


Abstract
Curcumin (diferuloylmethane) is one of the phytophenolic compounds found in the turmeric plant with anti-inflammatory and anticarcinogenic activities. One possible mechanism for these activities is the inhibition of prostaglandin (PG) E2 formation. In this study and other reports, curcumin suppresses interleukin-1β-induced formation of prostaglandin E2 in a concentration-dependent manner. Interleukin-1β-induced microsomal prostaglandin E synthase 1 (mPGES-1) and cyclooxygenase-2 were attenuated by curcumin at the protein and mRNA levels, but a more dramatic inhibition of mPGES-1 expression was observed at lower concentrations of curcumin in A549 human lung epithelial cells. The inhibition of mPGES-1 expression by curcumin shifted the arachidonic acid profile from PGE2 to PGF2α and 6-keto-PGF1α as major metabolites. The expression of early growth response gene 1 (EGR-1), a key transcription factor of cytokine-induced mPGES-1, was inhibited by curcumin. Incubation with siRNA for EGR-1 inhibited interleukin (IL)-1β-induced mPGES-1, and the controlled expression of EGR-1 increased the mPGES-1 expression. Several proinflammatory signaling molecules, such as nuclear factor κB (NF-κB) and mitogen-activated protein kinases, are also known to affect curcumin-regulated gene expression. Curcumin inhibited IκBα phosphorylation and degradation and thus reduced the expression of mPGES-1. Curcumin suppressed cytokine-induced mPGES-1 by inhibiting phosphorylation of Jun N-terminal kinase (JNK)1/2. However, EGR-1 expression was suppressed by lower concentrations of curcumin, as compared with JNK1/2 and IκBα. These results indicate that curcumin inhibits IL-1β-induced PGE2 formation by inhibiting the expression of mPGES-1 that is mediated by suppression of EGR-1 expression as well as NF-κB and JNK1/2.
The biosynthesis of prostaglandin (PG) E2 from arachidonic acid requires two enzymatic activities. First, cyclooxygenase, the rate-limiting enzyme, converts arachidonic acid into PGH2. The second enzyme, prostaglandin E synthase (PGES), then converts PGH2 into PGE2. Two isoforms of COX, COX-1 and COX-2, are well characterized (Tanabe and Tohnai, 2002). Generally, COX-1 is constitutively expressed in many human tissues and is involved in physiological functions. In contrast, COX-2 is the inducible form, and it is expressed in response to inflammatory stimuli. At least three PGES isoforms have been identified, including microsomal PGES-1 (mPGES-1), which was originally named MGST1-l-1 (membrane-bound glutathione S-transferase-1-like-1), mPGES-2, and cytosolic PGES (cPGES, or the heat shock protein-associated protein p23). Cytosolic PGES is constitutively and ubiquitously expressed and is preferentially coupled with COX-1 to promote immediate secretion of PGE2. In contrast, mPGES-1 is markedly up-regulated by proinflammatory or mitogenic stimuli and is functionally coupled with COX-2 for the delayed PGE2 synthesis. mPGES-2 is ubiquitously expressed, but its role remains unclear (Murakami et al., 2002).
mPGES-1 is up-regulated in vivo in a rheumatoid arthritis model, endotoxin-mediated fever, and symptomatic atherosclerotic plaques (Cipollone et al., 2001; Ivanov et al., 2002; Claveau et al., 2003; Kojima et al., 2003). Recent studies also have observed expression of this enzyme in lung, colon, gastric, and endometrial adenocarcinoma (McGinty et al., 2000; Kurie and Dubois, 2001; Yoshimatsu et al., 2001; Kamei et al., 2003; van Rees et al., 2003). Moreover, genetic ablation of mPGES-1 in the animal model caused a significant reduction in inflammatory and carcinogenic processes, which implies mPGES-1 as a potential target for therapeutic intervention.
Curcumin (diferuloylmethane), from turmeric like Curcuma longa, has been extensively investigated as a phenolic antioxidant with anti-inflammatory or anticancer potential. These clinical properties of curcumin have been attributed, at least in part, to suppression of prostaglandin synthesis via regulating COX-2 expression (Ireson et al., 2001; Chun et al., 2003; Hong et al., 2004). Molecular action of curcumin to elucidate these therapeutic effects have been associated with a variety of signaling pathways such as NF-κB, AP-1, and the early growth response gene (EGR-1) (Huang et al., 1995; Pendurthi and Rao, 2000; Grandjean-Laquerriere et al., 2002).
The early growth response gene product (EGR-1) is a nuclear transcription factor and is implicated in the regulation of a number of genes involved in inflammation, differentiation, growth, and development. EGR-1 is rapidly and transiently expressed in a number of cells after stimulation with a variety of agents, including cytokines and growth factors (Thiel and Cibelli, 2002). Recent studies demonstrate that EGR-1 is a key transcription factor in regulating the inducible expression of mPGES-1 (Cheng et al., 2004; Subbaramaiah et al., 2004).
Here, we have investigated the effects of curcumin on interleukin-1β-induced mPGES-1 gene expression in A549 cells, to determine the underlying regulatory mechanisms. A549 lung epithelial cells have been a well established model of induction of mPGES-1 by proinflammatory cytokines (Thoren and Jakobsson, 2000; Catley et al., 2003). Curcumin was found to inhibit the IL-1β-induced expression of mPGES-1 and hence PGE2 formation, by altering EGR-1 expression as well as NF-κB and JNK1/2 signals.
