


Abstract
The novel nonpeptide orphanin FQ/nociceptin (OFQ/N) ligand {(1S,3aS)-8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one} (Ro 64-6198) was characterized in vitro and in vivo for its agonistic potential. Ro 64-6198 was 130- to 3500-fold selective for the OFQ/N receptor (ORL1) compared with opiate receptors. In the cAMP inhibition assay, Ro 64-6198 was a full agonist at the ORL1 and a partial agonist at the mu opiate receptor. When human embryonic kidney 293 cells stably expressing the human ORL1 receptor were pre-exposed (30 min) to either OFQ/N or Ro 64-6198, the ability of both agonists to inhibit forskolin-mediated cAMP accumulation was strongly reduced, indicating a functional desensitization of the second messenger cascade. However, acidic washes of OFQ/N-exposed cells fully restored the sensitivity of the ORL1 receptor for agonists. In contrast, the cAMP response in Ro 64-6198-exposed cells remained impaired after acidic washes, suggesting sustained receptor internalization at 30 min. In agreement with this finding, the number of cell-surface ORL1 receptors was significantly reduced after Ro 64-6198 pre-exposure, and this effect could be blocked with high sucrose concentrations. When Ro 64-6198 was chronically administered to rats, no signs of tolerance to its anxiolytic-like effects were detected following 15 days of daily drug exposure. In agreement with the behavioral results, Ro 64-6198 was able to reduce brain ORL1 binding sites in both acutely and chronically treated rats. Full recovery of ORL1 binding sites was observed 24 h after Ro 64-6198 administration with at1/2 of ∼5.5 h. These data show that nonpeptide agonists at the ORL1 receptor have a good clinical potential as anxiolytics without causing tolerance.
The ORL1 receptor (the nomenclature is based on the recommendation ofDhawan et al., 1996) was cloned from human, mouse, and rat because of its high homology (∼50%) to the closely related delta (OP1), kappa (OP2), and mu (OP3) opiate receptors (reviewed in Meunier et al., 2000). However, the classical opiate ligands do not bind and activate ORL1, and until the discovery of its cognate ligand, the 17-amino acid peptide orphanin FQ/nociceptin (OFQ/N) (Meunier et al., 1995; Reinscheid et al., 1995), the receptor was considered an orphan opiate-like receptor. ORL1, like OP1, OP2, and OP3, is mainly coupled to the inhibitory G protein. Thus, activation of ORL1 by OFQ/N results in the inhibition of cAMP production. Besides its inhibitory G protein coupling, ORL1 has also been demonstrated to modulate hippocampal Ca2+ channels (Knoflach et al., 1996) and to increase a K+ conductance in rat dorsal raphe, locus coeruleus, and hypothalamic neurons (Connor et al., 1996;Vaughan and Christie, 1996; Wagner et al., 1998).
Potent nonpeptide ORL1 agonists that mimic the effects of the endogenous peptide OFQ/N have recently been reported (Wichmann et al., 2000). The most potent compound, Ro 64-6198 {(1S,3aS)-8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one} (Fig. 1A), when given intraperitoneally, was shown to be active, like the benzodiazepine alprazolam, in several models of anxiety (Jenck et al., 2000; Wichmann et al., 2000). However, unlike benzodiazepine anxiolytics, Ro 64-6198 was devoid of antipanic, anticonvulsant, sedative, and amnestic activity at anxiolytic doses in the rat.

Structure of Ro 64-6198 (A), binding of Ro 64-6198 to membranes prepared from cells transfected with cDNAs encoding human OP1, OP2, OP3, or ORL1 receptors (B), and inhibition of forskolin-mediated cAMP inhibition by Ro 64-6198 in cells stably expressing human ORL1 (C) or OP3 (D). Competitive binding and cAMP inhibition experiments were performed as described under Experimental Procedures. Data represent triplicates of one experiment repeated at least three times.
A purpose of this study was to determine whether tolerance develops to the anxiolytic-like effects of Ro 64-6198 following chronic parenteral treatment for 2 weeks. Tolerance is a common problem seen with several alkaloid agonists at the OP3 receptor (Keith et al., 1996; Blake et al., 1997) and with benzodiazepine receptor ligands (Rundfeldt et al., 1995). For example, morphine and heroin, both alkaloid OP3 agonists (Blake et al., 1997), cause tolerance, while methadone, a synthetic OP3agonist, is devoid of such activity (Blake et al., 1997). Recently it has been shown that in contrast to methadone and [d-ala2,N-methyl-phe4, glyol5][tyrosyl-3,5-3H]-enkephalin (DAMGO), morphine administration to cells expressing the recombinant OP3 receptor does not induce its desensitization and internalization (Whistler and von Zastrow, 1998; Zhang et al., 1998). It is now generally accepted that the lack of acute OP3 desensitization and internalization accounts for cellular adaptation, which as a long-term consequence results in a loss of cell-surface OP3 binding sites (Whistler et al., 1999). Because Ro 64-6198 is a nonpeptide agonist at the ORL1 and because ORL1 is closely related to the opiate receptors, a detailed pharmacological characterization of Ro 64-6198's action at ORL1 in vitro and in vivo was initiated.
In this study, the potency of Ro 64-6198 to desensitize the recombinant ORL1 receptor stably expressed in human embryonic kidney 293 (HEK293) cells was investigated and compared with that of the natural peptide ligand OFQ/N. We also provide evidence that chronic treatment with Ro 64-6198 does not produce tolerance in a rat model of anxiety and that in the brains of rats exposed chronically or acutely to Ro 64-6198, the ORL1 receptor undergoes a similar cycle of receptor desensitization and resensitization.
Experimental Procedures
Materials, Peptides, and Reagents.
All cell culture reagents were purchased from Life Technologies (Basel, Switzerland). Bovine serum albumin (BSA, fraction V) was obtained from Sigma (Munich, Germany). OFQ/N, DAMGO, and naloxone were obtained from Calbiochem (San Diego, CA). The purity of these peptides and reagents was greater than 95%. Ro 64-6198 (purity >99%) was synthesized in-house (see Jenck et al., 2000; Wichmann et al., 2000).
Radioligands.
