


Abstract
Receptor binding was characterized for [3H](1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azoniabicyclo[2.2.1]heptane ([3H]A-585539), a selective high-affinity α7 nicotinic acetylcholine receptor (nAChR) agonist with rapid kinetics, low nonspecific binding, and high specific activity. At 4°C, the association was monophasic and rapid (t½ = 8.0 min); dissociation was slower (t½ = 64.2 min). The Kd in rat brain at 4°C was 0.063 nM, whereas at 22 and 37°C, the Kd values were 0.188 and 0.95 nM, respectively. In contrast, the Bmax (34 fmol/mg protein) was unaffected by temperature. In human cortex, [3H]A-585539 bound with a Kd of 0.066 nM and a Bmax of 5.8 fmol/mg protein at 4°C, whereas under similar conditions, specific [3H]methyllycaconitine ([3H]MLA) binding was not measurable. A number of agonist and antagonist nAChR ligands displaced binding to rat brain membranes with rank order of affinity similar to that for [3H]MLA, and in general, a 5 to 10-fold higher affinity was observed for [3H]A-585539 binding. There was also a good correlation of Ki values between [3H]A-585539 binding to rat brain and human cortex. The use of a α7/5-hydroxytryptamine type-3 chimera revealed that the N-terminal domain of α7 nAChR was sufficient to faithfully reproduce the pharmacology of [3H]A-585539 binding. Autoradiographic studies comparing [3H]A-585539 and [125I]α-bungarotoxin revealed a similar pattern of labeling in the rat. In summary, [3H]A-585539 was shown to have excellent binding characteristics in rat and human brain and represents the first high-affinity α7 agonist radioligand with utility in the characterization of this important nAChR subtype that is targeted toward ameliorating cognitive deficits underlying neuropsychiatric and neurodegenerative disorders.
The α7 nicotinic acetylcholine receptor (nAChR) is a pentameric, rapidly activating and desensitizing ligand-gated ion channel expressed in the mammalian central nervous system. The α7 subunit is widely expressed in the brain, especially in regions associated with cognitive processing, autonomic ganglia, and adrenal chromaffin cells, and in non-neuronal cell types. The α7 subtype is distinguished from other nAChRs by its relatively high permeability to Ca2+, rapid desensitization, and sensitivity to antagonists, such as α-bungarotoxin (α-Bgt) and methyllycaconitine (MLA) (Ward et al., 1990). Unlike other nAChR subtypes, the functional significance of the α7 nAChR is not only attributable to its electrogenic properties (i.e., modulation of neuronal excitability and neurotransmitter release) but also to its high Ca2+-permeability and association with biochemical signaling pathways (reviewed in Role and Berg, 1996; Berg and Conroy, 2002; Dajas-Bailador and Wonnacott, 2004). At the cellular level, activation of α7 nAChRs can regulate interneuron excitability, modulate the release of excitatory and inhibitory neurotransmitters, and contribute to neuroprotective effects (for review see, Dani and Bertrand, 2007).
Recent studies with antisense, gene knockout, and subtype selective ligands have provided evidence that targeting the α7 nAChRs can lead to improvements in cognitive performance and sensory gating deficits in vivo. For example, α7 nAChR genetic knockout mice have shown impaired performance in ethanol-induced contextual fear conditioning (Wehner et al., 2004) and showed further deterioration in hippocampus-selective associative learning and memory when crossed with Tg2576 animals (Dineley et al., 2002). Selective α7 nAChR agonists, such as PNU-282987 (Bodnar et al., 2005; Hajos et al., 2005), PHA-543613 (Wishka et al., 2006), AR-R17779 (Levin et al., 1999; Van Kampen et al., 2004), SSR180711 (Biton et al., 2007), and A-582941 (Bitner et al., 2007), improve performance in models thought to capture sensory-gating deficit, short-term working memory, and memory consolidation domains of cognitive function. Moreover, sensory gating and cognitive performance are known to be impaired in rodents with decreased expression or following pharmacological blockade of α7 nAChRs (Luntz-Leybman et al., 1992; Felix and Levin, 1997; Stevens et al., 1998; Levin and Rezvani, 2002). Consistent with this finding, the expression of the α7 protein is reduced in the brains of patients with Alzheimer's disease (Burghaus et al., 2000) and schizophrenia (Freedman et al., 1995). The latter phenomenon has been correlated with polymorphisms in the promoter region of the α7 gene in schizophrenic patients where such polymorphisms have been shown to result in decreased gene transcription (Freedman et al., 1997; Leonard et al., 2002). These and other observations have attracted considerable attention to the α7 nAChR as a drug target in recent years, the hypothesis being that augmenting α7 nAChR function could ameliorate the cognitive deficits associated with neuropsychiatric and neurodegenerative diseases (Rezvani and Levin, 2001; Martin et al., 2004). Accordingly, selective agonists and positive allosteric modulators of α7 nAChRs are being discovered and developed for the treatment of cognitive deficits associated with neuropsychiatric and neurodegenerative disease, such as schizophrenia and Alzheimer's disease.
