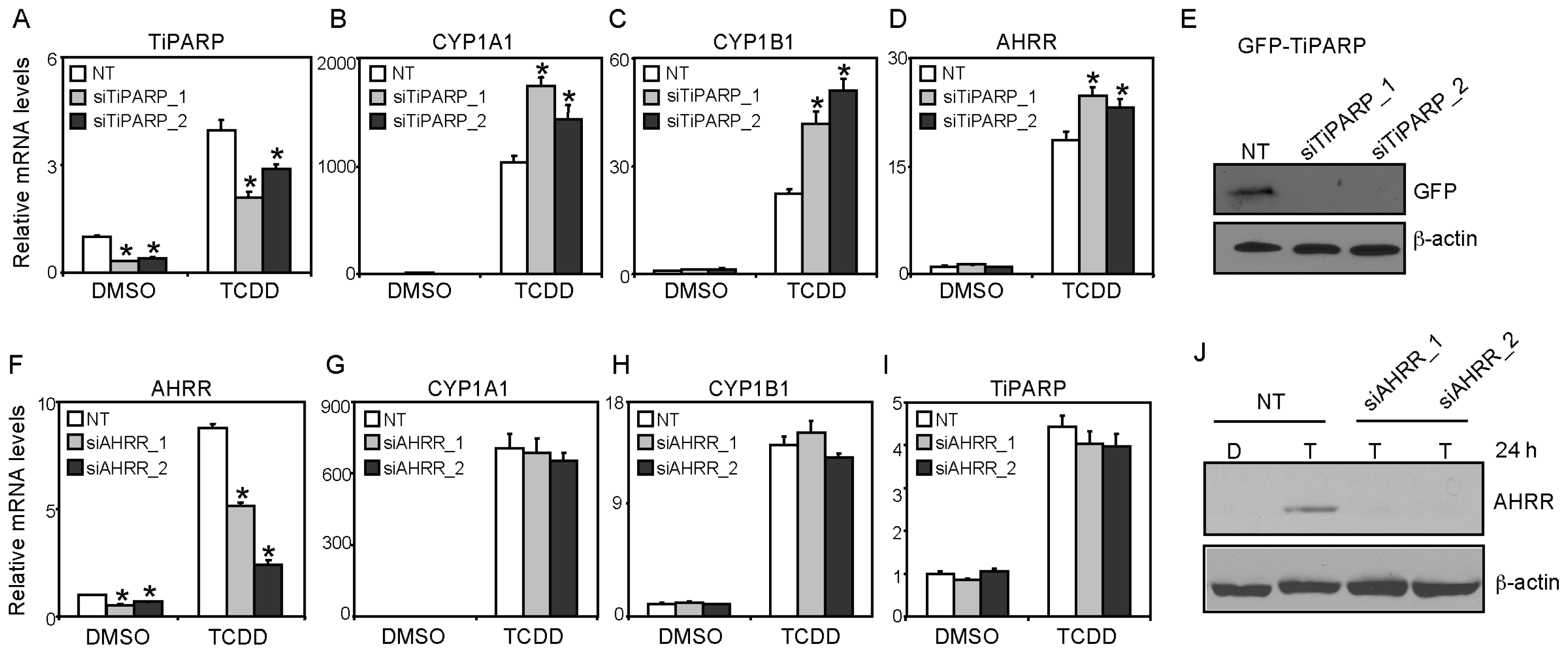
2.4. AHRR Knockdown did not Affect TCDD-Mediated AHR Target Transactivation or AHR/ARNT Protein Levels
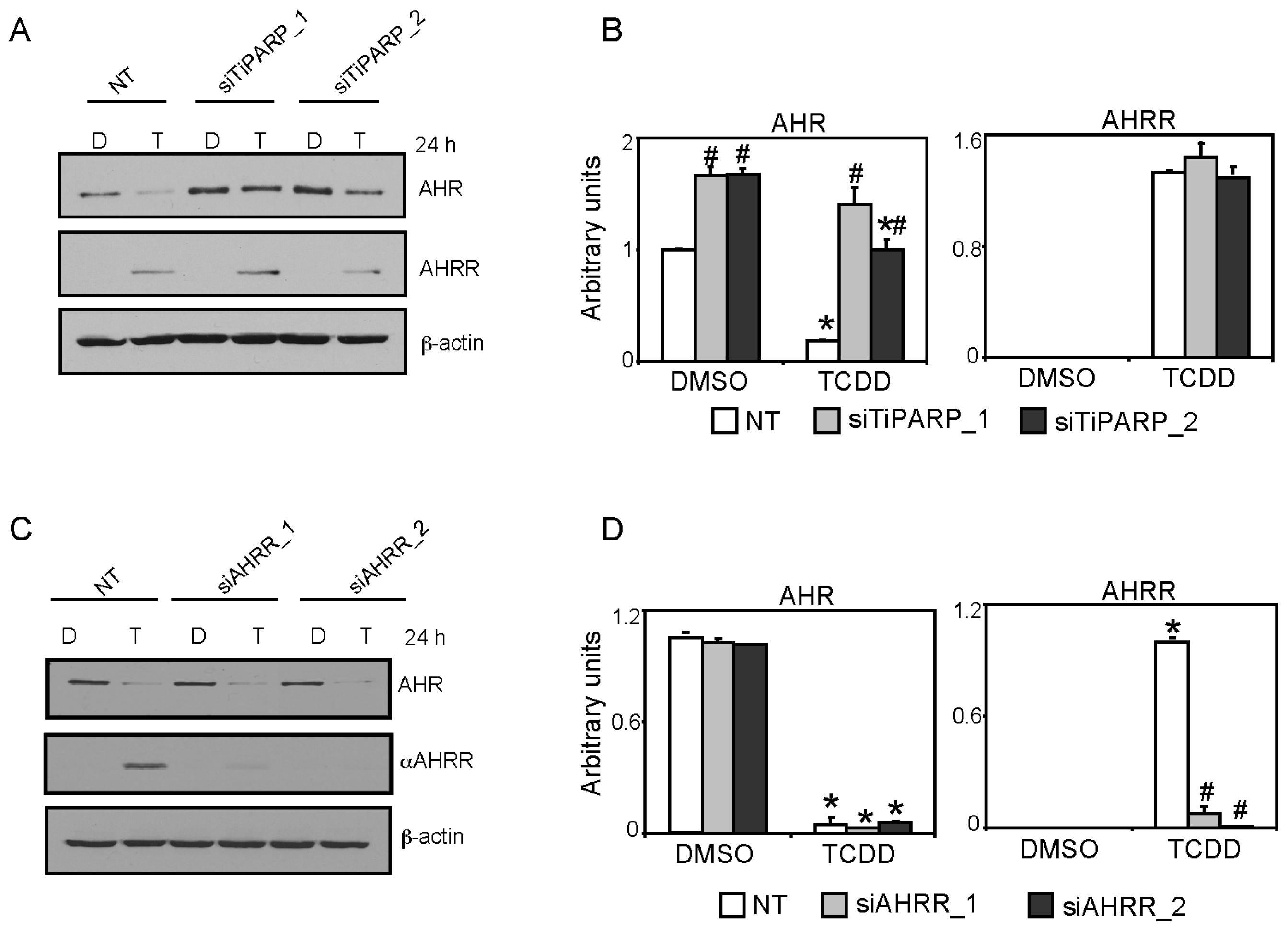
2.5. TiPARP and AHRR Differentially Affect AHR Protein Levels
2.6. Immortalized AHRR−/− MEFs Exhibit Increased AHR Transactivation but not Increased AHR Protein Levels
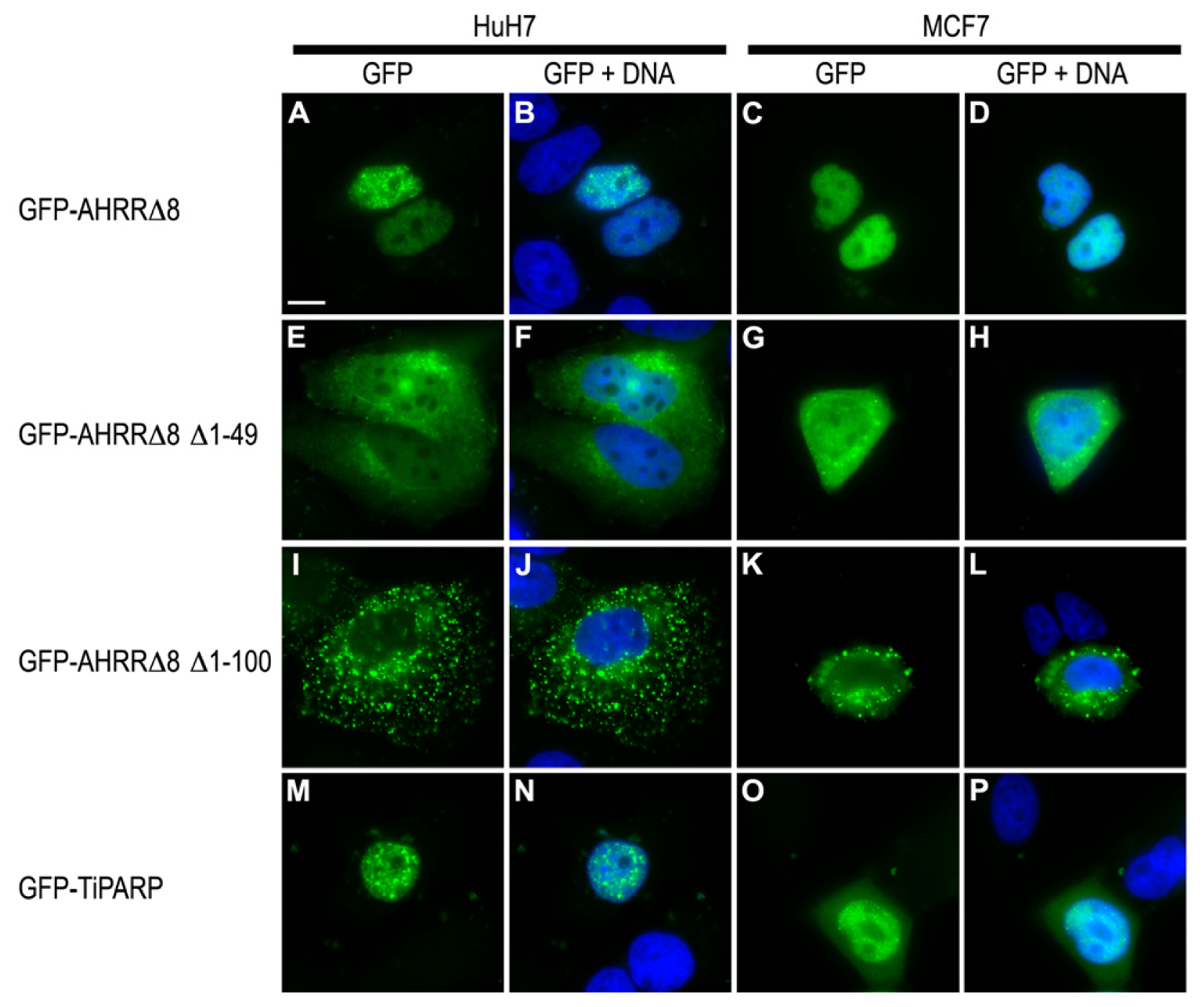
2.7. AHRR and TiPARP Are Predominantly Nuclear Proteins
3. Experimental Section
3.1. Chemicals and Biological Reagents
3.2. Plasmids
3.3. Cell Culture
3.4. Transient Transfection, Reporter Gene and RNAi Studies
3.5. RNA Isolation and qPCR
3.6. Western Blot
3.7. Chromatin Immunoprecipitation
3.8. Indirect Immunofluorescence
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsLaura MacPherson, Shaimaa Ahmed, and Laura Tamblyn performed the experiments. Jean Krutmann, Irmgard Förster, and Heike Weighardt contributed reagents and materials. Laura MacPherson, Shaimaa Ahmed, and Jason Matthews designed the studies and wrote the paper.
References
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol 2003, 43, 309–334. [Google Scholar]
- McMillan, B.J.; Bradfield, C.A. The aryl hydrocarbon receptor sans xenobiotics: Endogenous function in genetic model systems. Mol. Pharmacol 2007, 72, 487–498. [Google Scholar]
- Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol 1995, 35, 307–340. [Google Scholar]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev 1999, 13, 20–25. [Google Scholar]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci 2010, 35, 208–219. [Google Scholar]
- Hankinson, O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys 2005, 433, 379–386. [Google Scholar]
- Pollenz, R.S.; Buggy, C. Ligand-dependent and -independent degradation of the human aryl hydrocarbon receptor (hAHR) in cell culture models. Chem. Biol. Interact 2006, 164, 49–59. [Google Scholar]
