Article Text

Abstract
Objective: B lymphocytes have been implicated in the pathogenesis of lupus and other autoimmune diseases, resulting in the introduction of B cell-directed therapies. Epratuzumab, a humanised anti-CD22 monoclonal antibody, is currently in clinical trials, although its effects on patients’ B cells are not completely understood.
Methods: This study analysed the in vivo effect of epratuzumab on peripheral B cell subsets in 12 patients with systemic lupus erythematosus, and also addressed the in vitro effects of the drug by analysing anti-immunoglobulin-induced proliferation of isolated B cells obtained from the peripheral blood of 11 additional patients with lupus and seven normal subjects.
Results: Upon treatment, a pronounced reduction of CD27– B cells and CD22 surface expression on CD27– B cells was observed, suggesting that these cells, which mainly comprise naïve and transitional B cells, are preferentially targeted by epratuzumab in vivo. The results of in vitro studies indicate additional regulatory effects of the drug by reducing the enhanced activation and proliferation of anti-immunoglobulin-stimulated lupus B cells after co-incubation with CD40L or CpG. Epratuzumab inhibited the proliferation of B cells from patients with systemic lupus erythematosus but not normal B cells under all culture conditions.
Conclusions: Epratuzumab preferentially modulates the exaggerated activation and proliferation of B cells from patients with lupus in contrast to normal subjects, thus suggesting that epratuzumab might offer a new therapeutic option for patients with systemic lupus erythematosus, as enhanced B cell activation is a hallmark of this disease.
Statistics from Altmetric.com
B cell directed treatment of autoimmune diseases has gained considerable interest recently, in particular since specific antibodies have been made available for clinical investigation and use, such as the chimeric anti-CD20 monoclonal antibody (mAb), rituximab (Rituxan, Genentech, San Francisco, CA, USA; Mabthera, Roche, Basle, Switzerland), and the humanised anti-CD22 mAb, epratuzumab (hLL2, Immunomedics, Inc., Morris Plains, NJ, USA). In contrast to rituximab, epratuzumab does not drastically deplete circulating B cells, but reduces this population by about 30–45%.1–5 As CD22 is a new target of B cell directed treatment in autoimmune diseases, such as systemic lupus erythematosus (SLE),5 and primary Sjögren’s syndrome,4 further delineation of the effects of epratuzumab is of great interest.
CD22 is a B cell specific adhesion molecule and a co-receptor of the B cell antigen receptor (BCR). During B cell development, surface expression of CD22 appears at the stage of antigen responsiveness. Several ligands containing α2,6-linked sialic acid have been identified.6–12 As a co-receptor of the BCR, CD22 is able to attenuate BCR-mediated signalling by recruiting the inhibitory tyrosine phosphatase, SHP-1 (Src homology 2 domain-containing tyrosine phosphatase-1), or interacting with SHIP (Src homology 2 domain-containing inositol polyphosphate 5′ phosphatase)13 14 upon phosphorylation of its cytoplasmic tyrosine residues. The attenuation of calcium flow by CD22 is mediated by a SHP-1-dependent activation of plasma membrane calcium ATPase.15
Although considerable knowledge about the role and function of CD22 has been gained from animal models, few studies have examined human B cells. This situation has changed only recently, as CD22, a negative regulator of BCR signalling, became an interesting therapeutic target in diseases associated with B cell hyperactivity or malignancy, such as SLE, Sjögren’s syndrome and non-Hodgkin lymphoma. Thus, mAbs against CD22, such as epratuzumab, were developed and studied.2 4 16
Epratuzumab is a humanised CD22 IgG1(κ) mAb, which does not bind to the ligand-binding domain but induces a rapid CD22 internalisation and phosphorylation.17 Recently, the mechanism of epratuzumab was described as an immunomodulatory agent, in contrast to rituximab being a B cell depleting antibody.1 When immobilised, epratuzumab, but not rituximab, inhibited cell growth of Burkitt lymphoma cell lines. Unlike rituximab, epratuzumab showed no complement-dependent cytotoxicity, but did demonstrate modest yet significant antibody-dependent cellular cytotoxicity. Thus, these data indicate a different mode of action for this CD22 mAb.
