


Abstract
A novel mibefradil derivative, NNC55-0396, designed to be hydrolysis-resistant, was shown to be a selective T-type Ca2+ channel inhibitor without L-type Ca2+ channel efficacy. However, its effects on cytochromes P450 (P450s) have not previously been examined. We investigated the inhibitory effects of NNC55-0396 toward seven major recombinant human P450s—CYP3A4, CYP2D6, CYP1A2, CYP2C9, CYP2C8, CYPC19, and CYP2E1—and compared its effects with those of mibefradil and its hydrolyzed metabolite, Ro40-5966. Our results show that CYP3A4 and CYP2D6 are the two P450s most affected by mibefradil, Ro40-5966, and NNC55-0396. Mibefradil (IC50 = 33 ± 3 nM, Ki = 23 ± 0.5 nM) and Ro40-5966 (IC50 = 30 ± 7.8 nM, Ki = 21 ± 2.8 nM) have a 9- to 10-fold greater inhibitory activity toward recombinant CYP3A4 benzyloxy-4-trifluoromethylcoumarin-O-debenzylation activity than NNC55-0396 (IC50 = 300 ± 30 nM, Ki = 210 ± 6 nM). More dramatically, mibefradil (IC50 = 566 ± 71 nM, Ki = 202 ± 39 nM) shows 19-fold higher inhibition of CYP3A-associated testosterone 6β-hydroxylase activity in human liver microsomes compared with NNC55-0396 (IC50 = 11 ± 1.1 μM, Ki = 3.9 ± 0.4 μM). Loss of testosterone 6β-hydroxylase activity by recombinant CYP3A4 was shown to be time- and concentration-dependent with both compounds. However, NNC55-0396 (KI = 3.87 μM, Kinact = 0.061/min) is a much less potent mechanism-based inhibitor than mibefradil (KI = 83 nM, Kinact = 0.048/min). In contrast, NNC55-0396 (IC50 = 29 ± 1.2 nM, Ki = 2.8 ± 0.3 nM) and Ro40-5966 (IC50 = 46 ± 11 nM, Ki = 4.5 ± 0.02 nM) have a 3- to 4-fold greater inhibitory activity toward recombinant CYP2D6 than mibefradil (IC50 = 129 ± 21 nM, Ki = 12.7 ± 0.9 nM). Our results suggest that NNC55–0396 could be a more favorable T-type Ca2+ antagonist than its parent compound, mibefradil, which was withdrawn from the market because of strong inhibition of CYP3A4.
Voltage-gated Ca2+ channels are transmembrane proteins involved in the regulation of cellular excitability and intracellular Ca2+ signaling (Huang et al., 2004). They are divided into two main types: the high voltage–activated channels (L-, N-, P/Q-, and R-types) and the low voltage-activated or T-type channels (Armstrong and Matteson, 1985; Perez-Reyes et al., 1998). During the past three decades, Ca2+ channel antagonists belonging to many structurally diverse classes, such as dihydropyridines, phenylalkylamines, and benzothiazepines, have been developed for the treatment of hypertension and chronic stable angina pectoris (Oparil and Calhoun, 1991). Their mode of action is to inhibit the inward current of Ca2+ through the slow L-type Ca2+ channels (Triggle, 1991). Mibefradil was reported in 1989 as a novel Ca2+ antagonist whose structure belongs to a new class, containing a tetraline ring linked to a benzimidazole group via an aliphatic tertiary amine (Fig. 1) (Clozel et al., 1989). Mibefradil induces coronary and peripheral vasodilation through a direct effect on smooth muscle via blockade of T-type and L-type Ca2+ channels (Massie, 1997). Although mibefradil binds to a unique receptor site that overlaps with that of verapamil (Rutledge and Triggle, 1995), it does not depress myocardial contractility (Clozel et al., 1990), and it is not associated with negative inotropism (Portegies et al., 1991), which represents a therapeutic advantage for mibefradil.
Mibefradil was marketed by Roche (Basel, Switzerland) as Posicor after Food and Drug Administration approval in June 1997 for hypertension and chronic stable angina pectoris. About 200,000 American patients, and double that number worldwide, took the drug (SoRelle, 1998). Soon after its release, a number of case reports showed the dangers of mibefradil drug interactions, including rhabdomyolysis and renal failure with simvastatin (Schmassmann-Suhijar et al., 1998) and symptomatic bradycardia with β-blockers (Rogers and Prpic, 1998). Mibefradil is a strong inhibitor of CYP3A4, CYP2D6, and P-glycoprotein (Ernst and Kelly, 1998; Wandel et al., 2000). It irreversibly inhibits CYP3A4 (Prueksaritanont et al., 1999), which is a serious problem as this cytochrome P450 (P450) is responsible for the metabolism of more drugs than any other P450. Coadministration of mibefradil with terfenadine, cyclosporine A, or quinidine, for example, results in significant increases in their plasma concentrations; coadministration also leads to serious adverse effects with other drugs, including verapamil and diltiazem (Ernst and Kelly, 1998; Prueksaritanont et al., 1999; Varis et al., 2000). Because these drugs are all substrates for CYP3A4, it appears that inhibition of drug metabolism by mibefradil was the main cause for the adverse effects that led to the drug being withdrawn in June 1998 (SoRelle, 1998; http://www.fda.gov/medwatch/SAFETY/1997/posico.htm).