- Hahn, M.E.; Allan, L.L.; Sherr, D.H. Regulation of constitutive and inducible AHR signaling: Complex interactions involving the AHR repressor. Biochem. Pharmacol 2009, 77, 485–497. [Google Scholar]
- Karchner, S.I.; Jenny, M.J.; Tarrant, A.M.; Evans, B.R.; Kang, H.J.; Bae, I.; Sherr, D.H.; Hahn, M.E. The active form of human aryl hydrocarbon receptor (AHR) repressor lacks exon 8, and its Pro 185 and Ala 185 variants repress both AHR and hypoxia-inducible factor. Mol. Cell. Biol 2009, 29, 3465–3477. [Google Scholar]
- Evans, B.R.; Karchner, S.I.; Allan, L.L.; Pollenz, R.S.; Tanguay, R.L.; Jenny, M.J.; Sherr, D.H.; Hahn, M.E. Repression of aryl hydrocarbon receptor (AHR) signaling by AHR repressor: Role of DNA binding and competition for AHR nuclear translocator. Mol. Pharmacol 2008, 73, 387–398. [Google Scholar]
- Bernshausen, T.; Jux, B.; Esser, C.; Abel, J.; Fritsche, E. Tissue distribution and function of the Aryl hydrocarbon receptor repressor (AhRR) in C57BL/6 and Aryl hydrocarbon receptor deficient mice. Arch. Toxicol 2006, 80, 206–211. [Google Scholar]
- Tigges, J.; Weighardt, H.; Wolff, S.; Gotz, C.; Forster, I.; Kohne, Z.; Huebenthal, U.; Merk, H.F.; Abel, J.; Haarmann-Stemmann, T.; et al. Aryl Hydrocarbon Receptor Repressor (AhRR) Function Revisited: Repression of CYP1 Activity in Human Skin Fibroblasts Is Not Related to AhRR Expression. J. Investig. Dermatol 2013, 133, 87–96. [Google Scholar]
- Hosoya, T.; Harada, N.; Mimura, J.; Motohashi, H.; Takahashi, S.; Nakajima, O.; Morita, M.; Kawauchi, S.; Yamamoto, M.; Fujii-Kuriyama, Y. Inducibility of cytochrome P450 1A1 and chemical carcinogenesis by benzo[a]pyrene in AhR repressor-deficient mice. Biochem. Biophys. Res. Commun 2008, 365, 562–567. [Google Scholar]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci 2008, 13, 3046–3082. [Google Scholar]
- MacPherson, L.; Tamblyn, L.; Rajendra, S.; Bralha, F.; McPherson, J.P.; Matthews, J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acids Res 2013, 41, 1604–1621. [Google Scholar]
