


Abstract
Nateglinide (A-4166) is an amino acid derivative with insulinotrophic action in clinical development for treatment of type 2 diabetes. The aim of this study was to determine whether nateglinide's interaction at the KATP channel/sulfonylurea receptor underlies its more rapid onset and shorter duration of action in animal models. Binding studies were carried out with membranes prepared from RIN-m5F cells and HEK-293 cells expressing recombinant human sulfonylurea receptor 1 (SUR1). The relative order for displacement of [3H]glibenclamide in competitive binding experiments with RIN-m5F cell membranes was glibenclamide > glimepiride > repaglinide > glipizide > nateglinide >l-nateglinide > tolbutamide. The results with HEK-293/recombinant human SUR1 cells were similar with the exception that glipizide was more potent than repaglinide. Neither nateglinide nor repaglinide had any effect on the dissociation kinetics for [3H]glibenclamide, consistent with both compounds competitively binding to the glibenclamide-binding site on SUR1. Finally, the inability to measure [3H]nateglinide binding suggests that nateglinide dissociates rapidly from SUR1. Direct interaction of nateglinide with KATPchannels in rat pancreatic β-cells was investigated with the patch-clamp method. The relative potency for inhibition of theKATP channel was repaglinide > glibenclamide > nateglinide. Kinetics of the inhibitory effect onKATP current showed that the onset of inhibition by nateglinide was comparable to glibenclamide but more rapid than that of repaglinide. The time for reversal of channel inhibition by nateglinide was also faster than with glibenclamide and repaglinide. These results suggest that the unique characteristics of nateglinide are largely the result of its interaction at theKATP channel.
The maintenance of blood glucose concentration is an integrated process regulated primarily by the antihyperglycemic hormone insulin. When blood glucose rises, uptake of glucose into the β-cell leads to an elevation in the ATP/ADP ratio and closure of theKATP channels. The closure ofKATP channels and the resultant membrane depolarization lead to the increase of Ca2+ influx through voltage-gated Ca2+ channels, which triggers exocytosis and insulin release. In addition to glucose, many agents are capable of blocking KATP channels in pancreatic β-cells, thus inducing insulin secretion and having an antidiabetic effect. Among them, glibenclamide, a sulfonylurea, has been used for >30 years in the treatment of type 2 diabetes (Loubatiéres, 1977; Sturgess et al., 1985; Dunne et al., 1987). Repaglinide, a benzoic acid derivative of the meglitinide family, is reportedly a more potent insulinotropic agent than glibenclamide and other sulfonylureas (Frøkjær-Jensen et al., 1992; Gromada et al., 1995; Malaisse, 1995;Fuhlendorff et al., 1998). Nateglinide (N-[(trans-4-isopropylcyclohexyl)-carbonyl]-d-phenylalanine; A-4166) is a phenylalanine derivative (nonsulfonylurea) reported to have a similar mechanism of action to glibenclamide and repaglinide (Akiyoshi et al., 1995; Fujita et al., 1996; Ikenoue et al., 1997). Glibenclamide and repaglinide cause long-lasting hypoglycemic action under both normoglycemic and hyperglycemic conditions in animal models (Mark and Grell, 1997; DeSouza et al., 1997). Nateglinide, although less potent, appears to differ from the other two agents in several respects: 1) preferential first-phase insulin release effect due to rapid onset, 2) no sustained hypoglycemia and reduced total insulin secretion due to its short duration of action, and 3) enhanced activity under hyperglycemic conditions due to glucose-sensitive action (Akiyoshi et al., 1995; Sato et al., 1995; Ikenoue et al., 1997; DeSouza et al., 1997). Although these in vivo differences may be due to differential effects on absorption, distribution, and elimination of the various compounds, we have carried out receptor-binding andKATP channel activity measurements to distinguish the molecular mechanism of nateglinide from the mechanism of the sulfonylureas and other agents such as repaglinide. The results of these experiments indicate that the interactions of nateglinide with its receptor are unique compared with glibenclamide and repaglinide. Moreover, the nature of this interaction at theKATP channel also may be relevant to its shorter duration of action, reduction in excessive insulin release, and reduced risk of hypoglycemia.
Materials and Methods
Cell Culture and Preparation of RIN-m5F Membranes.
RIN-m5F cells (passages 23–32) [CRL-2058; American Type Culture Collection, Manassas, VA; (Bhathena et al., 1984)] were cultured in T175 flasks in RPMI 1640 (Life Technologies, Rockville, MD) supplemented with 10% fetal bovine serum (BioWhittaker, Walkersville, MD) and penicillin (100 U/ml)/streptomycin(100 μg/ml) (Life Technologies). Cells, at 85% confluence, were washed once with PBS, then scraped into 10 ml of PBS. Cells were collected and stored as a pellet at −80°C. Membranes were prepared by the method of Müller et al. (1994) and stored at −80°C until use. Protein determinations were made with the Coomassie stain method (Pierce, Rockford, IL) with BSA as the protein standard.
The human sulfonylurea receptor (SUR1) gene, contained in the pECE vector (pECESUR1.hum), was provided by Dr. Joseph Bryan of the Baylor College of Medicine. The excised human SUR1 gene was recloned into an appropriate vector developed in-house (pOriP/Zeo). Human embryonic kidney (HEK)-293 cells containing the Epstein Barr virus nuclear antigen 1 gene (EBNA-1; Invitrogen, San Diego, CA) were transfected by calcium phosphate precipitation and subsequently selected for stable expression by addition of zeocin (100 μg/ml) to the culture medium 48 h after transfection. A recombinant cell pool expressing high levels of human SUR1 (HEK.EBNA[human SUR1]) was cultivated in batch mode. Cells expressing the receptor were harvested and stored at −80°C. Membrane preparations and protein determinations were made as described above.
Binding Assays.