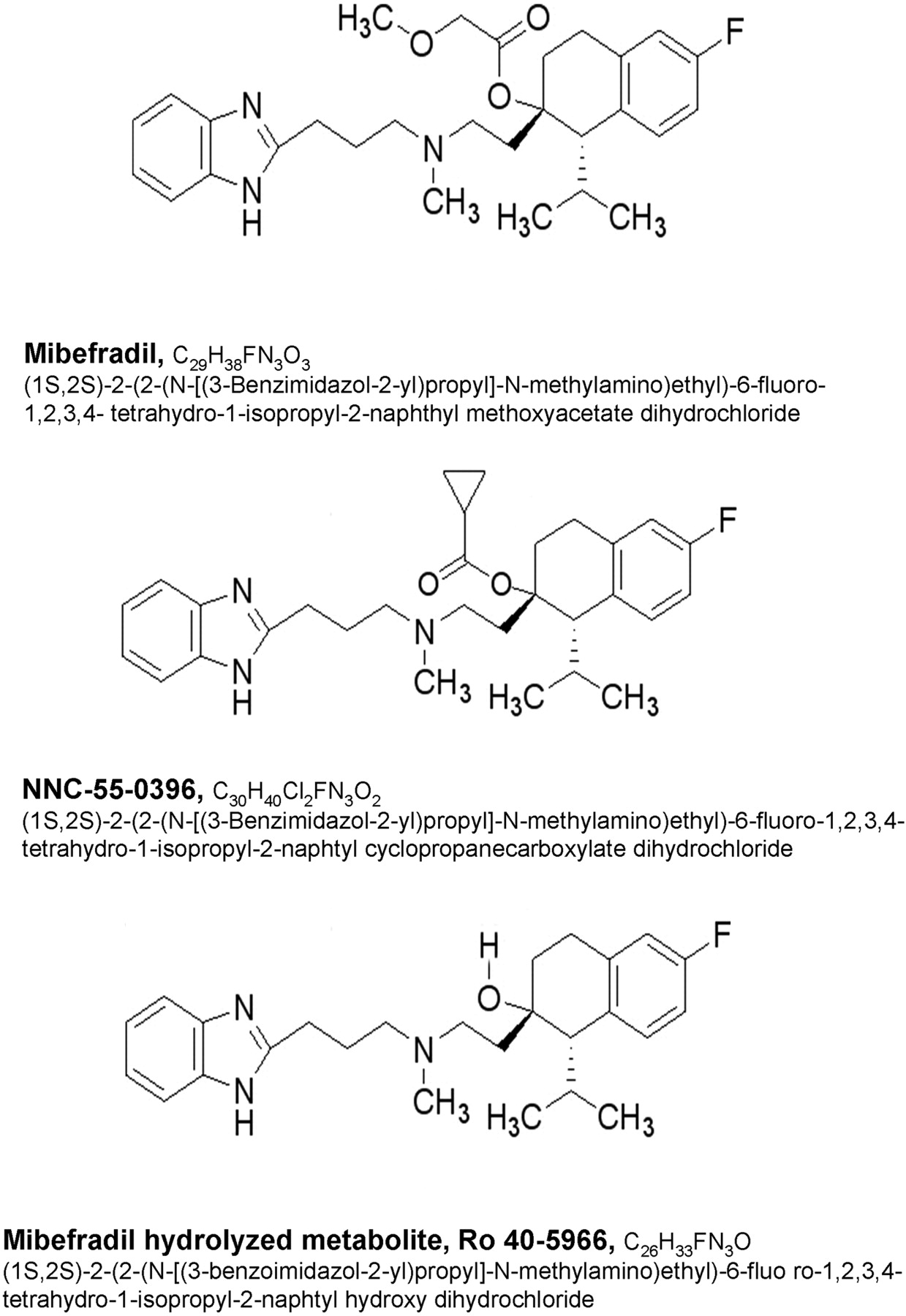
Chemical structures of mibefradil, NNC55-0396, and the hydrolyzed metabolite of mibefradil, Ro 40-5966.
Mibefradil is metabolized mainly in the liver, producing as many as 30 metabolites (Wiltshire et al., 1997a,b). Metabolism consists of a combination of P450-mediated oxidation, hydrolysis of the ester side chain by CYP3A4 and esterases, and conjugation with glucuronic acid by UDP glucuronosyltransferases (UGTs) (Wiltshire et al., 1997b; Ernst and Kelly, 1998). One of the major hydrolyzed metabolites, Ro 40-5966 (Fig. 1), was shown to strongly bind to L-type Ca2+ channels (Wu et al., 2000). Thus, L-type activity is a nonspecific feature of mibefradil administration (Li et al., 2005) and may account for other adverse effects associated with mibefradil. Recently, several analogs of mibefradil have been synthesized and tested for effects against T-type and L-type Ca2+ channels (Huang et al., 2004; Li et al., 2005). These analogs were designed to achieve more selectivity toward T-type Ca2+ channels. One of these is NNC55-0396, which has been shown to be equipotent as mibefradil in antagonizing the T-type Ca2+ channel while exhibiting no L-type Ca2+ channel inhibition. NNC55-0396 was developed to be resistant to hydrolysis by substituting the methoxyacetyl side chain of mibefradil with cyclopropanecarboxylate (Fig. 1) (Huang et al., 2004; Li et al., 2005). This compound is not able to generate the hydrolyzed L-type Ca2+ channel blocker metabolite, Ro 40-5966, thus rendering NNC55-0396 selective to T-type Ca2+ channels (Huang et al., 2004; Li et al., 2005). Such a selective T-type Ca2+ channel antagonist, if found to possess a favorable safety profile, would represent a desirable therapeutic for chronic hypertension (Li et al., 2005), a disorder characterized by T-type Ca2+ channel up-regulation in animals (Nuss and Houser, 1993).
A critical safety issue is whether NNC55-0396 has a more favorable P450 inhibition profile than mibefradil because such information would predict its potential for drug interactions. After preliminary observations indicated differences in behavioral drug interactions involving mibefradil and NNC55-0396 in mice (our unpublished data), we conjectured that NNC55-0396 may have, despite similarities in structure, a more benign P450 inhibition profile than mibefradil. In this article, we compared NNC55-0396's P450 inhibition profile with that of mibefradil and with that of the latter's hydrolyzed metabolite, Ro 40-5966, using both recombinant human P450s and human liver microsomes. These in vitro studies suggest that NNC55-0396 may be a more beneficial therapeutic agent for T-type Ca2+ channel antagonist.
Materials and Methods
Chemical and Enzymes. Bacterial membranes containing P450s and NAPDH-P450-reductase (CPR), and human liver microsomes were obtained from BD Gentest (Franklin Lakes, NJ). Crude bacterial lysate containing the recombinant human CYP3A4 + CPR and P450 reaction buffer mixtures containing the NADP(+)/H regeneration enzymes were obtained from Bio-Catalytics, Inc. (Pasadena, CA). Porcine liver esterase was obtained from Sigma-Aldrich (St. Louis, MO). The fluorescent substrates 3-[2-N,N-diethyl-N-methylamino]-7-methoxy-4-methylcoumarin (AMMC), 7-benzyloxy-4-trifluoromethylcoumarin (BFC), 7-methoxy-4-trifluoromethylcoumarin (MFC), and 7-hydroxy-4-trifluoromethylcoumarin (HFC) were purchased from BD Gentest. Testosterone, 6β-hydroxytestosterone, harmaline, harmalol, mibefradil, and NNC55-0396 were obtained from Sigma-Aldrich. All the high-performance liquid chromatography (HPLC) solvents (HPLC grade) were obtained from Sigma-Aldrich.
Km Determination for Probe Substrates. To determine the Km of BFC and AMMC for recombinant CYP3A4 and CYP2D6, respectively, seven different concentrations of substrates were incubated in 200-μl reactions with 30 nM recombinant P450 + CPR and 1× reaction buffer. The reactions were terminated after 10 min at 37°C by the addition of 200 μl of ice-cold acetonitrile. A similar procedure was used to determine the Km of testosterone and harmaline for human liver microsomes with the final concentration of microsomes being 0.13 mg/ml and time of incubation being 5 min at 37°C. All the samples were analyzed in duplicate. The reactions were frozen in dry-ice methanol for 5 min to precipitate proteins, followed by centrifugation at 13,000g for 15 min to remove proteins. The supernatants were analyzed by HPLC with diode array and fluorescence detectors. The Km of each substrate was then calculated with Prism software (GraphPad Software Inc., San Diego, CA) using nonlinear regression with the Michaelis-Menten equation.