Although the α7 nAChR continues to be explored as a target for a number of central nervous system indications, developments in high-affinity pharmacological tools have largely been antagonists, such as the snake toxin peptide, α-Bgt, and the alkaloid MLA and the corresponding radioligands [125I]α-Bgt and [3H]MLA. α-Bgt has been an invaluable tool in the characterization of nAChRs in skeletal muscle for functional studies as a pseudo-irreversible antagonist and for labeling, purifying, and visualizing native nAChRs. Of the cloned neuronal subunits, the α7, α8, and α9 subunits are unique in that they are all sensitive to α-Bgt when expressed in Xenopus laevis oocytes. MLA is a natural norditerpenoid alkaloid, identified as the principal toxic agent in Delphinium brownie (Ward et al., 1990), [3H]MLA binds to rat brain membranes with an affinity, pharmacological profile and regional distribution characteristic of α-Bgt-sensitive, putative α7 subunit-containing nAChRs (Davies et al., 1999). Although [3H]MLA displays rapid binding kinetics, it suffers from a relatively high degree of nonspecific binding. Unlike the heteromeric nAChRs where a number of agonist ligands (e.g., A-85380) have become available to characterize receptors in animals and humans under various drug treatment and disease conditions, high affinity α7 nAChR agonist ligands have not yet emerged.

Structures of [3H]A-585539 and [3H]MLA.
During the course of our in-house efforts, we discovered several bicycloheptane analogs with high-binding affinities toward displacement of [3H]MLA. In this study, we describe [3H]A-585539 as the first high-affinity agonist radioligand with excellent binding characteristics across both rodent and human brain.
Materials and Methods
Synthesis of [3H]A-585539. [3H]Methyl iodide in toluene (250 mCi in 0.1 ml, 85 Ci/mmol; American Radiolabeled Chemicals, Inc., St. Louis, MO) was combined with a solution of (1S,4S)-2-methyl-5-(6-phenylpyridazin-3-yl)-2,5-diazabicyclo[2.2.1]heptane (Schrimpf et al., 2007) in dichloromethane (0.788 mg, 2.96 micromole in 0.45 ml). The vial was capped, and the mixture was allowed to react overnight at room temperature. Methanol was added, and the solvents were evaporated to yield [3H]A-585539 (Schrimpf et al., 2007) at a total activity of 42 mCi. The product was taken up in methanol for high-performance liquid chromatography (HPLC) purification. The structure of [3H]A-585539 along with that of [3H]MLA is illustrated in Fig. 1.
For purification by HPLC, ∼7 mCi of [3H]A-585539 was evaporated to dryness, and the residue was dissolved in 4.5 ml of acetonitrile/water/trifluoroacetic acid (15:85:0.1). This solution was injected (0.9 ml per run) onto a Phenomenex Luna C18(2) column (5 μm, 250 mm × 4.6 mm i.d.; Phenomenex, Torrance, CA) using an Agilent HPLC system (Agilent Technologies, Palo Alto, CA). [3H]A-585539 was eluted by a gradient mobile phase from 10 to 20% B in 20 min where mobile phase A = 0.1% trifluoroacetic acid in water and mobile phase B = 0.1% trifluoroacetic acid in acetonitrile at a flow rate of approximately 1 ml/min. Peak detection and chromatograms were obtained with an Agilent variable wavelength UV detector set at 275 nm. The fractions containing [3H]A-585539 were collected at approximately 14 min using an Agilent fraction collector. The fractions were combined, and the solvents were evaporated in vacuo. The residue was dissolved in absolute ethanol (2 ml) to give 0.7 mCi.

Saturation binding isotherms of [3H]A-585539. Binding of [3H]A-585539 to membrane-enriched fractions from rat brain (A) and human frontal cortex (B). Membranes were incubated for 90 min at 4°C with a range of 0.01 to 2 nM [3H]A-585539. Shown are specific binding (quadruplicates) and nonspecific binding (duplicates), which was measured in the presence of 10 μM MLA. Figures shown are one of eight independent experiments for each tissue. Data from saturation binding experiments were analyzed by nonlinear regression, and the Kd and Bmax values are shown in Table 1.
[3H]A-585539 was assayed using an Agilent 1100 series HPLC system consisting of a quaternary pump, an autosampler, and a photodiode array UV detector. A Packard Radiomatic A 500 radioactivity detector (PerkinElmer Life and Analytical Sciences, Waltham, MA) was connected to the HPLC system. For radiodetection, a 500-μl flow cell and a 3:1 ratio of Ultima-Flo M scintillation cocktail to HPLC mobile phase were used. The analyses were performed using a Phenomenex Luna C18(2) column (5 μm, 250 mm × 4.6 mm i.d.). The mobile phase consisted of a gradient starting with 10% B and ramping to 20% B in 20 min followed by ramping to 90% B in 1 min and hold at 90% B for 9 min, where mobile phase A = 0.1% trifluoroacetic acid in water and mobile phase B = 0.1% trifluoroacetic acid in acetonitrile. The flow rate was set at approximately 1 ml/min, and the UV detection was set at 275 nm. The radiochemical purity of [3H]A-585539 was found to be >98%. The specific activity was determined to be 62.78 Ci/mmol by mass spectroscopy.
