


Abstract
CB1 cannabinoid (CB1) and D2 dopamine (D2) receptors are known to couple to the G protein Gαi/o. It has been reported that concurrent activation of D2 receptors and CB1 receptors, in primary striatal neuronal culture, promotes functional CB1 receptor coupling to Gαs resulting in elevations in intracellular cyclic AMP levels. We now report that in the absence of D2 receptors, acute activation of CB1 receptors inhibits cyclic AMP accumulation, whereas the presence of D2 receptors promotes CB1-stimulated cAMP accumulation, presumably through Gαs. This Gαs subunit switching was not prevented by pertussis toxin treatment and occurred in the presence and absence of D2 receptor activation. Thus, coexpression of the D2 receptor with the CB1 receptor was sufficient to switch the coupling of the CB1 receptors from Gαi/o to Gαs. Persistent activation of D2 receptors resulted in heterologous sensitization of adenylate cyclase to subsequent stimulation by forskolin, whereas the persistent activation of CB1 receptors did not. Additional studies in human embryonic kidney cells cotransfected with D2 and CB1 receptors revealed that persistent activation (18 h) of D2 receptors induced a switch of CB1 receptor coupling from Gαs to Gαi/o. This D2 receptor-induced effect allowed for CB1 receptor-mediated inhibition of cyclic AMP accumulation. The present studies suggest D2 receptors may have a significant modulatory role in determining the G protein coupling specificity of CB1 receptors.
The CB1 cannabinoid (CB1) receptor is expressed primarily in the central nervous system, especially in the basal ganglia and cortex (Herkenham et al., 1990; Matsuda et al., 1993; Tsou et al., 1998), with some expression occurring in peripheral tissues such as the uterus, testes, and ileum (Pacheco et al., 1991; Pertwee et al., 1992; Das et al., 1995). This distribution pattern in the brain suggests that the cannabinoid system and the ascending dopamine pathways may interact with one another. Indeed, functional links between dopaminergic and cannabinoid systems have been reported (Mailleux and Vanderhaeghen, 1993; Glass and Felder, 1997; Giuffrida et al., 1999). Microdialysis experiments have demonstrated that activation of D2 dopamine receptors (D2) promotes elevations in extracellular concentrations of the endocannabinoid anandamide (Giuffrida et al., 1999). Chronic treatment with D2 receptor antagonists causes up-regulation of CB1 mRNA in striatum (Giuffrida et al., 1999).
When expressed individually, activation of either the D2 or CB1 receptor inhibits cAMP accumulation. Several studies have shown that the CB1 receptor inhibits adenylate cyclase activity through coupling with a pertussis toxin-sensitive Gαi/o-protein (for review, see Howlett, 1995). The D2 receptor also inhibits adenylate cyclase via pertussis toxin-sensitive Gαi/o-proteins (Sibley and Monsma, 1992). Our interest in dopamine-cannabinoid interactions stems from reports that concurrent activation of D2 and CB1 receptors alters CB1 receptor coupling to signal transduction mechanisms. Specifically, in primary striatal cultured neurons, D2 receptor activation shifts CB1 receptor coupling from an inhibitory effect on the formation of the signaling molecule cAMP to a stimulatory effect on cAMP formation (Glass and Felder, 1997). Glass and Felder (1997) have suggested this increase in cAMP is brought about by the CB1 receptor switching from Gαi/o to Gαs linkage. Additional studies demonstrating the Gαs linkage of CB1 receptors were carried out in Chinese hamster ovary (CHO) cells stably expressing only the human CB1 receptors (Glass and Felder, 1997; Bonhaus et al., 1998; Felder et al., 1998). In the CB1 receptor/CHO cells augmentation of forskolin-stimulated cAMP accumulation upon CB1 receptor activation was observed only after pertussis toxin pretreatment. Together, the studies described above indicate that CB1 receptors can couple to multiple G proteins (i.e., Gαs and Gαi/o) after acute activation.
Acute activation of Gαs-coupled receptors enhances adenylate cyclase activity, which increases cAMP levels, whereas acute activation of Gαi/o-coupled receptors decreases adenylate cyclase activity, which results in decreased cAMP levels. However, long-term activation of Gαi/o-coupled receptors enhances subsequent stimulation of adenylate cyclase, a pharmacological phenomena known as heterologous sensitization (for review, see Watts, 2002). Although the exact mechanisms are not yet fully elucidated, persistent activation of a Gαi/o-coupled receptor induces heterologous sensitization via a pertussis toxin-sensitive G protein (Watts, 2002). Chronic activation of Gαi/o-coupled receptors such as D2 and CB1 receptors has been reported to potentiate adenylate cyclase responsiveness upon subsequent drug-stimulated cAMP accumulation (Watts and Neve, 1996; Rhee et al., 2000).
Because cultured cells have a unique composition of G proteins and adenylate cyclase isoforms, our initial experiments have focused on asking the question how generalized is the D2 receptor effect on CB1 receptor coupling? Do the D2 receptors promote the CB1 receptors to switch from Gαi/o to Gαs in transfected systems? Moreover, does the influence of the D2 receptor also affect the ability of the CB1 receptor to sensitize adenylate cyclase and vice versa? What are the effects of persistent activation of the D2 and CB1 receptor on adenylate cyclase? To address these questions, we investigated D2 and CB1 receptor signaling in HEK-293 cells. Our studies showed that the D2 receptor dramatically influences CB1 receptor coupling to Gα subunits. We determined that coexpression of the two receptors induces the CB1 receptor to switch to Gαs coupling, although activation of the D2 receptor is not necessary. Furthermore, overexpression of Gαi1 or persistent activation of the D2 receptor seemed to facilitate the reestablishment of Gαi/o coupling for the CB1 receptor. We asked questions seeking to further refine the relationship between D2 and CB1 receptor signaling. In the present study, we provide data that demonstrate the D2 receptor's ability to regulate the G protein coupling specificity of CB1 receptors.
