


Abstract
The metabolism of the antidepressant drug trazodone to its active metabolite, m-chlorophenylpiperazine (mCPP), was studiedin vitro using human liver microsomal preparations and cDNA-expressed human cytochrome P450 (P450) enzymes. The kinetics of mCPP formation from trazodone were determined, and three in vitro experiments were performed to identify the major P450 enzyme involved. Trazodone (100 μM) was incubated with 16 different human liver microsomal preparations characterized for activities of 7 different P450 isoforms. The production of mCPP correlated significantly with activity of cytochrome P4503A4 (CYP3A4) only. Trazodone (100 μM) was then incubated with microsomes from cells expressing human CYP1A1, CYP1A2, CYP2C8, CYP2C9arg, CYP2C9cys, CYP2C19, CYP2D6, or CYP3A4. Only incubations with CYP3A4 resulted in mCPP formation. In the third experiment, the CYP3A4 inhibitor ketoconazole was found to inhibit mCPP formation concentration dependently in both human liver microsomes and in microsomes from cells expressing human CYP3A4. The present results indicate that trazodone is a substrate for CYP3A4, that CYP3A4 is a major isoform involved in the production of mCPP from trazodone, and that there is the possibility of drug-drug interactions with trazodone and other substrates, inducers and/or inhibitors of CYP3A4.
Adverse pharmacokinetic drug interactions may occur when drugs that are substrates, inducers and/or inhibitors of the same cytochrome P450 (P450)2 enzymes are co-administered, potentially altering the expected rate of metabolism of one or both compounds. The clinical consequences can range from a lack of therapeutic efficacy to severe toxicity and, in extreme cases, fatality. Therefore, it is important to identify the major enzymes involved in the metabolism of a drug so that such interactions can be predicted and avoided.
Trazodone is a triazolopyridine antidepressant drug (fig.1), which is thought to act through combined 5-HT2 antagonism and 5-HT reuptake blockade (Haria et al., 1994). It is often co-prescribed with other antidepressants as a sleep-inducing agent because of its sedative side effects (Fabre, 1990; Jacobsen, 1990, Nierenberg et al., 1994) or as an augmentation strategy (Maes et al., 1997). This co-prescription introduces the potential for metabolic drug interactions.
The chemical structures of trazodone and mCPP.
Trazodone is extensively metabolized in the liver by hydroxylation, dealkylation, and N-oxidation (Baiocchi et al., 1974; Yamato, 1974a). The active metabolite mCPP is formed byN-dealkylation of the piperazinyl nitrogen (Melzackaet al., 1979; Yamato et al., 1974b). The metabolite mCPP is of interest because it has 5-HT2C agonistic and 5-HT2Aantagonistic properties (Conn and Sanders-Bush, 1987; Fiorella et al., 1995), as well as behavioral effects that are consistent with 5-HT agonistic properties such as worsening of psychoses in humans and anxiogenesis and anorexia in animals and humans (Kahn and Wetzler, 1991). It has also been suggested by some that mCPP may contribute to the antidepressant efficacy of trazodone (Maes, 1997). Therefore, a drug interaction that alters the production of mCPP could have clinically significant effects.
Current information available on the metabolism of trazodone by the P450 enzymes comes mainly from drug interaction studies, which provide only suggestive evidence of the enzymes involved and do not examine specific metabolic pathways, whether it is the parent compound or a metabolite causing the interaction or whether the interaction is competitive or non-competitive. For example, thioridazine, a CYP2D6 inhibitor, increases plasma concentrations of both trazodone and mCPP, suggesting that both are substrates for CYP2D6 but providing no information as to which metabolic pathways are involved (Yasui et al., 1995). Plasma levels of trazodone, but not mCPP, are lower in smokers than in non-smokers, suggesting a possible role of the smoking-inducible CYP1A2 in trazodone, but not mCPP, metabolism (Ishidaet al., 1995). Carbamazepine, a CYP3A4 inducer and substrate, decreases plasma concentrations of both trazodone and mCPP, but mCPP to a lesser extent (Otani et al., 1996). Clinical interactions between trazodone and fluoxetine have been reported in the form of adverse side effects such as headaches, dizziness, and excessive sedation (Metz and Shader, 1990; Nierenberg et al., 1992), as well as increased plasma levels of trazodone (Aranow et al., 1989; Maes et al., 1997) and mCPP (Maes et al., 1997). However, the causes of the interactions cannot easily be determined, as both fluoxetine and its main metabolite norfluoxetine are inhibitors of both CYP2D6 and CYP3A4 (Crewe et al., 1992; Greenblatt et al., 1996).