The radioligands [leucyl-3H]OFQ/N (specific activity 150 Ci/mmol), [3H]DAMGO (specific activity 78 Ci/mmol), [N-allyl-2--3-3H]naloxone (specific activity 54.5 Ci/mmol), and [Ile5,6-3H]deltorphin (specific activity 72 Ci/mmol) were from Amersham (Little Chalfont, UK).
Transfection of ORL1 and Opiate Receptors.
The cDNAs encoding human OP3 and ORL1 were inserted into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA), and the cDNAs coding for OP1 and OP2 were inserted into the pSFV2 vector. Recombinant Semliki Forest virus stocks were generated as described previously (Wichmann et al., 2000). Baby hamster kidney (BHK) cells were infected and harvested 24 h after infection. ORL1 was stably transfected into HEK293 cells, while OP3 was stably transfected into Chinese hamster ovary (CHO) cells using the Transfectam reagent (BioSepra Inc., Villeneuve La Garenne, France) as described previously (Dautzenberg et al., 2000). Two days after transfection with 10 μg of plasmid DNA, geneticin selection (1 mg/ml) was initiated and stable clones were selected.
Membrane Isolation and Radioligand Binding Assays.
Membranes from cells expressing hOP1, hOP2, hOP3, or hORL1 or from rat brain were isolated as described previously (Dautzenberg et al., 1998). In the case of the rat brain membrane homogenates, the membrane preparations were washed once with acidified phosphate-buffered solution (PBS, pH 4.0) to ensure that membrane-bound Ro 64-6198 was washed off. Afterward, the homogenates were washed once more with a PBS solution of physiological pH (pH 7.4), centrifuged at 39,000g, then processed further.
Competitive binding displacement analysis was performed with membranes prepared from permanently transfected HEK293 cells expressing hORL1 (10 μg membrane protein) and 0.4 nM [leucyl-3H]OFQ/N. Competitive binding displacement analyses for hOP3 were performed with membranes prepared from permanently transfected CHO cells (30 μg of membrane protein) and 0.5 nM [3H]DAMGO. Competitive binding displacement analyses for hOP1 and hOP2 receptors were performed with membranes prepared from BHK cells transiently expressing hOP1 or hOP2 (5 μg of membrane protein each) and 0.3 nM [Ile5,6-3H]deltorphin (hOP1) or 1.5 nM [N-allyl-2-3-3H]naloxone (hOP2).
The binding assays for all four receptors were performed in 96-well plates (Beckman Coulter, Inc., Fullerton, CA) using a scintillation proximity assay as described previously (Dautzenberg et al., 2000). Briefly, membranes from the receptor-expressing cells were combined with wheat germ agglutinin Scintillation Proximity Assay beads (0.5–1 mg, Amersham) and the indicated radioligands. Reactions were performed in 0.25 ml of binding buffer (50 mM Tris-HCl, pH 7.8, 1 mM EGTA, 5 mM MgCl2, 0.1% BSA) for 60 min at 22°C with shaking, and radioactivity was measured in a TopCount scintillation counter (Packard, Meriden, CT). Nonspecific binding was defined in the presence of 1 μM unlabeled OFQ/N (hORL1), 1 μM DAMGO (hOP3), 10 μM naloxone (hOP2), or 10 μM deltorphin (hOP1).
Saturation binding assays with rat brain membrane homogenates were performed in 1 ml of binding buffer with 10 μg of homogenate and increasing concentrations (25–800 pM) [leucyl-3H]OFQ/N. Incubation was performed for 60 min at 22°C. At the end of the incubation, the reaction mixture was filtered through GF/C filters (Amersham) that had been precoated (0.3% polyethyleneimine plus 0.1% BSA). Filters were washed three times with 0.5 ml of cold wash buffer (50 mM Tris-HCl, pH 7.5). Finally, 60 μl of scintillation fluid (MicroScint, Canberra Packard, Mississauga, ON, Canada) was added, and samples were counted in a TopCount scintillation counter. Nonspecific binding was defined in the presence of 1 μM OFQ/N.
cAMP Inhibition Assay.
The inhibition of forskolin-mediated cAMP accumulation by OFQ/N and Ro 64-6198 was determined in 96-well plates. Briefly, 20,000 cells were incubated in Krebs-Ringer-HEPES-buffered solution (124 mM NaCl, 5 mM KCl, 1.25 mM MgSO4, 1.5 mM CaCl2, 1.25 mM KH2PO4, and 25 mM HEPES, pH 7.4) in the presence of 100 μM Rolipram and 1 μM forskolin (both from Sigma) with increasing concentrations of agonists (10 pM to 100 nM) for 15 min at 37°C. Reactions were stopped by the addition of 0.12 ml of ice-cold ethanol and stored at −80°C for at least 4 h. The cAMP content was determined from the supernatant using the Biotrak nonradioactive cAMP kit (Amersham) according to the manufacturer's instructions.
Desensitization Protocol.
HEK293 cells stably expressing the hORL1 receptor were incubated with 100 nM OFQ/N or Ro 64-6198 for 30 min at 37°C. At the end of the incubation period, cells were washed three times with 40 volumes of ice-cold PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4; pH 7.4) as described previously (Dautzenberg and Hauger, 2000) or once with acidified PBS (PBS, pH 4.0) followed by two final washes with PBS, pH 7.4. Next, cells were resuspended in Krebs-Ringer-HEPES-buffered solution and stimulated with 1 μM forskolin and increasing concentrations of either OFQ/N or Ro 64-6198, as described above.
In Vivo Investigations and Chronic Administration.
Ro 64-6198 or vehicle (0.3% Tween 80 in physiological saline) was administered chronically to male Sprague-Dawley rats (150–200 g) for 21 days. A dose of 3.2 mg/kg Ro 64-6198 was chosen to induce a robust anxiolytic-like effect in the elevated plus-maze test, without significantly impairing sensorimotor functions in the rat (Jenck et al., 2000). Daily intraperitoneal administration was performed at variable abdominal injection sites (Ro 64-6198 has low oral bioavailability) between 8:00 and 9:00 AM. A preliminary subchronic observation study (5 days, 3 mg/kg i.p., b.i.d.) confirmed that the drug was well tolerated by rats when given repeatedly and did not accumulate to toxic levels as predicted by its acute pharmacokinetic profile. The experimental procedures used in this study received approval from a local governmental committee based on adherence to international guidelines and Swiss federal regulations on animal experimentation.