[3H]A-585539 Binding. [3H]A-585539 (62.8 Ci/mmol) binding was determined using membrane-enriched fractions (Sullivan and Anderson, 1998) from rat brain (minus cerebellum) or human cortex (ABS Inc., Wilmington, DE). Pellets were thawed at 4°C, washed, and resuspended with a Polytron homogenizer at a setting of 7 in 30 volumes of BSS-Tris buffer (120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 50 mM Tris-Cl, pH 7.4, 4°C). For saturation binding isotherms, 16 concentrations of [3H]A-585539 (0.01–2 nM) in quadruplicate and homogenate containing 100 to 200 μg of protein were incubated in a final volume of 500 μl for 90 min at 4°C. Nonspecific binding was determined in the presence of 30 μM MLA. For concentration-inhibition assays, seven log-dilution concentrations of test compounds in duplicate, homogenate containing 100 to 200 μg of protein, and 0.5 nM [3H]A-585539 were incubated in a final volume of 500 μl for 60 min at 4°C. Nonspecific binding in quadruplicate was determined in the presence of 10 μM MLA. Bound radioactivity was collected by vacuum filtration onto Millipore MultiScreen harvest plates FB (Millipore Corporation, Billerica, MA) presoaked with 0.3% polyethyleneimine using a Packard cell harvester. The filters were then rapidly rinsed with 2 ml of ice-cold BSS. PerkinElmer MicroScint-20 scintillation cocktail (40 μl) was added to each well, and bound radioactivity was determined using a PerkinElmer TopCount instrument. Binding studies were also performed using human embryonic kidney-293 cells expressing a α7/5-hydroxytryptamine type-3 (5-HT3) chimera containing ligand-binding domain of α7 nAChR and the transmembrane/pore-forming region of 5-HT3 receptor. Using reverse transcriptase-polymerase chain reaction, the coding sequence for the N-terminal 224 amino acids of human α7 nAChR (protein AAA83561) and that for the C-terminal 242 amino acids of human 5-HT3 (5-hydroxytryptamine; serotonin) receptor (protein AAP35868) were amplified with overlapping ends. Recombinant polymerase chain reaction using these two overlapping fragments yielded the open-reading frame of the chimeric receptor. Human embryonic kidney-293 cells were transfected, and stable transfectants were maintained using standard procedures. To harvest, the cells were rinsed with phosphate-buffered saline, scraped from flasks, and centrifuged at 1000g. Pellets were homogenized with a Polytron homogenizer at a setting of 7 for 30 s in 30 volumes of BSS-Tris buffer and diluted to yield approximately 5 μg of protein per well. Binding conditions were similar to those described above.
[3H]MLA Binding. Assay conditions were similar to those for [3H]A-585539 binding. Membrane-enriched fractions from rat brain were thawed, washed, and resuspended in 30 volumes of BSS-Tris buffer (120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 50 mM Tris-Cl, pH 7.4, 22°C). Samples containing 100 to 200 μgof protein, 5 nM [3H]MLA (25 Ci/mmol; Tocris Bioscience, Ellisville, MO), and 0.1% bovine serum albumin (BSA; Sigma, St. Louis, MO) were incubated in a final volume of 500 μl for 60 min at 22°C. Seven log-dilution concentrations of each compound were tested in duplicate. Nonspecific binding was determined in the presence of 10 μM MLA. Bound radioactivity was isolated by vacuum filtration onto Millipore MultiScreen harvest plates FB presoaked with 2% BSA. The plates were rinsed and counted as above.
Light Microscopic Autoradiography. Two 2-month-old adult rats of the Sprague-Dawley strain were killed by decapitation under pentobarbital anesthesia (50 mg/kg body weight i.p., following Ethical Rules of Animal Rights, Geneva, Switzerland); brains were dissected out and frozen by immersion in 2-methylbutane at –25°C. Serial sections (14 μm) were cut through the diencephalons in a cryostat, thaw-mounted on gelatin-chrome-alum-coated slides, and stored at –80°C until use. Two series of sections were incubated with [3H]A-585539 at 25 and 50 nM respectively. Four series of adjacent sections were incubated with [3H]A-585539 at the same concentrations together with either nicotine tartrate (1 mM) or unlabeled A-585539 (500 μM). Remaining series were incubated with [125I]α-bungarotoxin at 1 nM either alone or together with 300 μM nicotine tartrate. The binding procedure was conducted as described previously (Tribollet et al., 2004). Sections incubated with [3H]A-585539 were apposed for 3 weeks to a tritium-sensitive storage phosphorimaging screen. Digital images were obtained with a phosphorimaging system (Cyclone storage Phosphor System; PerkinElmer Life and Analytical Sciences) and Optiquant (PerkinElmer Life and Analytical Sciences), a Windows-based software. Sections incubated with [125I]α-bungarotoxin were placed in x-ray cassettes in contact with βmax hyperfilms (Amersham, Buckinghamshire, UK) for 3 days. Films were developed in Kodak D19 (Kodak SA, Lausanne, Switzerland) and photographed with a digital camera.