Materials and Methods
Cell Culture and Reagents. Nonsmall cell lung cancer A549 cells were purchased from American Type Culture Collection (Manassas, VA) and maintained in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Hyclone, South Logan, UT), 50 units/ml penicillin (Sigma-Aldrich, St. Louis, MO), and 50 μg/ml streptomycin (Sigma-Aldrich) in a 5% CO2-humidified incubator at 37°C. Cell number and viability were assessed by trypan blue (Sigma-Aldrich) dye exclusion using a hemacytometer. During incubation with chemicals or interleukin-1β, cells were cultured in Dulbecco's modified Eagle's medium without serum and antibiotics. Curcumin (diferuloylmethane), 6-amino-4-(4-phenoxyphenylethyl amino) quanazoline, and SP600125 were purchased from Calbiochem (San Diego, CA). Recombinant human interleukin-1β was from R&D Systems (Minneapolis, MN).
Cellular Viability Assay. Colorimetric analysis of cell growth was performed with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) according to the manufacturer's protocol (Promega, Madison, WI). Cells (5 × 104/well) were cultured in 96-well plate for each time, and the MTS (50 μl) was treated onto cells for 2 h. The absorbance at 490 nm of the culture media was measured.
Construction of Plasmids. The full-length EGR-1 cDNA in the pcDNA3.1/neo expression vector (pEgr-1) was described previously (Baek et al., 2003). The luciferase constructs containing the COX-2 (-1475/+59), mPGES-1 (-650/-15), and EGR-1 5′-untranscribed region (-1260/+35) in pGL3 basic vector were generated after PCR of each promoter region with PfuTurbo DNA polymerase (Stratagene, La Jolla, CA). The fragment was cloned into the TA vector (Invitrogen), sequenced, and further cloned into the pGLBasic3 vector.
Western Immunoblot Analysis. Levels of protein expression were compared using Western immunoblot using polyclonal anti-mPGES-1 antibody (Oxford Biomedical, Oxford, MI), monoclonal anti-human COX-2 antibody (Cayman Chemical, Ann Arbor, MI), goat polyclonal anti-human actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA), polyclonal anti-human IκBα antibody (Cell Signaling Technology, Beverly, MA), polyclonal anti-human JNK1/2 antibody (Cell Signaling Technology), and polyclonal anti-human EGR-1 antibody (Santa Cruz Biotechnology). Cell lysate was prepared in radioimmunoprecipitation assay buffer containing 1× phosphate-buffered saline, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/ml phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, and Protease Inhibitor Cocktail (Sigma-Aldrich). After brief sonication of samples, lysate protein was quantified using bicinchoninic acid protein assay kit (Pierce, Rockford, IL), and 50 μg of protein was separated by NuPAGE Novex Bis-Tris gel electrophoresis (Invitrogen). Protein was transferred onto a nitrocellulose membrane (Invitrogen), and the blots were blocked for 1 h with 5% skim milk in Tris-buffered saline plus Tween 0.05% (TBST) and probed with each antibody for 2 h at room temperature or overnight at 4°C. After washing three times with TBST, blots were incubated with horseradish-conjugated secondary antibody for 1 h and washed with TBST three times. Protein was detected by enhanced chemiluminescence detection system (GE Healthcare, Little Chalfont, Buckinghamshire, UK) according to the manufacturer's instruction.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR). RNA was extracted with RNeasy kit (QIAGEN, Valencia, CA) according to the manufacturer's instructions. RNA (100 ng) from each sample was transcribed to cDNA by Superscript II RNase H- Reverse-transcriptase (Invitrogen). The amplification was performed with Takara ExTaq DNA Polymerase (Chemicon International, Temecula, CA) in GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA) using the following parameters: denaturation at 94°C for 2 min and 25 cycles of reactions of denaturation at 98°C for 10 s, annealing at 59°C for 30 s, and elongation at 72°C for 45 s. An aliquot of each PCR product was subjected to 1.2% (w/v) agarose gel electrophoresis and visualized by staining with ethidium bromide. The 5′ forward and 3′ reverse-complement PCR primers for amplification of each gene were as follows: human COX-2, 5′-TATACTAGAGCCCTTC CTCCTGTGCC-3′ and 5′-ACATCGCATACTCTGTTGTGTTCCC-3′; human mPGES-1, 5′-CACAGCCTGGTGATGAG C-3′ and 5′-CCGCTTCCCAGAGGATCT-3′; human EGR-1, 5′-CAGTGGCC TAGTGAGCATGA-3′ and 5′-CCGCAAGTGGAT CTTGGTAT-3′; and human GAPDH, 5′-TCAACGGATTTGGTCGTATT-3′ and 5′-CTGTGGTCATGAGTCCTTCC-3′.