Recently, the safety, pharmacokinetics, and therapeutic efficacy of epratuzumab were analysed in 14 patients with SLE in an open-label, single-centre, clinical trial.5 Epratuzumab was administered once every other week for four treatments, at a dose of 360 mg/m2 body surface area. Serum levels were measurable for at least 10 weeks post-treatment, with median values of 120 µg/ml (range, 49–350 µg/ml) at week 6, 48 µg/ml (range, 31–138 µg/ml) at week 10, and 8.3 µg/ml (range, 1.82–25 µg/ml) at week 18. B cell counts and disease activity scores declined moderately over time. The first part of the current study revealed changes in B cell subsets and CD22 surface expression in patients with SLE receiving epratuzumab in this clinical trial.
B cell activation in SLE is very complex and comprises T cell dependent and T cell independent pathways. Based on this knowledge, the second part of the study investigated the anti-immunoglobulin-induced in vitro proliferation of normal and lupus B lymphocytes after co-incubation with CpG or CD40L and identified significant differences, which were shown to be influenced by epratuzumab.
MATERIAL AND METHODS
Whole blood was collected from 12 patients with SLE enrolled in an open-label, single-centre, clinical trial.5 Patients were analysed immediately before each infusion (weeks 0, 2, 4, 6) and at weeks 10, 18 and 32. This study was approved by the local ethics committee of the Charité University Hospitals Berlin, and participants gave written informed consent.
In order to analyse the in vitro proliferation of peripheral blood B cells, 40 ml of whole blood was drawn from a second group of patients (n = 11) and normal healthy subjects (NHS) (n = 7) at the Department of Medicine/Rheumatology and Clinical Immunology. All patients fulfilled the ACR criteria, revised in 1982.18 Patients (nine females, two males) were 42.45 (8.82) years old. The majority of these patients had low disease activity (SLEDAI between 2 and 12 with a median value of 2). Further details of anti-dsDNA antibodies, organ manifestations, and treatment are listed in table 1. Normal human subjects (four females, three males) were 31.71 (8.71) years old.
Peripheral blood mononuclear cells (PBMCs) were prepared for flow cytometric analysis by density gradient centrifugation. Subsequently, B cells for cell cultures were isolated using CD19-microbeads and LS columns (Miltenyi Biotech, Bergisch Gladbach, Germany). After two washing steps (phosphate-buffered saline/0.5% bovine serum albumin/5 mM EDTA and phosphate-buffered saline), B cells were incubated for 20 min at 37°C with carboxy–fluorescein–succinimidyl ester (1 µM) (CFSE, Invitrogen, Carlsbad, CA, USA). Cells were then washed in complete RPMI 1640 medium (Life Technologies, Paisley, UK) supplemented with 10% v/v fetal calf serum, penicillin (100 U/ml) and streptomycin (100 µg/ml) (all Invitrogen), and incubated in medium at 37°C for 1 h. Thereafter, B cells (1–2×105 cells/well) were cultured using a 96-well round bottom plate (Greiner bio-one, Kremsmuenster, Austria), with medium only or goat F(ab′)2-fragments against human IgG and IgM (2 µg/ml, Jackson ImmunoResearch Laboratories, West Grove, PA, USA), and human recombinant interleukin (IL)-2 and IL-10 (both 20 ng/ml, ImmunoKontact, Abingdon, UK) alone and either in combination with CD40L (50 ng/ml, R&D Systems, Minneapolis, MN, USA) or CpG oligodeoxynucleotide (sequence: 5′-TsCsg sTsCsg sTsTsT sTsgsT sCsgsT sTsTsT sgsTsC sgsTsT-3′, 2.5 µg/ml, TIB MolBiol, Berlin, Germany).