Materials and Methods
Materials. [3H]Cyclic AMP (32 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). Forskolin, pertussis toxin, and (–)-quinpirole were purchased from Sigma/RBI (Natick, MA). CB1 receptor cDNA was a gift from Dr. Tom Bonner (National Institute of Mental Health, Bethesda, MD). The G protein α subunit cDNAs were purchased from the Guthrie Research Institute (Sayre, PA). CP55,940 was a generous gift of the Research Triangle Institute (Research Triangle Park, NC). All other drugs and chemicals were of the highest grade possible and were purchased from standard commercial sources.
Cell Culture. HEK-293 wild-type cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum supplemented with 1% penicillin/streptomycin, and 1% l-glutamine. HEK-293 cells stably transfected with the D2 receptor (HEK/D2) were maintained in Dulbecco's modified Eagle's medium with 5% fetal bovine serum, 5% bovine calf serum supplemented with 2 mg/ml purinomycin, 1% penicillin/streptomycin, and 1% l-glutamine. The cells were grown in a 37°C humidified environment with 5% CO2. Where indicated, cells were transfected using hCB1pRcCMV, Gαi1pcDNA3.1, GαopcDNA3.1, and LipofectAMINE 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Cyclic AMP Accumulation Assay. Cells were plated at a density of 50,000 cells/well in 24-well culture plates. Cells were transfected 72 h after plating. Experiments were performed 48 h after the transfection. The cells were washed once with warm (37°C) Krebs-Ringer-HEPES buffer (120 mM NaCl, 4.7 mM KCl, 2.2 mM CaCl2, 10 mM HEPES, 1.2 mM KH2PO4, 1.2 mM MgSO4, pH 7.4). The indicated drugs were added, and the cells were incubated in a 37°C water bath for 15 min. Where required, cells were treated for 18 h with 5 ng/ml pertussis toxin. Where indicated, cells were incubated with 1 μM SR141716A for 10 min before other drug stimulation. After the incubation, the stimulation media was aspirated, and the reaction was terminated with 500 μl/well of ice-cold 3% trichloroacetic acid. The 24-well culture plates were stored at 4°C for up to 1 week before analysis. Cyclic AMP accumulation was quantified using a competitive binding assay (Nordstedt and Fredholm, 1990) with minor modifications (Watts and Neve, 1996). Samples of the cell lysate (10 μl) were added to reaction tubes. [3H]Cyclic AMP (∼1 nM final concentration) and cyclic AMP binding protein (∼150 μg) were diluted in cyclic AMP assay buffer (100 mM Tris/HCl, pH 7, 100 mM NaCl, 5 mM EDTA) and then added to each well for a total volume of 550 μl. The tubes were incubated on ice for 2 to 3 h and were harvested by filtration (PerkinElmer Unifilter GF/C) using a 96-well PerkinElmer Filtermate Cell harvester (PerkinElmer Life Sciences, Boston, MA). The filters were allowed to dry and Microscint o scintillation fluid was added. Radioactivity on the filters was determined using a PerkinElmer TopCount scintillation/luminescence detector.
