


Abstract
The capacities to inhibit coumarin 7-hydroxylase activity of human cytochrome P450 2A6 (CYP2A6) by organosulfur compounds were evaluated. Five dialkyl sulfides and five dialkyl disulfides, with alkyl chains from methyl to amyl, were examined. In addition to these chemicals, diallyl sulfide, diallyl disulfide, allyl methyl sulfide, allyln-propyl sulfide, allyl phenyl sulfide, diphenyl sulfide, diphenyl disulfide, difurfuryl disulfide, phenyl cyclopropyl sulfide, 2,2′-dipyridyl disulfide, 4,4′-dipyridyl sulfide, and 4,4′-dipyridyl disulfide were also examined for their capacity to inhibit CYP2A6. The membrane fraction of genetically engineeredEscherichia coli cells expressing CYP2A6 together with NADPH-cytochrome P450 reductase was used as an enzyme source. Dialkyl disulfides inhibited CYP2A6 more strongly than did dialkyl sulfides. Among dialkyl disulfides examined, di-n-propyl disulfide, contained in onion oil, was the most potent competitive inhibitor of CYP2A6, with a Ki value of 1.73 μM. Diallyl disulfide, present in garlic oil, inhibited CYP2A6 activity in a competitive/noncompetitive mixed manner, with theKi value of 2.13 μM. Among all of the organosulfur compounds tested, 4,4′-dipyridyl disulfide was the most potent inhibitor of CYP2A6, with a Ki value of 60 nM, followed by 4,4′-dipyridyl sulfide, with aKi value of 72 nM. These chemicals inhibited CYP2A6 in a competitive manner. The preincubation time did not affect the inhibitory effects of di-n-propyl disulfide, diallyl disulfide, 4,4′-dipyridyl disulfide, and 4,4′-dipyridyl sulfide on CYP2A6, indicating that these chemicals were not mechanism-based inhibitors of CYP2A6. 4,4′-Dipyridyl disulfide also inhibited midazolam 1′-hydroxylase activity of CYP3A4. We discovered 4,4′-dipyridyl disulfide to be a potent and relatively selective inhibitor of CYP2A6.
Cytochrome P450 (CYP1) is a heme-containing enzyme responsible for the metabolism of exogenous compounds such as drugs, environmental pollutants, and dietary chemicals, and endogenous compounds such as steroids, fatty acids, and prostaglandins (Nelson et al., 1996). Among the forms of CYP, CYP2A6 is known to be involved in coumarin 7-hydroxylation and is a predominant catalyst of (−)-nicotine oxidation to form (−)-cotinine (Berkman et al., 1995; Messina et al., 1997). Our studies have shown that CYP2A6 also catalyzes (−)-cotinine 3′-hydroxylation (Nakajima et al., 1996) and SM-12502 S-oxidation (Nunoya et al., 1996). CYP2A6 is also capable of metabolically activating genotoxins includingN-nitrosamines (Yamazaki et al., 1992; Patten et al., 1997). Previously, we examined the roles of CYP2A6 in the metabolic activation of promutagens using Salmonella cells expressing CYP2A6 together with OR (Kushida et al., 2000a,b), and we clarified that CYP2A6 was responsible for the mutagenic activation of tobacco-relatedN-nitrosamines, such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone andN-nitrosonornicotine.
In addition to the function of CYP2A6, the genetic polymorphism of theCYP2A6 gene (Fernandez-Salguero et al., 1995; Nunoya et al., 1998) has been noted as a factor modifying its role in vivo. Individuals showing no catalytic activity toward SM-12502 were found to possess the gene homozygous for CYP2A6*4C (Nunoya et al., 1998). Since the presence of this variant gene clearly shows poor metabolic capacity toward the drug, it seems possible that the polymorphism of CYP2A6 also affects the capacity to activate carcinogens including tobacco-related N-nitrosamines. Therefore, it was of interest to investigate the relationship between CYP2A6 polymorphism and cancer risk in humans. In accordance with this idea, Miyamoto et al. (1999) reported that CYP2A6 genetic polymorphism could be an important factor affecting individual susceptibility to lung cancer. The frequency of subjects homozygous forCYP2A6*4C was lower in the cancer patients than in the healthy control subjects, suggesting that the subjects carryingCYP2A6*4C alleles are resistant to carcinogenesis caused byN-nitrosamines because of the poor metabolic activation capacity.
Judging from these results, we hypothesized that the administration of chemicals that strongly and specifically inhibited the CYP2A6 activity might result in a reduction of the risk of promutagens in the body.