Incubations of Bacterially Recombinant Human P450s with Substrates and Inhibitors. Assays were conducted in 1.5-ml Eppendorf tubes in duplicate. Two hundred-microliter reactions contained 1× reaction buffer mixture (pH 7.5, NADPH/NADP+ regenerating enzymes), 30 nM recombinant P450 + CPR, 100 μM BFC or MFC, or 10 μM AMMC, and various concentrations of inhibitors. One hundred-micromolar MFC was used as a probe substrate for CYP1A2, CYP2C9, CYP2C19, CYP2C8, and CYP2E1; 100 μM BFC was used for CYP3A4, and 10 μM AMMC was used for CYP2D6. The substrates and the inhibitors (mibefradil, Ro 40-5966, and NNC55-0396) were all dissolved in acetonitrile. One hundred five microliters of an enzyme buffer (EB) was prepared first that contained the 2× reaction buffer and 60 nM P450 enzymes. The EB was prewarmed for 3 min at 37°C. The reaction was initiated by the addition of 95 μl of substrate and inhibitor mixture containing substrate and the inhibitor or vehicle (acetonitrile). One hundred nanomolar (low concentration) and 10 μM (high concentration) inhibitors were used in the preliminary inhibition studies. Ten micromolar inhibitors and eight successive 3:1 dilution concentrations were used in IC50 value determinations. The reactions were terminated after 20 min at 37°C by the addition of 200 μl of ice-cold acetonitrile. The reactions were frozen in dry-ice methanol for 5 min to precipitate proteins, followed by centrifugation at 13,000g for 15 min to remove proteins. The supernatants were analyzed by HPLC with diode array and fluorescence detectors.
Incubations of Human Liver Microsomes with Testosterone, Harmaline, and Inhibitors. Incubations were conducted in duplicate with a 200-μl volume in Eppendorf tubes. An EB was prepared that contained 0.13 mg/ml human liver microsomes and 2× reaction mixture buffer (pH 7.5, NADPH and NADP+/H regenerating enzymes and proprietary stabilizers, purchased from BioCatalytics, Inc.). One hundred five microliters of EB was prewarmed for 3 min at 37°C. The reaction was initiated by adding 95 μl of substrate and inhibitor mixture containing 200 μM testosterone or 20 μM harmaline and various concentrations of inhibitors. Ten micromolar inhibitors and eight successive 3:1 dilution concentrations were used in IC50 value determinations. Acetonitrile was used as vehicle. The reactions were stopped after 5-min incubation at 37°C with 200 μl of acetonitrile (2% acetic acid). The samples were frozen in dry-ice methanol for 5 min, followed by centrifugation at 13,000g for 15 min to remove proteins. The supernatant was analyzed by HPLC with diode array and fluorescence detectors.
Hydrolysis of Mibefradil and NNC55-0396 Using Pig Liver Esterase and Alkaline Hydrolysis. Forty micromolar NNC55-0396 or mibefradil was incubated with 27 unit/ml pig liver esterase for 1 h at 37°C. The reactions were stopped with 1× volume acetonitrile, and the incubation mixtures were centrifuged at 13,000g for 20 min. Supernatants were analyzed by HPLC. Ro 40-5966 was synthesized using alkaline hydrolysis as described previously with some modifications (Wu et al., 2000) as follows: 500 μl of 4 mM mibefradil in acetonitrile was added to 125 μl of 10 N NaOH; the mixture was incubated for 12 min in a boiling water bath and then neutralized with 125 μl of 5 M hydrochloric acid. Because of the evaporation of acetonitrile, the volume was adjusted with acetonitrile to the starting volume of 500 μl. Complete hydrolysis was checked by HPLC. All the samples were analyzed with a C18 reverse-phase column (150 × 4.6 mm, 5 μm, Vydac; Grace, Deerfield, IL). Mibefradil, NNC55-0396, and Ro 40-5966 were monitored with fluorescence at 270/300 nm (excitation/emission λ) and with UV absorbance at 275-nm wavelength. The HPLC condition was set at 70% B isocratic for 30 min (solvent A: H2O, 0.1% acetic acid, and 5 mM potassium phosphate, pH 7.5; solvent B: acetonitrile).
Time- and Concentration-Dependent Inhibition of Recombinant CYP3A4 by NNC55-0396 and Mibefradil. Studies on the mechanism-based inhibition of CYP3A4 were performed using the testosterone 6β-hydroxylase assay at a high concentration of testosterone (500 μM) to minimize the possibility of competitive inhibition by the inhibitors. Instead of bacterial membrane containing CYP3A4 + CPR, crude bacterial lysate containing CYP3A4 + CPR was used. The crude lysate was shown not to have more than 15% autoinactivation over the preincubation period in the absence of the inhibitors. All the reactions were carried out at 37°C in duplicate. One hundred nanomolar human recombinant CYP3A4 + CPR was preincubated with NAPDH (1 mM) in the presence of 0, 133, 400, 1200, and 3600 nM NNC55-0396 or 44, 133, and 400 nM mibefradil, in a final volume of 120 μl containing 0.1 M potassium phosphate buffer, pH 7.5. At 0, 4, 8, 12, and 16 min, 20-μl aliquots of the reaction mixture were transferred into 180 μl of secondary reactions containing 500 μM testosterone in the reaction buffer mixture, pH 7.5 (NADPH and NADP+/H regenerating enzymes). CYP3A4 testosterone 6β-hydroxylase activity in the secondary reactions was then determined using HPLC. The kinetics of CYP3A4 inactivation were determined as follows: the initial rate constant for inactivation (Kobs) at each concentration of inhibitor was estimated from the slope of the linear regression line (by Prism software) from the log of CYP3A4 remaining activity versus the preincubation time semilog graph. Kinetic parameters (Kinact and KI) for the inactivation of CYP3A4 testosterone 6β-hydroxylase activity were then estimated from the reciprocal plot of the rate constant (Kobs) versus the inhibitor concentrations.
Analytical Procedures. All the samples were analyzed using HPLC. The HPLC system consisted of the Shimadzu (Kyoto, Japan) prominence series, including the LC-20 AT prominence LC pump, DGU-20A5 degasser, CBM-20 prominence communications bus module, SPD-20A prominence UV-visible detector, and RF-10AXL fluorescence detector (Shimadzu). A C18 reversed-phase column (4.6 × 150 mm, 5 μm; Waters, Milford, MA) was used to detect and quantify metabolite products of MFC, BFC, and AMMC. The separation was achieved with 20% B from 0 to 5 min, linearly increased to 90% B from 5 min to 15 min, and was held until 20 min before returning to the starting condition (solvent A: 5 mM potassium phosphate, pH 7.5; solvent B: 100% acetonitrile; flow rate 1 ml/min). HFC, a product of MFC and BFC, and 3-[2-N,N-diethyl-N-methylamino]-7-hydroxy-4-methylcoumarin, a product of AMMC, were monitored by fluorescence detection with the setting at 410/510 nm (excitation/emission λ) and 390/460 nm (excitation/emission λ), respectively. For samples of testosterone and harmaline, a C18 reversed-phase column (4.6 × 250 mm, 5 μm; DuPont, Wilmington, DE) was used. The separation of testosterone samples was performed using 40% B from 0 to 5 min, increasing to 80% B from 5 min to 12 min, and then holding for another 17 min before returning to the starting condition (solvent A: H2O, 0.1% trifluoric acid; solvent B: 100% acetonitrile; flow rate 1 ml/min). The testosterone metabolite, 6β-hydroxytestosterone, was monitored at a wavelength of 240 nm and eluted at 5.7 min. For harmaline, solvents were held isocratic at 30% B from 0 to 8 min. The gradient was linear from 30% B to 70% B from 8 min to 12 min and held at 70% until 17 min; before returning to starting conditions (solvent A: H2O, 0.1% trifluoroacetic acid; solvent B: 100% acetonitrile; flow rate 1 ml/min). The harmaline metabolite, harmalol, was monitored with the fluorescence detector setting at 340/495 nm (excitation/emission λ) and eluted at 4.9 min.