Cells were cultured as described, with and without epratuzumab (10 µg/ml), for 6 days. The concentration chosen has previously been shown to induce saturation of surface CD22 and efficient internalisation of CD22/anti-CD22 mAb complexes.17 Furthermore, it was similar to the serum levels still present 18 weeks after the last application of the drug.5
After 6 days of culture, the cells were harvested and washed once in phosphate-buffered saline/0.5% bovine serum albumin/5 mM EDTA. Immunofluorescent labelling for flow cytometric analysis was performed by incubating B cells with a non-competing phycoerythrin-labelled anti-human CD22 mAb (SHCL-1, BD PharMingen, San Diego, CA, USA), which does not interfere with epratuzumab binding,17 and Cy5-labelled anti-human CD27 (2E4; a kind gift from R. van Lier, Amsterdam, The Netherlands). Incubation with mAbs was performed in phosphate-buffered saline/0.5% bovine serum albumin/5 mM EDTA at 4°C for 10 min. 4′,6-diamidino-2-phenylindole dihydrochloride (Invitrogen) was added immediately before flow cytometric analysis (final concentration 200 nM) to identify dead cells. Flow cytometric analysis was performed using an LSRII (Becton Dickinson, Franklin Lakes, NJ, USA) and FlowJo software (Treestar Inc, Ashland, OR, USA). For each analysis, all events available were collected.
PBMC of patients participating in the clinical study were additionally labelled with anti-CD19 (HD37, DAKOCytomation, Hamburg, Germany) and anti-CD20 (7H2, BD Pharmingen) mAb and 100 000 events were acquired per sample.
The frequencies of B cell subpopulations were calculated using FlowJo software (TreeStar). Differences between patients and NHS were compared using the non-parametric Mann–Whitney U test, and mean (SD) were determined if the data showed a Gaussian approximation. Otherwise, the median value and the range were used. To compare certain B cell subpopulations after and without incubation with epratuzumab, the Wilcoxon matched pairs test was applied. p<0.05 was considered as statistically significant. Data were analysed using the GraphPad Prism4 software (GraphPad, San Diego, CA, USA).
RESULTS
In vivo effects of epratuzumab on B cell subsets
Changes of B cell subsets (CD27–, CD27+ memory B cells, and CD27++, CD20– plasmablasts) were monitored in 12 patients with SLE enrolled in an open-label, single-centre, clinical trial.5 Treatment with this antibody led to a decrease of peripheral B cells, which was mainly due to a significant decrease of circulating CD27– B cells as early as at week 2 (p = 0.047), with a minimum at week 10 (p = 0.008) lasting until week 18 (p = 0.042) (fig 1B). Thus, there was a persistent decrease of CD27– B cells that was already evident prior to the second infusion. As the composition of the CD27– B cell subset varied among individual patients (transitional, naïve but also memory B cells), the decrease in this B cell subset was variable as well (fig 1B). The absolute numbers of circulating CD27+ memory B cells were, however, less affected by the drug (fig 1B), and no significant changes of this subset were observed over time. A slight increase of CD27high plasmablasts after the first infusion, which was, however, not statistically significant, was observed (p = 0.062, fig 1B).
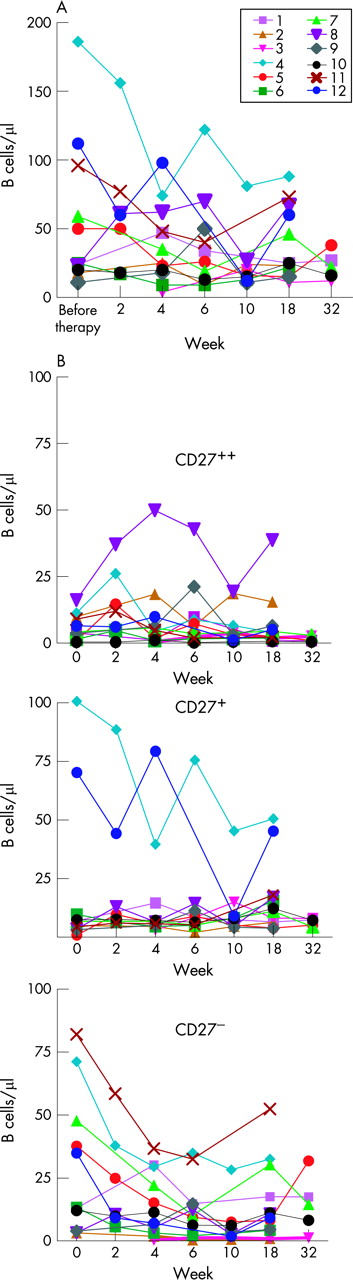
Analysis of the CD22-surface expression of all B cell subsets revealed a higher baseline CD22-expression on CD27– B cells in most patients, followed by CD27+ memory cells and plasmablasts, which were basically CD22– (fig 2A,B). After administration of epratuzumab, we observed an immediate decrease in CD22 surface expression on circulating CD27– and CD27+ memory B cells. In the majority of patients, we found a higher surface expression of CD22 on memory B cells during epratuzumab treatment compared with CD27– B cells (fig 2A, SLE1 and SLE2). The decrease in CD22 surface expression lasted until week 18 in all patients except SLE1 (fig 2A), a time when the concentration of the study drug was 8.3 µg/ml (range, 1.82–25 µg/ml). It was accompanied by a clinical improvement and by changes in B cell subsets, as documented in fig 1.