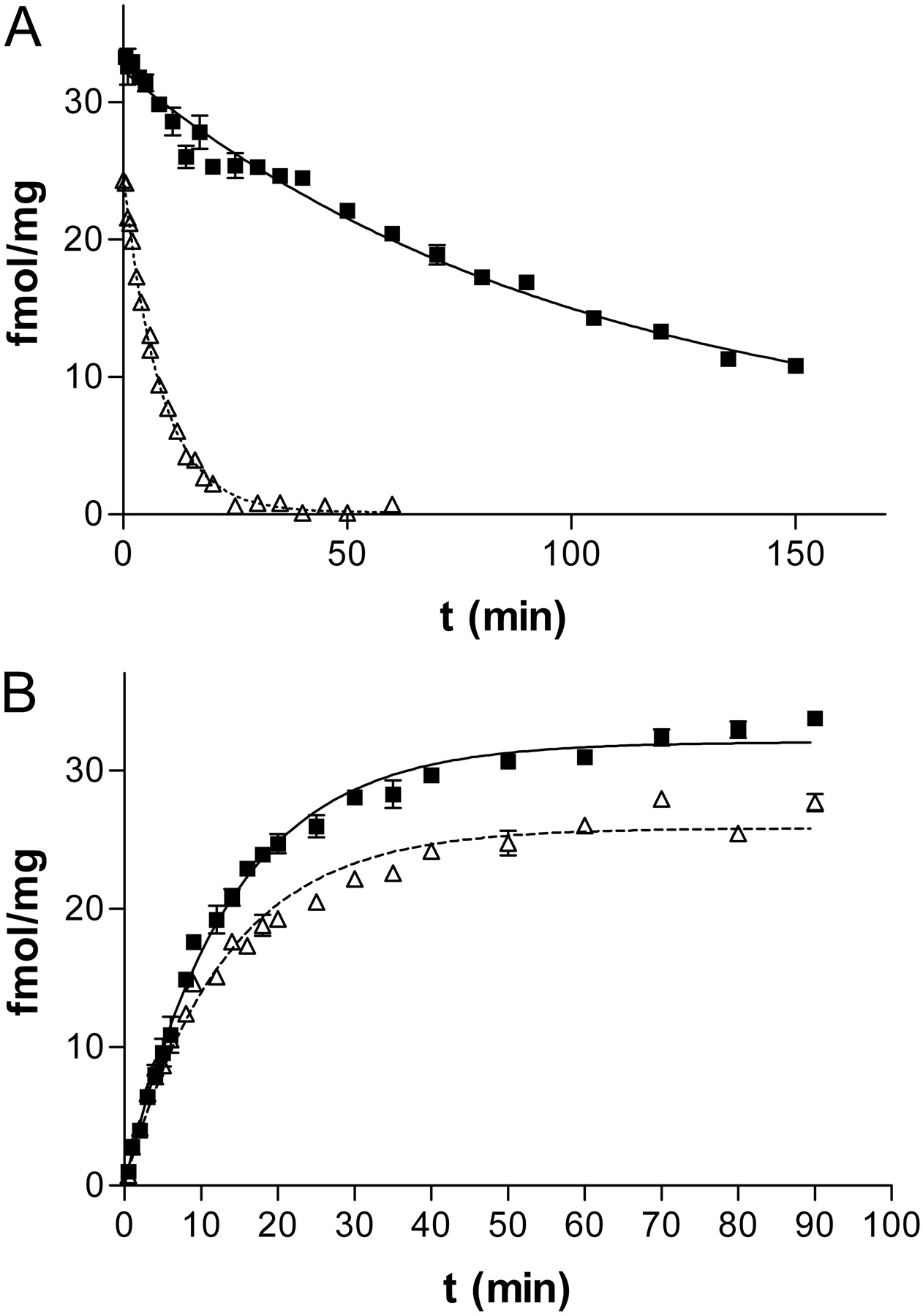
Kinetics of [3H]A-585539 binding to rat brain membranes. A, dissociation of [3H]A-585539. Samples in quadruplicate were equilibrated with 0.1 nM [3H]A-585539 for 90 min at 4°C (closed symbols, solid line) or at 22°C (open symbols, dotted line). The radioligand then was displaced with 10 μM unlabeled A-585539. Dissociation was terminated at various time intervals, as indicated, by rapid filtration onto 96-well glass fiber filter plates. Depicted is one experiment at each temperature for which the t1/2 values of dissociation for [3H]A-585539 binding were 70.1 min at 4°C and 5.7 min at 22°C. The average t1/2 values for dissociation were 64.2 ± 6.1 min at 4°C (n = 5) and 7.3 ± 0.5 min at 22°C (n = 4). B, association of [3H]A-585539. Samples in quadruplicate were incubated with 0.1 nM [3H]A-585539 at 4°C (closed symbols, solid line) or at 22°C (open symbols, dotted line) for various time intervals as indicated. Association was terminated with rapid filtration onto 96-well glass fiber filter plates. One experiment is shown at each temperature for which the t1/2 values of association for [3H]A-585539 binding were 9.3 min at 4°C and 9.0 min at 22°C. The average t1/2 values for association were 8.0 ± 0.8 min at 4°C (n = 5) and 7.4 ± 0.7 min at 22°C (n = 4).
Data Analysis. Dissociation constant (Kd) and maximal binding (Bmax) values from saturation binding and observed association rate (kob) and dissociation rate (koff) from kinetic experiments were determined using GraphPad Prism (GraphPad Software, San Diego, CA). The Kd was determined from the equation Kd = koff/kon, where kon = (kob – koff)/L, where L is the concentration of radioligand. The IC50 values were determined by nonlinear regression in Microsoft Excel or Assay Explorer. Ki values were calculated from the IC50 values using the Cheng-Prusoff equation, where Ki = IC50/(1 + [Ligand]/Kd).