Data Analysis. The P450 activities with and without inhibitors were measured by the area of products from the HPLC chromatograms. The activities of P450s in the presence of the inhibitors are addressed as the percentage activity of uninhibited P450s (area of product of the inhibited samples divided by the uninhibited samples times 100%). The IC50 values were determined by Prism software using the nonlinear regression mode with one site competition. The equilibrium dissociation constant of inhibitor, Ki, was calculated by Prism software using the IC50 values, the given concentration and the Km of the respective probe substrate, and the equation, Ki = (IC50)/[1 + (substrate)/Km] (Linden, 1982). Calculations of the Km for probe substrates and the kinetics of enzyme inactivations were described previously above. The t test was used for statistical analysis (Prism software).
Results
Comparison of NNC55-0396's P450 Inhibition Profile with That of Mibefradil. Seven major recombinant human P450s— CYP3A4, CYP1A2, CYP2E1, CYP2D6, CYP2C8, CYP2C19, and CYP2C9 —were chosen for inhibition studies with mibefradil and NNC55-0396. It has been shown that these P450s participate in the metabolism of approximately 80% of therapeutic drugs (Wienkers and Heath, 2005). To obtain a preliminary look at the P450 inhibitory profiles for mibefradil and NNC55-0396, two concentrations of the inhibitors were chosen, 100 nM and 10 μM. These two concentrations represent the low and the high concentrations and cover the range of therapeutic plasma concentrations of the inhibitors because it has been reported that the human therapeutic plasma concentration of mibefradil ranges from 300 to 1000 ng/ml after doses of 50 to 100 mg/day, which correspond to 0.5 to 1.8 μM mibefradil (Wiltshire et al., 1997a; Welker and Banken, 1998). At 100 nM, mibefradil exhibited more than a 2-fold greater inhibition than NNC55-0396 for CYP3A4 (63 and 27%, respectively, p < 0.001) (Fig. 2F). At 10 μM, both mibefradil and NNC55-0396 inhibited more than 90% of CYP3A4 activity (Fig. 2F). Mibefradil and NNC55-0396 had opposite inhibitory effects to those above for CYP2D6. At 100 nM, NNC55-0396 exhibited more than 2-fold greater inhibition of CYP2D6 compared with mibefradil (55 and 23%, respectively, p < 0.001) (Fig. 2G). At 10 μM, both compounds inhibited CYP2D6 > 95% (Fig. 2G). Our mibefradil data are in agreement with previous reports showing that mibefradil strongly inhibits CYP3A4 and CYP2D6 (Ernst and Kelly, 1998; Prueksaritanont et al., 1999; Stresser et al., 2000). Mibefradil at 100 nM slightly inhibited CYP2C9 (11%, p < 0.05) (Fig. 2C), whereas NNC55-0396 at 100 nM failed to do so (Fig. 2C). Neither CYP2C8 nor CYP2C19 was inhibited by 100 nM mibefradil or NNC55-0396, and 10 μM concentrations of these compounds inhibited CYP2C9, CYP2C8, and CYP2C19 considerably less than they inhibited CYP3A4 and CYP2D6 (Fig. 2, C–E). CYP2E1 and CYP1A2 were not inhibited by either compound, even at 10 μM (Fig. 2, A and B).

Inhibition profile of the activities of various human P450s by 100 nM and 10 μM mibefradil (Mib) or NNC55-0396 (NNC). The inhibition activities of CYP1A2 (A), CYP2E1 (B), CYP2C9 (C), CYP2C19 (D), CYP2C8 (E), CYP3A4 (F), and CYP2D6 (G) are expressed relative to the activities of the no inhibitor control (ACN) as the percentage of the inhibited activity over the uninhibited one. Thus, the percent of inhibition is calculated as 100% minus the percent listed on the graph. Data are represented as the mean ± S.D. of duplicate determinations. * and ***, p < 0.05 and p < 0.001, respectively, for comparison between control (ACN) and inhibitor added samples by an unpaired t test.
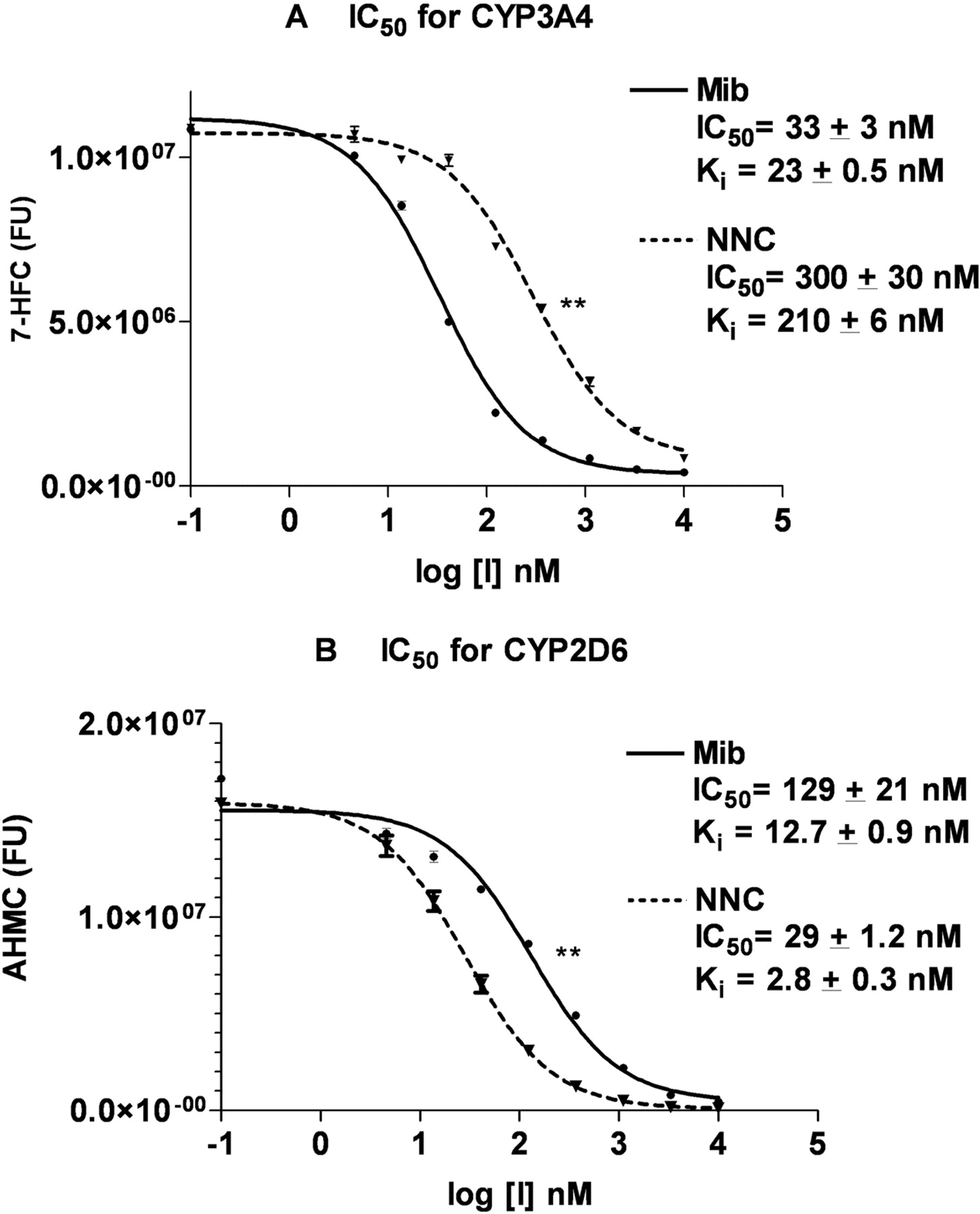