Data Analysis. Experiments were performed in triplicate and repeated in three separate assays. Analyses of data were done using GraphPad Prism 3.0 (GraphPad Software, Inc., San Diego, CA).
Results
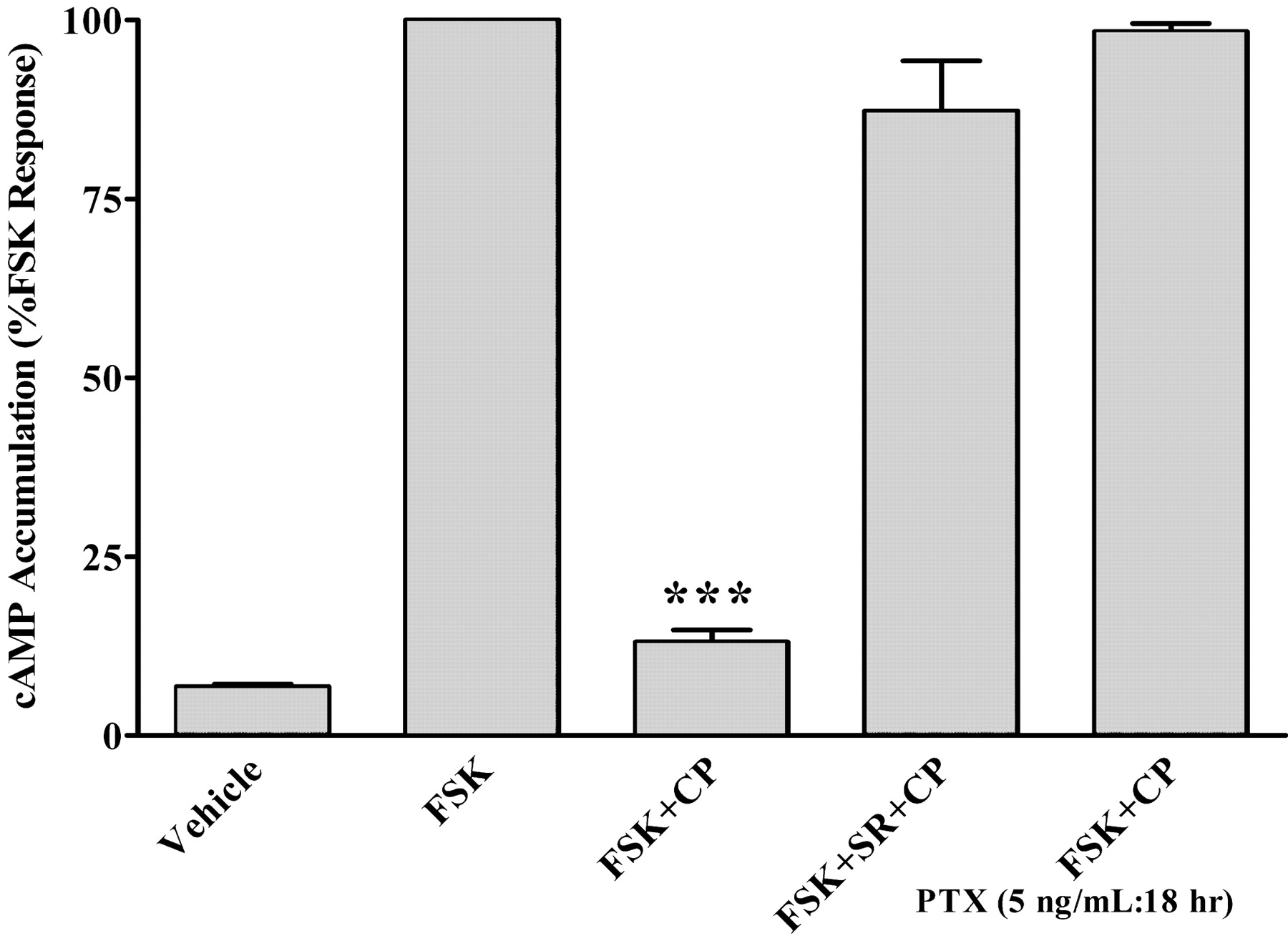
Acute Activation of the CB1 Receptor Inhibits cAMP Accumulation. HEK-293 cells were transiently transfected with the CB1 receptor (HEK-293 CB1 cells). Forskolin (10 μM) alone increased the cAMP accumulation severalfold as expected when compared with the vehicle-treated cells. The addition of CP55,940 (a CB1 receptor agonist) inhibited forskolin-stimulated cAMP accumulation by >95% (Fig. 1). The ability of CP55,940 to inhibit cAMP accumulation was prevented by the addition of 1 μM SR141716A (a CB1 receptor antagonist). The results of these experiments indicated that the lower levels of cAMP brought about by CP55,940 were a CB1 receptor-mediated response. CP55,940-mediated inhibition of cAMP was also examined in cells that were pretreated with 5 ng/ml pertussis toxin for 18 h. Pretreatment with pertussis toxin, which ADP-ribosylates Gαi/o and prevents the G protein heterotrimers from interacting with the receptor, blocked the effects of CP55,940 (Fig. 1). The pertussis toxin pretreatment data confirm that the CB1 receptor is Gαi/o-coupled in HEK-293 CB1 cells. CP55,940 did not inhibit forskolin-stimulated cAMP accumulation in wild-type HEK-293 cells (data not shown), indicating that there are no functional CB1 receptors in HEK-293 wild-type cells.

Effect of CB1 receptors on cAMP levels in HEK-293 CB1 cells, under acute stimulation conditions. cAMP accumulation in the presence of 10 μM forskolin (FSK), 10 μM CP55,940 (CP), 1 μM SR141716A (SR), and 5 ng/ml pertussis toxin (PTX). cAMP accumulation assays were performed using a standard competitive cAMP binding assay as described under Materials and Methods, using HEK-293 cells transiently transfected with the CB1 receptor. Data are mean values of three experiments performed in triplicate. Vertical lines represent SEM values. ***, significantly different from the FSK, FSK + SR + CP, and FSK + PTX + CP-treated cells, with p < 0.001 using analysis of variance with Bonferroni post-test.