Results
Saturation Binding. Specific binding of [3H]A-585539 to rat brain membranes was saturable, rapid, and represented 80 to 95% of total binding over the concentration range (0.01 to 2 nM) examined (Fig. 2A; Table 1). In saturation-binding isotherms, nonlinear regression analysis of specific binding revealed an apparent Kd of 0.063 ± 0.004 nM and a Bmax of 33.7 ± 1.1 fmol/mg protein (n = 8) at 4°C. Nonspecific binding was less than 10% of total binding at concentrations up to ten times the Kd. At 22°C, the Kd was 0.188 nM, and the Bmax was 31.7 fmol/mg protein (n = 5). In contrast, the binding of [3H]methyllycaconitine to rat membranes at 22°C was found to have a Kd of 1.26 ± 0.33 nM and a Bmax of 32.7 ± 5.8 fmol/mg protein (n = 3). However, nonspecific binding of [3H]MLA ranged from 20 to 50% of total binding, even with the addition of 0.1% BSA to the incubation.
Kd and Bmax values from [3H]A-585539 saturation binding analysis
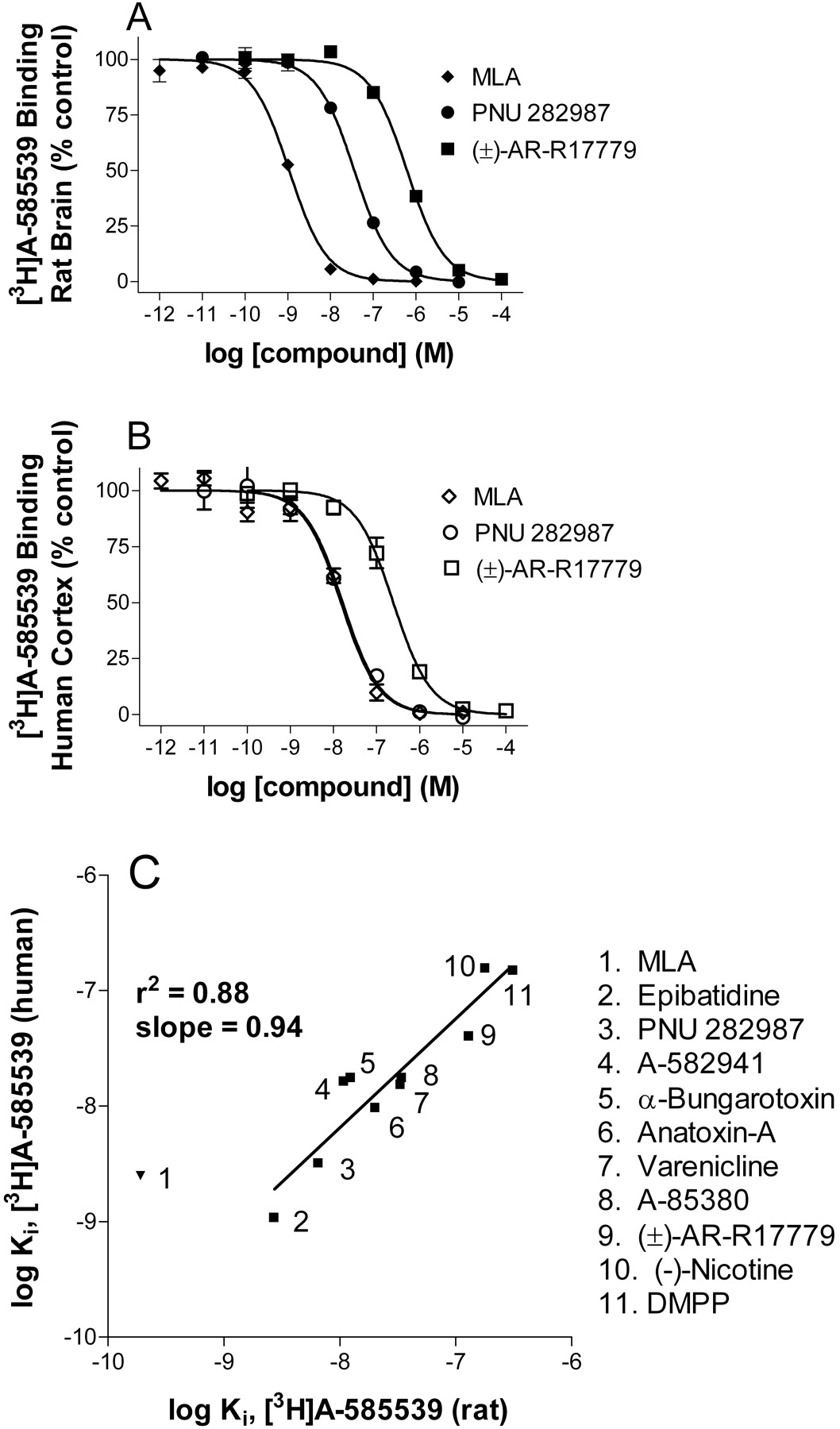
Displacement of [3H]A-585539 binding. Concentration-inhibition curves for the displacement of [3H]A-585539 binding to rat brain (A) and human cortex (B) membranes by three α7 nAChR compounds: MLA, PNU 282987, and racemic AR-R17779. Data points are the mean data from three independent experiments. The Ki values are summarized in Table 2. C, correlation plot for log Ki values of displacement of [3H]A-585539 binding from rat brain versus human frontal cortex. Excluding MLA, there was good correlation with a slope of 0.94 and a coefficient of correlation of 0.88. Data points are the mean of 3 to 10 independent determinations.