Real-Time RT-PCR with SYBR Green Detection. Real-time RT-PCR was performed using an ABI Prism 7700 (Applied Biosystems). Real-time RT-PCR fluorescence detection was performed in 96-well plates using Quantitect SYBR Green buffer (QIAGEN). Each 50-μl PCR reaction contained cDNA, 0.5 unit of Amp Erase uracil-N-glycosylase (UNG; PerkinElmer Life and Analytical Sciences, Boston, MA), forward and reverse primers, and the Passive Reference dye (ROX) to normalize the SYBR Green/double-stranded DNA complex signal during analysis to correct for well to-well variations. Primer concentrations were optimized to yield the lowest concentration of primers that yielded the same Ct values as recommended by Applied Biosystems. A no-RT control RNA sample was used with each real-time RT-PCR experiment containing human GAPDH primers to verify no genomic DNA contamination. Amplification parameters were UNG incubation, for one cycle at 50°C for 2 min to prevent amplification of carryover DNA; denaturation/UNG inactivation at 94°C for 10 min; and amplification, 40 cycles of 95°C/15 s and 60°C/30 s. Amplification products using SYBR Green detection were routinely checked using dissociation-curve software (PerkinElmer Life and Analytical Sciences) and by gel electrophoresis on a 1% agarose gel that was then visualized under UV light following staining with 0.05% ethidium bromide to confirm the size of the DNA fragment and that only one product was formed. Samples were compared using the relative (comparative) Ct method. The Ct value, which is inversely proportional to the initial template copy number, is the calculated cycle number where the fluorescence signal emitted is significantly above background levels. The -fold induction or repression by real-time RT-PCR was measured in triplicate relative to time-matched vehicle-treated controls and calculated after adjusting for GAPDH using 2-ΔΔCt, where ΔCt = target gene Ct - GAPDH Ct, and ΔΔCt = ΔCt control - ΔCt treatment.
Transfection and Stable Cell Line. Cells were transfected with mixture of plasmids using FuGENE 6 transfection reagent (Roche, Indianapolis, IN) according to the manufacturer's protocol. For transfection of the luciferase reporter gene, a mixture of 2.8 μg of firefly luciferase reporter and 0.2 μg of Renilla luciferase, pRL-null vector (Promega, Madison, WI) per 9 μl of FuGENE 6 reagent was applied for a 6-mm tissue culture dish. At 24 h after transfection, cells were exposed to interleukin-1β or chemicals for the next 24 h and lysed for dual-luciferase reporter assay system (Promega). All transfection efficiency was maintained at around 50 to 60%, which was confirmed with pMX-enhanced GFP vector. To create pEgr-1-expressing A549 stable cell line, A549 was transfected with the pEgr-1 or pCDNA3.1-neo using FuGENE 6 reagent. After 48 h, the cells were subjected to selection for stable integrants by exposure to 1000 μg/ml G418 (Geneticin; Invitrogen) in complete medium containing 10% fetal bovine serum. Selection was continued until monolayer colonies formed. The transfectants were then maintained in medium supplemented with 10% fetal bovine serum and 500 μg/ml G418.
Treatment with siRNA. Cells were transfected with EGR-1 siRNA (Dharmacon, Lafayette, CO), targeting the sequence, AAGTTACTACCTC TTATCCAT. Synthetic RNA transfection was performed with LipofectAMINE 2000 according to the manufacturer's protocol (Invitrogen).
Luciferase Assay. Cells were washed with ice-cold phosphate-buffered saline, lysed with lysis buffer (25 mM Tris-H3PO4, pH 7.8, 2 mM trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid, 2 mM dithiothreitol, 10% (v/v) glycerol, and 1% (v/v) Triton X-100), and then centrifuged at 12,000g for 4 min. The supernatant was collected, isolated, and stored at -80°C until assessment of luciferase activity. Luciferase activity was measured with a dual-mode luminometer (model TD-20/20; Turner Designs Co., Sunnyvale, CA) after briefly mixing the supernatant (20 μl) with 100 μl of firefly luciferase assay substrate solution, followed with 100 μl of stopping Renilla luciferase assay solution (Promega). The firefly luciferase activity was normalized against Renilla luciferase activity using the following formula: firefly luciferase activity/Renilla luciferase activity.
Analysis of Arachidonic Acid Metabolism. A549 cells were grown to near confluence in 75-cm2 flasks and then washed with serum-free media. Cells were treated with ±25 μM curcumin for 2 h prior to incubation with IL-1β (1 ng/ml) and [3H]arachidonic acid (3 μCi, final concentration, 100 μM; 5 ml total volume) for 1 h at 37°C. The radiolabeled [5,6,8,9,11,12,14,15-3H]arachidonic acid was purchased from PerkinElmer Life and Analytical Sciences with specific activity of 65.9 Ci/mmol. After completion of the incubations, reactions were quenched by addition of an equal volume (5 ml) of methanol followed by acidification of the media to pH 3.5 with glacial acetic acid. Arachidonic acid metabolites were extracted by application of samples to a C18-PrepSep column (solid-phase extraction) preconditioned with 10 ml of methanol followed by 10 ml of water. After the application of samples and washing of a column with an additional 10 ml of water, samples were eluted with 2 ml of methanol and evaporated to dryness under argon. Samples were reconstituted in HPLC solvent prior to analysis by reverse-phase HPLC.
Reverse phase-HPLC analyses were conducted with an ODS Ultrasphere column (5 μm; 4.6 × 250 mm). Separation of eicosanoids was achieved by elution with a stepwise mobile phase methanol gradient (55 to 100%, pH 5.0) at a flow rate of 1.0 ml/min. Eluted fractions were monitored with a variable wavelength UV detector and a Flo-One detector (PerkinElmer Life and Analytical Sciences) for radioactive fractions. Elution times of sample fractions were compared with elution times of authentic standards, and integrated peak areas were normalized to a PGB2 internal standard.
Statistics. Data were analyzed using SigmaStat for Windows (Jandel Scientific, San Rafael, CA). For comparison of two groups of data, Student's two-tailed t test was performed. For the comparison of multiple groups, data were subjected to ANOVA, and pairwise comparisons made by the Student-Newman-Keuls (SNK) method. Data not meeting normality assumption were subjected to Kruskal-Wallis ANOVA on ranks and then pairwise comparisons made by the SNK method.