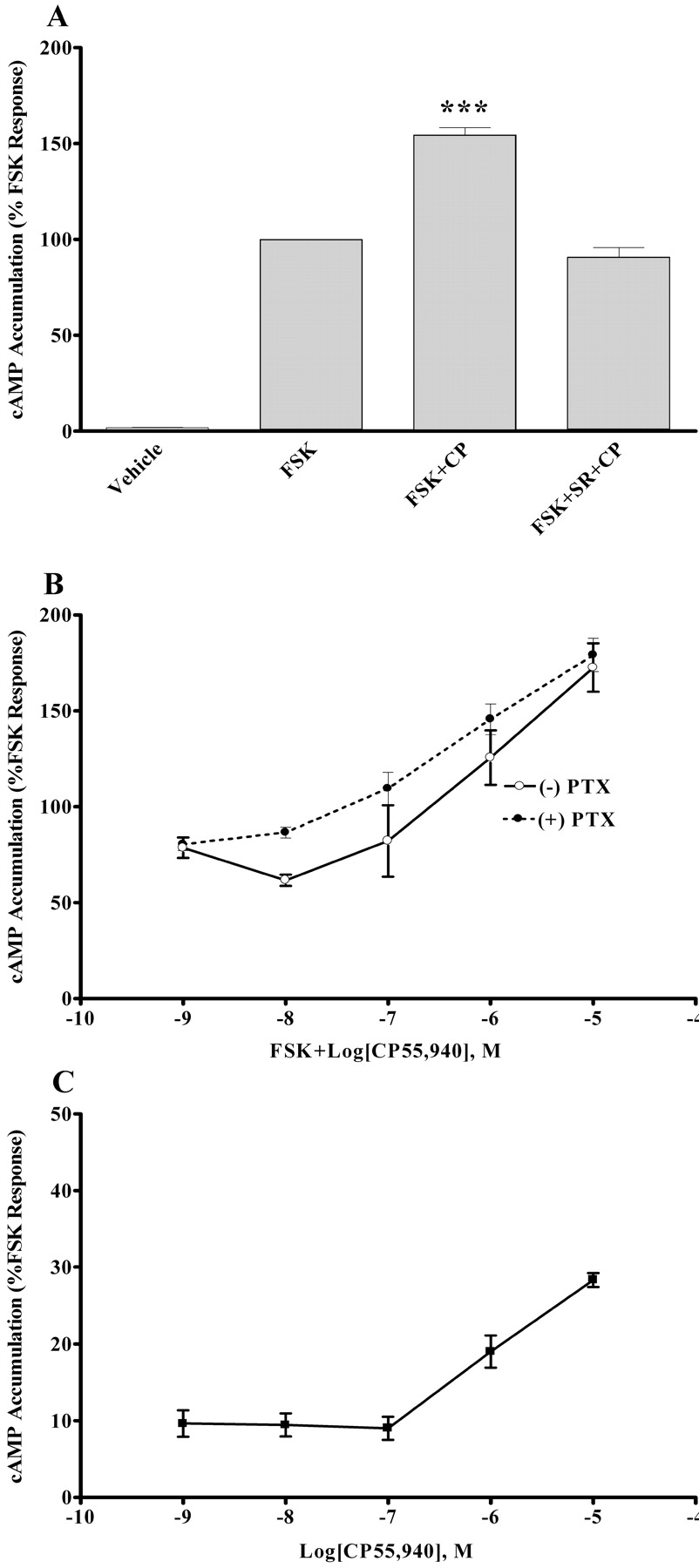
D2 Receptor Expression Alters CB1 Receptor Signaling. These experiments were performed in a HEK-293 cell line that expressed both the D2 and CB1 receptors; the D2 was stably expressed and the CB1 receptor was transiently transfected into the cell line (HEK-293/D2 CB1 cells). The addition of forskolin (10 μM) induced a marked increase in cAMP; however, this increase was not inhibited by CP55,940. Surprisingly, the addition of CP55,940 to HEK-293/D2 CB1 cells increased forskolin-stimulated cAMP accumulation to nearly 150% compared with forskolin alone (Fig. 2A). This increase was antagonized when SR141716A was present in the incubation (Fig. 2A), which indicated that the CB1 receptor was responsible for the increase in cAMP levels. CP55,940 concentration dependently enhanced forskolin-stimulated cAMP accumulation (Fig. 2B, open circles). Additional experiments examined this concentration-dependent increase in cAMP in HEK-293/D2 CB1 cells that were pretreated with pertussis toxin (5 ng/ml; 18 h). Pretreatment with pertussis toxin did not alter the elevated cAMP response (Fig. 2B, closed circles), suggesting that the CP55,950-induced elevation in cAMP was not mediated via activation of Gαi/o proteins. These data suggest that in HEK-293 cells expressing D2 receptors, CB1 receptors couple to stimulatory Gαs and not Gαi/o. Forskolin acts in concert with Gαs to increase adenylate cyclase activity, an action that may allow the CB1 receptor to stimulate cAMP production. Thus, additional experiments examined the effect of CP55,940 on cAMP accumulation in the absence of forskolin stimulation. These studies demonstrate that CP55,940 stimulated cAMP accumulation in the absence of forskolin. In the absence of forskolin, 10 μM CP55,940 resulted in cAMP levels that were approximately 30% of the forskolin-stimulated cAMP accumulation (Fig. 2C). This CP55,940-mediated increase in cAMP is approximately 8-fold higher than the basal response, indicating that CP55,940 is able to increase cAMP accumulation in the absence of forskolin. Coexpression of D2 and CB1 receptors in HEK-293 cells resulted in increased levels of cAMP after CB1 receptor activation, suggesting that in the presence of the D2 receptor, the CB1 receptor can switch coupling from Gαi/o to Gαs proteins.

Effect of coexpression of D2 and CB1 receptors on cAMP levels, under acute stimulation conditions. cAMP accumulation in the presence of 10 μM forskolin (FSK), 10 μM CP55,940 (CP), and 1 μM SR141716A (SR) (A), or 10 μM FSK, 1 nM to 10 μM CP, and 5 ng/ml pertussis toxin (PTX) (B), or 1 nM to 10 μM CP alone (C). cAMP accumulation assays were performed using a standard competitive cAMP binding assay as described under Materials and Methods, using HEK-293/D2 cells transiently transfected with the CB1 receptor. Data are mean values of three experiments performed in triplicate. Vertical lines represent SEM values. ***, significantly different from the FSK and FSK + SR + CP-treated cells, with p < 0.001 using analysis of variance with Bonferroni post-test.