Some chemicals such as methoxsalen, (R)-(+)-menthofuran, and tranylcypromine were reported to inhibit CYP2A6 (Draper et al., 1997; Khojasteh-Bakht et al., 1998). However, methoxsalen and tranylcypromine inhibited other forms of CYP. Methoxsalen inhibited CYP1A2, CYP2B6, CYP3A4, and CYP3A5 activities to extents similar to CYP2A6 (Ono et al., 1996). Tranylcypromine inhibited CYP2C19 (Inaba et al., 1985; Wienkers et al., 1996). It has not been clarified whether or not (R)-(+)-menthofuran is a specific inhibitor of CYP2A6. Thus, there is no chemical so far that specifically inhibits CYP2A6 activity.
Some of the sulfide or disulfide derivatives contained in foods inhibit CYP activity. For example, diallyl sulfide, a component of garlic oil (Brodnitz et al., 1971), inhibited mouse CYP2A5 activity toward acetaminophen metabolism (Genter et al., 1998). Diallyl disulfide, which is also a component of garlic oil, shows protective effects against carcinogenesis induced by N-nitrosodiethylamine in mice and rats (Wattenberg et al., 1989; Takahashi et al., 1992). These lines of evidence may indicate that diallyl disulfide shows chemoprevention toward nitrosamine-induced carcinogenesis by inhibition of CYP2A.
In the present study, we examined 22 structurally related organosulfur compounds for the inhibition of CYP2A6 by determining their ability to inhibit coumarin 7-hydroxylase activity. CYP2A6 expressed in the membrane fraction of Escherichia coli cells together with OR was used as an enzyme source.
Materials and Methods
Chemicals.
G6P, G6P dehydrogenase, and NADP+were obtained from Oriental Yeast (Tokyo, Japan). Allyl methyl sulfide, allyl phenyl sulfide, allyl n-propyl sulfide,p-aminophenol hydrochloride, diallyl sulfide, diallyl disulfide, di-n-amyl sulfide, di-n-amyl disulfide, di-n-butyl sulfide, di-n-butyl disulfide, diethyl sulfide, diethyl disulfide, difurfuryl disulfide, dimethyl sulfide, dimethyl disulfide, diphenyl sulfide, diphenyl disulfide, di-n-propyl sulfide, di-n-propyl disulfide, 2,2′-dipyridyl disulfide, 4,4′-dipyridyl sulfide, 4,4′-dipyridyl disulfide, and phenyl cyclopropyl sulfide were purchased from Tokyo Chemical Industry (Tokyo, Japan). 4′-Hydroxymephenytoin and (S)-mephenytoin were obtained from Sumitomo Chemicals (Osaka, Japan). 7-Ethoxycoumarin was purchased from Aldrich Chemical (Milwaukee, WI). (−)-Cotinine, diclofenac sodium salt, 7-ethoxyresorufin, (−)-nicotine, propranolol hydrochloride, resorufin, paclitaxel (Taxol), and tranylcypromine were obtained from Sigma (St. Louis, MO). Aniline hydrochloride, clonazepam, coumarin, dextromethorphan hydrobromide, 7-hydroxycoumarin, isopropyl β-d(−)-thiogalactopyranoside, and phenobarbital sodium were purchased from Wako Pure Chemicals (Osaka, Japan). Dextrorphan, 4′-hydroxydiclofenac, 1′-hydroxymidazolam, 6α-hydroxypaclitaxel, and midazolam were purchased from Daiichi Pure Chemicals (Tokyo, Japan). Emulgen 911, a nonionic detergent, was kindly provided by Kao (Tokyo, Japan). All other chemicals and solvents were of the highest grade commercially available.
E. coli Strains.
Nine strains of E. coli DH5α expressing each form of human CYP (CYP1A1, CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4), together with OR, were used. These strains of E. coli were established by Iwata et al. (1998). E. coliDH5α strains harboring CYP1B1 or CYP3A5 together with OR were established in our laboratory according to the methods of Shimada et al. (1998) or Gillam et al. (1995).
Culture Conditions for the Expression of CYP and OR in E. coli DH5α Cells.