A detailed in vitro investigation is thus necessary to identify the individual enzymes involved in the various interactions of trazodone. In particular, the pathway leading to the formation of mCPP from trazodone is of interest given the psychopharmacological effects of this metabolite. Several in vitro methods are routinely used to identify the P450 enzymes involved in the oxidation of compounds (e.g.Guengerich, 1996; Iwatsubo et al., 1997). The current experiments were designed to directly identify the major P450 enzymes involved in the metabolism of trazodone to mCPP using human liver microsomal preparations and cDNA-expressed human P450 enzymes.
Materials and Methods
Chemicals.
Trazadone and mCPP were purchased from RBI (Natick, MA) and Sigma (St. Louis, MO), respectively. The HCl salt of the internal standardp-chlorophenylethylamine was synthesized in our laboratory. Pentafluorobenzoyl chloride was purchased from Aldrich (Milwaukee, WI), glass-distilled toluene from BDH (Toronto), and potassium carbonate from Fisher Scientific (Nepean, Ontario). The components of the NADPH-generating system, namely β-nicotinamide adenine dinucleotide phosphate, glucose 6-phosphate, glucose 6-phosphate dehydrogenase, and MgCl2, were all obtained from Sigma. Potassium phosphate monobasic and potassium phosphate dibasic (J.T. Baker) were used to prepare a solution of 0.1 M potassium phosphate buffer (pH 7.4).
Microsomal Preparations.
Microsomal preparations from metabolically competent cell lines expressing human CYP1A1, CYP1A2, CYP2C8, CYP2C9arg, CYP2C9cys, CYP2C19, CYP2D6, or CYP3A4 were purchased from Gentest (Woburn, MA).
Human liver microsomes characterized for protein content and enzyme activities were obtained from the International Institute for the Advancement of Medicine (Exton, PA).
Incubation Conditions.
The drug metabolism experiments were carried out in a volume of 100 μl in 1.5-ml polypropylene microcentrifuge tubes (Fisher). The incubation medium consisted of 25 μl of an NADPH-generating system [final concentration in 100 μl of 1 mg/ml β-nicotinamide adenine dinucleotide phosphate, 1 mg/ml glucose 6-phosphate, 0.4 U/ml glucose 6-phosphate dehydrogenase, and 0.66 mg/ml MgCl2in 0.1 M potassium phosphate buffer (pH 7.4), 10 μl of microsomal enzyme preparation (1.5 mg microsomal protein/ml incubation mixture), 50 μl of trazodone solution in 0.1 M potassium phosphate buffer, and 15 μl 0.1 M potassium phosphate buffer (pH 7.4)]. The tubes were incubated for 10 min at 37°C in a water bath (Fisher Isotemp hot water bath). The incubation time was chosen based on preliminary experiments showing that the formation of mCPP was linear within the first 20 min of incubation time. Following the incubation period, the tubes were placed on ice, and 100 μl of ice-cold 25% potassium carbonate solution was added to terminate metabolism.
Assay Procedure for mCPP.
To the basified incubation mixture, 300 μl of double distilled H2O (ddH2O) and 1000 ng of the internal standard (p-chlorophenylethylamine, in 100 μl ddH2O) were added. The incubation mixtures were then transferred to screw cap culture tubes (Fisher, 160 mm × 15 mm), and mCPP was extracted and derivatized by shaking the tubes for 15 min on an Ika Vibrex VXR vortex mixer (Janke & Kunkel, Staufen, Germany) with 2 ml of a solution of toluene and pentafluorobenzoyl chloride in a ratio of 100:1. The tubes were then centrifuged at 1000g for 5 min in a benchtop centrifuge (Sorvall GLC-2B general laboratory centrifuge, DuPont, Wilmington, DE). The organic phase was pipetted to 100 × 13-mm screw cap culture tubes and taken to dryness in a Savant evaporator (Speed Vac SC 110, Fisher). The residue was reconstituted in 150 μl of toluene for gas chromatographic analysis.
Instrumental Analysis.
A 1-μl aliquot of the solution in toluene was injected on a Hewlett-Packard (HP) model 5890 gas chromatograph equipped with a nitrogen-phosphorus detector and linked to an HP 3392A integrator. A 15-m fused silica capillary column (internal diameter of 0.25 mm) coated with a 0.25-μm film thickness of 5% phenylmethyl polysiloxane was used. The carrier gas was helium at a flow rate of 3.5 mL/min, and the make-up gas was helium at a flow rate of 30 mL/min. Hydrogen and air were used at flow rates of 4 and 80 mL/min, respectively. The oven temperature was set at 105oC for an initial time of 0.5 min and was then set to increase at a rate of 12oC/min to a final temperature of 295oC. The injection port temperature was set at 270oC, and the detector temperature was 325oC. All injections were in the splitless mode with a purge off time of 0.5 min.