Results
Effect of Curcumin on Inteleukin-1β-Mediated PGE2 Production and mPGES-1/COX-2 Expression. The increase in prostaglandin E2 formation in response to the inflammatory stimuli is dependent on the induction of cyclooxygenase-2 and microsomal prostaglandin E synthase 1. Interleukin-1β induced the expression of mPGES-1 and COX-2 in A549 lung alveolar epithelial cells, with maximal expression observed at 1 ng/ml interleukin-1β (data not shown). Thus, this concentration of interleukin-1β was used for subsequent experiments. A549 cells were treated with proinflammatory interleukin-1β, with or without curcumin, and PGE2 was measured using enzyme-linked immunosorbent assay. The cytokine-induced PGE2 production was significantly suppressed by curcumin treatment in a concentration-dependent manner (Fig. 1A). Since PGE2 reduction could be derived from the chemical-induced cytotoxicity, cellular viability was measured using MTS reagent (as described under Materials and Methods). Significant different viability (around 80% of vehicle control group) was observed only at the treatment with 50 μM curcumin (Fig. 1B). Thus, most part of reduction in PGE2 by curcumin was due to the other mechanism than cytotoxicity.
To determine the responsible mechanisms, expressions of COX-2 and mPGES-1 were measured. Treatment with curcumin suppressed the interleukin-1β-mediated induction of both COX-2 and mPGES-1 at both the protein and mRNA levels (Fig. 2, A and C). The inhibition of the expression of mPGES-1 was more dramatic at lower concentrations than the inhibition of COX-2 expression, both at the protein and mRNA levels (Fig. 2, B and D). Taken together, mPGES-1 and COX-2 was involved in curcumin-mediated inhibition of PGE2 production in interleukin-1β-activated A549 cells. In addition, mPGES-1 was more susceptibly affected by curcumin treatment, and the following studies will be focused on the gene regulation of mPGES-1 by curcumin.
Arachidonic Acid Metabolism. Since curcumin suppressed the expression of mPGES at lower concentrations than the expression of COX-2, incubation of cells with curcumin may result in a shift in the arachidonic acid profile. Cells were incubated with 25 μM curcumin, and arachidonic acid metabolism was examined by HPLC analysis as described under Materials and Methods. The major metabolite formed by interleukin-1β-activated A549 cells was PGE2. With addition of curcumin in the incubation, the major metabolites were PGF2α and 6-keto-PGF1α as shown in Table 1.
Effects of curcumin on production of major arachidonate metabolites in A549 cells Results shown represent three separate experiments.

Curcumin reduces prostaglandin E2 production. A, A549 cells were cultured with or without 1 ng/ml interleukin-1β in the presence of each dose of curcumin (0, 5, 10, 25, and 50 μM) for 24 h. Culture media was harvested and directly analyzed by PGE2 enzyme-linked immunosorbent assay. All asterisks indicate significant difference (p < 0.05) from the IL-1β alone (control). B, A549 cells were incubated with each dose of curcumin for 24 h, and MTS reagent was applied 2 h before the end of the curcumin incubation to measure the cellular viability. Asterisk indicates significant difference (p < 0.05) from the vehicle control group. All results are representative of three experiments.
Regulation of EGR-1 Expression by Curcumin. EGR-1 is a critical transcription factor in regulating cytokine-mediated induction of mPGES-1 (Cheng et al., 2004; Subbaramaiah et al., 2004). Some reports demonstrated curcumin can suppress EGR-1 expression in monocytes and endothelial cells (Pendurthi and Rao, 2000; Giri et al., 2004). To determine the effects of curcumin on EGR-1 expression in lung epithelial cell A549, cells pretreated with curcumin were incubated in the presence of interleukin-1β, and the cell lysate was analyzed by Western blotting. Interleukin-1β alone enhanced EGR-1 protein, with the maximum expression observed at 1 h, and then the expression declined to basal levels. In contrast, levels of EGR-1 protein were significantly reduced in cells at all time points by pretreatment with curcumin (Fig. 3A). Additionally, treatment with curcumin also decreased interleukin-1β-mediated activation of the EGR-1 promoter reporter in a concentration-dependent manner (Fig. 3B). These results suggest that curcumin inhibits IL-1β-induced expression of EGR-1 at the transcriptional level and thereby could inhibit the expression of mPGES-1.

Curcumin-mediated reduction of mPGES-1/COX-2 expression. A, A549 cells were cultured with or without 1 ng/ml interleukin-1β in the presence of each dose of curcumin (0, 10, 25, and 50 μM) for 24 h. Total cell lysates were subjected to Western blot analysis. B, the ratio of band density per actin was calculated at each treatment in A. C, cells were cultured with or without 1 ng/ml interleukin-1β in the presence of each dose of curcumin (0, 10, 25, and 50 μM) for 2 h. Total RNA was analyzed by RT-PCR. D, from the same RNA sample in C, mRNA levels were compared using real-time RT-PCR. All results are representative of three experiments.