CYP and the OR were expressed in a culture containing the genetically engineered E. coli cells according to the method reported byIwata et al. (1998). Briefly, 20 μl of bacterial stock solution was inoculated into 2 ml of a Luria-Bertani medium supplemented with ampicillin (100 μg/ml). Cultures were grown overnight with shaking at 37°C. One milliliter of the culture was inoculated into 100 ml of modified Terrific Broth (Gillam et al., 1995) and grown with shaking at 30°C for 8 h before induction by addition of 1.5 mM isopropyl β-d(−)-thiogalactopyranoside. The expression of recombinant proteins was achieved by a further incubation at 30°C for 12 h with shaking. The membrane fraction of E. coli cells was prepared according to the method reported by Sandhu et al. (1993). The CYP content in the bacterial membrane fraction was determined according to the method of Omura and Sato (1964).
Inhibition of Catalytic Activities of CYP by Organosulfur Compounds.
All assays were carried out with the membrane fraction of E. coli cells expressing both CYP and the OR. A typical incubation mixture consisted of 100 mM sodium potassium phosphate buffer (pH 7.4), 50 μM ethylenediamine tetraacetic acid, an NADPH-generating system (0.5 mM NADP+, 5 mM MgCl2, 5 mM G6P, and 1 unit/ml G6P dehydrogenase), and 10 to 50 pmol of CYP in a final volume of 1 ml. Inhibition of CYP activity by organosulfur compounds was examined using substrate concentrations about 2 times theKm value for each reaction. The inhibitor concentrations were 0.1, 1, and 10 μM for inhibition assays with CYP2A6, CYP2C8, CYP2C9, and CYP2C19, 1 and 10 μM for CYP1A1, CYP1A2, CYP1B1, CYP2D6, CYP2E1, and CYP3A5, 0.1 and 1 μM for CYP3A4. An inhibitor was added to the incubation mixture and preincubated for 5 min before the reaction was started by the addition of a substrate. Metabolites were produced linearly within an incubation time and a CYP content in the reaction mixture described below.
Coumarin 7-hydroxylase activity was assayed by fluorometric determination of a metabolite (Pearce et al., 1992). An incubation mixture consisted of a 2.5 μM substrate and 10 pmol of CYP. Incubations were performed at 37°C for 10 min. To determine kinetic parameters for the inhibition of coumarin 7-hydroxylase activity of CYP2A6 by organosulfur compounds, the coumarin concentration ranged from 0.313 to 5.0 μM. Kinetic parameters were calculated from Lineweaver-Burk plots by linear least-squares regression analysis with Microsoft Excel 2000.
7-Ethoxycoumarin O-deethylase activity (Greenlee and Porland, 1978) and 7-ethoxyresorufin O-deethylase activity (Lake, 1987) were determined fluorometrically. The concentrations of 7-ethoxycoumarin were 40 μM for the assay of CYP1A1 activity and 20 μM for CYP1B1 activity, and the concentration of 7-ethoxyresorufin was 2.5 μM. The incubation mixture contained 50 pmol of CYP. Incubations were carried out at 37°C for 10 min for the assay of 7-ethoxycoumarin O-deethylation of CYP1A1 and CYP1B1 and 7 min for the assay of 7-ethoxyresorufin O-deethylation of CYP1A2.
Paclitaxel 6α-hydroxylation was assayed as described by Cresteil et al. (1994), with minor modifications. Briefly, the reaction mixture consisted of 150 mM sodium potassium phosphate buffer (pH 7.4), the NADPH-generating system described above, 16 μM paclitaxel, and 10 pmol of CYP2C8 in a final volume of 0.5 ml. Reactions were carried out at 37°C for 5 min. Analysis of a metabolite, 6α-hydroxypaclitaxel, was performed by HPLC using a computerized HPLC system (HITACHI model L-7000 series, HITACHI, Tokyo, Japan) equipped with a Capcell Pak C18 analytical column (4.6 × 250 mm; SG120Å; 5 μm; Shiseido, Tokyo Japan). The metabolite was separated with 40% acetonitrile as a solvent system at a flow rate of 1.0 ml/min. Quantification of the metabolite was achieved by comparing the peak area of the metabolite in a chromatogram with that of an internal standard, taxotere.
The assay of diclofenac 4′-hydroxylase activity was performed according to the protocol of GENTEST with minor modifications (GENTEST, Woburn, MA) (http://www.gentest.com/hlm_meth.htm/). Briefly, the incubation mixture consisted of 100 mM Tris-HCl buffer (pH 7.4), the NADPH-generating system described above, 8 μM diclofenac, and 10 pmol of CYP in a final volume of 0.25 ml. Incubations were performed at 37°C for 15 min. Metabolite was analyzed by HPLC equipped with a Capcell Pak C18 analytical column. The mobile phase consisted of 12.5 mM Tris-HCl buffer (pH 7.4), methanol, and acetonitrile (80:15:5, v/v; solvent A) and methanol (solvent B). The metabolite, 4′-hydroxydiclofenac, was separated using a solvent system: 100 to 0% solvent A linear gradient, 0 to 20 min, at a flow rate of 1.0 ml/min. The metabolite was quantified by comparing the peak area of the metabolite with that of standard curve with 4′-hydroxydiclofenac.