Three groups of 15 rats each were included in the study: Veh-Veh = chronic vehicle treatment and effect of vehicle injection tested on day 15; Veh-Ro = chronic vehicle treatment and effect of Ro 64-6198 (3 mg/kg, i.p.) injection tested on day 15; and 3) Ro-Ro = chronic Ro 64-6198 (3 mg/kg) treatment and effect of Ro 64-6198 (3 mg/kg, i.p.) injection tested on day 15.
Body weight was recorded daily before injections and rectal body temperature was measured twice a week as an additional marker for general health. An interim analysis of sensorimotor functions was also performed at 7 days. Animals were evaluated for their muscular strength, motor performance, and nociceptive reactivity as described in detail elsewhere (Jenck et al., 1997, 2000). Briefly, forelimb grip strength was quantitatively assessed using a digital strain gauge, motor performance was measured in a body traction test situation, and thermal nociception was assessed using a standard tail-flick assay. Since rats needed to be naive to the elevated plus-maze procedure, no interim analysis was performed at 7 days, but all animals were exposed to the plus-maze on day 15, 30 min following drug or vehicle administration. This test is based on the natural aversion of rodents for open spaces and heights and uses an elevated maze with two open and two closed arms. The time spent and the number of entries into open arms are indices of neophobic anxiety in animals (details in Jenck et al., 2000). Directly following plus-maze exposure, animals were tested again for muscular strength, motor performance, and nociceptive reactivity. Chronic drug or vehicle administration continued for an additional week, and all animals were sacrificed on day 21 for brain and liver dissection. Brain samples were used for receptor assays (see above), and liver samples were analyzed for hepatic enzyme induction using a standard cytochrome P450 activity assay.
Data Reduction and Statistical Analyses.
OFQ/N- or Ro 64-6198-stimulated cAMP inhibition data were calculated as percentage of control or percentage of cAMP values, as previously described (Hauger et al., 1997; Dautzenberg and Hauger, 2000). One-way analysis of variance (ANOVA) across experimental groups was performed using StatView Student (Abacus Concepts, Inc., Berkeley, CA) and Xlfit software (F. Hoffmann-La Roche AG, Basel, Switzerland). If the one-way ANOVA was statistically significant, planned post hoc analyses were performed using Bonferroni's multiple comparison tests to determine individual group differences.
Results
Pharmacological Characterization of Ro 64-6198 at Human OP1, OP2, OP3, and ORL1 Receptors.
The binding characteristics of cells transiently expressing hOP1 and hOP2receptors or stably producing hOP3 and hORL1 receptors were assessed. Saturation binding assays were performed with the four receptor preparations and radiolabeled deltorphin (OP1), naloxone (OP2), DAMGO (OP3), and OFQ/N (ORL1) as ligands. High-affinity binding of the radioligands to OP1(Kd = 0.09 ± 0.02 nM;Bmax = 1.9 ± 0.3 pmol/mg), OP2 (Kd = 0.6 ± 0.1 nM; Bmax = 1.7 ± 0.4 pmol/mg), OP3(Kd = 0.2 ± 0.04 nM;Bmax = 0.2 ± 0.02 pmol/mg), and ORL1 (Kd = 0.12 ± 0.03 nM;Bmax = 1.3 ± 0.2 pmol/mg) receptors was observed (not shown).
In competition binding assays, Ro 64-6198 displayed high affinity to the ORL1 receptor only. The compound competed for binding to the ORL1 receptor with subnanomolar affinity (Ki = 0.39 ± 0.05 nM,n = 10), while 130- to 250-fold higher ligand concentrations were needed to compete for binding to the OP3 (Ki = 52.4 ± 5.5 nM, n = 9) and OP2 (Ki = 106 ± 9 nM, n = 7) receptors (Fig. 1B). Finally, Ro 64-6198 displayed very low affinity to the OP1receptor (Ki = 1.4 ± 0.1 μM,n = 7) and thus differed in its affinity compared with the ORL1 receptor by a factor of 3500 (Fig. 1B).
In another set of experiments, the ability of Ro 64-6198 to inhibit forskolin-stimulated cAMP accumulation in cells stably expressing the ORL1 or the OP3 receptor was studied. Like the natural agonist OFQ/N (EC50 = 0.3 ± 0.1 nM,n = 12), Ro 64-6198 was a potent and full agonist (EC50 = 0.26 ± 0.08 nM, n = 10) at the ORL1 receptor (Fig. 1C). Both compounds inhibited forskolin-mediated cAMP accumulation by approximately 80 to 90% (Fig.1C). In contrast to its ability to potently and efficiently inhibit the forskolin response in ORL1-transfected cells, Ro 64-6198 was less potent and efficacious in OP3-transfected CHO cells. While DAMGO (EC50 = 6.3 ± 1.9 nM,n = 4) inhibited cAMP accumulation with high affinity, Ro 64-6198 (EC50 = 89.1 ± 12.4 nM,n = 3) was more than 15-fold less potent (Fig. 1C). Furthermore, Ro 64-6198 was ∼350-fold less potent at the OP3 receptor than at the ORL1 receptor. More important, Ro 64-6198 only inhibited forskolin-mediated cAMP inhibition in OP3-expressing cells to a smaller extent (∼30%) than DAMGO (Emax ∼65%) (Fig. 1C), a well known full agonist at the OP3receptor (Whistler et al., 1999). Thus, Ro 64-6198 behaves as a partial agonist at the OP3 receptor.
Differential Desensitization of ORL1 by OFQ/N and Ro 64-6198.
The ability of OFQ/N and Ro 64-6198 to functionally desensitize the cAMP response, and subsequently, the signaling response of the ORL1 receptor was assessed. In a first set of experiments, the effects of OFQ/N or Ro 64-6198 pre-exposure on forskolin-mediated cAMP accumulation was investigated. After incubation with 100 nM agonist, the cells were washed with either a pH 7.4 or a pH 4.0 buffered PBS solution, to ensure removal of the ligand, and then stimulated with forskolin. Pretreatment of ORL1-expressing cells with either 100 nM OFQ/N or Ro 64-6198 did not render the cells significantly more responsive to forskolin (not shown).