IC50 and Ki Values of Mibefradil and NNC55-0396 for Recombinant CYP3A4 and CYP2D6. Our preliminary look at the P450 inhibitory profiles for mibefradil and NNC55-0396 therefore showed that of the above P450s, only recombinant CYP3A4 and CYP2D6 exhibited marked inhibition by both compounds at low concentrations. Therefore, we went on to compare the inhibitory effects of the above compounds on CYP3A4 and CYP2D6 by estimating their IC50 and Ki values. We determined the IC50 values of mibefradil and NNC55-0396 using eight inhibitor concentrations generated by serial 1:3 dilutions from 10 μM. Although the IC50 value expresses the concentration of the inhibitor that competes for half the specific substrate binding sites, it does not quantify the affinity of the inhibitor for the enzyme. Ki, the equilibrium dissociation constant, is a measure of this last parameter. It is the concentration of the inhibitor that will bind to half the binding sites at equilibrium and is calculated based on the IC50 values of inhibitor, the substrate's Km, and the given substrate concentration (Linden, 1982) The Kms of BFC and AMMC, the substrates for CYP3A4 and CYP2D6, respectively, were determined to be 240 and 1.08 μM, respectively. For CYP3A4 inhibition, the IC50 and Ki values of mibefradil were 33 ± 3 and 23 ± 0.5 nM, whereas the IC50 and Ki values of NNC55-0396 were 300 ± 30 and 210 ± 6 nM (Fig. 3A), showing that NNC55-0396 is nine times less inhibitory than mibefradil toward recombinant human CYP3A4 (p < 0.001) (Fig. 3A). Our mibefradil IC50 value for recombinant CYP3A4 is similar to that reported by Stresser et al. (2000). For CYP2D6 inhibition, mibefradil had IC50 and Ki values of 129 ± 21 and 12.7 ± 0.9 nM, whereas NNC55-0396 had IC50 and Ki values of 29 ± 1.2 and 2.8 ± 0.3 nM, representing a 4-fold greater inhibition efficacy for NNC55-0936 compared with mibefradil (p < 0.005) (Fig. 3B).
Effects of Mibefradil and NNC55-0396 on P450 Activities in Human Liver Microsomes. To further verify mibefradil and NNC55-0396's inhibitory effects on human CYP3A4 and CYP2D6, we used human liver microsomes, which contain CYP3A4, CYP2D6, and other P450s. CYP3A4 is the main hepatic enzyme involved in testosterone 6β-hydroxylation (Gillam et al., 1993), whereas CYP2D6 is the main hepatic enzyme responsible for harmaline metabolism (Yu et al., 2003). Thus, testosterone and harmaline were used as substrates to determine the activities of CYP3A4 and CYP2D6 in human liver microsomes. The Kms of testosterone and harmaline were determined to be 111 and 74 μM, respectively. Eight concentrations of mibefradil and NNC55-0396 generated by serial 1:3 dilutions of 10 μM were used to determine their IC50 and Ki values for CYP3A4 and CYP2D6 in liver microsomes. The present data show that mibefradil was 19 times more inhibitory than NNC55-0396 for testosterone 6β-hydroxylation in the human liver microsomes because the former had IC50 and Ki values of 566 ± 71 and 202 ± 39 nM, respectively, whereas the latter had IC50 and Ki values of 11.0 ± 1.1 and 3.9 ± 0.4 μM, respectively (p < 0.001; Fig. 4A). In contrast, the data showed that NNC55-0396 was 15 times more inhibitory than mibefradil for harmaline metabolism in the human liver microsomes (p < 0.01) (Fig. 4B). NNC55-0396 had IC50 and Ki values of 72.6 ± 6 and 57.1 ± 5 nM, whereas mibefradil had IC50 and Ki values of 1111 ± 52 and 833 ± 39 nM for CYP2D6 in liver microsomes (Fig. 4B).
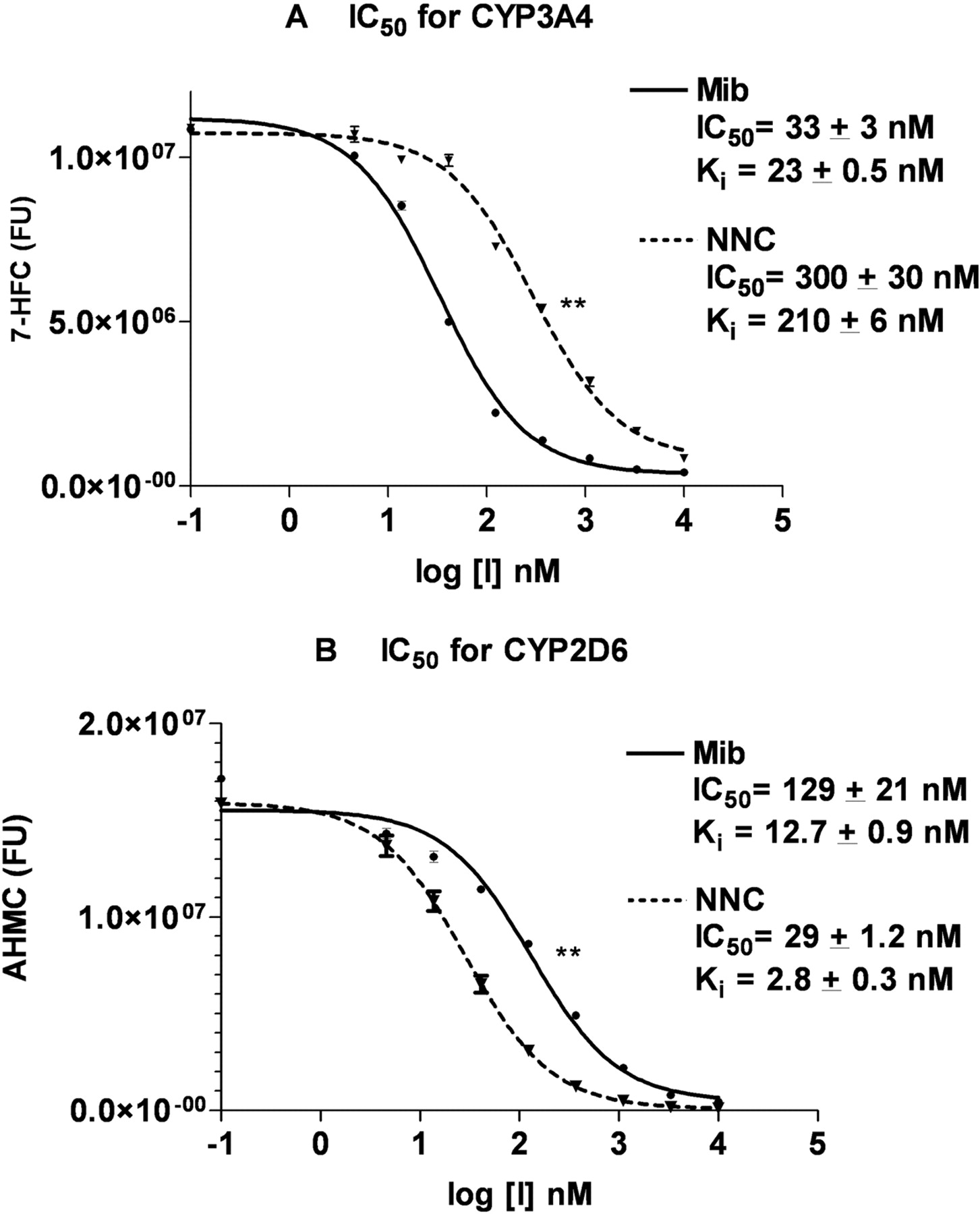
Comparison of mibefradil and NNC55-0936 inhibition of CYP3A4 and CYP2D6. A, the IC50 and Ki determination of mibefradil and NNC55 for BFC debenzylation by CYP3A4. B, the IC50 and Ki determination of mibefradil and NNC55-0396 for AMMC N-demethylation by CYP2D6. Data are represented as the averages ± S.D. of duplicate determinations. **, p < 0.005 comparing the mibefradil and NNC55-0396 samples by a paired t test.