Effect of D2 Receptor Activation on CB1 Signaling. In HEK-293/D2 CB1 cells, 1 μM quinpirole, a potent D2 receptor agonist, inhibited forskolin-stimulated cAMP accumulation by 90% (Fig. 3A). This quinpirole-mediated inhibition was prevented in cells that were pretreated with 5 ng/ml pertussis toxin (Fig. 3A), implicating D2 receptor coupling to Gαi/o in HEK-293/D2 CB1 cells. The effect of D2 receptor activation on CB1 receptor signaling was also examined in these cells. Concurrent activation of the D2 and CB1 receptors (10 μM forskolin + 1 μM quinpirole + 10 μM CP55,940) resulted in increased levels of cAMP compared with the activation of the D2 receptor alone (10 μM forskolin + 1 μM quinpirole; Fig. 3B). Addition of SR141716A prevented the increase in cAMP brought about by concurrent use of quinpirole and CP55,940 (Fig. 3B), indicating that the CB1 receptor was responsible for the increase in cAMP. The CP55,940-mediated increase in cAMP accumulation was dose-dependent with an approximate EC50 value of 300 nM (Fig. 3C).
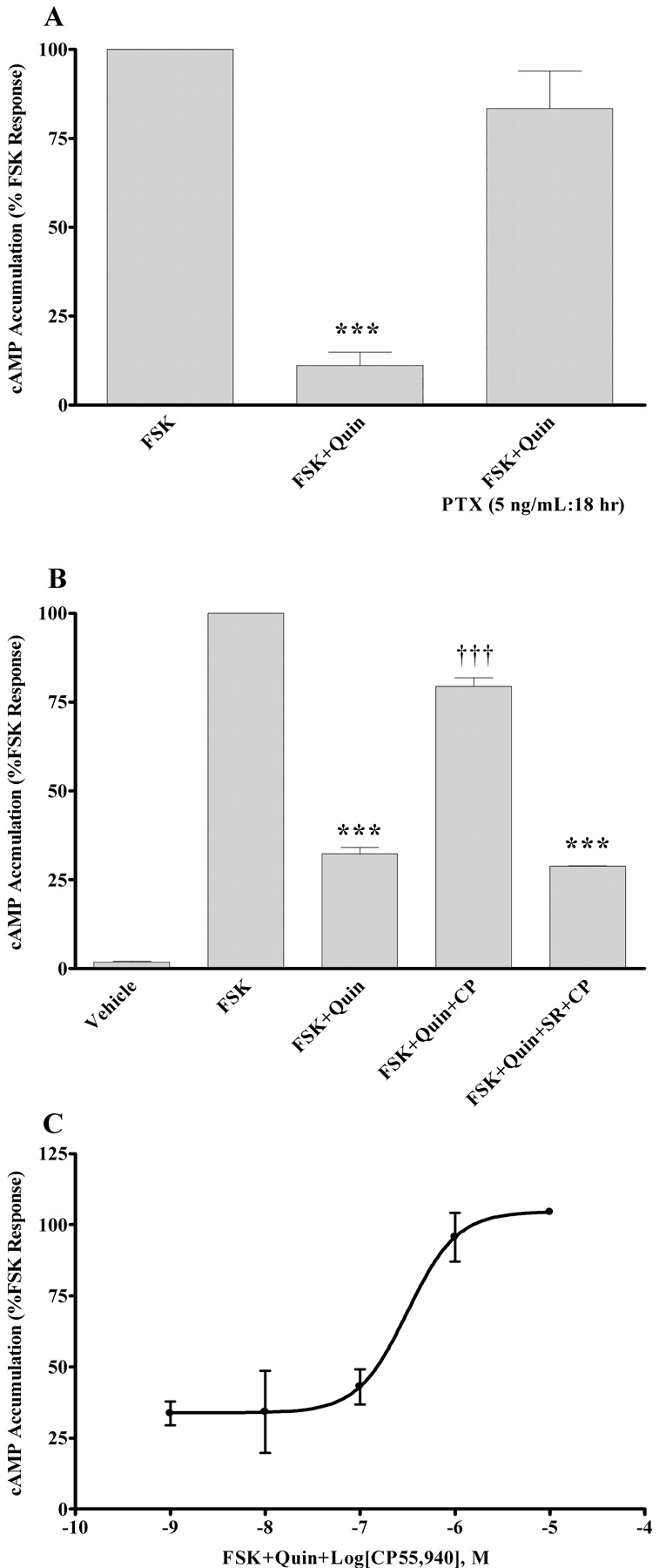
Effect of concurrent activation of D2 and CB1 receptors on cAMP levels, under acute stimulation conditions. cAMP accumulation in the presence of 10 μM forskolin (FSK), 1 μM quinpirole (Quin), and 5 ng/ml pertussis toxin (PTX) (A), or 10 μM FSK, 1 μM Quin, 10 μM CP55,940 (CP), and 1 μM SR141716A (SR) (B), or 10 μM FSK, 1 μM Quin, 1 nM to 10 μM CP (C). cAMP accumulation assays were performed using a standard competitive cAMP binding assay as described under Materials and Methods, using HEK-293/D2 cells transiently transfected with the CB1 receptor. Data are mean values of three experiments performed in triplicate. Vertical lines represent S.E.M. values. A, ***, significantly different from the FSK and FSK + PTX + Quin-treated cells, with p < 0.001 using analysis of variance with Bonferroni post-test. B, ***, significantly different from the FSK-treated cells, with p < 0.001 using analysis of variance with Bonferroni post-test. †††, significantly different from the FSK + Quin- and FSK + Quin + SR + CP-treated cells, with p < 0.001 using analysis of variance with Bonferroni post-test.