The assay of (S)-mephenytoin 4′-hydroxylation was performed as described by Yasumori et al. (1989), with minor modifications. Briefly, the substrate concentration was 70 μM. The incubation mixture contained 10 pmol of CYP in a final volume of 0.25 ml. Incubations were carried out at 37°C for 60 min. Analysis of the metabolite was performed by HPLC equipped with an analytical TSK-gel ODS-120A column (4.6 × 250 mm; 4 μm; TOSOH, Tokyo, Japan). The mobile phase consisting of acetonitrile, methanol, and 2.5 mM sodium perchlorate (pH 2.5) (6:25:69, v/v) was delivered at a flow rate of 1.0 ml/min. The quantification of the metabolite, 4′-hydroxymephenytoin, was performed by comparing the HPLC peak area with that of an internal standard, phenobarbital.
Dextromethorphan O-demethylation was performed according to the method described by Kronbach et al. (1987), with minor modifications. Briefly, 5 pmol of CYP was added to the reaction mixture. The substrate concentration was 4.0 μM, and the final volume was 0.25 ml. Incubations were carried out at 37°C for 5 min. Analysis of the metabolite was performed by HPLC equipped with a Capcell Pak C18 column. Elution was carried out with a solvent system consisting of acetonitrile and 2.5 mM sodium perchlorate (pH 2.5) (3:7, v/v), with a flow rate of 1.0 ml/min. The metabolite, dextrorphan, was quantified by comparing the peak area of the metabolite in a chromatogram with that of an internal standard, propranolol.
Aniline hydroxylation was assayed according to a colorimetric method described elsewhere (Imai et al., 1966), except that the substrate concentration was 1000 μM, the incubation mixture contained 25 pmol of CYP2E1, and the incubations were carried out at 37°C for 15 min.
The assay of midazolam 1′-hydroxylation was performed by the method described by Li et al. (1996), with modifications. A typical reaction mixture contained 30 pmol of CYP. The concentrations of midazolam were 20 and 7 μM for CYP3A4 and CYP3A5, respectively. After incubation for 5 min, 5 ml of ethyl acetate was added to stop the reaction. One nanomole of clonazepam as an internal standard was added to a tube after incubation. The mixture was extracted with ethyl acetate and centrifuged at 3,000 rpm for 10 min. The organic layer was transferred to a tube and evaporated. The residue was dissolved in 200 μl of a mixture containing 10 mM sodium acetate, methanol, and acetonitrile (9:1:1, v/v) and was subjected to HPLC equipped with an analytical TSK-gel ODS-120T column (4.6 × 150 mm; 4 μm; TOSOH). The mobile phase consisted of 10 mM sodium acetate, methanol, and acetonitrile (9:1:1, v/v; solvent A) and methanol and acetonitrile (2:1, v/v; for solvent B). The metabolite, 1′-hydroxymidazolam, was separated using 40% solvent A as a solvent system at a flow rate of 0.8 ml/min.
Determination of 4,4′-Dipyridyl Disulfide and 4,4′-Dipyridyl Sulfide.
Analysis of 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide was performed by HPLC using a computerized HPLC system (Agilent 1100 series, Agilent Technologies, Palo Alto, CA) equipped with an analytical YMC-Pack Pro C18 column (2.0 × 150 mm; 5 μm; YMC, Milford, MA). The organosulfur compounds were separated with 28% acetonitrile as a solvent system at a flow rate of 0.2 ml/min. The elutions of 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide were monitored at 250 nm. Quantification of the 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide was achieved by comparing the peak area of these chemicals in a chromatogram with those of respective internal standards, 4,4′-dipyridyl sulfide and 4,4′-dipyridyl disulfide.
Results
Inhibition of Coumarin 7-Hydroxylase Activity of CYP2A6 by Organosulfur Compounds.