The binding assays with [3H]glibenclamide (DuPont/NEN, Boston, MA) with RIN-m5F cell membranes were performed in buffer A [50 mM 3-(N-morpholino)propanesulfonic acid, pH 7.4, and 0.1 mM CaCl2] in a total volume of 1.0 ml/tube at 23–25°C. The RIN-m5F cell membranes (0.1 mg/ml) were incubated with [3H]glibenclamide at a concentration range of 0.2 to 30 nM. Nonspecific binding was determined in the presence of 2 μM unlabeled glibenclamide and was <20% of total binding. The protein concentration used in the binding assay was in the range in which there was a linear ligand-binding response with protein concentration. Glibenclamide and glipizide were obtained from Research Biochemicals International (Natick, MA). Tolbutamide was obtained from the Upjohn Company (Kalamazoo, MI). Nateglinide, [3H]nateglinide, repaglinide, and glimepiride were prepared in-house. The l-isomer of nateglinide was obtained from Ajinomoto Co. Inc. (Yokohama, Japan). Compound stock solutions were prepared in dimethyl sulfoxide. The final concentration of dimethyl sulfoxide (2%) did not affect binding. The binding assay proceeded for 2 h with orbital shaking and was terminated by rapid filtration through Whatman GF/F 25-mm-diameter glass microfiber filters (presoaked in buffer A) followed by 5 × 5-ml washes with cold 0.1 M NaCl. The 2-h incubation time was sufficient to ensure that a steady state in binding was achieved over the range of glibenclamide concentrations used in the assay. The wet filters were placed in 10 ml of Formula 989 scintillation fluid (Packard, Meriden, CT) and radioactivity was determined by liquid scintillation counting after overnight incubation. The binding assays with the HEK.EBNA[human SUR1] membranes were carried out in a 96-well format with Millipore multiScreen-FC opaque plates with 1.2-μm glass fiber type C filters. Binding assays with [3H]glibenclamide (DuPont/NEN) were performed in buffer A in a total volume of 250 μl at 23–25°C. HEK.EBNA[human SUR1] membranes (25 μg/well) were incubated with [3H]glibenclamide over a concentration range of 0.25 to 80 nM. The assay conditions and compound preparation were as described above. The assay was terminated by rapid filtration followed by 5 × 250-μl washes with ice-cold 0.1 M NaCl. Wallac Optiphase Hi Safe 3 scintillation cocktail was added per well (200 μl) and plates were counted in a Wallac Microbeta liquid scintillation counter. The competitive binding assays were carried out in the presence of 2.0 nM [3H]glibenclamide (Kd = 1.8 ± 0.002 nM for [3H]glibenclamide with human SUR1). The nonspecific binding was <5% of total binding under these assay conditions. All competitive binding experiments were repeated at least three times. Specific [3H]glibenclamide binding was not observed with membrane preparations from HEK-293 cells that were not transfected with the human SUR1 expression plasmid (pOriP/ZeoSUR1.hum).
[3H]Glibenclamide dissociation kinetics were determined as follows. RIN-m5F cell membranes were preincubated with 0.5 nM [3H]glibenclamide for 90 min at 25°C in a shaking water bath at 100 strokes/min. At the beginning of the kinetic assay, 2 μM unlabeled glibenclamide (or 10 μM repaglinide or 100 μM nateglinide) was added and portions (0.1 mg of membrane protein) of the incubation mixture were removed at various times and assayed by the filtration binding assay procedure previously described.
Centrifugation-binding assays were carried out with RIN-m5F cell membranes (Forget et al., 1993). The receptor preparation was layered on top of a solution whose density is greater than that of water but less than that of the membrane preparation. For these experiments, both sucrose and oil layers [silicon oil mixtures and mixtures of dibutyl phthalate/dinonyl phthalate (3:2) and dibutyl phthalate/dioctyl phthalate (1:1)] were used. Centrifugation (14,000 rpm for 2.5 min) through this layer separated the free (upper aqueous layer) and bound (lower pellet layer) ligand. The upper layers were carefully removed. Under these conditions, specifically bound radioactivity remains with the pellet. The pellet was dissolved in 100 μl of Soluene-350 (Packard) overnight and then added to 10 ml of Formula 989 scintillation fluid. After sitting overnight, the radioactivity was determined by liquid scintillation counting as described above.
Enzymatic Isolation of Rat Pancreatic β-Cells.
Male Sprague-Dawley rats weighing 250 to 275 g were anesthetized with sodium pentobarbital i.p. at 250 mg/kg before the operative procedure. After a midline abdominal incision was performed, the distal end of the bile duct was clamped with a hemostat to occlude it adjacent to the duodenum. The upper portion of the duct was nicked with a retina scissor and cannulated with a PE-50 polyethylene catheter near the hilus of liver. The acinar tissue was disrupted by injection of 20 ml of HEPES saline into the common bile duct and the pancreas was dissected from the stomach, duodenum, and spleen. The pancreas was cleaned free of fat, connective tissue, and blood vessels, and chopped into small pieces (1 × 1 mm). The pancreas slurry was transferred to a jar filled with 10 ml of HEPES saline containing librase at 0.5 mg/ml (Boehringer Mannheim, Indianapolis, IN) and placed on a submersible stirrer in a 37°C water bath for 25 min. The digest was then washed several times by centrifugation. The supernatant was discarded and the final sediment resuspended in HEPES saline for Ficoll (type 400 DL; Sigma Chemical Co., St. Louis, MO) gradient purification.
After the addition of 27% Ficoll to the pancreatic digest and thorough vortexing, solutions with 23, 20.5, and 11% Ficoll were sequentially layered on top of each other. The tube containing Ficoll and pancreatic digest was centrifuged at ∼2000 rpm for 10 min and the islets were largely present at the 11/20.5% Ficoll interface. The islets were washed multiple times until free of Ficoll and resuspended in HEPES saline in a black-bottom Petri dish. The islets were hand-picked by gentle suction through a large fire-polished pipette (∼400 μm i.d.) into a test tube. After washing twice with HEPES saline, islets were treated with 0.5 mg/ml protease (type IX; Sigma Chemical Co.) in the same buffer at 37°C for 20 min. At the end of the digestion, the mixture was washed several times with CMRL medium (Life Technologies) to inactivate the protease. The individual β-cells were seeded in CMRL medium and incubated at 37°C in an atmosphere of 95% air, 5% CO2 for 2 to 5 days before the electrophysiological recording.