Comparison of displacement of [3H]A-585539 and [3H]MLA binding from rat brain membranes by nAChR agonists and antagonists. Data points are the log Ki values from 3 to 11 independent determinations of data taken from Table 2. The coefficient of correlation and slope values are 0.96 and 0.95, respectively.
The high specific activity and excellent affinity of [3H]A-585539 made it possible to directly detect binding to human brain (Fig. 2B; Table 1). Human frontal cortex membranes bound [3H]A-585539 at 4°C with a Kd value of 0.066 ± 0.006 nM, comparable to that of rat brain, and a Bmax of 5.8 ± 0.5 fmol/mg protein (n = 8), which is lower than that observed in the rat brain. Nonspecific binding ranged from 15 to 50% of total binding for range of concentrations of [3H]A-585539. In contrast, specific binding of [3H]MLA to human frontal cortex membranes was not detectable due to low counts and high nonspecific binding levels.
Kinetics of [3H]A-585539 Binding. [3H]A-585539 binding was temperature-dependent. Kinetic analysis demonstrated temperature dependence for the dissociation rate but not for the association rate (Fig. 3, A and B). The association half-time (t1/2) for [3H]A-585539 binding at 4°C was 8.0 ± 0.8 min, and the dissociation t1/2 was 64.2 ± 6.1 min (n = 5). Calculation of the dissociation constant from kinetic rate constants yielded a Kd of 0.021 ± 0.003 nM, which is in reasonable agreement with the Kd value obtained from saturation binding isotherms. At 22°C, the association t1/2 (7.4 ± 0.7 min) was comparable to that obtained at 4°C, whereas the dissociation t1/2 was much faster (7.3 ± 0.5 min, n = 4). The rapid koff rate at 22°C precludes the determination of meaningful dissociation constant by typical filtration assays.
Displacement Profile of [3H]A-585539. The radioligand binding pharmacology of [3H]A-585539 was assessed by examining the displacement of specific binding by agonists, antagonists, and positive allosteric modulators from rat and human brain membranes (Figs. 4 and 5; Table 2). A number of α7-selective nAChR compounds, including unlabeled A-585539, MLA, PNU-282987, (±)-AR-R17779, α-bungarotoxin, and A-582941 were found to displace [3H]A-585539 and [3H]MLA binding with the similar rank order of affinity. Other nAChR ligands, such as (±)-epibatidine, (–)-nicotine, (–)-cytisine, (+)-anatoxin-A, varenicline, and A-85380 also exhibited a high degree of correlation (Fig. 5). In general, a shift in Ki values was observed between [3H]A-585539 and [3H]MLA binding displacement in rat brain membranes. Displacement of [3H]A-585539 binding to human cortex membranes yielded nearly identical Ki values as those for rat brain membranes (Fig. 4C). An interesting exception was MLA, which showed considerably lower apparent affinity for human cortex membranes. Positive allosteric modulators of α7 nAChRs, including 5-hydroxyindole and PNU-120596, neither displaced nor enhanced [3H]A-585539 binding under the present experimental conditions.
Displacement of [3H]A-585539 binding to rat and human brain: comparison with [3H]MLA
To determine specificity of A-585539 for the α7 nAChR, the compound was further evaluated in a radioligand binding screen panel containing representatives of multiple G-protein coupled receptors, and ligand- and voltage-gated ion channel binding sites at CEREP (receptor binding and enzyme profile; CEREP, Potiers, France). A-585539 at 10 μM did not show significant displacement of binding of over 75 targets, with the exception of 5-HT3 receptors, where 89% inhibition of [3H]BRL 43694 (granisetron) binding was observed with 10 μM A-585539. The 5-HT3 antagonist, MDL 72222, does not significantly inhibit [3H]A-585539 binding to rat brain at concentrations up to 10 μM. In contrast, MDL 72222 inhibited the binding of the 5-HT3 antagonist [3H]GR65630 to human 5-HT3 serotonin receptors with a Ki of 12 nM (95% CL 9–15 nM, n = 3), whereas A-585539 had a Ki of 1.4 μM (95% CL 1.2–1.7 μM, n = 3).
Localization of [3H]A-585539 Binding. To assess the regions on α7 nAChRs responsible for binding interactions, studies were conducted using a α7/5-HT3 chimera where the N-terminal domain of α7 nAChR was fused with the transmembrane/pore-forming region of the 5-HT3 receptor. [3H]A-585539 showed no detectable binding to 5-HT3 receptors (data not shown), whereas specific and saturable binding of [3H]A-585539 was detected in cells expressing the α7/5-HT3 chimera (Fig. 6A). Binding was rapid and represented >95% of total binding over the concentration range (0.05 to 5 nM) examined. Saturation binding isotherms revealed an apparent Kd of 0.801 ± 0.118 nM and a Bmax of 10,500 ± 1000 fmol/mg protein (n = 4). The pharmacology of [3H]A-585539 binding was found to be consistent with an interaction with α7 nAChRs. A number of α7 nAChR-selective compounds, including MLA, TC-5280, PNU-282987, α-bungarotoxin, and SSR180711 were found to displace [3H]A-585539 binding with the same rank order of affinity as that seen in rat brain. Non-α7 nAChR-selective nAChR compounds, such as (±)-epibatidine, (–)-nicotine, (+)-anatoxin-A, 1,1-dimethyl-4-phenylpiperazinium, varenicline, and A-85380, had a high degree of correlation for displacement between α7/5-HT3 chimera-expressing cells and rat brain (Fig. 6B). Overall, there was a coefficient of correlation of 0.94 and a slope of 1.0.