In vitro effects of epratuzumab
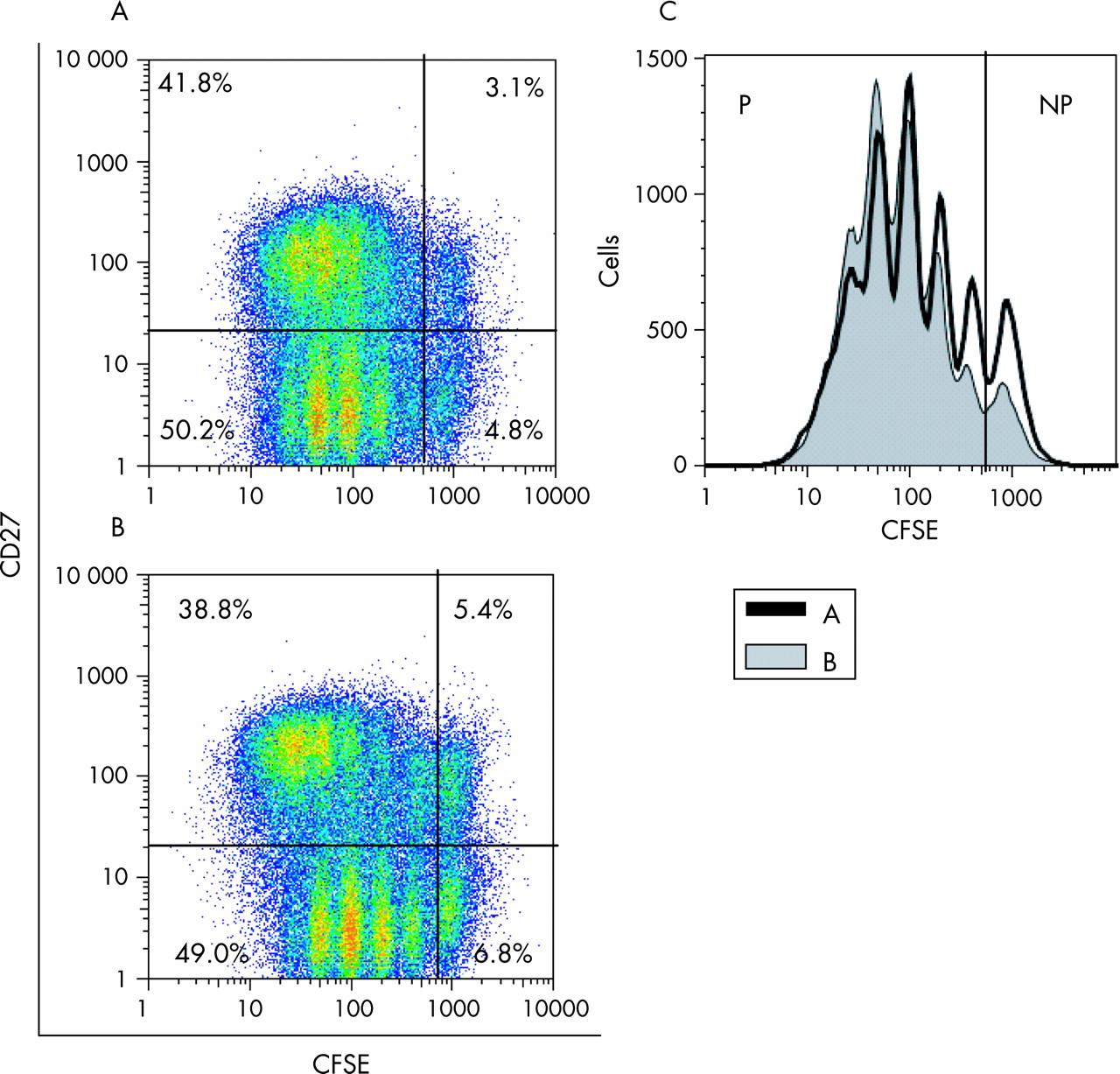
The proliferation of normal and SLE B cells under different culture conditions, including those mimicking T cell dependent (CD40L) and T cell independent (CpG) responses, was studied after day 6 of culture. Figure 3A shows a representative example of the influence of the anti-CD22 mAb, epratuzumab, on B cell proliferation after incubation with IL-2, IL-10, goat anti-human immunoglobulin F(ab′)2 fragments and CpG. As demonstrated in fig 3A, CFSE content and surface expression of CD27 were the parameters used to define four subpopulations by flow cytometric analysis of live B cells. The frequencies of cells with or without CD27 surface expression, which have (proliferating; CFSEdiluted) or have not yet undergone cell division (non-proliferating, CFSEhigh), were determined for all conditions shown in fig 3(A,B). In addition, all proliferating (CFSEdiluted) and non-proliferating cells (CFSEhigh) were analysed, as shown in fig 3C. Altogether, there was no difference in the frequency of live B cells after 6 days in cultures with versus without epratuzumab in patients with SLE or in normal controls under all conditions (fig 4A).


When B cell proliferation and the influence of epratuzumab were investigated after culturing B cells for 6 days without addition of stimuli, only a minor fraction of live cells proliferated in cultures of B cells from both patients with SLE and NHS, and the frequency of proliferating cells was not influenced by epratuzumab (fig 4B). Addition of IL-2, IL-10 and F(ab′)2 resulted in an increase in proliferating B cells in cell cultures from patients with SLE and NHS. Of note, addition of the anti-CD22 mAb was able to inhibit the observed proliferation significantly in SLE B cells (p = 0.027) and normal B cells (p = 