- Ma, Q.; Baldwin, K.T.; Renzelli, A.J.; McDaniel, A.; Dong, L. TCDD-inducible poly (ADP-ribose) polymerase: A novel response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Biophys. Res. Commun 2001, 289, 499–506. [Google Scholar]
- Dere, E.; Lo, R.; Celius, T.; Matthews, J.; Zacharewski, T.R. Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 2011, 12. [Google Scholar] [CrossRef]
- Pansoy, A.; Ahmed, S.; Valen, E.; Sandelin, A.; Matthews, J. 3-methylcholanthrene induces differential recruitment of aryl hydrocarbon receptor to human promoters. Toxicol. Sci 2010, 117, 90–100. [Google Scholar]
- Baba, T.; Mimura, J.; Gradin, K.; Kuroiwa, A.; Watanabe, T.; Matsuda, Y.; Inazawa, J.; Sogawa, K.; Fujii-Kuriyama, Y. Structure and expression of the Ah receptor repressor gene. J. Biol. Chem 2001, 276, 33101–33110. [Google Scholar]
- Haarmann-Stemmann, T.; Bothe, H.; Kohli, A.; Sydlik, U.; Abel, J.; Fritsche, E. Analysis of the transcriptional regulation and molecular function of the aryl hydrocarbon receptor repressor in human cell lines. Drug Metab. Dispos 2007, 35, 2262–2269. [Google Scholar]
- Hao, N.; Lee, K.L.; Furness, S.G.; Bosdotter, C.; Poellinger, L.; Whitelaw, M.L. Xenobiotics and loss of cell adhesion drive distinct transcriptional outcomes by aryl hydrocarbon receptor signaling. Mol. Pharmacol 2012, 82, 1082–1093. [Google Scholar]
- Lo, R.; Matthews, J. High-resolution genome-wide mapping of AHR and ARNT binding sites by ChIP-seq. Toxicol. Sci 2012, 130, 349–361. [Google Scholar]
- Chen, W.V.; Delrow, J.; Corrin, P.D.; Frazier, J.P.; Soriano, P. Identification and validation of PDGF transcriptional targets by microarray-coupled gene-trap mutagenesis. Nat. Genet 2004, 36, 304–312. [Google Scholar]
- Kininis, M.; Chen, B.S.; Diehl, A.G.; Isaacs, G.D.; Zhang, T.; Siepel, A.C.; Clark, A.G.; Kraus, W.L. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol. Cell. Biol 2007, 27, 5090–5104. [Google Scholar]