Inhibition of coumarin 7-hydroxylase activity of CYP2A6 by 22 structurally related organosulfur compounds was examined. The chemical structure of the chemicals is shown in Fig.1. The inhibition of CYP2A6 activity by these chemicals is summarized in Table 1. The inhibitory effects of dialkyl disulfides toward CYP2A6 activity were similar with or stronger than those of corresponding dialkyl sulfides (Fig. 2). The length of the alkyl chain was one of the determinants of the inhibitory effects of dialkyl disulfide toward CYP2A6. Among dialkyl disulfides tested, di-n-propyl disulfide, which is known to be contained in onion oil, inhibited CYP2A6 most potently (Fig. 2). The addition of 10 μM di-n-propyl disulfide to a reaction mixture resulted in the inhibition of coumarin 7-hydroxylase activity to about 40% of control without an inhibitor. Diallyl disulfide, present in garlic oil, di-n-butyl disulfide, allyl phenyl sulfide, diphenyl sulfide, and phenyl cyclopropyl sulfide also inhibited CYP2A6 to the same extent as did di-n-propyl disulfide (Table 1). Among these organosulfur compounds tested, 4,4′-dipyridyl disulfide was the most potent inhibitor of CYP2A6, followed by 4,4′-dipyridyl sulfide. The addition of 1 μM 4,4′-dipyridyl disulfide to a reaction mixture caused 84.1% inhibition of CYP2A6 activity. These organosulfur compounds inhibited the CYP2A6 activity more strongly than tranylcypromine, a known representative inhibitor of CYP2A6 (Table 1).

Structure of organosulfur compounds.
Inhibition of coumarin 7-hydroxylase activity of CYP2A6 by organosulfur compounds
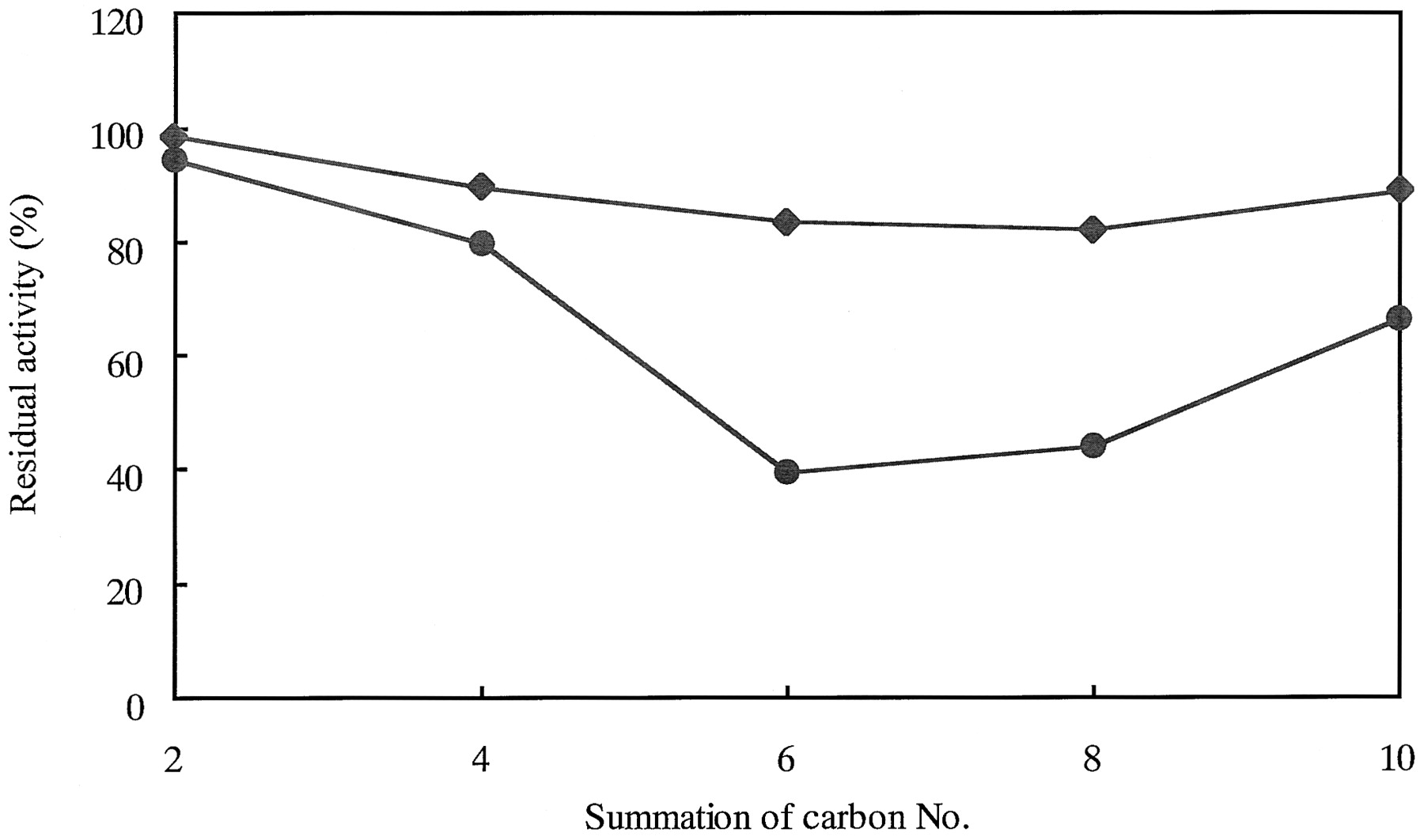
The relationship between the length of alkyl chains of dialkyl derivatives of sulfide and disulfide and the inhibition of coumarin 7-hydroxylase activity of CYP2A6.