Expression of Gαi1 Promotes CB1 Receptor-Mediated Inhibition of cAMP Accumulation. We considered the possibility that the presence of the D2 receptor along with the CB1 receptor in the HEK-293 cells might restrict the availability of the pool of Gαi/o proteins. When an equal amount of Gαi1 cDNA (1×) was cotransfected with the CB1 receptor into the HEK-293/D2 receptor cells, the addition of CP55,940 potentiated cAMP accumulation both in the presence and absence of D2 receptor activation (Fig. 4, A and C). This effect was consistent with previous observations (Figs. 2 and 3) and comparable with control transfected cells (Fig. 4, A and C, CB1 only). However, cotransfection of the Gαi1 cDNA with the CB1 receptor at a mass ratio of 2:1 (2×) in the HEK-293/D2 receptor cells, and subsequent stimulation of CB1 receptors with CP55,940, in the absence of D2 receptor activation, led to inhibition of forskolin-stimulated cAMP accumulation (Fig. 4A). Subsequent experiments revealed that in the presence of D2 receptor activation, overexpression of Gαi1 (2× cDNA) seemed to potentiate quinpirole-mediated inhibition of forskolin-stimulated cAMP accumulation (Fig. 4C). Additional studies examined the ability of overexpression of Gαo to modulate CB1 receptor-mediated effects on cAMP accumulation (Fig. 4, B and D). These results revealed that even at a cotransfection ratio of 2:1 (2× Gαo cDNA) Gαo did not alter CB1 receptor signaling. These results suggest that overexpression of Gαi1 in the HEK-293/D2 CB1 transfected cells promotes CB1 receptor coupling to inhibition of cAMP accumulation.

Effect of overexpression of Gαi1 and Gαo on cAMP levels in HEK-293/D2 CB1 cells, under acute stimulation conditions. cAMP accumulation in the presence of 10 μM forskolin (FSK) and 10 μM CP55,940 (CP) in HEK-293/D2 CB1 cells expressing Gαi1 (A), or Gαo (B). cAMP accumulation in the presence of 10 μM forskolin (FSK), 1 μM quinpirole (Quin), and 10 μM CP55,940 (CP) in HEK-293/D2 CB1 cells expressing Gαi1 (C), or Gαo (D). cAMP accumulation assays were performed using a standard competitive cAMP binding assay as described under Materials and Methods, using HEK-293/D2 cells transiently transfected with the CB1 receptor and an equal amount (1×, 0.9 μg) or twice the amount (2×, 1.8 μg) of the indicated Gα subunit. Data are mean of three experiments performed in triplicate. Vertical lines represent S.E.M. values.
Effect of Persistent Receptor Activation on cAMP Accumulation. HEK-293/D2 CB1-transfected cells were pretreated with either 2 μM quinpirole or CP55,940 for 18 h. When challenged with 10 μM forskolin, the cells pretreated with the D2 receptor agonist quinpirole exhibited a marked increase in cAMP production consistent with the development of heterologous sensitization (Fig. 5). In contrast, the cells pretreated with the CB1 receptor agonist CP55,940 exhibited a forskolin response similar to vehicle-pretreated cells (Fig. 5), i.e., they did not show heterologous sensitization to forskolin-stimulated cAMP accumulation. Persistent activation of Gαi/o-coupled receptors is known to sensitize adenylate cyclase, whereas persistent activation of Gαs-coupled receptors does not. Thus, this lack of sensitization after CP55,940 pretreatment is consistent with the hypothesis that in the presence of D2 receptors, the CB1 receptors are coupled to the stimulatory Gαs and not Gαi/o (Fig. 2 and 3B). Pretreatment with the CB1 receptor agonist CP55,940 did not lead to sensitization of adenylate cyclase, whereas pretreatment with the D2 receptor agonist quinpirole did result in sensitization of adenylate cyclase (Fig. 5).

Effect of forskolin on cAMP levels after chronic pretreatment with CP55,940 and quinpirole in HEK-293/D2 CB1 cells. cAMP accumulation in the presence of 10 μM forskolin (FSK). Cells were pretreated for 18 h with either 2 μM CP55,940 (CP) or 2 μM quinpirole (Quin). Basal levels of the vehicle, Quin, and CP pretreatments were 6.3 ± 1.1, 5.6 ± 0.7, and 13.3 ± 3.2 pmol/well, respectively. cAMP accumulation assays were performed using a standard competitive cAMP binding assay as described under Materials and Methods, using HEK-293/D2 cells transiently transfected with the CB1 receptor. Data are mean values of three experiments performed in triplicate. Vertical lines represent S.E.M. values. ***, significantly different from the vehicle pretreatment, with p < 0.001, using analysis of variance with Bonferroni post-test. ††, significantly different from the quinpirole pretreatment, with p < 0.01 using analysis of variance with Bonferroni post-test.