Electrophysiological Recording of KATPCurrents.
Experiments were performed at 22°C with the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981) in the primary culture of rat pancreatic β-cells. Whole-cell current was used instead of single-channel current in a membrane patch as the end product of the measurement was to maximally maintain the cell integrity and preserve cytosolic nucleotides.
Corning 35-mm culture dishes, in which β-cells were grown, were fitted with Sylgard O-rings to serve as recording chambers. Cells were perfused continuously with HEPES saline or other testing solutions at a constant rate of ∼1.5 ml/min. The volume of the chambers was maintained at ∼0.3 ml. Experiments were performed at a 600× magnification under a Nikon inverted microscope. Because β-cells were reported to have a volume usually 2- to 3-fold larger than that of α-cells (Pipeleers et al., 1985), only cells with a diameter >7 μm and well preserved granulation were used for recording currents.
The KATP current in β-cells was elicited by a voltage ramp ranging from −120 mV to +40 mV over a 1500-ms period from a holding potential of −80 mV. At very negative voltages where the voltage-dependent K+ channels and Ca2+-activated K+channels were all inactivated (Dunne and Petersen, 1991), the only remaining current component was the voltage-independentKATP current. In addition, the inwardKATP current was further magnified by using high K+ (140 mM) symmetrical bath and pipette solutions to shift the K+ reversal potential from the conventional −80 to 0 mV. Thus, theKATP currents at negative potentials can be measured in the absence of interference of any other ion current components. The currents recorded were amplified by a List EPC-7 amplifier (Adams & List Assoc., Darmstadt, Germany), digitized at 4 kHz with a TL-1-125 DMA interface (Axon Instruments, Foster City, CA), and stored on a Compaq microcomputer for later analysis with software pClamp version 6.03 (Axon Instruments). The junction potential between the electrodes and the bath solution was compensated by the DC offset on the amplifier. The capacitance of β-cells was 12.1 ± 0.5 pF (n = 35). No leak subtraction was applied. Patch-clamp electrodes were pulled from Kimax-51 capillary tubes. The resistance of electrodes after fire-polishing was between 3 and 4 MΩ. In the experiments measuring the time courses of drug effects, the voltage ramp was repetitively applied to cells under investigation with an interval of 1 min, and the current amplitude was therefore measured every minute to follow its change with the time.
Data Analysis.
IC50 values for the binding studies were calculated from a dose-response curve with the four-parameter logistic equation. F tests were used to compare models, i.e., single-site versus two-site binding models and single- versus double-exponential decay kinetics, for data fitting. For comparison of the Hill coefficients (slope values of the four-parameter logistic equation), statistical significance was determined with at test. Groups with P values <.05 were considered significantly different. TheKi values for the competitive ligands were estimated from the experimental IC50 values with the Cheng-Prusoff equation, i.e.,Ki = IC50/(1 + ([Glib]/KiGlib)) (Titeler, 1989).
In patch-clamp recordings, the KATPcurrent amplitude at 300 ms from the beginning of the voltage ramp pulse (approximately at −90 mV) was used as the current index. The magnitude of drug effects was evaluated by comparing the current after drug treatment to that before the treatment in the same β-cell. The remaining currents after blockade by drugs expressed as fractions of control at various drug concentrations were taken to form the concentration-response curve. The data were then fit to the logistic equation Y = 1/[1 + (X/a)b] with a general nonlinear, least-squares analysis. In the equation, X and Yrepresent the drug concentration and the remaining current after blockade, respectively. The a is the IC50 value (defined as the concentration for a half-maximal blockade) and b is the slope coefficient. Statistical significance was determined with a t test (single-tailed).
Results
Steady-State Binding of [3H]Glibenclamide to Membranes from RIN-m5F Cells and HEK.EBNA[Human SUR1] Cells.
The saturation-binding curve for glibenclamide with RIN-m5F cell membranes indicates that there are two binding sites present in the membrane preparations (data combined from seven separate experiments; data not shown). Both high-affinity (Kd = 0.27 ± 0.07 nM) and low-affinity (Kd = 25 ± 13 nM) binding sites were observed. In the competitive binding and dissociation kinetic experiments described below, low concentrations of [3H]glibenclamide are used such that only binding at the high-affinity site would be measured. The saturation-binding experiments also were carried out with membrane preparations from HEK.EBNA[humanSUR1] cells (data obtained from five separate experiments; data not shown). In this case, a single binding site was observed with a Kd = 1.8 ± 0.002 nM. This value compares favorably with the reported value of 2 nM for 5-iodo-2-hydroxyglibenclamide binding to recombinant rat-SUR1 transfected into COSm6 cells (Aguilar-Bryan et al., 1995). 5-Iodo-2-hydroxyglibenclamide binds with a 2-fold lower affinity than glibenclamide to HIT-T15 cell SUR1 (Aguilar-Bryan et al., 1990)
Competitive-Binding Experiments with [3H]Glibenclamide.
A series of compounds was tested for their ability to directly compete with 0.5 nM [3H]glibenclamide for binding to RIN-m5F cell membranes. Displacement of 0.5 nM [3H]glibenclamide should reflect binding primarily to the high-affinity sites, i.e., the sites involved withKATP channel activity and insulin release. Table 1 summarizes the IC50, Ki, and slope (Hill coefficients) values determined in these competitive binding experiments. These results indicate that the relative order of potency is glibenclamide > glimepiride > repaglinide > glipizide > nateglinide > l-isomer of nateglinide > tolbutamide. TheKi values were estimated from the Cheng-Prusoff equation (see Data Analysis).