0.016, fig 4B). However, a different situation was observed when CD40L or CpG were used in addition to anti-immunoglobulin, IL-2 and IL-10. As shown in fig 4B, CD40L induced B cell proliferation more effectively in patients with SLE compared with NHS (p = 0.09), and the percentage of CD27–CFSEdiluted B cells in culture was significantly higher in patients with SLE compared with NHS (17.3 (5.3)% vs 10.2 (6.1)%; p = 0.03, data not shown). Of interest, incubation with epratuzumab was able to decrease the enhanced proliferation induced by CD40L in SLE B cells (p = 0.002) without showing a significant influence on CD40L-induced proliferation in normal B cells (fig 4B). The percentage of CD27–CFSEdiluted B cells decreased to 10.5 (6.6)% by adding epratuzumab to cultures containing SLE B cells.
The most striking proliferation was observed after adding CpG to anti-immunoglobulin-stimulated normal or lupus B cells (figs 3 and 4B). In patients with lupus, the total number of B cells at day 6 was 8.5 times higher when cells were incubated with anti-immunoglobulin, IL-2, IL-10 and CpG compared with medium alone, whereas in NHS, we observed only a 3.5-fold difference, compared with medium (p = 0.09). In patients with SLE, only a minor fraction (4.98 (3.6)%) of B cells did not divide (CFSEhigh) after 6 days of culture, whereas 15.8 (9.5)% of normal B cells did not proliferate (p = 0.037, fig 4). The frequency of non-proliferating B cells was doubled (10.0 (9.7)%) by adding epratuzumab to lupus B cells (p = 0.0137 for all B cells in culture, p = 0.03 for CD27– B cells, and p = 0.002 for CD27+ B cells), whereas the proportion of non-proliferating normal B cells remained unchanged. In contrast to normal B cells, epratuzumab was able to inhibit proliferation of B cells from patients with SLE under all culture conditions.