Determination of the Kinetic Constants for mCPP Formation from Trazodone.
The kinetic constants of KM andVmax were estimated for the formation of mCPP from trazodone by incubating varying concentrations of trazodone (450, 300, 200, 133, 88.89, 59.26, 39.51, 26.34, 17.56, 11.71, 7.80, 5.20, 3.47, 2.31, and 0 μM) with human liver microsomes under the conditions described above. The data were analyzed by iterative nonlinear least squares regression analysis (GraphPad Prism), fitting the data to the equation v = (Vmax·S)/(KM + S), where v is the reaction velocity corresponding to S, the substrate concentration (trazodone),Vmax is the maximal velocity, andKM is the substrate concentration at which the reaction velocity equals 50% of Vmax.
Correlations with P450 Enzyme Activities in a Panel of Human Liver Microsomal Preparations.
Trazodone (100 μM final concentration) was incubated with the NADPH-generating system and microsomes prepared from a panel of 16 human livers characterized for their catalytic activity for CYP1A2 (phenacetin O-deethylation), CYP2A6 (coumarin 7-hydroxylation), CYP2C19 (mephenytoin 4-hydroxylation), CYP2D6 (dextromethorphan O-demethylation), CYP2E1 (chlorzoxazone 6-hydroxylation), CYP3A4 (6-hydroxylation of [14C]testosterone), and CYP4A11 (omega-hydroxylation of [14C]-lauric acid). The rate of formation of mCPP was then correlated with the activities of the specific enzymes for each of the 16 human livers (GraphPad Prism).
Incubations with Single Expressed Enzymes.
Trazodone (100 μM final concentration, added in a volume of 50 μl) was incubated in the NADPH-generating system (25 μl), potassium phosphate buffer (15 μl), and 10 μl of a microsomal preparation (1 mg microsomal protein/ml incubation mixture) expressing CYP1A1, CYP1A2, CYP2C8, CYP2C9arg, CYP2C9cys, CYP2C19, CYP2D6, or CYP3A4 for 30 min. These incubations were repeated in four separate experiments.
Inhibition with Ketoconazole.
The CYP3A4 inhibitor ketoconazole (final concentrations in 100 μl of 6.4, 3.2, 1.6, 0.8, 0.4, 0.2, and 0.0 μM, added in 10 μl of buffer) was pre-incubated for 10 min with 25 μl of the NADPH-generating system, 5 μl of potassium phosphate buffer, and 10 μl of either human liver microsomes (1.5 mg of microsomal protein/ml incubation mixture) or microsomes from cells expressing human CYP3A4 (1 mg of microsomal protein/ml incubation mixture). Trazodone (100 μM final concentration, added in a volume of 50 μL) was then added, and the incubation was continued for a further 10 min. As a control, quinidine, a specific inhibitor of CYP2D6, was also incubated as described for ketoconazole, using concentrations of 6.0, 3.0, 1.5, 0.75, 0.375, and 0 μM. The inhibitions were repeated in three separate experiments.
Results
Kinetic Analyses.