Binding parameters with RIN-m5F cell membranes
These compounds also were tested in direct competition studies with 2 nM [3H]glibenclamide and membranes from HEK.EBNA[human SUR1] cells. The relative order of efficacy for displacement of [3H]glibenclamide in steady-state competitive binding experiments was glibenclamide > glimepiride > glipizide > repaglinide > nateglinide > l-isomer of nateglinide > tolbutamide (Table 2). In general, the inhibitors bind more weakly (5- to 17-fold for all compounds except repaglinide) to human SUR1 than to RIN-m5F SUR1. The greatest difference is seen with repaglinide, which binds ∼130-fold more weakly to human SUR1 than to RIN-m5F SUR1. Figure1 displays the relationship for the binding of the seven compounds to RIN-m5F SUR1 and human SUR1.
Binding parameters with HEK.EBNA[Human SUR1] cell membranes

Comparison of steady-state binding constants for various ligands that bind to RIN-m5F SUR1 and human SUR1. The steady-state binding constants were determined in competitive binding experiments with [3H]glibenclamide with cell membranes from either RIN-m5F β-cells or HEK.EBNA[human SUR1] cells. The line represents a linear regression fit of the data with all compounds except repaglinide. Data points were collected in duplicate and combined from three separate experiments.
[3H]Glibenclamide Dissociation Kinetics.
One approach to determine whether a displacing ligand, such as nateglinide, binds to the same or a different site as glibenclamide on SUR1 is to measure the effect of nateglinide on the dissociation kinetics of [3H]glibenclamide. If nateglinide binds to a site separate from glibenclamide (but prevents its binding through a conformation change on the receptor), a change in the rate of dissociation of [3H]glibenclamide should occur. Therefore, the rate of [3H]glibenclamide dissociation was determined by preincubating the RIN-m5F cell membranes with [3H]glibenclamide. An excess (2 μM) of unlabeled glibenclamide was then added and the amount of bound [3H]glibenclamide was determined at various time points over a 4-h period. Figure 2illustrates such an experiment. Note that the dissociation kinetics fit best to a double exponential decay model. It also should be noted that the observed biphasic kinetics occurred under conditions (low [3H]glibenclamide concentrations) that reflect only high-affinity glibenclamide binding. The concentration of [3H]glibenclamide that was used in the initial preincubation conditions was 2.0 nM [3H]glibenclamide. At this concentration, only the high-affinity sites should have [3H]glibenclamide bound. However, the experiments also were carried out at lower concentrations of [3H]glibenclamide during the preincubation phase of the experiment and in all cases biphasic dissociation kinetics were observed. Even varying the concentration of [3H]glibenclamide over the 20-fold concentration range had little effect on the dissociation kinetic parameters for [3H]glibenclamide (data not shown). Therefore, 0.5 nM [3H]glibenclamide was preincubated with the RIN-m5F cell membranes and then either nateglinide, repaglinide, or glibenclamide was added during the dissociation phase of the experiments. Table3 summarizes the results of these experiments. Note that neither nateglinide nor repaglinide had any effect on the dissociation kinetic parameters for [3H]glibenclamide. This result is consistent with nateglinide and repaglinide binding directly to the glibenclamide-binding site on the sulfonylurea receptor.

[3H]Glibenclamide dissociation kinetics. RIN-m5F cell membranes were preincubated with 0.5 nM [3H]glibenclamide for 90 min at 25°C. Att = 0, 1 μM unlabeled glibenclamide was added and portions of the incubation mixture were removed at various times and assayed by the filtration-binding assay procedure. Data were fit to single- and double-exponential decay kinetic equations. AnF test of these two models indicated that the double-exponential decay equation described the data significantly better than a single-exponential decay equation. The kinetic parameters are listed in Table 3. Data points were collected in duplicate and combined from three separate experiments.
Summary of [3H]glibenclamide dissociation kinetic experiments with various competitive ligands
It is of interest to note that biphasic release kinetics also was observed for [3H]glibenclamide with HEK.EBNA[human SUR1] cell membranes (data not shown). The dissociation half-lives for the fast and slow phases were 1.4 ± 0.16 min (koff = 0.49 ± 0.055 min−1; ∼75% of total release) and 40 ± 16 min (koff = 0.017 ± 0.007 min−1; ∼25% of total release), respectively. These values are comparable to the kinetic values observed in experiments with the RIN-m5F cell membranes (Table 3).
Attempts to Measure [3H]Nateglinide Binding to RIN-m5F Cell Membranes.
The experiments described thus far indicate that nateglinide can compete with [3H]glibenclamide binding to RIN-m5F and HEK.EBNA[human SUR1] cell membranes. This finding suggests that [3H]nateglinide should bind to RIN-m5F cell membranes and that glibenclamide should compete with the binding. However, all attempts to measure specific binding of [3H]nateglinide to β-cell membranes have been unsuccessful in our laboratory as well as in other laboratories (Fujita et al., 1996). A plausible explanation is that [3H]nateglinide dissociated from its binding site during the workup procedure of the filtration-binding assay. A method that has been used to measure weak binding ligands is a centrifugation assay (see Materials and Methods). However, specific [3H]nateglinide binding could not be detected with this assay, whereas specific [3H]glibenclamide binding was measured (data not shown). Consequently, it appears that even under conditions of rapid separation (∼5 s; Bennett, 1978), specific binding with [3H]nateglinide to the RIN-m5F cell membranes is not observed.
Glucose Dependence of KATP Currents in β-Cells.