The concentration of inhibitors and coumarin was 10 and 2.5 μM, respectively. An inhibitor was added to the incubation mixture and preincubated for 5 min before the reaction was started by the addition of a substrate. Incubations were performed at 37°C for 10 min. The control activity of CYP2A6 determined without an inhibitor was 3.86 nmol/min/nmol of CYP. ♦, dialkyl sulfide; ●, dialkyl disulfide. Each point represents the means of two independent experiments (n = 2).
Kinetic Analysis for the Inhibition of CYP2A6 Activity by Organosulfur Compounds.
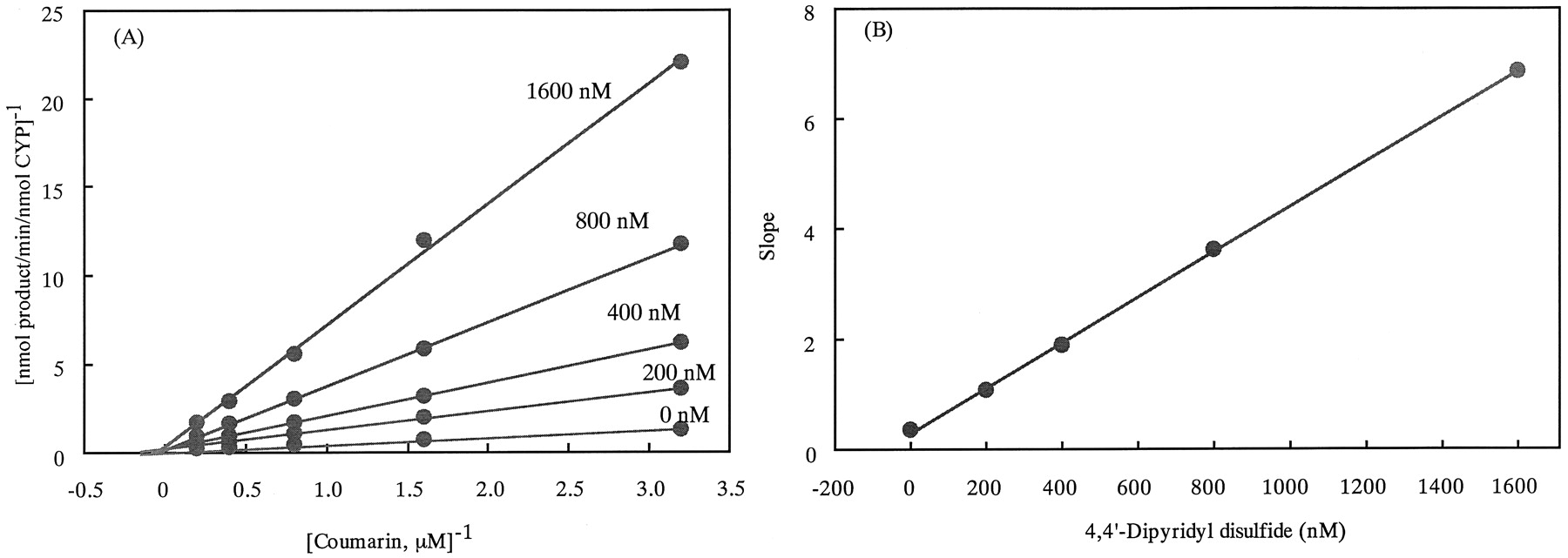
The kinetic parameters for the inhibition of coumarin 7-hydroxylase activity by 4,4′-dipyridyl disulfide, 4,4′-dipyridyl sulfide, di-n-propyl disulfide, and diallyl disulfide were examined. The results are shown in Fig. 3 and Table 2. Naturally occurring organosulfur compounds di-n-propyl disulfide and diallyl disulfide inhibited CYP2A6 less potently than did 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide. 4,4′-Dipyridyl disulfide, 4,4′-dipyridyl sulfide, and di-n-propyl disulfide inhibited the CYP2A6 activity in a competitive manner, whereas diallyl disulfide showed a mixed-type inhibition.