Binding of [3H]A-585539 to α7/5-HT3 chimera expressed in HEK-293 cells. A, saturation binding of [3H]A-585539 to α7/5-HT3 cell membranes. One of four independent experiments is represented. Specific binding was determined in the presence of [3H]A-585539 (0.05–10 nM; 90-min incubation at 4°C). Nonspecific binding was determined in the presence of 30 μM MLA. Data were analyzed by nonlinear regression, and the Kd and Bmax values were determined for each experiment. The mean ± S.E.M. estimates for Kd were 0.80 ± 0.12 nM and 10,500 ± 1000 fmol/mg protein for Bmax. B, comparison of displacement of [3H]A-585539 binding from rat brain membranes and α7/5-HT3 chimera expressing cell membranes by nAChR ligands. Data points are log Ki values of 3 to 10 independent determinations for rat brain and three to four determinations for α7/5-HT3 cell membranes. The coefficient of correlation and slope values are 0.94 and 1.02, respectively.
Regional Distribution of [3H]A-585539 Binding. Autoradiography studies using [3H]A-585539 in rat brain showed a distribution of binding comparable with that of [125I]α-bungarotoxin, with a high level of binding in hippocampus and the superficial gray layer of the superior colliculus (Fig. 7A). Binding was fully displaced by unlabeled A-585539 (Fig. 7C). Nicotine also effectively displaced binding, although some residual labeling was seen in the CA3 area of the hippocampus (Fig. 7B). In contrast, the intense labeling yielded by [125I]α-bungarotoxin, in particular in the hippocampus and in the superior colliculus, (Fig. 7D), was fully displaced (Fig. 7E).
Discussion
Selective modulation of α7 nAChRs is thought to regulate cellular processes impaired in schizophrenia, Alzheimer's disease, and other dementias. Our laboratory and others have previously reported on the synthesis of novel analogs derived from various diamines exemplified by SSR180711 (Biton et al., 2007), A-582941 (Bitner et al., 2007), and quinuclidine derivatives, such as PNU-282987 (Bodnar et al., 2005; Hajos et al., 2005), PHA-543613 (Wishka et al., 2006), and AR-R17779 (Levin et al., 1999; Van Kampen et al., 2004), as α7 nAChR agonists. Despite the heightened interest in this molecular target for a number of central nervous system indications, developments in high-affinity radioligands, especially agonists with which to study this important subtype, have been less forthcoming. In the course of our discovery efforts to develop novel selective α7 nAChR agents from a series of 2.2.1 diamines, we found the diazabicycloheptane analog A-585539 to have greater than 1000-fold selectivity for α7 nAChRs over α4β2 nAChRs in radioligand binding. Initial experiments in X. laevis oocytes expressing rat α7 nAChRs showed that A-585539 activated ionic currents with an EC50 of 0.61 μM (95% CI 0.29–1.2 μM) and efficacy of 94.0 ± 6.4% (n = 3). No agonist activity was detected at concentrations up to 100 μM in IMR-32 (ATCC, Manassas, VA) neuroblastomas or in transfected cell lines containing α3β2, α3β4, α4β2, or α4β4 nAChRs using fluorometric imaging plate reader-based calcium flux assays (J.-H. Gronlein, J. Malysz, and M. Gopalakrishnan, unpublished observations). Given that A-585539 could be modified using a tritium methylation, it was chosen as a suitable candidate for radiolabeling. Here, we describe the characterization of [3H]A-585539, one of the first selective high-affinity α7 agonist radioligand with rapid kinetics, low nonspecific binding, and high specific activity.
[3H]A-585539 bound to rat brain and to human frontal cortex membranes with high affinity and with a pharmacological profile akin to that of [3H]MLA, a well characterized α7 nAChR antagonist (Ward et al., 1990; Davies et al., 1999; Whiteaker et al., 1999). Saturation binding isotherms revealed comparable binding affinities in rat (63 pM) and human (66 pM), whereas analysis of receptor density demonstrated that human cortex expressed some 6-fold lower density of binding sites compared to the rat. A similar ratio and density were as previously observed with [125I]α-Bgt in rat P2 membranes (35 fmol/mg protein) and human cortex (5 fmol/mg protein) (Davies et al., 1999; Falk et al., 2003). The high affinity and high specific activity (63 Ci/mmol) of [3H]A-585539 allowed for reliable pharmacological analysis of the low density of receptors in the human brain. This is a distinct advantage over [3H]MLA, which with an affinity of 2 nM does not generate measurable labeling of α7 nAChRs in human membranes under standard experimental conditions. An additional attribute of [3H]A-585539 is its relatively low nonspecific binding. At concentrations near the Kd, the nonspecific binding accounts for only 5% of total binding in rat brain and approximately 20% in human brain. In contrast, with [3H]MLA binding, substantially higher levels of nonspecific binding (∼35% at Kd) were observed in rat brain, even with the addition of 0.1% BSA in the incubation. Another limitation with [3H]MLA binding is the existence of displaceable binding from glass fiber filters, which can be reduced to some extent by presoaking the filter plates with 2% BSA. Such limitations were not encountered with [3H]A-585539.