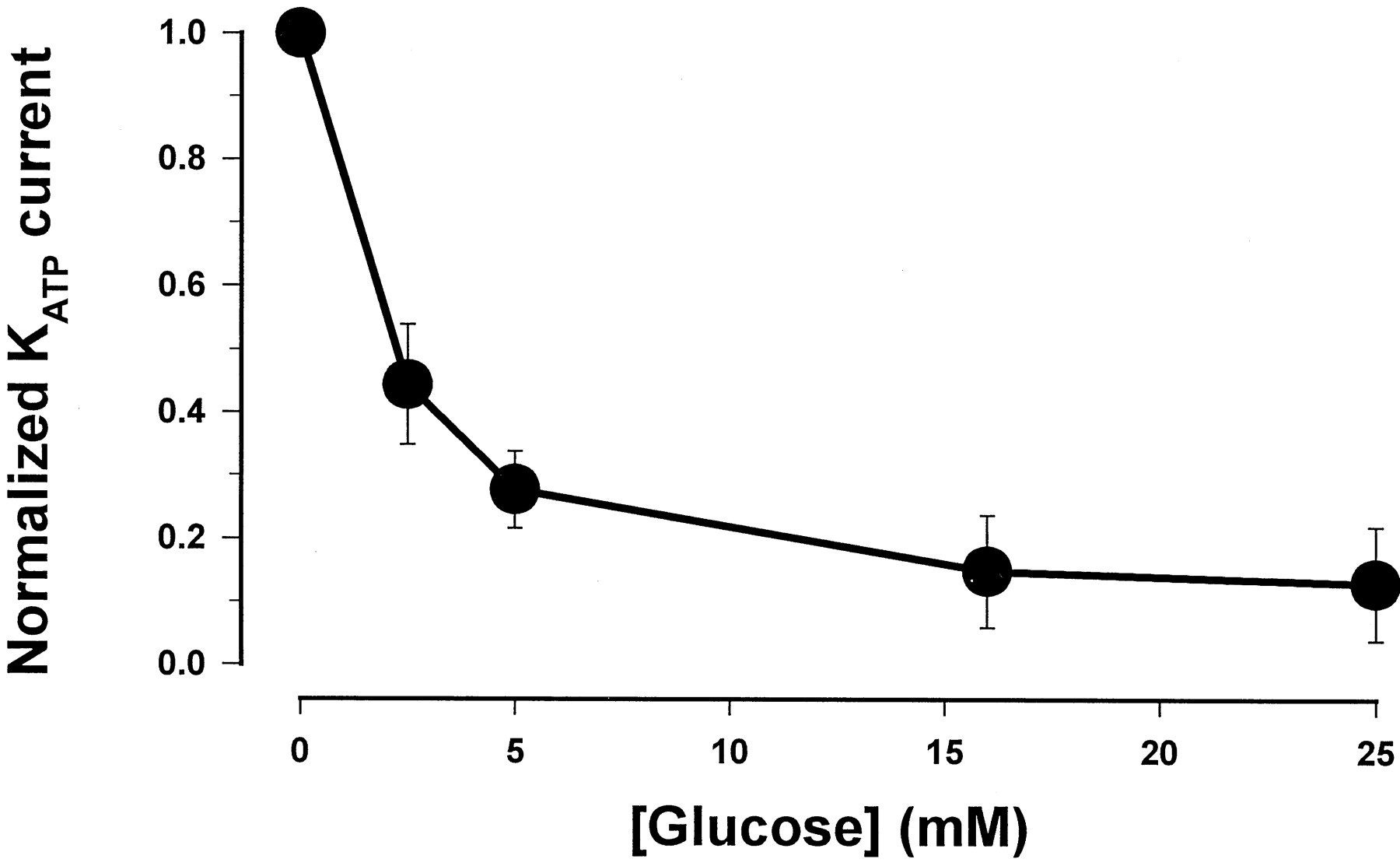
In pancreatic β-cells, the effect of glucose on insulin secretion is mediated via its influence on cellular ATP levels. A rise in intracellular ATP after hyperglycemia results in the closure of KATP channels, which initiates a sequence of electrical events to induce insulin release. We first assessed the influence of ambient glucose on whole-cellKATP current in pancreatic β-cells. Figure 3 shows that theKATP currents in β-cells were dependent on extracellular glucose concentration such that their amplitude reduced with increasing concentrations of glucose. Normalized with the maximal current without glucose, the magnitude of the currents at various glucose concentrations fit reasonably well with a single exponential equation, in which the amplitude ofKATP currents reduces by e-fold with every 3.7 mM increase in extracellular glucose. These results demonstrate that the pancreatic β-cells used in the electrophysiological experiments are metabolically active and respond appropriately to changes in external glucose levels.