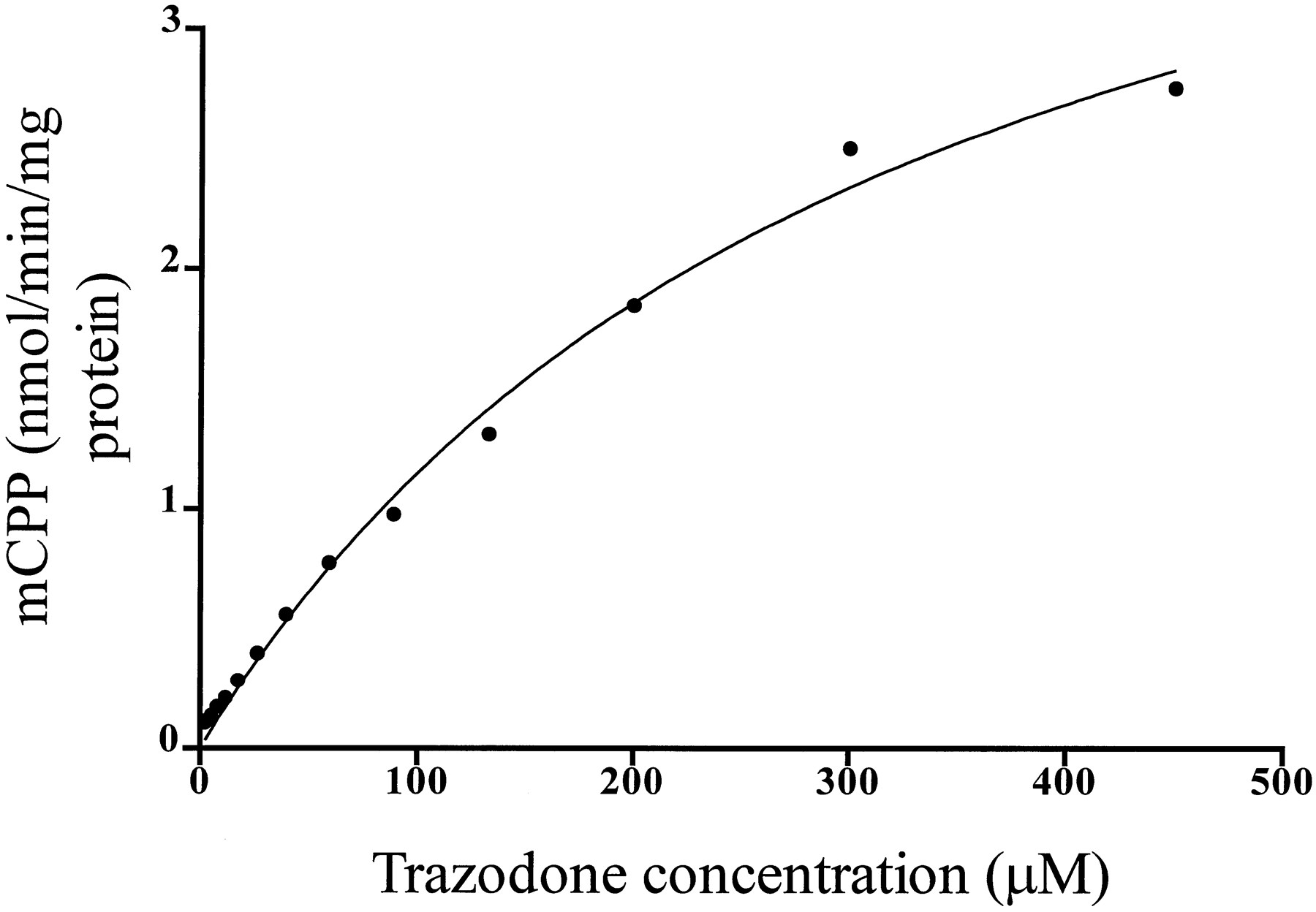
Incubations of various concentrations of trazodone with human liver microsomes resulted in a concentration-dependent formation of mCPP, as shown in fig. 2. The apparentKM was 311.3 ± 32.19 μM, and apparent Vmax was 4.95 ± 0.29 nmol/min/mg protein.
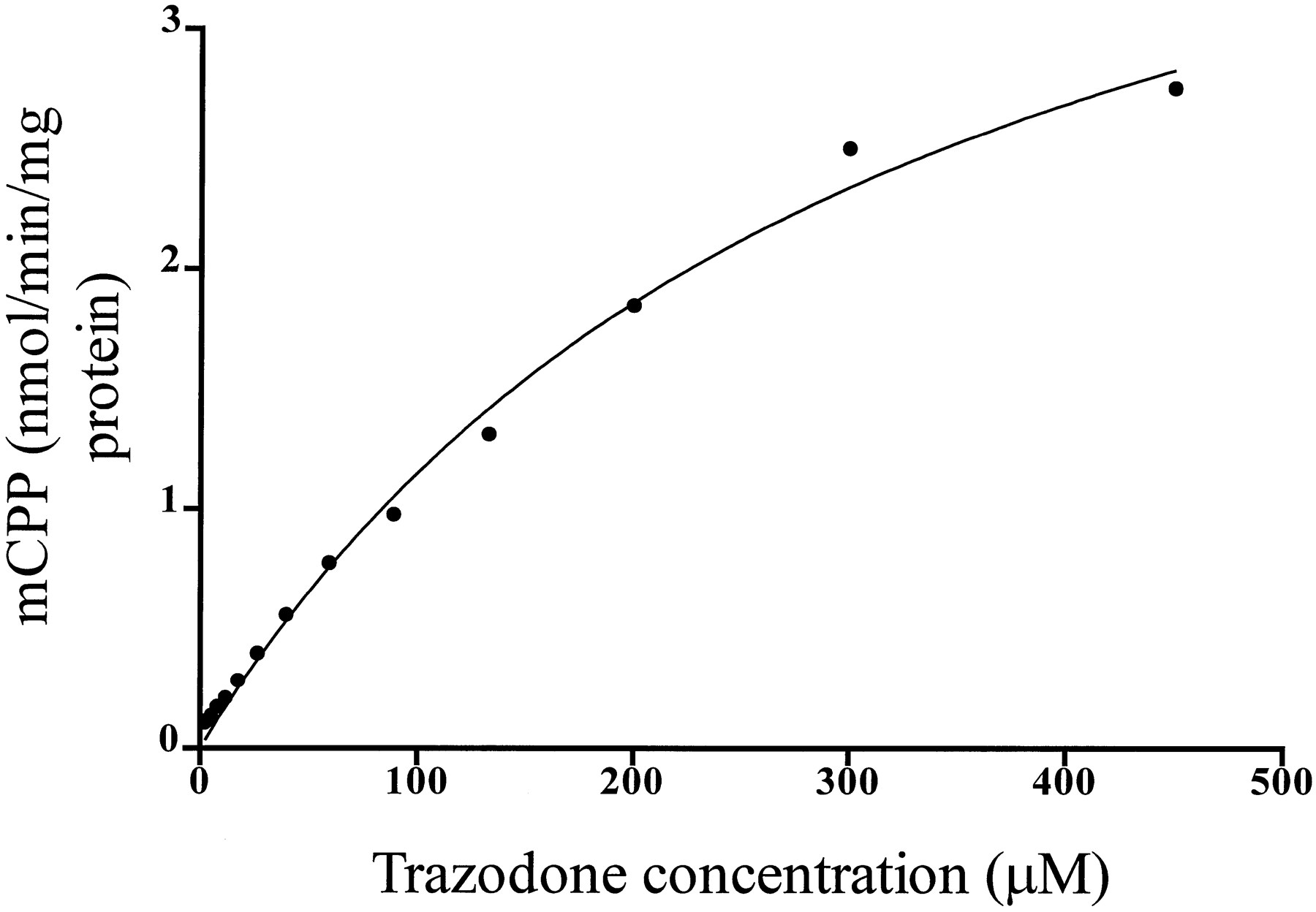
Formation of mCPP (y axis, nmol/min/mg protein) at various concentrations of the substrate trazodone (x axis, μM). The solid linewas determined by nonlinear least squares regression analysis.