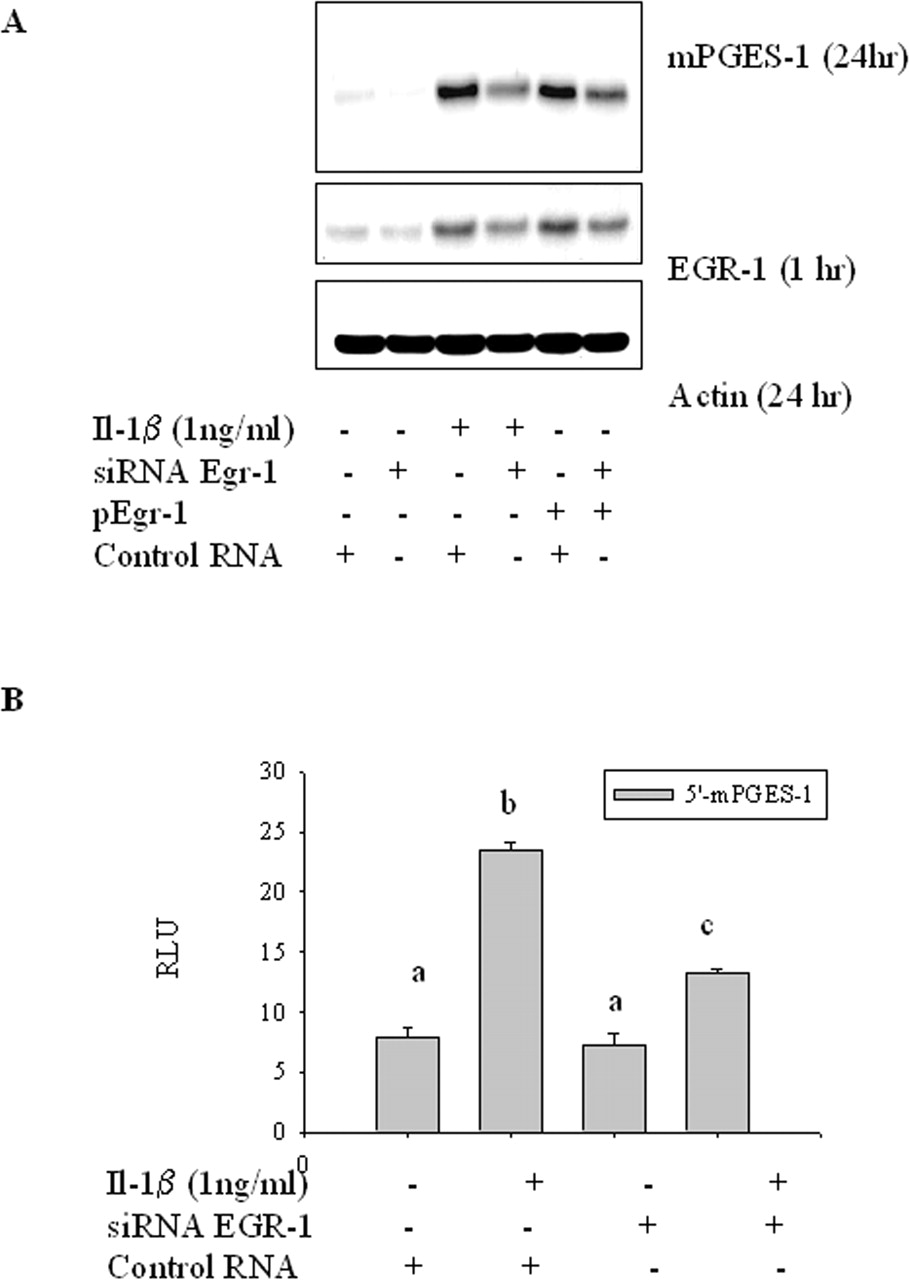
Inhibition of mPGES-1 Induction with EGR-1 siRNA. To confirm the involvement of EGR-1 in IL-1β-induced mPGES-1, cells were pretreated with siRNA for EGR-1 or with a control siRNA for 48 h. Interleukin-1β was then added, and cells were incubated for 1 or 24 h before EGR-1 and mPGES-1 were measured by Western blot analysis. EGR-1 siRNA reduced the EGR-1 protein level and the mPGES-1 levels induced by IL-1β. When cells were overexpressed with pEgr-1 plasmid, production of mPGES-1 was also enhanced. However, treatment with EGR-1 siRNA suppressed mPGES-1 as well as EGR-1 in the pEgr-1-transfected cells, which served as a positive control (Fig. 4A). Modulation of mPGES-1 expression by inhibiting EGR-1 gene expression was further confirmed for the transcriptional activity. Interleukin-1β-mediated increase in the promoter activity of mPGES-1 luciferase construct was significantly decreased by EGR-1 siRNA (Fig. 4B). This result supports the conclusion that curcumin can inhibit IL-1β-induced mPGES-1 expression by inhibiting the expression of EGR-1 at the promoter levels.

Curcumin-mediated reduction of EGR-1 expression induced by interleukin-1β in A549 A, cells were pretreated with 25 μM curcumin for 2 h, and then 1 ng/ml interleukin-1β was added to the cells. After each incubation time, cellular lysates were analyzed by Western blot analysis. Results are representative of three experiments. B, A549 cells were cotransfected with EGR-1 promoter luciferase plasmid and pRL-null vector for 24 h and treated with or without 1 ng/ml interleukin-1β in the presence of each dose of curcumin for another 24 h. Asterisks indicate significant difference (p < 0.05) from the IL-1β alone (control). Results are representative of three experiments.