Next, the magnitude of ORL1 desensitization by OFQ/N or Ro 64-6198 was investigated. ORL1-expressing HEK293 cells were preincubated at 37°C for 30 min with vehicle (PBS) or a maximally effective concentration of either 100 nM OFQ/N or Ro 64-6198. After the cells had been washed extensively with a pH 7.4 buffered PBS solution, increasing doses (1 pM to 100 nM) of OFQ/N or Ro 64-6198 were incubated in the presence of forskolin (1 μM), and the cAMP content of the cells was determined. In control cells exposed to vehicle, OFQ/N and Ro 64-6198 potently inhibited forskolin-mediated cAMP accumulation (Fig.2A) at low agonist concentrations (OFQ/n = 0.19 ± 0.08 nM, n = 3; Ro 64-6198 = 0.22 ± 0.06, n = 3). Furthermore, OFQ/N and Ro 64-6198 produced maximal inhibitions of forskolin-mediated cAMP production by 81 ± 3% (OFQ/N) and 84 ± 4% (Ro 64-6198) that did not differ significantly from one another. In contrast, pre-exposure of ORL1-transfected cells with either OFQ/N or Ro 64-6198 significantly shifted the dose-response curves for both agonists to the right (p < 0.01) and in addition, desensitized their maximal response by 44 ± 3% (OFQ/N) and 50 ± 6% (Ro 64-6198) (p < 0.0001).

Desensitization of the ORL1 receptor-mediated cAMP responses by pretreatment with OFQ/N (A) or Ro 64-6198 (B) and washing with either pH 7.4 or pH 4.0 buffers. HEK293 cells stably expressing the ORL1 receptor were incubated with agonists (100 nM) for 30 min at 37°C and then washed with physiological pH (pH 7.4) or acidified (pH 4.0) PBS solution. Afterward, cells were stimulated with forskolin (1 μM) in the presence of increasing concentrations (10−12to 10−7 M) of OFQ/N (A) or Ro 64-6198 (B). Data are triplicates of one experiment repeated at least four times.
Because of the hydrophobic nature of Ro 64-6198, we wondered whether the desensitization of the ORL1 receptor, especially by Ro 64-6198, might be due to occupancy rather than functional desensitization. A ligand-occupied receptor, although still expressed on the cell surface, would mimic the behavior of a sequestered or internalized receptor in a functional assay. Thus, a set of experiments was conducted in which ORL1-expressing cells pretreated with OFQ/N or Ro 64-6198 were either washed with a buffer of physiological pH (pH 7.4, see above) or with an acidified (pH 4.0) buffer. In ORL1-expressing cells pre-exposed to Ro 64-6198, only small differences in the magnitude of desensitization were obtained when the cells had been washed with a pH 7.4 or pH 4.0 buffer (Fig. 2B). Restimulation of Ro 64-6198-desensitized and pH 4.0 washed cells resulted in a ∼55% desensitization of the second messenger signal of the ORL1 receptor (Fig. 2D), which was comparable to the results obtained with pH 7.4 washed cells.
Surprisingly, strongly differing results were obtained when ORL1-expressing cells were preincubated with 100 nM OFQ/N, then washed with either pH 7.4 or pH 4.0 buffered saline. In contrast to the ∼45% desensitization of the cAMP responses after OFQ/N pre-exposure and pH 7.4 wash, a complete restoration of both the potency (OFQ/N: EC50 = 0.14 ± 0.02 nM; Ro 64-6198: EC50 = 0.16 ± 0.03 nM) and efficacy of OFQ/N agonists was observed (Fig. 2A).
Saturation binding studies with [3H]OFQ/N and membranes isolated from control cells, OFQ/N-desensitized cells washed with either pH 7.4 or pH 4.0 buffer, and Ro 64-6198-treated cells washed with pH 4.0 buffer were performed. The Ro 64-6198-mediated functional desensitization of the ORL1 receptor was accompanied by a net loss of membrane binding sites (Table1). When compared with control membranes, desensitization by 100 nM Ro 64-6198 lowered the number of ORL1 receptors significantly, by ∼39% (Table 1). In addition to the reduced number of ORL1 receptors, theKd for OFQ/N was shifted ∼5-fold to the right in membranes isolated from Ro 64-6198-desensitized cells. In contrast, a reduced number (∼28%) of ORL1 binding sites, which was accompanied by a 4-fold lower apparent affinity for OFQ/N agonists, was only seen after pretreatment with OFQ/N and pH 7.4 washes (Table 1). However, acidic washes completely restored the amount and high-affinity state of ORL1 receptors in membranes from OFQ/N-exposed cells (Table1).
Kd and Bmax values of HEK293 cells stably expressing the human ORL1 receptor after different treatments
To ensure that the reduced responsiveness of ORL1-expressing cells and number of binding sites after incubation with Ro 64-6198 followed by acidic washes was not due to a strong residual binding of the compound to the ORL1 receptor, we performed additional experiments with intact cells. ORL1-expressing cells were incubated with either OFQ/N or Ro 64-6198 in the presence or absence of 0.5 M sucrose. High sucrose concentrations completely prevent receptor internalization (Keith et al., 1996; Smalley et al., 2001). After preincubation with OFQ/N or Ro 64-6198, cells were washed either with pH 7.4 or pH 4 buffer and finally, binding of [3H]OFQ/N to the intact cells was determined (Fig. 3). Only in ORL1-expressing cells that had been desensitized with OFQ/N and washed with pH 7.4 buffer was a reduced number of binding sites and a rightward shift of the binding curve observed. However, OFQ/N-desensitized cells that had been washed with pH 4 buffer or that had been incubated in the presence of high sucrose concentrations showed a binding profile similar to that of the control cells (Fig.3A). In contrast, a pre-exposure with Ro 64-6198 followed by pH 7.4 or pH 4 washes reduced the number of cell-surface binding sites as well as the affinity of the ORL1 receptor (pH 7.4 wash: IC50 ∼15 nM; pH 4 wash: IC50 ∼17 nM). However, a full restoration of the maximal binding capacities and affinity for OFQ/N ligands (IC50 ∼2.5 nM) was observed when the cells were pretreated with Ro 64-6198 in the presence of high sucrose concentrations. Thus, we conclude that the reduced binding sites of ORL1 cells after Ro 64-6198 treatment reflect an internalization of the ORL1 receptor rather than residual receptor occupancy.