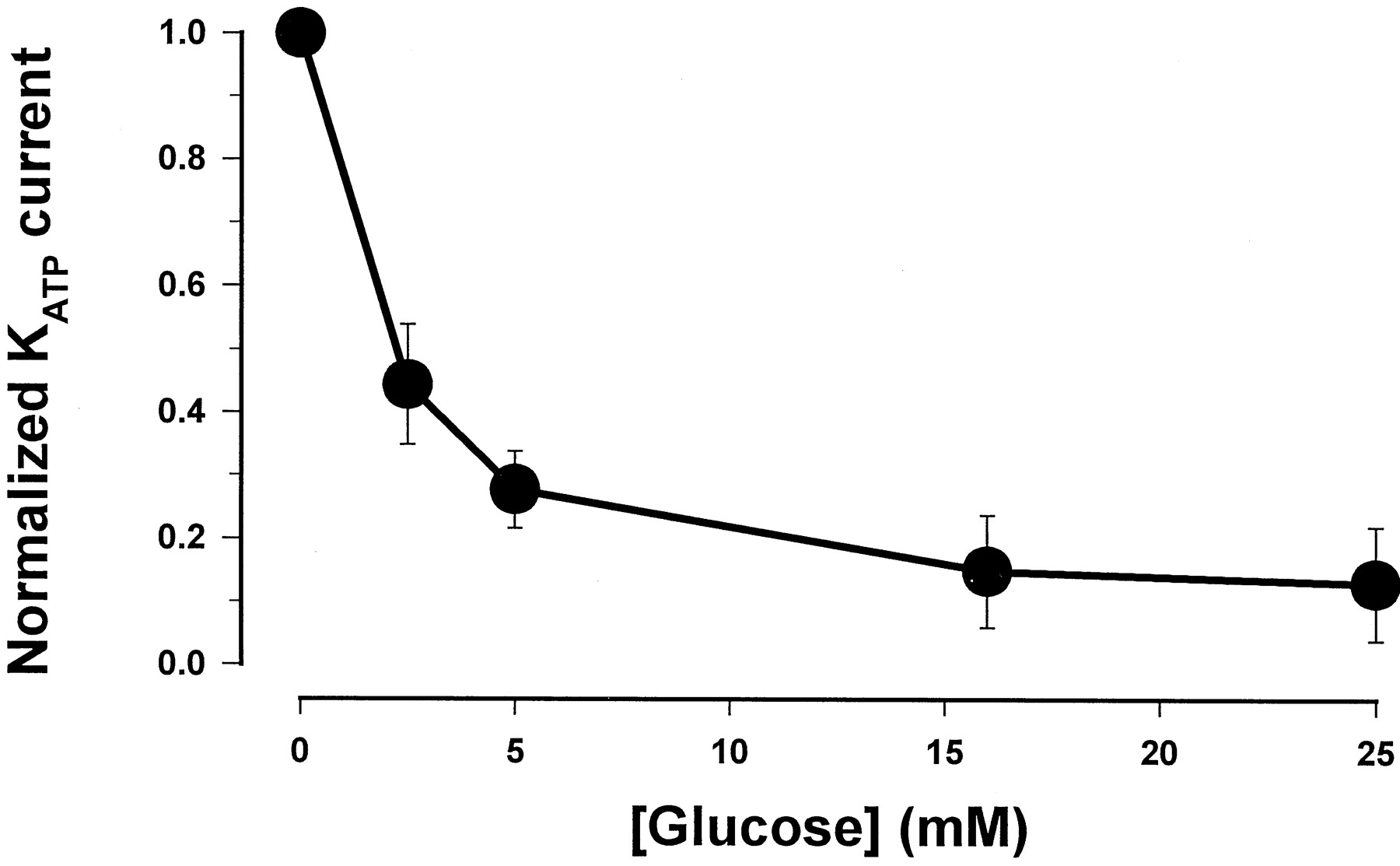
Concentration-response curve for glucose-dependence of the KATP current in β-cells. The data were fit with a single exponential equation: Y = 0.98 exp[(−X)/3.7 mM]. Y is the normalized KATP currents andX is the glucose concentration (millimolar). The points are the mean of five separate experiments.
Inhibition of KATP Currents by Nateglinide, Glibenclamide, and Repaglinide.
The ability to block the KATP current in β-cells by the antidiabetic agents nateglinide, glibenclamide, and repaglinide was evaluated. Given that the basal level ofKATP currents in β-cells is rather low, we applied 100 μM diazoxide, a known opener of theKATP channels in insulin-secreting cells, to activate KATP currents before the administration of the blockers. The data in Fig.4A demonstrate that nateglinide inhibitedKATP currents in a concentration-dependent manner. Glibenclamide and repaglinide produced inhibitory effects on KATP currents in a manner similar to that observed for nateglinide (Fig. 4, B and C). The effects of all agents were examined in 5 mM extracellular glucose to mimic a normoglycemic condition and concentration-response curves were formed as shown in Fig. 5. The IC50 values and the slope coefficients were obtained from the nonlinear fitting of the data with least-squares analysis. The IC50 values forKATP-blocking effect in normal glucose are 5.0 ± 1.4 nM for repaglinide, 16.6 ± 0.3 nM for glibenclamide, and 7.4 ± 0.2 μM for nateglinide. In a rank order of repaglinide > glibenclamide > nateglinide, the potencies for closure of KATP channels were commensurate with those shown to stimulate insulin release from isolated islets (Malaisse, 1995; Ikenoue et al., 1997).

Representative recordings of the inhibition ofKATP currents by nateglinide (NAT), glibenclamide (GLY), and repaglinide (REP). Experiment was performed in 5 mM glucose. KATP currents are shown in control, in 100 mM diazoxide (DIA), and in diazoxide with nateglinide at various concentrations as indicated. Dotted lines indicate zero current levels. The amplitude of current at 300 ms from the beginning of the pulse (∼−90 mV) was measured to performed the quantitative analysis. All currents were recorded from the same cell.

Concentration-response curves for the inhibition ofKATP currents by nateglinide (▪), glibenclamide (●), and repaglinide (▴) in 5 mM glucose. Points are the amplitudes of KATP currents at −90 mV, averaged from six to eight experiments. The y-axis values are the KATP currents in the presence of drugs relative to the maximal values activated by diazoxide (as percentages). The x-axis indicates the concentration of the compounds (molar concentration) presented in a logarithmic scale. The slope coefficients were 0.7, 0.9, and 0.7, respectively, for repaglinide, glibenclamide, and nateglinide.
Time Course of KATP Channel-Blocking Actions.
We studied the time-dependence of theKATP channel-blocking effects of nateglinide and compared it with that of glibenclamide and repaglinide. All compounds were tested at an equipotent concentration, i.e., 2-fold of their respective IC50 values. To circumvent cell-cell variability, the time courses of nateglinide and one of the other two agents were investigated sequentially in the same cell. Figure 6, A and B, show, respectively, the time course of nateglinide with glibenclamide and nateglinide with repaglinide. The effect by nateglinide onKATP channels had a rapid onset and was largely or completely reversed shortly after the drug was removed. In addition, the action of nateglinide could be seen with repeated application without a sign of desensitization. In contrast, the duration of KATP channel blocking action by glibenclamide and repaglinide was longer lasting. This was especially true with repaglinide, whose action often outlasted the duration of drug presence by severalfold. In most cases, a complete recovery of repaglinide's effect was not seen within ∼3 h (Fig. 6B). A summary of the time to a half-maximal inhibition [t1/2(on)] and the time to a half-recovery from maximal inhibition [t1/2(off)] is given in Table4.