Lineweaver-Burk plots for the inhibition of the coumarin 7-hydroxylase activity of CYP2A6 by 4,4′-dipyridyl disulfide.
A, Lineweaver-Burk plots for the inhibition of CYP2A6 activity by 4,4′-dipyridyl disulfide. B, the relationship between slopes (ratio of apparent Km to apparentVmax) of the Lineweaver-Burk plots and the inhibitor concentrations. Each point represents the means of two independent experiments (n = 2).
Kinetic parameters for inhibition of coumarin 7-hydroxylase activity of CYP2A6 by organosulfur compounds
Examination of Organosulfur Compounds as Mechanism-Based Inhibitors.
We examined whether 4,4′-dipyridyl disulfide, 4,4′-dipyridyl sulfide, di-n-propyl disulfide, and diallyl disulfide inhibit CYP2A6 in a mechanism-based mechanism. If these organosulfur compounds were mechanism-based inhibitors of CYP2A6, we would expect the inhibition to be enhanced by preincubation of the inhibitor with the enzyme preparation. Results are shown in Fig.4. The inhibition potency of 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide was not enhanced in a preincubation time-dependent manner, suggesting that these organosulfur compounds are not mechanism-based inhibitors of CYP2A6. We measured the concentrations of 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide in the reaction mixture before and after the 15 min of preincubation and the 10 min of incubation with coumarin. When 1.0 μM 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide were added to the reaction mixture, the final concentrations of these chemicals were 1.03 and 1.05 μM, respectively, indicating that these chemicals were stable in the reaction mixture and were not metabolized by CYP2A6. Diallyl disulfide, diallyl sulfide, and 2,2′-dipyridyl disulfide were also not suicidal inhibitors of CYP2A6. Interestingly, the inhibition of CYP2A6 activity by di-n-propyl disulfide decreased with the increase of preincubation time. The reason for this phenomenon is not known at present.

The effect of preincubation time on the inhibition of coumarin 7-hydroxylase activity by organosulfur compounds.
The concentration of inhibitors was 10 μM, except that the concentration of 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide was 1 μM. Coumarin concentration was 2.5 μM. An inhibitor was added to the incubation mixture and preincubated for indicated periods before the reaction was started by the addition of a substrate. Incubations were performed at 37°C for 10 min. The control activity of CYP2A6 measured in the absence of an inhibitor was 3.77 nmol/min/nmol of CYP. ♦, 4,4′-dipyridyl disulfide; ▪, 4,4′-dipyridyl sulfide; ●, diallyl disulfide; ▴, di-n-propyl disulfide; ○, 2,2′-dipyridyl disulfide; ▵, diallyl sulfide. Each point represents the means of two independent experiments (n = 2).
The Inhibitory Effects of 4,4′-Dipyridyl Disulfide on Activity of Other CYPs.
To determine whether 4,4′-dipyridyl disulfide inhibits the activity of CYP2A6 selectively, the inhibitory effects of 4,4′-dipyridyl disulfide toward catalytic activities of ten other CYPs were examined. A representative substrate for each form of CYP was used. The results are shown in Table 3. 4,4′-Dipyridyl disulfide did not inhibit catalytic activities of other CYP forms, except for CYP3A4. The addition of 1 μM 4,4′-dipyridyl disulfide to an incubation mixture caused the inhibitions of midazolam 1′-hydroxylase activity of CYP3A4 by 80%, paclitaxel 6α-hydroxylase activity of CYP2C8 by 34%, and (S)-mephenytoin 4′-hydroxylase activity of CYP2C19 by 31%.
Inhibition of enzyme activities of CYP forms for typical substrates by 4,4′-dipyridyl disulfide
Judging by the inhibitory effects of 4,4′-dipyridyl disulfide on the activities of human CYP, we conclude that 4,4′-dipyridyl disulfide is a potent and relatively selective inhibitor of CYP2A6 and CYP3A4.