Effects of acidic washes and blockade of receptor internalization by high sucrose concentrations on OFQ/N (A)- and Ro 64-6198 (B)-mediated reduction of ORL1 binding sites in intact ORL1-expressing cells. HEK293 cells (50,000 cells/well) stably expressing the ORL1 receptor were incubated with agonists (100 nM) in the presence or absence of 0.5 M sucrose for 30 min at 37°C and then washed with physiological pH (pH 7.4) or acidified (pH 4.0) PBS solution. Afterward, cells were incubated with 1 nM [3H]OFQ/N for 60 min at 37°C. At the end of the incubation, wells were rinsed twice with 500 μl of ice-cold PBS, pH 7.4, then lysed with 200 μl of 1 M NaOH. After addition of 60 μl of scintillation fluid (MicroScint), the bound radioactivity was counted in a TopCount. Data are triplicates of one experiment repeated at least twice.
In Vivo Effects Following Acute or Chronic Exposure of Rats to Ro 64-6198.
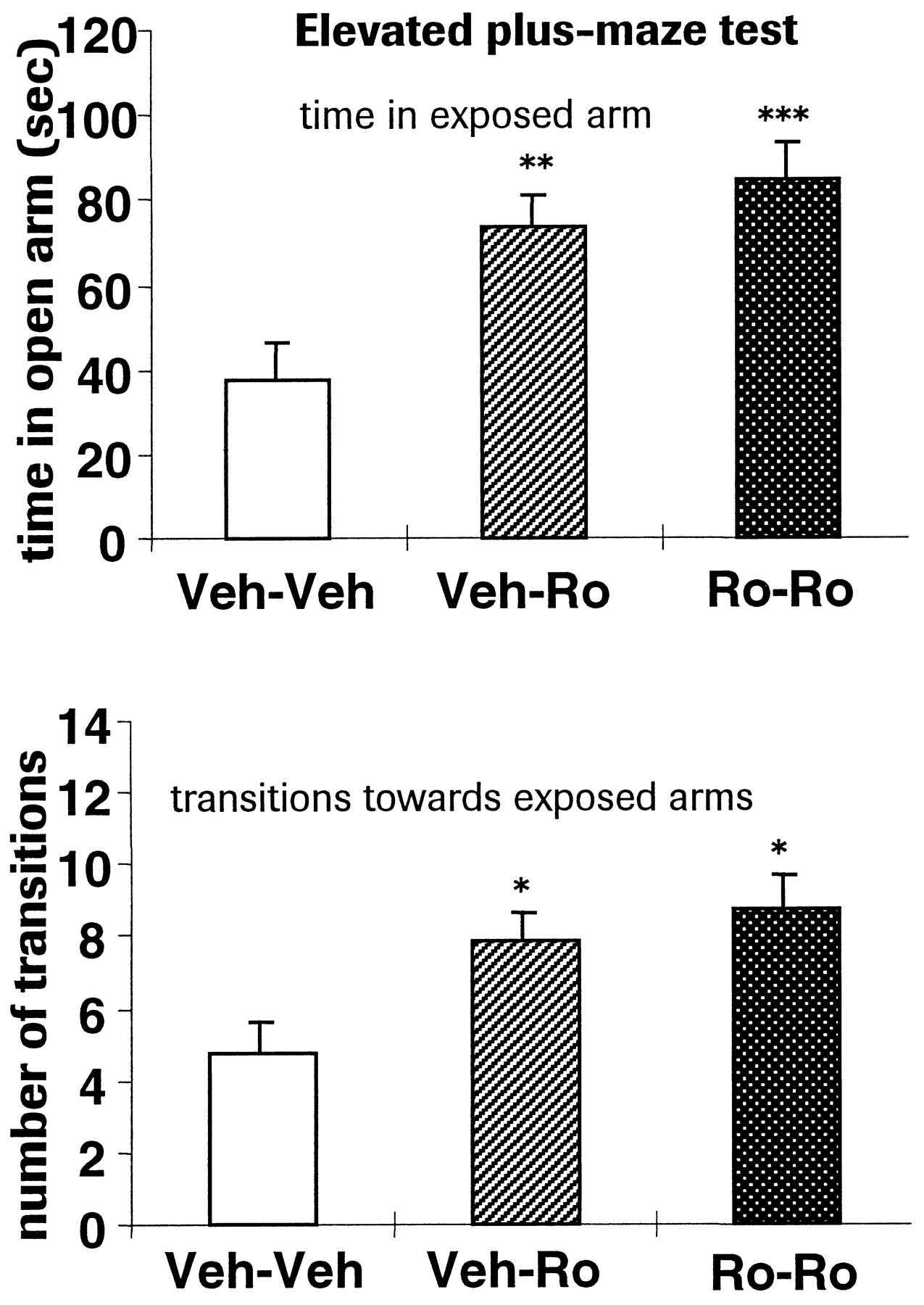
Average body weight gain and rectal temperatures were not significantly different among the three treatment groups (single-factor ANOVA, p > 0.05 in all cases, data not shown). The 7-day interim analysis of muscular strength, motor performances, and nociceptive reactivity revealed normal sensorimotor functions at day 7 in Ro 64-6198-treated compared with vehicle-treated animals (unpaired Student's t test, p > 0.05). In the elevated plus-maze test on day 15, Ro 64-6198 elicited significant anxiolytic-like effects as measured in time spent and transitions to the open arms (Fig. 4) in both groups exposed to the drug (post hoc Bonferroni, p < 0.01 in Veh-Ro and p < 0.001 in Ro-Ro versus Veh-Veh group). Subsequent analysis of muscular strength, motor performances, and nociceptive reactivity revealed no differences in sensorimotor functions at day 15 following Ro 64-6198 or vehicle treatment (single-factor ANOVA, p > 0.05 in all cases; Fig.5). Liver weights and cytochrome P450 enzyme activity was not different in Ro 64-6198-treated compared with vehicle-treated animals (1.56 nmol of cytochrome P450/mg of protein for vehicle and 1.26 for Ro 64-6198, unpaired Student'st test, p > 0.05).