We analysed CD22-surface expression to address whether the differences in B cell proliferation observed after co-incubation with epratuzumab were related to this parameter. The change in CD22 surface expression after 6 days of incubation with epratuzumab was expressed as mean fluorescence intensity (MFI) (in percentage change of the MFI of B cells cultured without epratuzumab; fig 5). A markedly decreased CD22 surface expression was observed consistently after incubation with epratuzumab. When lupus or normal B cells were incubated with the anti-CD22 mAb, the CD22 surface expression was comparably reduced after 6 days (−29.4 (14.22)% vs −32.48 (18.73)%, respectively). After stimulation with F(ab′)2, IL-2 and IL-10 with or without additional CD40L, however, B cells obtained from NHS showed a more marked but not significantly different decrease compared with those from patients with lupus when incubated with epratuzumab (−46.87 (17.34)% vs −27.40 (15.76)% and −50.10 (12.00)% vs −29.70 (22.20), p = 0.070 and p = 0.085, respectively; fig 5). In B cell cultures stimulated with CpG instead of CD40L, we also observed a similar reduction of CD22 surface expression by epratuzumab in patients with SLE (−50.26 (14.28)%) and NHS (−60.84 (7.18)%, p = 0.085).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Of note, epratuzumab co-incubation led to a smaller reduction of CD22 expression (expressed by ΔMFI) in patients with SLE versus NHS, especially when the B cells were stimulated.
DISCUSSION
In this study, our aim was to further evaluate the mechanism of action of the humanised anti-CD22 mAb, epratuzumab, using in vivo and in vitro approaches. Epratuzumab has shown indications of clinical efficacy in patients with SLE5 and caused an average reduction of peripheral B cells of 30% that lasted up to 12 weeks after the last infusion. In contrast to the chimeric anti-CD20 mAb, rituximab, which induces a complement-dependent cytotoxicity- and antibody-dependent cellular cytotoxicity-mediated B cell depletion, the underlying mechanism of action of epratuzumab remains unclear. Previous results by Carnahan et al suggested a modest role of antibody-dependent cellular cytotoxicity in the depletion of B cells and an additional modulatory effect of epratuzumab on BCR signalling.1 17
Summarising the results of B cell monitoring by flow cytometry, we observed a pronounced reduction of CD27– B cells expressing high surface levels of CD22; these cells appear to be preferentially targeted by epratuzumab in vivo. Compared with CD27– B cells, CD27+ memory B cells were not reduced significantly. Altogether, moderate changes of B cell subsets were observed in patients with SLE enrolled in the clinical study of epratuzumab. However, these changes do not sufficiently explain the clinical effects observed. Moreover, binding of epratuzumab to CD22 can induce rapid internalisation of CD22 followed by tyrosine phosphorylation.17 Therefore, we anticipated that the drug exerts additional regulatory effects on BCR-mediated activation of B cells from patients with lupus.
BCR engagement leads to activation of tyrosine kinases, resulting in tyrosine phosphorylation of numerous signalling proteins; phosphatidylinositol turnover mediated by phosphoinositol-3-kinase (PI3K) or phospholipase Cγ (PLC γ); influx of calcium and the activation of serine/threonine kinase cascades, such as the extracellular signal-regulated kinase (ERK), Jun N-terminal kinase (JNK), and p38 pathways.19 CD22 engagement has been shown to attenuate BCR-mediated signalling by recruiting the inhibitory tyrosine phosphatase, SHP-1, or interacting with SHIP13 14 upon phosphorylation of its cytoplasmic tyrosine residues. Calcium flow appears to be attenuated by CD22, as B cells from mice lacking CD22 have exaggerated calcium responses after BCR engagement.20 21 The modulation of the BCR signal strength by CD22 might not only influence B cell maturation and selection, but also activation and proliferation, and is therefore an important key regulator of B cell fate. In contrast, co-stimulatory signals provided by T cells or T cell-independent toll-like receptor TLR-mediated signals are able to increase B cell responses to foreign and self-antigen.22 23