Correlations with P450 Enzyme Activities in a Panel of Human Liver Microsomal Preparations.
The rate of formation of mCPP showed significant correlation (r = 0.81, p < 0.0001) with CYP3A4 activity (fig. 3). Correlations with all other P450 enzymes failed to reach significance (CYP1A2,r = 0.27, p = 0.32; CYP2A6,r = 0.47, p = 0.07; CYP2C19,r = 0.16, p = 0.55; CYP2D6,r = 0.41, p = 0.11; CYP2E1,r = 0.40, p = 0.13; CYP4A11,r = 0.33, p = 0.21).
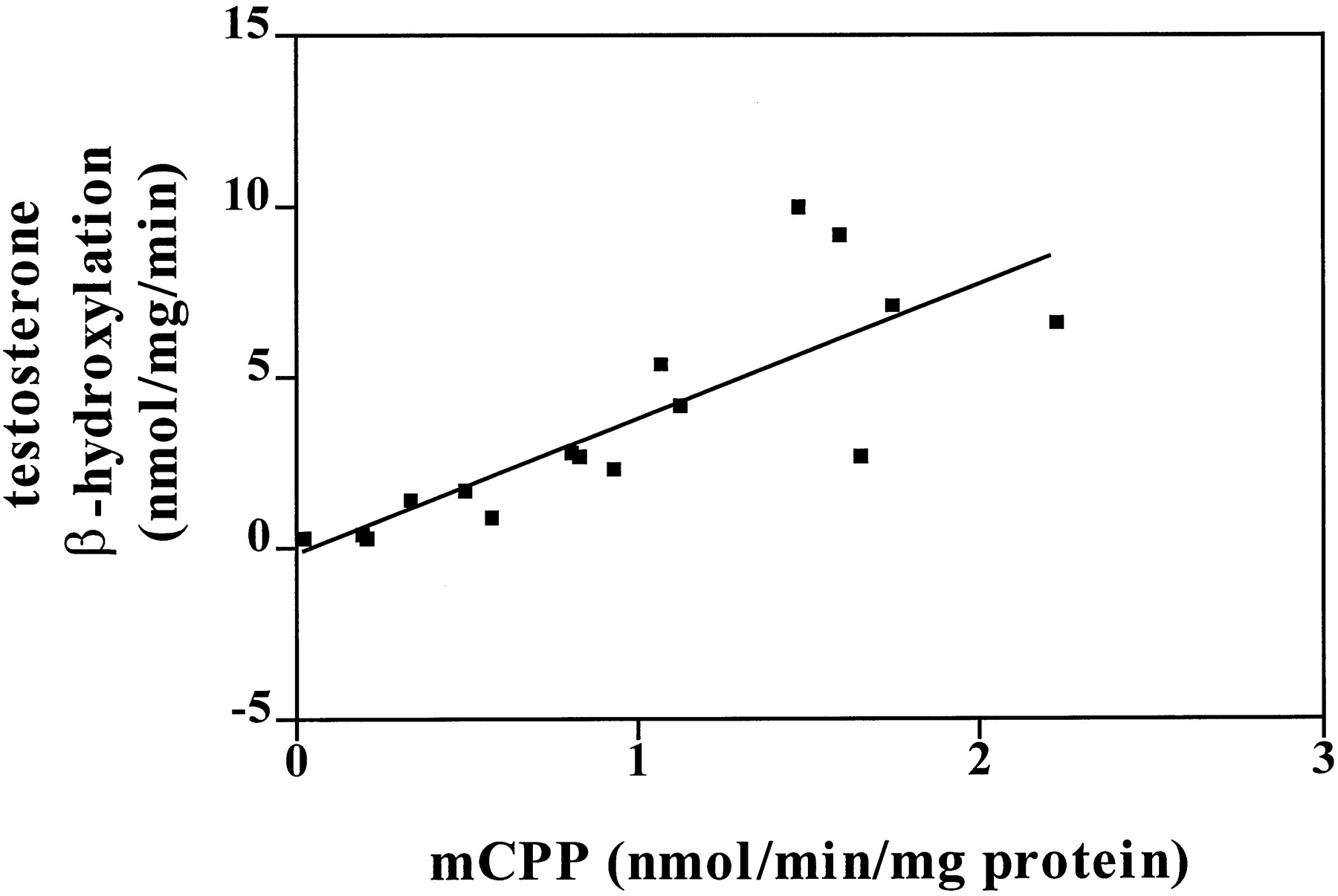
Correlation (r = 0.81,p < 0.0001) of the activity of CYP3A4 in 16 individual human liver samples (y axis) with the rate of mCPP production from trazodone (100 μM, 10-min incubation) (x axis) in the same samples. Correlations with other P450 isoform activities failed to reach significance.
Incubations with Single Expressed Enzymes.
Trazodone incubations with microsomes from cells expressing only CYP3A4 resulted in mCPP production (0.3903 ± 0.0631 nmol/min/mg protein,N = 4), whereas incubations with microsomes from cells expressing only CYP1A1, CYP1A2, CYP2C8, CYP2C9arg, CYP2C9cys, CYP2C19, or CYP2D6 did not result in detectable mCPP formation.
Inhibition with Ketoconazole.