Effects of chronic administration (vehicle or Ro 64-6198, 3 mg/kg, i.p.) on the anxiolytic-like activity of Ro 64-6198 (3 mg/kg, i.p., 30 min) measured versus vehicle on day 15 in the elevated plus-maze test. Time spent on the exposed arm (top) and transitions from closed to open arms (bottom) are represented. Veh-Veh = chronic vehicle treatment and effect of vehicle injection tested on day 15, Veh-Ro = chronic vehicle treatment and effect of RO 64-6198 (3 mg/kg, i.p.) injection tested on day 15, Ro-Ro = chronic Ro 64-6198 (3 mg/kg) treatment and effect of Ro 64-6198 (3 mg/kg, i.p.) injection tested on day 15.n = 15 rats per group, *p < 0.05, **p < 0.01, ***p < 0.001 versus vehicle, post hoc Bonferroni test.
No effect of chronic administration (vehicle or Ro 64-6198, 3 mg/kg, i.p.) on motor functions (top and middle) and nociception (bottom) measured following a dose of 3 mg/kg of Ro 64-6198 versus vehicle on day 15. Veh-Veh = chronic vehicle treatment and effect of vehicle injection tested on day 15, Veh-Ro = chronic vehicle treatment and effect of Ro 64-6198 (3 mg/kg i.p.) injection tested on day 15, Ro-Ro = chronic Ro 64-6198 (3 mg/kg) treatment and effect of Ro 64-6198 (3 mg/kg, i.p.) injection tested on day 15.
Transient Loss of ORL1 Binding Sites in Acutely and Chronically Ro 64-6198-Treated Rats.
Membrane homogenates from the brains (n = 2–3/time point) of rats treated acutely (1 dose) or chronically (1 dose/day for 25 days) with Ro 64-6198, and brains from the control groups, were prepared and tested for their affinity to bind [3H]OFQ/N. To eliminate unspecific binding of Ro 64-6198 to the brains, the homogenates were washed once with acidified PBS solution (pH 4.0).
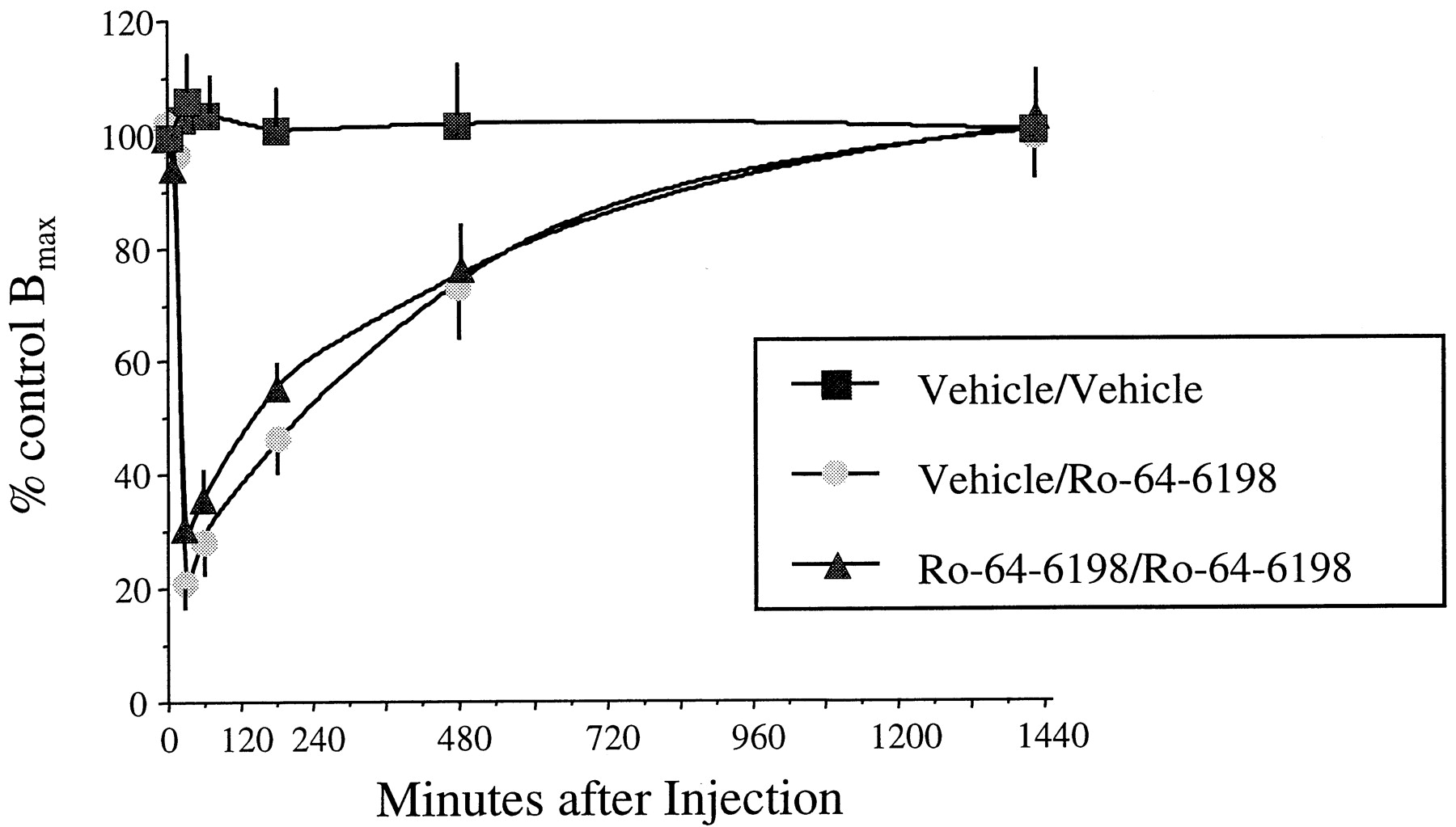
Vehicle-treated animals showed no alterations of brain ORL1 binding sites across all time points analyzed (Fig.6; Table2). Marked differences to the vehicle group, however, were observed when rat brain homogenates from animals that had been treated with Ro 64-6198 acutely or chronically (25 days) were assessed for their ORL1 binding sites. In both groups similar amounts of ORL1 receptors (∼150 fmol/mg) were calculated before Ro 64-6198 injection, and no significant differences were observed 10 min after injection (Table 2). The number of binding sites in the two Ro 64-6198 treatment groups also did not differ from the values of the control group (Fig. 6; Table 2). However, 30 min post-Ro 64-6198 injection, a dramatic loss of ORL1 binding sites was detected (77% in the acute group and 71% in the chronic group) (Fig. 6; Table 2). The number of binding sites remained strongly reduced (71% in the acute group and 66% in the chronic group) after 60 min and then slowly started to recover (Fig. 6). Three hours after Ro 64-6198 injection, the number of binding sites was back to 47 to 55% of the original number, and after 8 h, a recovery to >70% was detected. Finally, 24 h after Ro 64-6198 injection, a full recovery of the ORL1 binding sites in both treatment groups was noted (Fig. 6; Table 2). For both treatment groups a t1/2 of ∼5.5 h for the recovery of ORL1 binding sites was calculated.