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 2009, 19, 2163–2171. [Google Scholar]
- Kanno, Y.; Takane, Y.; Takizawa, Y.; Inouye, Y. Suppressive effect of aryl hydrocarbon receptor repressor on transcriptional activity of estrogen receptor alpha by protein-protein interaction in stably and transiently expressing cell lines. Mol. Cell. Endocrinol 2008, 291, 87–94. [Google Scholar]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar]
- Ahmed, S.; Valen, E.; Sandelin, A.; Matthews, J. Dioxin increases the interaction between aryl hydrocarbon receptor and estrogen receptor alpha at human promoters. Toxicol. Sci 2009, 111, 254–266. [Google Scholar]
- Ahmed, S.; Wang, A.; Celius, T.; Matthews, J. Zinc finger nuclease-mediated knockout of AHR or ARNT in human breast cancer cells abolishes basal and ligand-dependent regulation of CYP1B1 and differentially affects estrogen receptor alpha transactivation. Toxicol. Sci 2014. [Google Scholar] [CrossRef]
- Kanno, Y.; Miyama, Y.; Takane, Y.; Nakahama, T.; Inouye, Y. Identification of intracellular localization signals and of mechanisms underlining the nucleocytoplasmic shuttling of human aryl hydrocarbon receptor repressor. Biochem. Biophys. Res. Commun 2007, 364, 1026–1031. [Google Scholar]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun 2013, 4. [Google Scholar] [CrossRef]
- Partch, C.L.; Gardner, K.H. Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc. Natl. Acad. Sci. USA 2011, 108, 7739–7744. [Google Scholar]
- Macpherson, L.; Matthews, J. Inhibition of aryl hydrocarbon receptor-dependent transcription by resveratrol or kaempferol is independent of estrogen receptor alpha expression in human breast cancer cells. Cancer Lett 2010, 299, 119–129. [Google Scholar]
- Matthews, J.; Wihlen, B.; Thomsen, J.; Gustafsson, J.A. Aryl hydrocarbon receptor-mediated transcription: Ligand-dependent recruitment of estrogen receptor alpha to 2,3,7,8-tetrachlorodibenzo-p-dioxin-responsive promoters. Mol. Cell. Biol 2005, 25, 5317–5328. [Google Scholar]