Discussion
CYP2A6 has been recognized to be involved in the mutagenic activation of promutagens such as tobacco-relatedN-nitrosamines (Kushida et al., 2000a,b). Epidemiological studies have demonstrated a close association between lung cancer risk and the CYP2A6 genotypes (Miyamoto et al., 1999). If one of the causes of lung cancer is related to the metabolic activation of tobacco-related N-nitrosamines by CYP2A6, the inhibition of CYP2A6 activity by chemicals that strongly and specifically inhibit the CYP activity may result in the chemoprevention of lung cancer. In individuals possessing CYP2A6*1A, the inhibition of CYP2A6 activity by specific inhibitors may prevent the mutagenic activation of tobacco-related N-nitrosamines, resulting in the chemoprevention of lung cancer.
If 4,4′-dipyridyl disulfide is a potent inhibitor of coumarin 7-hydroxylase activity of CYP2A6 in vitro, it is necessary to investigate the in vivo inhibition of CYP2A6 activity and the toxicity of the chemical.
Comparing the inhibition of CYP2A6 activity by dialkyl disulfide with that by dialkyl sulfide possessing the same length of alkyl chains as dialkyl disulfide, the dialkyl disulfide was a more potent inhibitor of CYP2A6 activity than dialkyl sulfide (Fig. 2). The data suggest that the sulfur atom may play important roles in the inhibition of CYP2A6 activity. The exact mechanism for the role of the sulfur atom in these organosulfur compounds on the inhibition of CYP2A6 activity remains to be elucidated at present.
4,4′-Dipyridyl disulfide and 4,4′-dipyridyl sulfide strongly inhibited the coumarin 7-hydroxylase activity of CYP2A6. These organosulfur compounds possess two pyridine rings. Metyrapone is known to be an inhibitor of CYP (Testa and Jenner, 1981), possessing two pyridine rings. One of the mechanisms for the inhibition of CYP by metyrapone is thought to be the interaction of the nitrogen atom of pyridine rings to heme iron of CYP (Testa and Jenner, 1981). Therefore, it may be possible that nitrogen atoms of the pyridine rings of 4,4′-dipyridyl disulfide and 4,4′-dipyridyl sulfide bind to the heme iron atom of CYP2A6 to inhibit the CYP2A6 activity. On the other hand, 2,2′-dipyridyl disulfide did not inhibit the CYP2A6 activity, even though the chemical possessed two pyridine rings. The distance between nitrogen atoms of the two pyridine rings may determine the inhibitory effects of pyridine derivatives of organosulfur compounds toward CYP2A6 activity.
Interestingly, 4,4′-dipyridyl disulfide inhibited the midazolam 1′-hydroxylase activity of CYP3A4 but did not inhibit the midazolam 1′-hydroxylase activity of CYP3A5. The amino acid sequence of CYP3A5 is about 85% identical to that of CYP3A4. The change of these amino acid residues of CYP3A4 to those of CYP3A5 by the site-directed mutagenesis method may result in the clarification of amino acid residues responsible for the substrate binding of CYP3A4 and CYP3A5. CYP3A5 is known to be expressed in the adult liver of 20 to 30% of the Caucasian population (Aoyama et al., 1989; Wrighton et al., 1989). Thus, 4,4′-dipyridyl disulfide may be a useful tool to screen the expression of CYP3A5 in the adult liver at a holo-protein level. Midazolam 1′-hydroxylation is catalyzed by both CYP3A4 and CYP3A5. If CYP3A5 is not expressed in the liver microsomes, then the addition of 4,4′-dipyridyl disulfide is expected to inhibit midazolam 1′-hydroxylase activity, while the midazolam 1′-hydroxylase activity may remain if CYP3A5 is expressed in the liver microsomes.
The results obtained in the present study provide insights into the structural and chemical properties of organosulfur compounds as inhibitors of CYP2A6 activity as well as in vivo chemoprevention by organosulfur compounds.
Footnotes
-
This study was supported in part by a grant-in-aid from the Ministry of Education, Science, Sports and Culture of Japan, by a grant (99-2) from the Organization for Pharmaceutical Safety and Research (OPSR), by grants-in-aid for Cancer Research from the Ministry of Health and Welfare of Japan, and by a fund under a contract with the Environment Agency of Japan.
- Abbreviations used are::
- CYP
- cytochrome P450
- G6P
- glucose 6-phosphate
- HPLC
- high-performance liquid chromatography
- OR
- NADPH-cytochrome P450 reductase
- SM-12502
- (+)-cis-3, 5-dimethyl-2-(3-pyridyl) thiazolin-4-one hydrochloride
- Received January 16, 2001.
- Accepted April 16, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}