Quantification of ORL1 binding sites in rat brain after acute i.p. challenge with Ro 64-6198 in rats treated with vehicle or Ro 64-6198 for 15 days. Rats received a final injection of either vehicle or Ro 64-6198. After different time points (10 min to 24 h) animals were decapitated and brain membrane homogenates were prepared as described under Experimental Procedures. Saturation binding experiments were performed with 10 μg of membrane homogenates and increasing concentrations (25–800 pM) of [3H]OFQ/N. Data represent the average of several experiments (n >5) performed in triplicate.
Time course of [3H]OFQ/N binding to rat brain membrane homogenates of animals treated with either vehicle or acutely or chronically with Ro-64-6198
Discussion
This study provides a full characterization of the novel nonpeptide OFQ/N agonist Ro 64-6198, a ligand binding the ORL1 receptor with subnanomolar affinity but showing considerably lower affinity to the OP3 (130-fold), OP2(250-fold), and OP1 (3500-fold) receptors. Thus, Ro 64-6198 displays a selectivity profile similar to that of OFQ/N (Meunier et al., 1995; Reinscheid et al., 1995). Because of the high sequence homology (∼50%) between the ORL1 and the opiate receptors (Meunier et al., 2000), such a selectivity profile is important for an ORL1 ligand to avoid undesired side effects mediated by the activation of opiate receptors. Besides their importance in pain transmission, opiate receptors, especially the OP3 receptor, are responsible for many features of drug addiction (Nestler, 1996). Despite the high sequence similarity, the physiological function of OFQ/N and its receptor differs considerably from that of the opiate ligands and receptors. OFQ/N has been reported to facilitate pain transmission, but the precise effects of OFQ/N on nociceptive sensitivity remain unclear (reviewed in Meunier et al., 2000). On the other hand, OFQ/N has been shown to be a potent anxiolytic in vivo (Jenck et al., 1997) but also promotes memory impairments (Manabe et al., 1998). Thus, the development of nonpeptide antagonists of the ORL1 receptor have a good clinical potential for the treatment of Alzheimer's disease, while OFQ/N agonists are beneficial for the treatment of stress disorders and depression (Jenck et al., 2000;Wichmann et al., 2000).
In the cAMP inhibition assay, Ro 64-6198 was a full agonist at the ORL1 receptor, and its potency did not differ from that of the natural ligand OFQ/N. As expected from the strong differences in the binding assays, Ro 64-6198 was >300-fold less potent at inhibiting forskolin-mediated cAMP inhibition in OP3- than in ORL1-expressing cells. Surprisingly, Ro 64-6198 was only a partial agonist at the OP3 receptor, displaying less than 50% of the efficacy of DAMGO, a full agonist at this receptor. This result contrasts with our original finding that Ro 64-6198 is a full agonist at the OP3 receptor in the GTPγS binding assay (Jenck et al., 2000), a functional assay that is conducted with membrane preparations (Röver et al., 2000). However, in this study we used cells expressing significantly lower amounts of the OP3 receptor (200 fmol/mg versus >2 pmol/mg) than in our previous studies (Wichmann et al., 1999, 2000;Röver et al., 2000). Furthermore, because the cAMP assay is performed with intact cells, it may be more predictive for the functional properties of a ligand than an assay conducted with isolated membranes. Indeed, the central nervous system effects elicited by Ro 64-6198 cannot be blocked by the high-affinity OP3 receptor antagonist naloxone (unpublished observations). Thus, it is likely that Ro 64-6198 does not activate the OP3 receptor in vivo.
To gain more insights into the functional properties of Ro 64-6198, we performed a series of desensitization experiments to study the functional regulation of the ORL1 receptor by this compound. We compared the following parameters: the effect of agonist activation of the ORL1 receptor on maximal forskolin-mediated cAMP production, the effects of either physiological (pH 7.4) or acidic (pH 4.0) pH washes on OFQ/N- or Ro 64-6198-mediated functional desensitization of the ORL1 receptor, and finally, the effects of physiological or acidic pH washes on the numbers of ORL1 binding sites.
In the recombinant expression system, the ORL1 receptor, like the opiate receptor, inhibits cAMP production (Reinscheid et al., 1995;Blake et al., 1997). Thus, cAMP inhibition assays are performed in the presence of forskolin, a direct activator of adenylyl cyclase (Hauger et al., 1997). Because none of the pretreatments produced elevated cAMP levels, we assumed that the different magnitudes of agonist-induced cAMP inhibition represented true desensitization of the signaling responses.