- Lo, R.; Celius, T.; Forgacs, A.; Dere, E.; MacPherson, L.; Zacharewski, T.; Matthews, J. Identification of aryl hydrocarbon receptor binding targets in mouse hepatic tissue treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol 2011, 15, 38–47. [Google Scholar]
- Diani-Moore, S.; Ram, P.; Li, X.; Mondal, P.; Youn, D.Y.; Sauve, A.A.; Rifkind, A.B. Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem 2010, 285, 38801–38810. [Google Scholar]
- Diani-Moore, S.; Zhang, S.; Ram, P.; Rifkind, A.B. Aryl hydrocarbon receptor activation by dioxin targets phosphoenolpyruvate carboxykinase (PEPCK) for ADP-ribosylation via 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-inducible poly(ADP-ribose) polymerase (TiPARP). J. Biol. Chem 2013, 288, 21514–21525. [Google Scholar]








© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MacPherson, L.; Ahmed, S.; Tamblyn, L.; Krutmann, J.; Förster, I.; Weighardt, H.; Matthews, J. Aryl Hydrocarbon Receptor Repressor and TiPARP (ARTD14) Use Similar, but also Distinct Mechanisms to Repress Aryl Hydrocarbon Receptor Signaling. Int. J. Mol. Sci. 2014, 15, 7939-7957. https://doi.org/10.3390/ijms15057939
MacPherson L, Ahmed S, Tamblyn L, Krutmann J, Förster I, Weighardt H, Matthews J. Aryl Hydrocarbon Receptor Repressor and TiPARP (ARTD14) Use Similar, but also Distinct Mechanisms to Repress Aryl Hydrocarbon Receptor Signaling. International Journal of Molecular Sciences. 2014; 15(5):7939-7957. https://doi.org/10.3390/ijms15057939
Chicago/Turabian StyleMacPherson, Laura, Shaimaa Ahmed, Laura Tamblyn, Jean Krutmann, Irmgard Förster, Heike Weighardt, and Jason Matthews. 2014. "Aryl Hydrocarbon Receptor Repressor and TiPARP (ARTD14) Use Similar, but also Distinct Mechanisms to Repress Aryl Hydrocarbon Receptor Signaling" International Journal of Molecular Sciences 15, no. 5: 7939-7957. https://doi.org/10.3390/ijms15057939