Interference of NF-κB Signals by Curcumin in Interleukin-1β-Induced mPGES-1 Expression. NF-κB is a well known target of curcumin. Interleukin-1β stimulates phosphorylation of IκB, which is degraded by proteasome complex, and then NF-κB is allowed to translocate into the nucleus to promote gene transcription. However, pretreatment with curcumin before stimulation with interleukin-1β reduced degradation and phosphorylation of IκB (Fig. 5A). To confirm the involvement of NF-κB signals in interleukin-1β-induced mPGES-1 expression, cells were administered with a specific inhibitor of NF-κB signals, 6-amino-4-(4-phenoxyphenyl-ethyl amino) quanazoline, which retarded interleukin-1β-induced mPGES-1 expression efficiently (Fig. 5B). Moreover, the NF-κB inhibitor also reduced on interleukin-1β-activated mPGES-1 transcriptional activity, confirmed using reporter of mPGES-1 promoter (Fig. 5C). Taken together, the NF-κB signal pathway was one of targets of curcumin in regulating mPGES-1 expression.

Effect of EGR-1 siRNA on interleukin-1β-induced mPGES-1. Cells were transfected with EGR-1 siRNA, control siRNA, or LipofectAMINE 2000 reagent only. A, after 48 h of incubation with siRNA, cells were treated with 1 ng/ml interleukin-1β or vehicle for 24 or 1 h. B, after 24-h incubation, cells were transfected with PGES-1 reporter and then incubated with 1 ng/ml interleukin-1β (IL-1β) or vehicle for additional 24 h. Groups with different letters are significantly different (p < 0.05) by the SNK method. Results are representative of three experiments.
Effect of Curcumin on Interleukin-1β-Activated c-Jun N-Terminal Kinase 1/2 (JNK MAP Kinase) Cascade. Active form of JNK1/2 phosphorylates c-Jun protein that can dimerize into AP-1 complex. Interleukin-1β enhanced phosphorylated JNK1/2 in A549 cells, but this active form of JNK1/2 was dramatically suppressed by pretreatment with curcumin (Fig. 6A). When JNK activity was blocked with a specific JNK inhibitor, SP600125, the expression of interleukin-1β-mediated mPGES-1 was also suppressed (Fig. 6B). Moreover, SP600125 also reduced promoter activity of mPGES-1 in response to interleukin-1β (Fig. 6C). Thus, JNK cascade as well as NF-κB signal and EGR-1 was a target of curcumin, which contributed to the reduced production of mPGES-1 in A549 cells.
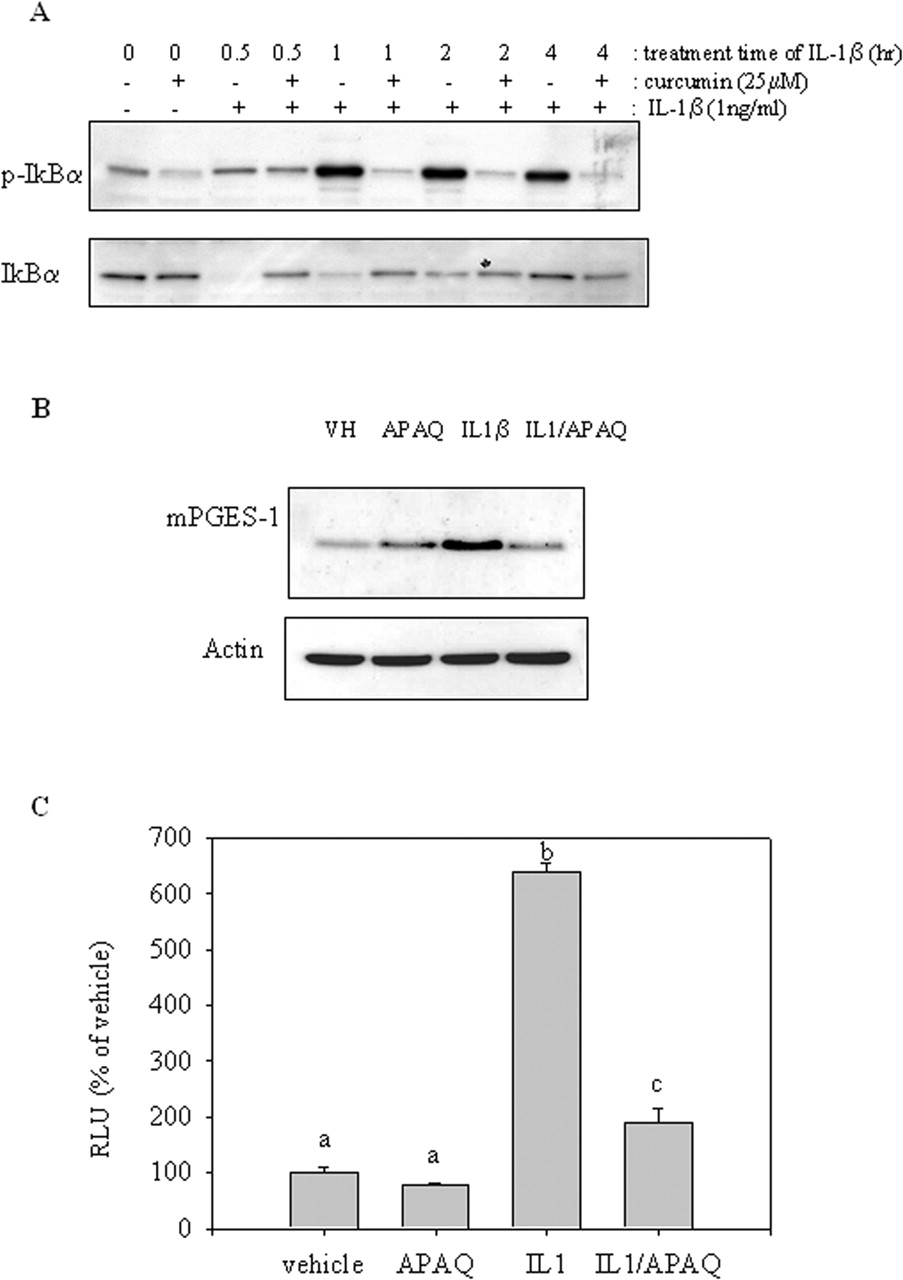
Curcumin-mediated suppression of interleukin-1β-activated NF-κB signals in A549. A, cells were pretreated with 25 μM curcumin or vehicle for 2 h, and then 1 ng/ml interleukin-1β was added to the cells. Total cellular lysate was applied for the Western blot analysis. B, cells were cotreated with 1 ng/ml interleukin-1β [or vehicle (VH)] and 2 μM 6-amino-4-(4-phenoxyphenylethyl amino) quanazoline (APAQ or vehicle) for 24 h. Total cellular lysate was applied for the Western blot analysis. C, A549 cells were cotransfected with mPGES-1 promoter luciferase plasmid plus pRL-null vector for 24 h and treated with 1 ng/ml interleukin-1β (or vehicle) in the presence of 2 μM 6-amino-4-(4-phenoxyphenylethyl amino) quanazoline (or vehicle) for another 24 h. Groups with different letters are significantly different (p < 0.05) by the SNK method. Results are representative of three experiments.