Effects of Esterase on Mibefradil and NNC55-0396. Mibefradil is easily hydrolyzed by enzymes after it enters the cell (Wiltshire et al., 1992; Wu et al., 2000). Its hydrolyzed metabolite, Ro 40-5966, has been shown to block L-type Ca2+ channels (Wu et al., 2000). NNC55-0396 has more specific inhibitory activity toward T-type Ca2+ channels than mibefradil (Huang et al., 2004; Li et al., 2005), most likely because of its resistance to hydrolysis conferred by the replacement of the methoxy acetate chain with cyclopropanecarboxylate. To test whether NNC55-0396 is more resistant to hydrolysis by esterase than mibefradil, both were incubated with pig liver esterase. Pig liver esterases are commonly used for in vitro study because human esterases are not available. Our data show that after 1 h at 37°C, 10% of mibefradil was hydrolyzed by esterase to yield a product peak at 15.5 min that corresponds to the Ro 40-5966 standard (Fig. 5B), whereas no hydrolyzed product was detected in NNC55-0396 sample incubated with esterase (Fig. 5A). Thus, mibefradil is susceptible to hydrolysis by the esterase yielding Ro 40-5966, whereas NNC55-0396 is not. However, whether NNC55-0396 is resistant to hydrolysis in humans needs to be examined.

Inhibition profiles for testosterone and harmaline metabolism in human liver microsomes by mibefradil and NNC55-0936. A, IC50 and Ki determination of mibefradil and NNC55-0396 for testosterone metabolism mainly by CYP3A in human liver microsomes. ***, p < 0.001 comparing the mibefradil and NNC55-0396 samples by a paired t test. B, IC50 and Ki determination of mibefradil and NNC55-0396 for harmaline metabolism mainly by CYP2D6 in human liver microsomes. **, p < 0.01 for comparing NNC55-0396 to mibefradil by a paired t test.
P450 Inhibition Profile of Mibefradil's Hydrolyzed Metabolite, Ro 40-5966, in Comparison with Mibefradil and NNC55-0396. According to Wiltshire et al. (1992, 1997a), 20 to 32% of mibefradil is converted to Ro 40-5966 after clinical administration. Although it has been shown here and by others that mibefradil inhibits several P450s, it has not previously been investigated whether Ro 40-5966 can also inhibit P450s. Therefore, we tested Ro 40-5966's inhibition toward recombinant CYP2D6, CYP3A4, CYP2C9, CYP1A2, CYP2C19, and CYP2C8 (Fig. 6, A–F). As with mibefradil and NNC55-0396, only recombinant CYP3A4 and CYP2D6 showed strong inhibition by Ro 40-5966 (Fig. 6, A and B). Our results show that Ro 40-5966 has IC50 and Ki values of 30 ± 7.8 and 21 ± 2.8 nM for CYP3A4, which are comparable with mibefradil's values of 33 ± 3 and 23 ± 0.5 nM (Figs. 3A and 6B). These IC50 and Ki values indicate that like mibefradil, Ro 40-5966 is 9 to 10 times more inhibitory than NNC55-0396 toward recombinant CYP3A4, which has IC50 and Ki values of 300 ± 30 and 210 ± 6 nM (Fig. 3A). For recombinant CYP2D6, Ro 40-5966 was determined to have IC50 and Ki values of 46 ± 11 and 4.5 ± 0.02 nM (Fig. 6A), which are comparable with the values for NNC55-0396 (IC50 = 29 ± 1.2 and Ki = 2.8 ± 0.3 nM) (Fig. 3B). Comparison of the inhibition values of Ro 40-5966 with that of mibefradil (IC50 = 129 ± 21 and Ki = 12.7 ± 0.9 nM) (Fig. 3B) indicates a 3-fold greater inhibition efficacy of Ro 40-5966 toward CYP2D6 (p < 0.05) (Figs. 3B and 6A). Inhibition of CYP2C19 and CYP1A2 by Ro 40-5966 was observed only at the supraoptimal concentration of 10 μM(p < 0.001) (Fig. 6, D and E). The IC50 for CYP2C9 was estimated to be 823 ± 390 nM (Fig. 6C), whereas there was no significant inhibition of CYP2C8 even at 10 μM concentration (Fig. 6F).

Hydrolysis of mibefradil but not NNC55-0396 by pig liver esterase. A, HPLC chromatograms of 40 μM NNC55-0396 incubated with pig liver esterase (solid line) and of a standard for Ro 40-5966 (broken line). The 14- and 15.5-min peaks correspond to NNC55-0396 and Ro 40-5966, respectively. B, HPLC chromatograms of 40 μM mibefradil incubated with pig liver esterase (solid line) and of standard Ro 40-5966 (broken line). The 11.5- and 15.5-min peaks correspond to mibefradil and Ro 40-5966, respectively. Open arrow indicates the presence of Ro 40-5966 at 15.5 min generated by hydrolysis in the mibefradil sample.