Persistent Activation of the D2 Receptor Changes the Coupling of the CB1 Receptor to Gαi/o. The effects of persistent activation of the D2 receptor on the subsequent activation of the CB1 receptor were also examined in HEK-293/D2 CB1 cells. Cells were pretreated with either vehicle or 2 μM quinpirole for 18 h. In vehicle-pretreated cells, forskolin alone stimulated cAMP production and CP55,940 enhanced forskolin-stimulated cAMP accumulation (Fig. 6), consistent with a Gαs-coupled CB1 receptor. Persistent D2 receptor activation (quinpirole-pretreated cells) enhanced forskolinstimulated cAMP accumulation by greater than 4-fold consistent with the development of heterologous sensitization; surprisingly, the addition of CP55,940 inhibited cAMP accumulation (Fig. 6). That CP55,940 inhibits forskolin-stimulated cAMP accumulation in quinpirole pretreated cells is consistent with a Gαi/o-coupled receptor. These data suggest that persistent activation of the D2 receptor reestablishes coupling of the CB1 receptor to Gαi/o.

Effect of forskolin and CP55,940 on cAMP levels after chronic pretreatment with quinpirole in HEK-293/D2 CB1 cells. cAMP accumulation in the presence of 10 μM forskolin (FSK) and 10 μM CP55,940 (CP). Cells were pretreated for 18 h with 2 μM quinpirole. cAMP accumulation assays were performed using a standard competitive cAMP binding assay as described under Materials and Methods, using HEK-293/D2 cells transiently transfected with the CB1 receptor. Data are mean values of three experiments performed in triplicate. Vertical lines represent S.E.M. values.
Discussion
The initial studies demonstrating the Gαs-linkage of CB1 receptors were carried out in striatal neurons in primary culture (Glass and Felder, 1997). Both the CB1 and D2 receptor agonists inhibited forskolin-stimulated cAMP accumulation when applied separately. When the two receptor agonists were added concurrently, there was an enhancement of forskolin-stimulated cAMP accumulation. We obtained similar results in HEK-293/D2 CB1 cells. Glass and Felder (1997) also showed that in striatal neurons in primary culture, the CB1 receptor agonist alone was only able to elicit a concentration-dependent increase in forskolin-stimulated cAMP accumulation after pertussis toxin pretreatment. Other studies demonstrating the Gαs linkage of CB1 receptors were carried out in CHO cells stably expressing human CB1 receptors (Glass and Felder, 1997; Bonhaus et al., 1998; Felder et al., 1998). In these studies, various CB1 cannabinoid receptor agonists inhibited forskolin-stimulated cAMP accumulation. After pertussis toxin pretreatment, the CB1 receptor agonists were able to concentration dependently increase forskolin-stimulated cAMP accumulation. Pertussis toxin pretreatment ADP-ribosylates Gαi/o and prevents the G protein heterotrimers from interacting with the receptor. Thus, there are no Gαi/o-subunits left to interact with the CB1 cannabinoid receptor. Essentially, pertussis toxin pretreatment promotes CB1 cannabinoid receptor binding to the Gαs subunits available in the cell.
We showed that in HEK-293/D2 CB1 cells, CP55,940 alone elicited a concentration-dependent increase in forskolin-stimulated cAMP accumulation both with and without pertussis toxin pretreatment. Our studies are the first to demonstrate that coexpression of the D2 and CB1 receptors is sufficient to alter coupling. Moreover, we also showed that in HEK-293/D2 CB1 cells, CP55,940 is able to increase cAMP levels without forskolin stimulation. Heretofore, this finding was only demonstrated in rat globus pallidus slices (Maneuf and Brotchie, 1997).
Activation of the CB1 receptor, which is normally coupled to inhibitory G proteins (Gαi/o), should result in an inhibition of adenylate cyclase and subsequent reduction in cellular levels of cAMP (for review, see Howlett 1995). Under acute conditions, D2 receptor activation is not necessary for the switch of the CB1 receptor from Gαi/o to Gαs proteins. The coexpression of the D2 receptor with the CB1 receptor was adequate to promote the coupling of CB1 receptors to Gαs. Concurrent activation of the CB1 and D2 receptors, with or without pertussis toxin, again points to a non-Gαi/o process, implicating that the CB1 receptor is coupled to Gαs instead of Gαi/o. We suggest that the D2 receptor may sequester the Gαi/o pool, preventing the binding of the CB1 receptor to Gαi/o, promoting interactions with Gαs. It has been shown that the human CB1 receptor can sequester Gαi/o protein from a common pool and prevent other pertussis toxin-sensitive Gαi/o receptors from signaling (Vasquez and Lewis, 1999). It has also been shown that cannabinoid and opioid receptors share a common pool of GTP-binding proteins in cotransfected cells (Shapira et al., 2000). It is likely that in our receptor-transfected cell line, the D2 receptor, not the CB1 receptor, sequesters the shared Gαi/o pool. Our experiments with overexpression of the Gα subunits are consistent with this idea. Overexpression of Gαi1, but not Gαo, promoted CB1 receptor-mediated inhibition of cAMP accumulation. Our findings suggest that if the D2 receptor is indeed sequestering Gαi subunits, then this effect can be overcome by overexpressing the Gαi1 subunit. The ability of CB1 receptors to couple to Gαi1 for inhibition of adenylate cyclase is consistent with previous findings suggesting that the effectors of Gαi are adenylate cyclase as well as potassium and calcium channels (Ross, 1992).