Time course of theKATP-blocking effects by nateglinide (NAT) versus glibenclamide (GLY) (A) and nateglinide versus repaglinide (REP) (B). In each figure, data were recorded from the same cell and expressed as the fraction of their maximal values activated by 100 μM diazoxide. The duration of compound applications are marked at the top of the figures.
Time course of the KATP channel activity-blocking effects
Discussion
The high-affinity glibenclamide binding site (Kd = 0.27 ± 0.07 nM) is most likely involved in controlling KATPchannel activity and insulin release because the effect on insulin release by glibenclamide occurs at low nanomolar concentrations after correction for ligand binding to BSA (Gaines et al., 1988). Moreover,Aguilar-Bryan et al. (1992) have observed a correlation between the number of high-affinity receptors andKATP channel density with HIT-T15 cells at low- and high-passage number. The low-affinity sites (Kd = 25 ± 13 nM) may have resulted from damage to the receptor during the membrane isolation procedure because binding experiments with intact RIN-m5F β-cells reveal only a single receptor population (data not shown;Kd = 2.4 ± 0.27 nM). With intact HIT-T15 β-cells, a single binding site with aKd = 0.29 nM for [3H]glibenclamide has been reported (Ikenoue et al., 1997). Gaines et al. (1988) also reported a single high-affinity binding site with HIT-T15 cell membranes (Kd = 0.76 ± 0.04 nM). However,Niki et al. (1989) observed both high- and low-affinity binding sites (Kd = 1.1 and 140 nM) with membrane preparations from HIT-T15 cells. The relative magnitude of the differences between the high- and low-affinity sites is similar, i.e., 130-fold difference versus the 90-fold difference observed in the present experiments.
As shown in Table 1 glibenclamide is 440-fold more potent than nateglinide and nateglinide is 160-fold more potent than tolbutamide in binding to RIN-m5F cell membranes. In addition, nateglinide is 25-fold more potent than the l-isomer of nateglinide. These results compare favorably with the results of Fujita et al. (1996) and Ikenoue et al. (1997) with HIT cell membranes and intact HIT cells. The 25-fold reduced potency of the l-isomer in the membrane-binding assay is also consistent with the 60-fold reduced in vivo biological activity for the l-isomer of nateglinide (Shinkai et al., 1989). The relative order of efficacy for displacement of [3H]glibenclamide is consistent with a mode of action involving competitive binding at SUR1. Moreover, there is also a correlation between the relative potencies for binding to SUR1 and the ability of the compounds to inhibit theKATP channel and/or stimulate insulin secretion (Sugita et al., 1981; Schmid-Antomarchi et al., 1987; Gaines et al., 1988).
It is of interest to note that all of the compounds tested with the RIN-m5F cell membranes, with the exception of repaglinide and glimepiride, have Hill coefficients near unity (>0.85). The Hill coefficient for repaglinide is 0.60 ± 0.009 and for glimepiride is 0.58 ± 0.001. These values are significantly different from the Hill coefficients of the other compounds tested based ont tests of the Hill coefficients determined in the individual competitive binding experiments. A plausible interpretation of this result is that there is heterogeneity in the high-affinity receptor population (McPherson, 1989), i.e., there are two forms of the high-affinity SUR1 in RIN-m5F membranes or SUR1 can exist in two interchangeable states. Repaglinide and glimepiride (but not the other compounds) bind to the two forms or states of the receptor with different affinities. Additional experiments, described below, in which [3H]glibenclamide dissociation kinetics is measured also support a two-state/two-receptor model. It should be noted that an analog of repaglinide, AZ-DF 265, also exhibits a reduced Hill coefficient (0.51 ± 0.04) in competitive binding experiments with RIN-m5F cells (Ronner et al., 1992). The reduced Hill coefficients for repaglinide and glimepiride in the competitive binding experiments with RIN-m5F cell membranes (Table 1) distinguish the interactions of nateglinide, repaglinide, and glimepiride because nateglinide binds to both putative receptor states with equal affinity (Hill coefficient = 0.98 ± 0.06), whereas repaglinide and glimepiride bind with different affinity (Hill coefficients = 0.60 ± 0.009 and 0.58 ± 0.001, respectively). However, these differences in Hill coefficients are not observed with membranes from HEK.EBNA[human SUR1] cells (Table 2). This would suggest that the heterogeneity observed with RIN-m5F cell membranes, i.e., reduced Hill coefficients, is either artifactual, e.g., some of the receptor was “damaged” during its preparation, or that another membrane component (such as the inward rectifier component of the β-cellKATP channel, Kir6.2, which is present in RIN-m5F cells but not in the HEK-293[human SUR1] cells) is required to observe the two putative states of SUR1.
These compounds also were tested in direct competition studies with 2 nM [3H]glibenclamide and membranes from HEK.EBNA[human SUR1] cells. In general, the inhibitors bind more weakly (5- to 17-fold for all compounds except repaglinide) to human SUR1 than to RIN-m5F SUR1. The greatest difference is seen with repaglinide, which binds ∼130-fold more weakly to human SUR1 than to RIN-m5F SUR1. Figure 1 displays the relationship for the binding of the seven compounds to RIN-m5F SUR1 and human SUR1. These results indicate that although the ligand-binding site is similar for RIN-m5F SUR1 and human SUR1, there are differences that become most apparent with repaglinide binding. Substantial differences in the receptor/enzyme ligand binding sites of human and other species is often observed (LoGrasso et al., 1994).