Inhibition Mechanism of Mibefradil and NNC55-0396 toward Recombinant CYP3A4. Mibefradil has been shown to be a powerful mechanism-based inhibitor of CYP3A4 (Prueksaritanont et al., 1999). Our preliminary data indicated that NNC55-0396 also exhibited time-dependent inactivation of CYP3A4. To further investigate the inhibition mechanism of NNC55-0396 and to compare it with that of mibefradil, mechanism-based inhibition assays were performed using recombinant human CYP3A4-catalyzed testosterone 6β-hydroxylase activity. Crude lysate containing CYP3A4 was used instead of a membrane fraction containing the enzyme because the latter was shown to have a 50% decrease in activity during the period of incubation even in the absence of inhibitor, whereas the former showed no more than 15% autoinactivation. The kinetic parameters, such as the concentration required for half-maximal inactivation (KI) and the rate constant of maximal inactivation at saturation (Kinact), were determined. These numbers were used to compare the inactivation efficiency of mibefradil and NNC55-0396 toward the CYP3A4 enzyme. Preincubation of recombinant CYP3A4 with various concentrations of mibefradil and NNC55-0396 in the presence of NADPH resulted in a time- and concentration-dependent loss of testosterone 6β-hydroxylase activity. However, NNC55-0396 significantly caused time-dependent inactivation of testosterone 6β-hydroxylase only at the high concentrations 1.2 and 3.6 μM compared with the control (p < 0.05) (Fig. 7C). The KI and Kinact of NNC55-0396 for CYP3A4 were estimated to be 3.87 μM and 0.061/min, respectively (Fig. 7D).

Inhibition profiles of the activities of various human P450s by Ro 40-5966. The IC50 and Ki determinations of Ro 40-5966 for recombinant CYP2D6 (A) and CYP3A4 (B), and the IC50 determinations for CYP2C9 (C) and CYP1A2 (D). The relative activities of 100 nM and 10 μM Ro 40-5966 samples to a control (ACN) catalyzed by recombinant CYP2C19 (E) and CYP2C8 (F). IC50 determinations for CYP2C19 and CYP2C8 were not necessary because they were not strongly inhibited by Ro 40-5966 even at 10 μM. Data are represented as the mean ± S.D. of duplicate determinations. *, p < 0.05 comparing control (ACN) and Ro 40-5966 samples by an unpaired t test.
In contrast, mibefradil significantly caused time-dependent loss of testosterone 6β-hydroxylase activity even at 44 nM, the lowest concentration tested (p < 0.01) (Fig. 7A). The estimated KI and Kinact of mibefradil were 83 nM and 0.048/min, respectively (Fig. 7B). Therefore, the KI of mibefradil is 47 times lower than that of NNC55-0396. From the kinetics values, the inactivation efficiency [Einact = Kinact/(KI + I)] can be estimated (Zhou et al., 2005). Einact of mibefradil is at least 3.3-fold higher than that of NNC55-0396 for inactivating CYP3A4 testosterone 6β-hydroxylase, when the inhibitor concentrations are at 1 μM or less. The lower the inhibitors' concentrations, the greater the difference in inactivation efficiency between mibefradil and NNC55-0396.
Discussion
Mibefradil, a potent T-type Ca2+ channel inhibitor, was marketed clinically for cardiac indications but was withdrawn after serious drug-drug interactions emerged, particularly because of CYP3A4 inhibition and inactivation. It also has the disadvantage of L-type Ca2+ channel antagonism contributed by its hydrolyzed metabolite, Ro 40-5966. Recently, a mibefradil derivative, NNC55-0396, designed to be hydrolysis-resistant, was shown to be a selective T-type Ca2+ channel inhibitor without L-type Ca2+ channel efficacy (Li et al., 2005). Whether NNC55-0396 will deliver a clinical benefit with minimized drug-drug interactions is not known. However, potential adverse effects can often be predicted. In the preclinical drug discovery and development phase, studies of P450 enzyme inhibition and inactivation have become one of the major approaches for the in vitro assessment of the risk associated with new molecular entities as precipitants of drug-drug interactions (Venkatakrishnan et al., 2007). Thus, this report is the first in vitro study of NNC55-0396's effects on human P450s in which its effects are also being compared with mibefradil and its hydrolyzed metabolite, Ro 40-5966. The study will provide crucial information about the possible risk associated with NNC55-0396.

Mechanism-based inactivation of recombinant human CYP3A4 by mibefradil and NNC55-0396. Time- and concentration-dependent loss of recombinant CYP3A4 testosterone 6β-hydroxylase activity with mibefradil and NAPDH (A) or NNC55-0396 and NAPDH (C). The double reciprocal plot of the rates of inactivation as a function of the mibefradil (B) and NNC55-0396 (D) concentration. The values for 1/Kobs (/min) were calculated from the slopes of inactivation experiments by mibefradil or NNC55-0396 in A or C, respectively. All the slopes were normalized to the slope of no inhibitor experiment. The KI and Kinact values are estimated by 1 divided by the x- and y-intercepts, respectively. * and **, p < 0.05 and 0.01, respectively, comparing with control (0 nM) samples by an unpaired t test.
Using seven major recombinant human P450s and human liver microsomes, and HPLC as a detection and analytical method, we showed that compared with mibefradil, NNC55-0396 has a significantly less inhibitory profile for the CYP3A4 activity of recombinant CYP3A4, as well as CYP3A4-dependent activity in human liver microsomes. The IC50 and Ki of NNC55-0396 are both 9 to 10 times higher than those of mibefradil for recombinant CYP3A4, suggesting that the former compound is 9 to 10 times less inhibitory than the latter. The differences in these values are more dramatic when inhibition of testosterone 6β-hydroxylase activity of CYP3A was examined in human liver microsomes. The IC50 and Ki of NNC55-0396 are 19-fold higher than that of mibefradil. The higher IC50 and Ki values observed for CYP3A4 in human liver microsomes compared with the values for recombinant CYP3A4 may result from the presence of other microsomal proteins that bind to the inhibitors, or to the presence of other enzymes that can metabolize the inhibitors, thus making them less available to inhibit the targeted enzyme (Wiltshire et al., 1997a). Moreover, the difference could have been contributed by different substrates being used.
Previous evidence suggested that mibefradil metabolites also inhibit CYP3A4 (Prueksaritanont et al., 1999; Ma et al., 2000), but the degree of inhibition has not previously been investigated. Mibefradil is hydrolyzed by esterase and CYP3A4 to generate a hydroxyl mibefradil, Ro 40-5966 (Wiltshire et al., 1997a), one of many mibefradil metabolites shown to contribute to the L-type Ca2+ channel nonspecific inhibition of mibefradil (Wu et al., 2000). Our results show that Ro 40-5966 inhibits CYP3A4 with an efficacy comparable with mibefradil and thus is 10 times more inhibitory than NNC55-0396. This result indicates that the Ro 40-5966 metabolite may also contribute to mibefradil's drug interactions.
When using a time- and concentration-dependent inhibition assay, we found that NNC55-0396 also caused time-dependent inactivation of CYP3A4 but was less potent in this regard than mibefradil. The concentration of NNC55-0396 required for half-maximal inactivation (KI) of CYP3A4 is 3.4 μM, which is 47-fold higher than that of mibefradil. Its t1/2inact (0.693/Kinact) is just slightly shorter (11.3 min) than that of mibefradil (14.4 min). KI values can be used to classify inhibitory potency of compounds (Zhou et al., 2005). The KI value of NNC55-0396 is comparable with the KI values of verapamil (1.7 μM) and diltiazem (2.0 μM), which are the Ca2+ channel antagonists being used as antihypertensive drugs and are currently available on the market (Zhou et al., 2005). In addition, verapamil and diltiazem even have shorter t1/2inact (7.70 and 6.30 min, respectively) (Zhou et al., 2005) than that of NNC55-0396.