Concentration-Dependent Inhibition of Signaling Pathways. Since curcumin inhibits several proinflammatory signaling pathways of EGR-1, NF-κB, and MAP kinases, the concentration-dependent inhibition of each pathway was examined to determine the sensitivity to curcumin inhibition. Cells were exposed to different concentrations of curcumin, and the three signaling molecules, phospho-JNK1/2, phospho-IκBα, and EGR-1, were measured by Western analysis in IL-1β-activated cells. Inhibition of EGR-1 expression was dependent on the concentration of curcumin, and inhibition was observed at 2.5 μM with complete inhibition observed at 10 μM. Compared with the other two kinases, the inhibition of EGR-1 expression was detected at concentrations lower than required for the other two pathways (Fig. 7). Significant inhibition of NF-κB and MAP kinases was not observed until higher concentrations, 25 μM curcumin. Thus, EGR-1 expression was the most sensitive to curcumin's effect and could be an important modulator of curcumin-mediated inhibition of mPGES-1 gene expression.

Curcumin-mediated suppression of interleukin-1β-activated JNK1/2 signals in A549. A, cells were pretreated with 25 μM curcumin or vehicle for 2 h, and then 1 ng/ml interleukin-1β was added to the cells. Total cellular lysate was applied for the Western blot analysis. B, cells were cotreated with 1 ng/ml interleukin-1β (or vehicle) and 5 μM SP600125 (SP) (or vehicle) for 24 h. Total cellular lysate was applied for the Western blot analysis. C, cells were cotransfected with mPGES-1 promoter luciferase plasmid plus pRL-null vector for 24 h and then treated with 1 ng/ml interleukin-1β (or vehicle) in the presence of 5 μM SP600125 (or vehicle) for another 24 h. Groups with different letters are significantly different (p < 0.05) by the SNK method. Results are representative of three experiments.
Discussion
The anticancer activity of curcumin is well established and recognized as an effective anti-inflammatory agent (Aggarwal et al., 2003). The anti-inflammatory and antitumorigenic activities could be mediated by inhibition of the formation of prostaglandins. In this report, we first confirmed that curcumin inhibits PGE2 production and then investigated possible mechanisms in an attempt to understand how this inhibition occurred. The results presented here indicate that curcumin attenuates PGE2 biosynthesis by inhibiting the expression of mPGES-1 that occurs by several mechanisms. Incubation with IL-1β increased the expression of mPGES-1 and COX-2 as reported previously by other investigators (Thoren and Jakobsson, 2000; Catley et al., 2003; Moore et al., 2004). As a result, arachidonic acid incubated with IL-1β-treated cells is extensively metabolized into PGE2, but the inclusion of curcumin inhibited the formation of PGE2. Moreover, the suppression of mPGES-1 altered arachidonic acid metabolism from PGE2 to PGF2α and 6-keto-PGF1α as the major prostaglandins formed by these cells. Curcumin inhibited the IL-1β-induced expression of COX-2 and mPGES-1, with mPGES-1 being more susceptible to the effects of curcumin. These findings indicated a target for the inhibition of PGE2 by curcumin was mPGES-1 with more preference to COX-2.

Concentration-dependent suppression of interleukin-1β-signaling pathways by curcumin. Cells were pretreated with each concentration of curcumin for 2 h, and then 1 ng/ml interleukin-1β (or vehicle) was added to the cells for 45 min. After each incubation time, cellular lysate was analyzed by Western blot analysis. Results are representative of three experiments.