Striking differences in the desensitization potencies of OFQ/N and Ro 64-6198 were observed when ORL1-expressing cells were washed with either pH 7.4 or pH 4.0 buffer. No differences in Ro 64-6198's desensitizing properties were observed with both buffers, and Ro 64-6198 potently desensitized the maximal responses of the ORL1 receptor and significantly shifted the agonist-dose response curves to the right. In agreement with the functional studies, we observed a strong reduction of ORL1 binding sites after pre-exposure with Ro 64-6198, and this reduction was also independent of the washing conditions. Furthermore, when the desensitization experiments with Ro 64-6198 were conducted in the presence of high sucrose concentrations, no reduction of ORL1 binding sites after acidic washes was observed. Since high sucrose concentrations prevent receptor internalization (Keith et al., 1996; Smalley et al., 2001), we conclude that acidic washes quantitatively remove the nonpeptide agonist from its receptor, and that the reduced responsiveness of the ORL1 receptor after Ro 64-6198 challenge reflects desensitization of the second messenger cascade and receptor internalization. Surprisingly, this seems not to be the case for OFQ/N. We observed a functional desensitization of the ORL1 receptor and a reduction of receptor sites after OFQ/N exposure only when the cells had been washed with pH 7.4 buffer. However, after acidic washes of OFQ/N-exposed cells, the functional response of the ORL1 receptor was completely restored, and a full recovery of the number of OFQ/N binding sites was observed. These results indicate that OFQ/N occupies the ORL1 receptor and thereby blocks it from being stimulated again, without promoting receptor sequestration and internalization (Freedman and Lefkowitz, 1996; Carman and Benovic, 1998). Future experiments using a tagged ORL1 receptor should further aid insight into the differential regulation of the ORL1 receptor by its natural ligand OFQ/N or by the synthetic compound Ro 64-6198.
Another important part of our study addressed the question whether chronic Ro 64-6198 administration induces tolerance in vivo (Matthes et al., 1996). Tolerance is characterized as a loss of efficacy of a drug during chronic treatment (Harrison et al., 1998). This is exemplified by morphine, a high-affinity agonist at the OP3receptor (Whistler et al., 1999). Usually during chronic treatment, morphine doses have to be gradually increased to maintain its analgesic properties (Mestek et al., 1996; Raynor et al., 1996). Similarly, other opiate alkaloids and drugs of abuse, like heroin, also cause tolerance in drug addicts (Mestek et al., 1996; Nestler, 1996; Raznor et al., 1996). While in recombinant in vitro systems morphine seems not to mediate OP3 receptor internalization (Whistler and von Zastrow, 1998; Zhang et al., 1998; Whistler et al., 1999), it has been speculated that the lack of internalization might account for maladaptive processes which finally might cause tolerance in vivo. However, in a recent study it was shown that morphine is capable of mediating OP3 receptor internalization in vivo, while in animals being deficient for β-arrestin-2, an important mediator of G protein-coupled receptor internalization, no tolerance to morphine developed (Bohn et al., 2000). Thus, it seems likely that internalization plays an important role in the development of opiate receptor tolerance and that the potency of an agonist to mediate internalization in vitro is not predictive for its potential to cause tolerance in vivo.
However, no tolerance developed to the anxiolytic-like effects of Ro 64-6198 following 2 weeks of daily administration in rats. In addition, Ro 64-6198 was well tolerated at anxiolytic doses administered chronically, did not interfere with sensorimotor functions at this dosage, and did not affect hepatic function as no enzyme induction was observed in the animals. These results provide further evidence that the selective ORL1 agonist Ro 64-6198 may prove a useful anxiolytic compound. Preliminary studies from our laboratory indicate that the in vivo effects of Ro 64-6198 are exclusively mediated by the ORL1 receptor. In ORL1-deficient animals, the anxiolytic effects of Ro 64-6198 and its sedative side effects are abolished (G. A. Higgins and A. J. Grottick, unpublished observations). For OFQ/N, tolerance has been reported to the locomotor-inhibiting effects of OFQ/N given at high dosage (Devine et al., 1996), to its supraspinal antimorphine effects (Lutfy et al., 1999), and to its spinal antinociceptive action (Hao et al., 1997). These results suggest that differences exist between the endogenous ligand and exogenous synthetic agonists in their interactions with ORL1 receptors.
In agreement with the behavioral data, the number of ORL1 binding sites was not altered during chronic treatment with Ro 64-6198. However, we observed a strong and rapid reduction of ORL1 binding sites when Ro 64-6198 was given acutely. These effects were seen in the vehicle group and in animals that had been treated chronically with Ro 64-6198. In both groups a >70% reduction of ORL1 binding sites was monitored as rapidly as 30 min after i.p. injection of the compound. Afterward, the ORL1 receptor slowly reappeared with at1/2 of 5.5 h, and the number of OFQ/N binding sites was fully restored after 24 h. Thus, we conclude that the reduction of OFQ/N binding sites after acute Ro 64-6198 application represents a temporary and reversible down-regulation of the ORL1 receptor and that a once-daily dosage of a nonpeptide OFQ/N agonist is likely to prevent the development of tolerance.
In conclusion, we have characterized the novel nonpeptide OFQ/N agonist Ro 64-6198 as a selective full agonist at the ORL1 receptor in vitro. Pre-exposure of ORL1-expressing cells with Ro 64-6198 potently desensitized cAMP responses and down-regulated the number of cell-surface ORL1 receptors. In contrast, OFQ/N, despite its potency to desensitize the second messenger responses, was unable to down-regulate its receptor. In rats treated chronically with Ro 64-6198, no signs of tolerance were observed. ORL1 binding sites were unaltered by the chronic treatment, and only a transient decrease of the receptor was observed after acute Ro 64-6198 administration.
Acknowledgments
We thank Ivo Vogel for helping with the membrane preparations, Philipp Oberli for his help in synthesizing Ro 64-6198, and Martine Maco for skillful technical assistance. Finally, we thank Dr. Kenneth Lundstrom for the infection of BHK cells to obtain OP1 and OP2 membranes.
Footnotes
-
This study was supported by F. Hoffmann-La Roche AG.
- Abbreviations:
- ORL1
- OFQ/N receptor
- HEK293
- human embryonic kidney 293
- DAMGO
- [d-ala2,N-methyl-phe4, glyol5][tyrosyl-3,5]-enkephalin
- OFQ/N
- orphanin FQ/nociceptin
- OP1
- delta opiate receptor
- OP2
- kappa opiate receptor
- OP3
- mu opiate receptor
- Ro 64-6198
- {(1S,3aS)-8-(2,3,3a,4,5,6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one}
- BSA
- bovine serum albumin
- BHK
- baby hamster kidney
- CHO
- Chinese hamster ovary
- PBS
- phosphate-buffered saline
- ANOVA
- analysis of variance
- Received December 21, 2000.
- Accepted May 1, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}