CYP3A4 is responsible for metabolizing 50 to 60% of all the therapeutic drugs that are metabolized by P450s in humans (Thummel et al., 1996). Drugs that inhibit CYP3A4 such as ketoconazole, cisapride, ritonavir, and nefazodone have been found to cause deleterious side effects when coadministered with other drugs. Therefore, many drugs that strongly inhibit CYP3A4 such as mibefradil have been withdrawn from the market (SoRelle, 1998; Bohets et al., 2000; Watanabe et al., 2007; http://www.fda.gov/medwatch/SAFETY/1997/posico.htm). Mibefradil was shown to strongly inhibit CYP3A4 via mechanism-based inactivation at its relevant therapeutic concentration. Such inhibition is highly potent because it irreversibly reduces the amount of the enzyme available, so that restoration of activity requires the synthesis of new enzyme. The pharmacokinetics of NNC55-0396 need to be determined to get an accurate assessment of its clinical drug-drug interaction outcomes. However, if the pharmacokinetics of NNC55-0396 are the same as mibefradil, then NNC55-0396 would likely be a lesser or even nonpotent mechanism-based inhibitor of CYP3A4 in vivo. Given the total Cmax of mibefradil of around 0.5 to 1.8 μM (Wiltshire et al., 1997a; Welker and Banken, 1998) and the fact that more than 90% of mibefradil is protein-bound, the available plasma concentration of NNC55-0396 will be much below its apparent Ki (3.9 μM), and the Cmax/Ki ratio of NNC55-0396 would be less than 1 (based on the Ki derived from human liver microsomes). This ratio indicates that drug-drug interaction is unlikely, but possible, because according to the current Food and Drug Administration guidelines for predicting the likelihood of clinically relevant drug-drug interaction if Cmax/Ki > 1, a drug interaction is likely, if Cmax/Ki > 0.1, a drug interaction is possible, and if Cmax/Ki < 0.1, a drug interaction is unlikely. However, it is also possible that the NNC55-0396's Cmax will be higher in clinical settings. If this is the case, then the Cmax/Ki ratio may be higher than 1, and drug-drug interaction is likely.
CYP2D6 has been reported to be inhibited by mibefradil, but the IC50 and Ki values have not previously been determined. We are the first to report mibefradil's IC50 and Ki values for CYP2D6 and to show that Ro 40-5966 strongly inhibits CYP2D6 with relatively lower IC50 and Ki than its parent compound. This could have also contributed to the complexity of mibefradil's toxicity. Our results also show that NNC55-0396 inhibits CYP2D6 with relatively low IC50 and Ki values with both recombinant CYP2D6 and human liver microsomes. This strong inhibition of CYP2D6 could be a concern because CYP2D6, although playing a less important role than CYP3A4 in drug metabolism, is the second most significant P450 in this regard because it metabolizes 20 to 25% of clinically used drugs (Ingelman-Sundberg, 2004). However, many strong inhibitors of CYP2D6 with low IC50 values, such as terbinafine, dextromethorphan, quinidine, and propranolol, are currently on the market.
Although mibefradil, NNC55-0396, and Ro 40-5966 also inhibit CYP2C9, CYP2C8, and CYP2C19, they do so only at high concentrations (10 μM), indicating that relative high dosages of these drugs would need to be taken to reach sufficiently high plasma levels to inhibit these enzymes. In addition, CYP2C9, CYP2C8, and CYP2C19 have a less important role clinically than CYP3A4 in drug metabolism (Ingelman-Sundberg, 2004). However, they are not irrelevant in drug-drug interactions.
Clinically, T-type Ca2+ channels have represented important therapeutic targets in various types of diseases from cardiovascular diseases, diabetes, and neurological diseases to cancer (Li et al., 2005; Tanaka and Shigenobu, 2005; Panner and Wurster, 2006). Besides their strong association with cardiovascular disease, T-type Ca2+ channels are associated with cancer cell proliferation, and inhibiting T-type Ca2+ channels protects from delayed ischemia-induced damage (Nikonenko et al., 2005). T-type Ca2+ channels are also implicated in the pathogenesis of epilepsy and neuropathic pain (Tanaka and Shigenobu, 2005). Thus far, highly selective T-type Ca2+ channel antagonists have not been clinically available. Several candidate drugs that potently inhibit T-type Ca2+ channels unfortunately, like mibefradil, also strongly inhibit L-type Ca2+ channels. Interestingly, a methoxyacetyl group of mibefradil seems to play not only an important role in selectivity toward the Ca2+ channels but also in P450 inhibition. We and others (Huang et al., 2004; Li et al., 2005) found changes in inhibition specificities when this ester group is modified. Thus, it is probable that additional nonhydrolyzable analogs of mibefradil could be designed that exert less inhibition of CYP3A4 and CYP2D6 but still retain the same potency for T-type Ca2+ channel antagonism.
In summary, our data suggest that NNC55-0396 is a more favorable T-type Ca2+ channel antagonist than its parental compound, mibefradil, for the following reasons. 1) NNC55-0396 is a selective T-type Ca2+ channel blocker without L-type Ca2+ channel efficacy (Li et al., 2005). 2) It has a considerably weaker inhibitory profile for CYP3A4 than mibefradil. Such inhibition of CYP3A4 was an important problem that contributed to mibefradil's withdrawal from the market. 3) Unlike mibefradil, NNC55-0396 is not hydrolyzed by esterase to form Ro 40-5966, itself a very potent CYP3A4 and L-type Ca2+ antagonist (Wu et al., 2000). However, freedom from CYP3A4-based interactions will also depend on the pharmacokinetic properties of NNC55-0396. Predicting drug-drug interaction is even more challenging when it involves mechanism-based inactivation of enzyme because the clinical outcomes depend on a number of factors, including the pharmacokinetic effects (Ki, Kinact, and partition ratio), and the zero-order synthesis rate of new or replacement enzyme (Zhou et al., 2005). Nevertheless, our findings are highly suggestive for a weaker interaction CYP3A4 profile for NNC55-0396. More work is needed, however, to establish whether its differences from mibefradil may be clinically meaningful.
Acknowledgments
We thank Dr. Curt Eckhert for allowing us to use his HPLC equipment.
Footnotes
-
This research is supported by National Institute of Environmental Health Sciences Grant RO1ES015384 to Oliver Hankinson, by a fellowship to Peter Bui from the University of California Toxic Substances Research and Teaching Program, by the Department of Veterans Affairs, and by the Ralph M. Parsons Foundation.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.020115.
-
ABBREVIATIONS: P450, cytochrome P450; UGT, UDP glucuronosyltransferase; CPR, NAPDH-P450-reductase; AMMC, 3-[2-N,N-diethyl-N-methylamino]-7-methoxy-4-methylcoumarin; BFC, 7-benzyloxy-4-trifluoromethylcoumarin; MFC, 7-methoxy-4-trifluoromethylcoumarin; HFC, 7-hydroxy-4-trifluoromethylcoumarin; HPLC, high-performance liquid chromatography; EB, enzyme buffer.
- Received December 20, 2007.
- Accepted April 8, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}