Recent studies indicate EGR-1 as a critical regulator of mPGES-1 transcription (Naraba et al., 2002; Cheng et al., 2004; Subbaramaiah et al., 2004). Binding of EGR-1 transcription factor to GC box adjacent to the initiation site was demonstrated as a key step in the regulation of the expression of cytokine-induced mPGES-1. EGR-1 promoter site contains multiple potential regulatory sites such as four Sp1 sites, two AP-1 sites, two cAMP response elements, and six serum response elements (SRE). Cytokines enhance EGR-1 transcription by modulating serum response factors bound to SRE. IL-1β increased the expression of EGR-1 in confirmation of previous studies on a human chondrocyte cell line (Tan et al., 2003). The serum response factors bound to SRE are also modified through phosphorylation by kinases such as MAP kinases and calcium/calmodulin-dependent protein kinase to activate EGR-1 transcription. Early studies have shown the inhibitory action of curcumin on such kinase cascades (Aggarwal et al., 2003), which could be one of the possible mechanism for the reduced EGR-1 transcription by curcumin.
Curcumin inhibited IL-1β-induced expression of EGR-1 and inhibited the transcriptional activity of EGR-1. Furthermore, siRNA for EGR-1 attenuated the expression of mPGES-1, and the controlled expression of EGR-1 increased the expression of mPGES-1. Thus, these data support the hypothesis that curcumin inhibits the expression of mPGES-1 and thereby suppresses the formation of PGE2 from arachidonic acid by attenuation of the expression of the transcription factor EGR-1.
mPGES-1 is an important determinate in inflammation and the development of tumors. The expression of mPGES-1 is induced by several inflammatory cytokines and is overexpressed in inflammatory bowel disease (Subbaramaiah et al., 2004). Studies with mPGES-1-deficient mice showed impaired inflammatory and pain responses, suggesting mPGES-1 may be a critical determinate in inflammation and thus is a target for the development of new drugs for the treatment of human rheumatoid arthritis (Murakami et al., 2002; Kojima et al., 2003). In addition, mPGES-1 is overexpressed in human colorectal adenomas and cancer, although studies with cells in culture and nude mice tumor xenograft models suggest a potential role of mPGES-1 in tumorigenesis (Kamei et al., 2003). Thus, inhibition of mPGES-1 expression by curcumin may also provide addition information to explain the anticancer and anti-inflammatory activity of curcumin.
Enhanced production of EGR-1 protein was a critical mediator in the production of mPGES-1 in this study. Moreover, EGR-1 can play additional roles in lung carcinogenesis, such as angiogenesis and metastasis. EGR-1 has been associated with malignant metastasis in several tumor models, such as gastric and prostate cancers (Szabo et al., 2001; Kobayashi et al., 2002; Baron et al., 2003). A high level of EGR-1 in cultured vascular endothelial cells enhances cell division, migration, and tubule formation in vitro (Khachigian, 2004). For this reason, suppressed EGR-1 by curcumin also could contribute to the suppression of malignant processes. The concentration of curcumin required to suppress mPGES-1 agrees with previous reports on in vitro effect of curcumin (10–100 μM) to inhibit PGE2 level (Plummer et al., 1999; Aggarwal et al., 2003). However, the physiological concentration of curcumin in human and rodents is reported to be as low as 5 μM (Pan et al., 1999; Ireson et al., 2001), although concentrations are reported to be as high as 300 nmol/g tissue of the intestinal tract after i.p. administration of 100 mg/kg of curcumin. Therefore, there could be a limitation to elicit acute biological effects associated with chemoprevention in nongastrointestinal tissues when consumed as a food constituent. Here, we used A549 lung alveolar epithelial adenocarcinoma cell line as a well established model of mPGES-1 regulation (Thoren and Jakobsson, 2000; Catley et al., 2003) and it is thus warranted to address the chemopreventive effect of curcumin on other tumor models.
EGR-1, as well as JNK1/2 signals and NF-κB, contributed to mPGES-1 down-regulation by curcumin. The expression of EGR-1, a key transcriptional modulator of cytokine-induced mPGES-1, was regulated by the curcumin and resulted in decreased production of mPGES-1. PPARγ ligands inhibit inflammation in a number of animal models and inhibit tumor formation (Mendez and LaPointe, 2003; Cheng et al., 2004) and also inhibit the production of PGE2 by suppressing EGR-1-dependent expression of mPGES-1. Thus, both curcumin and the ligands for PPARγ may act by the similar mechanism, the reduced expression of the transcription factor EGR-1 to attenuate the expression of mPGES-1. This may represent a novel mechanism by which both curcumin and PPARγ ligands alter the expression of mPGES-1, an important gene associated with both inflammatory diseases and cancer.
Footnotes
-
This research was supported (in part) by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.084434.
-
ABBREVIATIONS: PG, prostaglandin; PGES, prostaglandin E synthase; COX, cyclooxygenase; mPGES-1, microsomal prostaglandin E synthase 1; NF-κB, nuclear factor κB; AP-1, activator protein-1; EGR-1, early growth response gene-1; IL, interleukin; JNK1/2, Jun N-terminal kinase 1/2; SP600125, anthra[1,9-cd]pyrazol-6(2H)-one 1,9-pyrazoloanthrone; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; PCR, polymerase chain reaction; TBST, Tris-buffered saline plus Tween 0.05%; RT-PCR, reverse transcription-polymerase chain reaction; UNG, uracil N-glycosylase; HPLC, high-performance liquid chromatography; ANOVA, analysis of variance; SNK, Student-Newman-Keuls; MAP, mitogen-activated protein; SRE, serum response element(s); PPARγ, peroxisome proliferator-activated receptor γ.
-
↵1 1-10 Department of Microbiology and Immunology, Pusan National University Medical School and Medical Research Institute, Busan Republic of Korea.
- Received February 2, 2005.
- Accepted August 1, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}