Neither nateglinide nor repaglinide had any effect on the dissociation kinetic parameters for [3H]glibenclamide (Table3). This is consistent with nateglinide and repaglinide binding directly to the glibenclamide-binding site on SUR1. The biphasic release kinetics observed with membranes from both RIN-m5F cells and HEK.EBNA[human SUR1] cells can be explained by heterogeneity in the high-affinity binding site. One possible explanation for the heterogeneity is a two-state model for SUR1, i.e., the binding site exists in two interconvertable states. Consistent with this possibility, Aguilar-Bryan et al. (1998) have proposed at least two states for SUR1 and suggest that SUR1 may function to regulate the transition between the silent and bursting states of theKATP channel. However, for human SUR1 this heterogeneous (biphasic) behavior in [3H]glibenclamide dissociation kinetics is not reflected in the Hill coefficients for the steady-state binding of the various ligands, all of which have values near unity (Table 2).
The inability to measure [3H]nateglinide binding in both the filtration and centrifugation binding experiments is consistent with a large koff value for [3H]nateglinide binding to SUR1. The estimated dissociation half-life for nateglinide is ∼1 s (koff = ∼34 min−1), which was calculated assuming that the konvalue is 2 × 108 min−1M−1. This is a reasonable estimate of thekon value because Müller et al. (1994) has reported kon values of 1.7 × 108 min−1M−1 for glibenclamide and 4.9 × 108 min−1 M−1 for glimepiride binding to RIN-m5F cell membranes. This value is also in the range of association rates reported for enzyme-substrate interactions. For example, the association rate of tyrosine with tyrosyl-tRNA synthetase is 1.4 × 108 min−1 M−1(Ferst, 1985). This rapid dissociation of nateglinide from its binding site would explain the inability to directly measure [3H]nateglinide binding with the filtration and centrifugation assay methods. Biphasic dissociation kinetics were observed for [3H]glibenclamide with dissociation half-lives of 2.9 min (koff = 0.23 min−1) and 63 min (koff = 0.011 min−1). The value for the fast phase is in good agreement with the value reported by Müller et al. (1994) for the rate of [3H]glibenclamide dissociation from RIN-m5F cell membranes (half-life = 2.8 min;koff = 0.25 min−1). The estimated dissociation half-life for repaglinide is ∼2 min (koff = ∼0.36 min−1; calculated assuming that kon = 2 × 108 min−1 M−1). The estimated dissociation rates for nateglinide, glibenclamide, and repaglinide are consistent with the kinetic effects observed onKATP channel activity after removal of the compounds in the whole-cell patch-clamp experiments (see below).
Table 4 summarizes the kinetic effects of the compounds onKATP channel activity. Thet1/2(on) of nateglinide (4.1 min) was similar to that of glibenclamide (4.2 min), although nateglinide exhibited a considerably earlier in vivo insulinotropic action (Ikenoue et al., 1997). The discrepancy is most likely attributed to their different pharmacokinetic profiles, with nateglinide being absorbed more rapidly than glibenclamide. In our experiments, the duration of compound application was in the range between 15 and 20 min because all three compounds reached their steady-state maximal effect in <20 min. The t1/2(off) values of glibenclamide and repaglinide, being considerably >20 min, suggested that a majority ofKATP channels remained in a closed state long after the compounds were removed. Consequently, these agents would cause a prolonged insulinotropic action in vivo. Thus, the mechanism-based slow recovery of the action by glibenclamide and repaglinide appears to be a crucial factor that contributes to the long-lasting hypoglycemic action of these compounds.
The kinetic data on the recovery ofKATP channel activity after removal of the compound is consistent with the reported data on the reversibility of the effect of these agents on insulin release with perifused pancreatic islets. Thus, the effect of nateglinide on insulin release with perifused islets is completely reversed within 10 min after removal of the compound (Jijakli et al., 1997). However, both glibenclamide and repaglinide show little or no change in insulin release after removal of the compounds (Lebrun and Malaisse, 1992;Jijakli et al., 1996).
Conclusions
Both nateglinide and repaglinide bind competitively with glibenclamide to rat (RIN-m5F) and human SUR1. Differences in binding are most apparent with repaglinide, which binds 130-fold more weakly to human SUR1 than to RIN-m5F SUR1. The six other ligands bind, on average, 9-fold more weakly to human SUR1 than to RIN-m5F SUR1. Nateglinide rapidly dissociates from the RIN-m5F SUR1 with an estimated half-life of ∼1 s, whereas the half-life for glibenclamide is 2.9 and 63 min (biphasic dissociation kinetics) and ∼2 min for repaglinide (estimated value). The effect of nateglinide onKATP channel activity with intact β-cells is more rapidly reversed compared with the effects of glibenclamide and repaglinide. Table 5compares nateglinide, glibenclamide, and repaglinide with respect to receptor binding, KATP channel activity and insulin secretion. Finally, the receptor-binding results and intact β-cell KATP channel activity measurements are consistent with the more rapid in vivo and ex vivo onset and shorter duration of action for nateglinide relative to repaglinide and sulfonylureas. These characteristics may contribute to the low hypoglycemic potential for this compound and reduction in excessive insulin release.
Comparison of nateglinide, glibenclamide, and repaglinide under various assay conditions
Acknowledgments
We thank Susan Cornell-Kennon, Michele Eckhardt, and Mei Dong for assistance with the RIN-m5F cell culture.
Footnotes
-
Send reprint requests to: Brian R. Boettcher, Metabolic and Cardiovascular Disease Department, Novartis Institute for Biomedical Research, 556 Morris Ave., Summit, NJ 07901. E-mail:brian.boettcher{at}pharma.novartis.com
- Abbreviations:
- SUR1
- sulfonylurea receptor 1
- HEK
- human embryonic kidney
- EBNA
- Epstein Barr nuclear antigen 1 gene
- HEK.EBNA[humanSUR1]
- HEK.EBNA cells expressing human SUR1 from the pOriP/Zeo expression plasmid
- RIN-m5F
- rat islet cell tumor β-cell line
- pOriP/Zeo
- expression plasmid containing the oriP origin of replication of the Epstein Barr virus and the zeocin resistance gene
- Received August 19, 1999.
- Accepted January 7, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}