HEK-293/D2 CB1 cells that were pretreated with quinpirole for 18 h and then stimulated with forskolin exhibited an amplified cAMP response. This is the phenomena of heterologous sensitization and is an expected response after persistent activation of Gαi/o-coupled receptors. HEK-293/D2 CB1 cells that were pretreated with CP55,940 for 18 h and then stimulated with forskolin did not have an amplified cAMP response. Initially, we hypothesized that this lack of sensitization was a result of the CB1 receptor now being coupled to Gαs. However, if the D2 receptor has caused the CB1 receptor to switch to Gαs, then chronic activation of the D2 receptor should result in a cAMP accumulation that is amplified when acutely stimulated with forskolin alone and heightened even more when stimulated with forskolin and a CB1 receptor agonist. We predicted this because chronic activation of the D2 receptor sensitizes adenylate cyclase, and the expression of this amplified cAMP response is thought to be a Gαs-mediated event (for review, see Watts, 2002). As expected quinpirole-pretreated cells showed an amplified cAMP response when challenged with forskolin. However, there was no heightened response for quinpirole-pretreated cells that were challenged with forskolin and CP55,940 concurrently. Not only was there no amplified cAMP response, CP55,940 markedly inhibited forskolin-stimulated cyclic AMP accumulation. If the CB1 receptor remained Gαs-coupled after sensitization, then we would have expected to see a sensitized response. In contrast, persistent activation of the D2 receptor seems to revert the CB1 receptor back to coupling with Gαi/o.
Our data led us to suggest the following model. When the CB1 receptor is expressed alone in HEK-293 cells, it is coupled to the Gαi/o subunit. Coexpression of D2 and CB1 receptors in HEK-293 cells, resulted in the coupling of the CB1 receptor to Gαs, as a result of Gαi/o sequestration by the D2 receptor. Overexpression of Gαi1 restores coupling of the CB1 receptor with Gαi, consistent with our sequestration hypothesis. Persistent activation of the D2 receptor also facilitates the reestablishment of Gαi/o coupling with the CB1 receptor. The mechanisms for G protein switching remain unknown; however, changes in membrane microdomain localization of Gα subunits may be involved. For example, chronic activation of Gαi/o-coupled receptors decreases detergent solubility of G protein subunits (i.e., Gαi and β), which is thought to correlate to compartmental alterations that the G protein α subunits undergo (Bayewitch et al., 2000). Alternatively, chronic drug treatment may alter the membrane microdomain localization of Gαs that influences receptor modulated adenylate cyclase activity (Ammer and Schulz, 1997; Ostrom et al., 2001).
Is there a physiological relevance for the coupling of the CB1 receptor to Gαs? A single receptor subtype can be affiliated with multiple signal transduction pathways. It is likely that different effectors may be activated by more than one G protein subtype. It is also likely that not all signaling pathways are active all the time. These and other data in the literature suggest that certain signaling pathways can be affected by interactions with other receptors, perhaps to provide a more specific regulation of these pathways. Is there a physiological relevance for the reestablishment of Gαi/o-coupling when the D2 receptor is chronically activated? The utility of this shutdown in cAMP production is very logical when examined in the context of the “dopamine hypothesis” of schizophrenia. In this hypothesis, schizophrenia patients have excessive dopaminergic activity that may be the underlying cause of schizophrenia (Seeman, 1987). The brains of schizophrenic patients have increased levels of adenylate cyclase activity (Memo et al., 1983; Kerwin and Beats, 1990), and there is evidence that indicates adenylate cyclase inhibitors may have a therapeutic role in the treatment of excited psychosis (Roitman et al., 1998). The switch of the CB1 receptor back to Gαi/o coupling under persistent D2 receptor activation may be the brain's compensatory attempt to ablate the increase in adenylate cyclase activity in the schizophrenic brain. Furthermore, the switching of the CB1 receptor to Gαi/o coupling after persistent D2 receptor activation may be a regulatory mode to attenuate CB1 receptor signaling. Giuffrida et al. (1999) have shown that in the rat striatum, anandamide release is stimulated when the D2 receptors are activated. Persistent activation of D2 receptors could increase anandamide levels to abnormally high levels. In turn, this could result in cannabinoid hyperactivity. It is conceivable that in addition to the transport process that inactivates anandamide's actions at the synapse, switching coupling back to Gαi/o after chronic activation of the D2 receptor is another way for the cell to modulate CB1 receptor signaling by essentially switching off cAMP production. These present studies confirm past G protein coupling studies and provide a novel mechanism for D2 receptor and CB1 receptor interactions that may play an important role in central nervous disorders associated with the regulation of dopaminergic signaling such as drug abuse and schizophrenia.
Acknowledgments
We thank Dr. Tom Bonner for providing the CB1 receptor cDNA and the Research Triangle Institute for providing CP55,940.
Footnotes
-
This work was supported by a National Association for Research in Schizophrenia and Depression Young Investigator Award (to E.L.B) and MH60397 (to V.J.W.). A preliminary report of these findings was made at the 2001 meeting of the Society for Neuroscience and the 2003 meeting of the Federation of American Societies for Experimental Biology.
-
DOI: 10.1124/jpet.103.057620.
-
ABBREVIATIONS: CB1, cannabinoid receptor; CHO, Chinese hamster ovary cell line; D2, type 2 dopamine receptor; HEK, human embryonic kidney; CP55,940, (–)-cis-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol; SR141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide.
-
↵1 Current address: Department of Pharmaceutical Sciences, Butler University College of Pharmacy and Health Sciences, 4600 Sunset Ave., Indianapolis, IN 46208. E-mail: ajarrahi{at}butler.edu
- Received July 28, 2003.
- Accepted November 14, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}