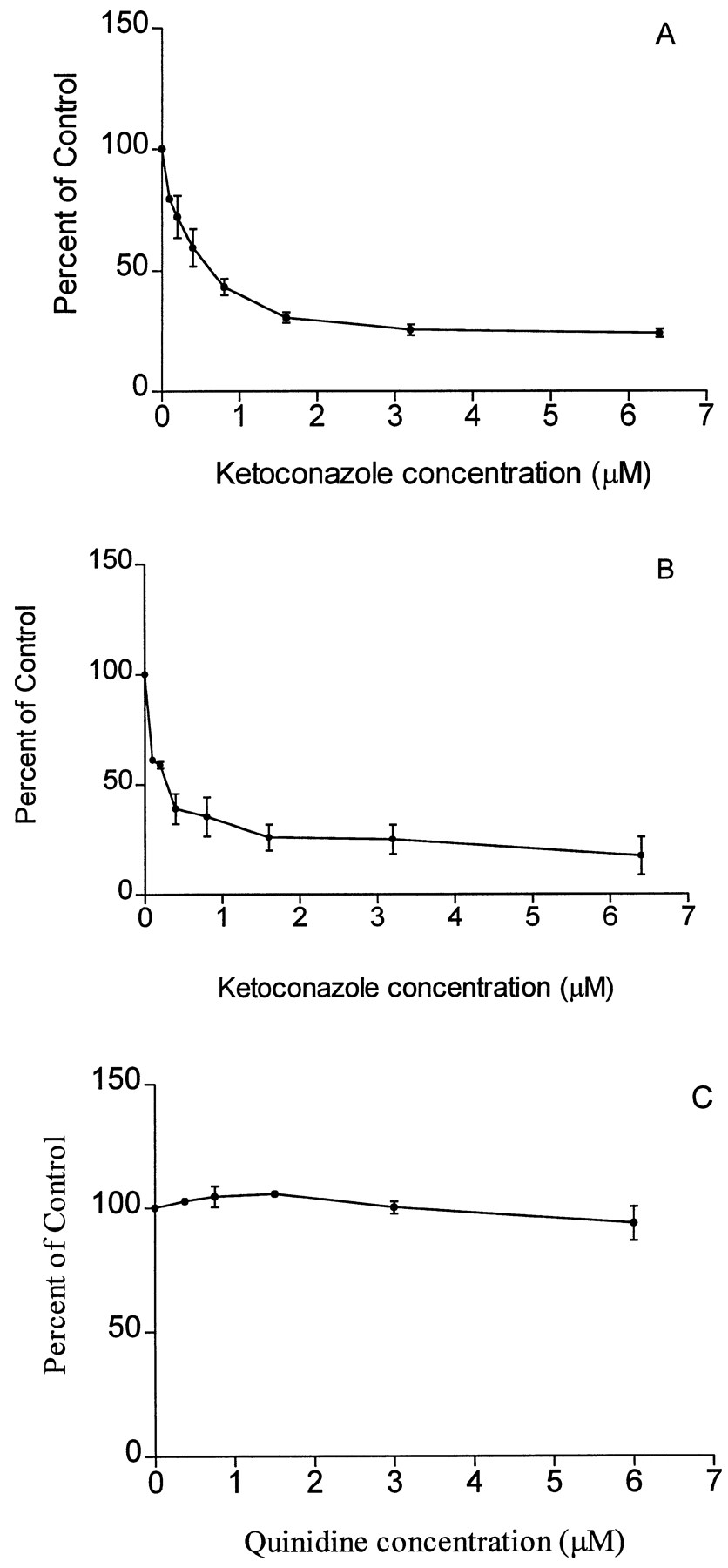
The CYP3A4 inhibitor ketoconazole resulted in a concentration-dependent inhibition of mCPP production in microsomes from both human liver and cells expressing human CYP3A4 (fig. 4). Incubations with quinidine did not inhibit mCPP formation (fig. 4).

Inhibition of mCPP production (expressed as % of control activity; y axis) from trazodone (100 μM) by ketoconazole in human liver microsomes (A) and in microsomes from cells expressing human CYP3A4 (B) and by quinidine in human liver microsomes (C). Eachpoint represents the average of three separate experiments (mean ± SEM).
Discussion
The present experiments showed that mCPP production from trazodone is correlated with CYP3A4 activity in human liver microsomes, is formed from incubations with microsomes from cells expressing CYP3A4 only, and is diminished in the presence of an inhibitor of CYP3A4. Taken together, these results indicate strongly that trazodone is a substrate of CYP3A4 and that this P450 enzyme is important in the formation of the metabolite mCPP. Therefore, there is the potential for drug-drug interactions with this drug and other substrates, inhibitors, and/or inducers of CYP3A4.
Plasma levels of trazodone show wide interindividual differences and typically range from about 0.38 to 5.8 μM (Vatassery et al., 1997). However, it is not the plasma concentration that is relevant but rather the concentration at the enzyme site, or the hepatic concentration, which is of importance with respect to drug interactions (Harvey and Preskorn, 1995; Preskorn, 1996; von Moltkeet al., 1996). Lipophilic drugs partition extensively into the liver, and liver/water partition ratios are typically used to estimate liver drug concentrations (Greenblatt et al., 1996). The hepatic extraction ratio for trazodone is not currently known, but if it is assumed to have a partition ratio similar to the selective serotonin reuptake inhibitors, which are also highly lipophilic and have hepatic extraction ratios of 12 to 26 (Harvey and Preskorn, 1995; Schmider et al., 1996), then hepatic concentrations of trazodone can be expected to range from 60 to 100 μM at a plasma concentration of 5 μM. Therefore, the concentration of trazodone used in the present experiments can be considered to be clinically relevant. Furthermore, as the concentration of 100 μM trazodone is within the linear range of metabolite formation, kinetic parameters should be constant and thus are applicable to lower trazodone concentrations (Iwatsubo et al., 1997).