This delicate balance of signals causing B cell activation versus inhibition seems to be disturbed in autoimmune disorders. B cells isolated from patients with SLE have been shown to undergo immunoglobulin class-switch and secrete immunoglobulin spontaneously.24 25 Several B cell extrinsic but also some intrinsic factors enhancing B cell survival, activation and proliferation have been identified in mouse models of lupus and in human SLE.26–37 In particular, a role of CD40/CD154 interactions has been suggested repeatedly in autoimmune mouse models and in patients with SLE.27 36 37 In addition, cell damage and impaired clearance can result in an increased load of chromatin-containing immune complexes, which were shown to stimulate autoreactive B cells in a TLR-dependent manner.30 In contrast, only few gene polymorphisms leading to intrinsic B cell defects have been described in patients with SLE,29 38 39 and CD22 expression was reported to be normal in patients with lupus.40
To mimic polyclonal T cell-independent B cell activation and also T cell-dependent costimulation, we incubated anti-immunoglobulin-stimulated B cells with either CpG or CD40L, respectively. The results of these experiments suggested that abnormally enhanced B cell proliferation following TLR- or CD40-engagement in lupus can be influenced significantly by epratuzumab, in line with a regulatory mode of action of this antibody. Comparing patients with SLE and normal controls, we observed a significant difference in the percentage of B cells that were unresponsive to CpG. In vivo priming by interferon-α might abrogate anergy by upregulating TLR expression, as reviewed recently.41 In addition, increased frequencies of naïve B cells reactive to nuclear antigens have been observed in patients with SLE, especially during active disease.42 43 In contrast to normal B cells, these cells may show a more effective direct internalisation of CpG by the BCR.41 However, the effect of epratuzumab on CpG-induced B cell proliferation appears to be more complex, and is probably not restricted to early mechanisms, such as the internalisation of the TLR-L. While the detailed mechanisms that contribute to the enhanced proliferation of lupus B cells require further investigation, nevertheless it is interesting to note that they are apparently alleviated by epratuzumab. Similarly, it has been reported that, in contrast to normal tonsillar B cells, B cell lymphoma cell lines show an impaired growth when incubated with epratuzumab.1
The CD22 surface expression was decreased by epratuzumab not only in vitro, as shown for cell lines and human B cells previously,17 but also in vivo during the clinical trial. The in vitro decrease was similar in patients and controls if cells were not stimulated. Activation led, however, to a more marked decrease in normal controls compared with patients with SLE. Potential explanations might be either an increased turnover of CD22 with a more rapid replacement of internalised molecules, or a less efficient internalisation by lupus B cells. The in vitro decrease of CD22 surface expression did not, however, translate directly into an inhibitory effect.
As CD27+ memory B cells experienced a less marked decrease of CD22 surface expression compared with CD27– B cells after infusion of epratuzumab, we cannot rule out that pre-existing differences in peripheral blood B cell subsets account for the less marked decrease of the CD22 surface expression observed in cultured lupus B cells, even though the ratio of CD27+:CD27– B cells was comparable in cell cultures obtained from patients with SLE and normal controls after 6 days.
Although CD22 is expressed on memory B cells and is also internalised after ligation, recent results of in vitro studies and animal models suggest that the inhibitory effects of CD22 on the Ca2+ signal and on B cell proliferation are more pronounced in IgM-expressing, antigen-naïve, B cells,44 45 whereas immunoglobulin class-switched post GC cells are regulated rather by the low-affinity Fc receptor for IgG (FcγRIIb).46–48 Consistent with this notion, we observed less pronounced in vivo and in vitro effects of epratuzumab on CD27+ memory B cells and CD27++ plasmablasts.
In summary, the results of the current study indicate differential effects of epratuzumab on B cells from patients with SLE and normal subjects. Although the pathways and mechanisms involved remain to be elucidated, we could demonstrate that epratuzumab is able to preferentially modulate the increased activation and proliferation of lupus B cells after stimulation with CpG or CD40L. This differential regulation of normal versus lupus B cells by epratuzumab supports the contention that this mAb may offer a new therapeutic option for patients with SLE, as enhanced B cell activation is a hallmark of lupus.
REFERENCES
Footnotes
Competing interests: DMG is an officer, director and shareholder of Immunomedics, Inc., which owns epratuzumab. GRB and TD were PIs of the clinical trial and received financial support by the sponsor for conducting the study. The other authors disclaim any potential financial conflicts.