The clinical significance of potential drug interactions with trazodone depends upon several factors. First, it is important to distinguish between interactions resulting from trazodone’s effects on other compounds and interactions resulting from the effects of other compounds on trazodone. When considering the effects of trazodone on other compounds, it is important to note that the present experiments showed that trazodone is a substrate of CYP3A4 and therefore may act as a competitive inhibitor of other CYP3A4 substrates. The consequences of this interaction will depend upon the relative affinities and concentrations of trazodone and the competing drug at the enzyme, as well as the therapeutic index of both drugs. The therapeutic index of the interacting drug is important, as clinically significant interactions with a CYP3A4 substrate with a narrow therapeutic index, such as terfenadine, could result (Wilkinson, 1996).
The second type of interaction that may occur is from the effects of other compounds on trazodone metabolism. Although trazodone has a relatively wide therapeutic index, it does have potentially bothersome or dangerous side effects such as excessive sedation, which could become a problem at higher plasma concentrations (Haria et al., 1994). However, the most important consideration with respect to trazodone metabolism is the ability to maintain therapeutic plasma concentrations. Clinical antidepressant response is significantly correlated with steady-state plasma trazodone concentrations, and a threshold concentration of 650 ng/ml is considered necessary for antidepressant response (Monteleone et al., 1989). Therefore, any factor that results in a lowering of plasma trazodone levels may interfere with the clinical efficacy of the drug. Because CYP3A4 levels vary 5–20-fold between individuals (Wilkinson, 1996) and because CYP3A4 is inhibited and induced by many commonly encountered drugs and environmental compounds (von Moltke et al., 1995;Wilkinson, 1996), it is important to be aware that trazodone is a substrate of CYP3A4 and thus subject to many factors that may alter its plasma concentration. The clinical significance of this potential interaction has already been noted with carbamazepine, a CYP3A4 inducer, which decreased plasma trazodone levels (Otani et al., 1996). The extent of any interaction with trazodone will of course depend upon individual differences in CYP3A4 activity, as well as plasma levels of both trazodone and the interacting drug. However, as therapeutic concentrations of trazodone are typically below itsKM , it is subject to first-order kinetics and as such is highly sensitive to changes in the concentration of enzyme or substrate (Iwatsubo et al., 1997). The highKM value found in the present experiments for trazodone transformation is consistent with the linear pharmacokinetics of trazodone and mCPP seen clinically (Nilsen et al., 1993). Therefore, the potential for interactions with CYP3A4 substrates and/or inducers is clinically significant.
The quantitative importance of CYP3A4 on the overall disposition of trazodone in man is not currently known, and such knowledge will depend upon the elucidation of the P450 enzymes involved in trazodone’s other metabolic pathways. However, because approximately 20% of a dose of trazodone is recovered in urine as triazolopropionic acid and its conjugates, which is the other fragment formed when trazodone isN-dealkylated to mCPP (Haria et al., 1994; Yamatoet al., 1974a, 1974b), it is reasonable to assume that 20% of the dose is also converted to mCPP. Furthermore, plasma levels of mCPP reach 1–20% those of the parent compound (Otani et al., 1996; Vatassery et al., 1997; Yasui et al., 1995); therefore, CYP3A4 is expected to play a significant role in trazodone’s metabolism.
The present experiments are a direct examination of trazodone metabolism to mCPP by the P450 enzymes and provide evidence that CYP3A4 is a major enzyme responsible for this biotransformation. This finding indicates that the potential for drug-drug interactions between trazodone and other substrates or inhibitors of CYP3A4 exists.
Acknowledgments
The authors are grateful to the Medical Research Council of Canada and the Alberta Heritage Foundation for Medical Research (Mental Health Research Fund) for funding support.
Footnotes
-
Send reprint requests to: Dr. G. B. Baker, Neurochemical Research Unit, Department of Psychiatry, 1E7.44 Mackenzie Health Sciences Ctr., University of Alberta, Edmonton, AB, T6G 2B7, Canada.
-
↵1 This work is in partial fulfillment for the degree of Doctor of Philosophy.
-
Portions of this work were presented at the Joint Meeting of the Canadian College of Neuropsychopharmacology and the British Association of Psychopharmacology in Cambridge, U.K., July, 1997. [Abstract published in J Psychopharmacol11:A27 (1997)]
- Abbreviations used are::
- mCPP
- m-chlorophenylpiperazine
- P450
- cytochrome P450
- Received October 15, 1997.
- Accepted March 2, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}