Comparison of labeling obtained with [3H]A-585539 and [125I]α-bungarotoxin in the rat brain. A to C, taken from three adjacent coronal sections cut through the diencephalon. Section A was incubated with 25 nM [3H]A-585539; sections B and C with 25 nM [3H]A-585539 together with 1 mM nicotine tartrate (B) or 500 μM unlabeled A-585539 (C). D and E are from two adjacent sections cut from another rat brain and incubated with 1 nM 125I-α-bungarotoxin either alone (D) or together with 300 μM nicotine tartrate (E). Note that [3H]A-585539 and [125I]α-bungarotoxin yielded a similar labeling of field CA3 of the hippocampus (CA3) and of the superficial gray layer of the superior colliculus (SuG). Note also that [3H]A-585539 binding in CA3 (A and B) is only partially displaced by nicotine tartrate (B), whereas it is not detectable in the presence of unlabeled A-585539 in excess (D). Bar = 5 mm.
The kinetics of binding of [3H]A-585539 at 4°C were rapid for association (t1/2 = 8.0 min) and slower for dissociation (t1/2 = 64 min), which allows for vacuum filtration assays. The Kd value determined from kinetic experiments (21 pM) is in reasonable agreement with values obtained from equilibrium binding experiments. At 22°C, the association rate was similar (t1/2 = 7.4 min), although the dissociation rate was more rapid (t1/2 = 7.3 min) compared to that observed at 4°C. From saturation binding isotherms, the estimated Kd was 188 pM, which is some 8-fold lower than the value measured at 4°C. Under similar conditions, the Kd for [3H]MLA binding did not show temperature dependence (D. J. Anderson, unpublished observations). The lower affinity of binding observed with [3H]A-585539 at increased temperatures is characteristic of thermodynamic discrimination of binding kinetics in which enthalpy-driven processes produce a temperature-dependent shift in apparent affinity, unlike entropy-driven processes that do not exhibit temperature-dependent shifts in affinity (Borea et al., 1998). These observations are consistent with thermodynamic analysis with [3H]cytisine binding that predominantly labels high-affinity α4β2 nAChRs, where it was shown that agonist binding was both enthalpy- and entropy-driven, whereas antagonist binding was totally entropy-driven (Borea et al., 2000).
Displacement of [3H]A-585539 binding by structurally diverse nAChR ligands, including agonists and antagonists, indicated a rank order of selectivity characteristic of α7-selective binding (Table 2; Fig. 4), with A-588539 showing highest affinity. In general, a good correlation between log Ki values for inhibition of [3H]A-585539 binding from both rat brain and human cortex membranes was observed. A notable exception was MLA, which interestingly had a 10-fold greater affinity for rat brain than for human cortex. This may be an additional reason why [3H]MLA binding was not measurable in human cortex in our hands. When displacement of [3H]A-585539 binding in rat brain membranes was compared to displacement of [3H]MLA binding in the same tissue (Table 2; Fig. 5), an excellent correlation was noted. However, compounds showed some 5 to 10-fold higher affinity in displacement of [3H]A-585539 binding relative to [3H]MLA, which may be related to the temperature-dependent shift in affinity with [3H]A-585539.
To further investigate the regions responsible for high-affinity interactions of [3H]A-585539 binding, we assessed its interaction with a chimeric α7 nAChR, the α7/5-HT3 chimera, where the N-terminal domain of the human 5-HT3 receptor was replaced with the N-terminal domain of the human α7 nAChR. Figure 6 shows [3H]A-585539 binding to the α7/5-HT3 chimera where high levels of expression of the chimeric receptor could be detected. The log Ki values for the displacement by a range of ligands at the α7/5-HT3 chimera were highly correlated to the log Ki values obtained in rat brain membranes. In general, incorporation of the N-terminal domain of α7 nAChR alone was sufficient to faithfully reproduce the pharmacology of [3H]A-585539 binding. Interestingly, even though the transmembrane/pore-forming region of 5-HT3 receptor has a similar protein structure and a relatively high degree of homology with the α7 nAChR, the Kd of 0.8 nM represents approximately 10-fold decrement in the affinity of [3H]A-585539 binding. This relatively lower affinity was also translated across the range of nAChR compounds tested, as reflected in the leftward shift of the correlation curve when comparing log Ki values. This implies that regions beyond the N-terminal domain of the α7 nAChR may also be involved in the formation of the binding pocket responsible for high-affinity binding interactions of [3H]A-585539.
The conventional target for α7 nAChR drug discovery has focused on the “agonist”-binding site of nAChRs, with the emergence of several agonist and competitive antagonists. More recently, ligands that are positive allosteric modulators of α7 nAChRs have been identified (Hurst et al., 2005; Gronlein et al., 2007; reviewed in Faghih et al., 2007). The positive allosteric modulators, 5-hydroxyindole and PNU-120596, failed to displace or increase [3H]A-585539 or [3H]MLA binding to brain membranes. This is consistent with the notion that, unlike agonists, positive allosteric modulators do not directly activate or desensitize receptors by interacting with orthosteric binding sites, but instead they enhance the sensitivity and/or efficacy of the receptor during agonist activation (Bertrand and Gopalakrishnan, 2007).
In autoradiography studies, [3H]A-585539 displayed a regional binding pattern similar to that observed with 125I-α-bungarotoxin (Fig. 7). Both ligands labeled comparable regions of the rat brain with highest density of binding in the CA3 area of the hippocampus (CA3) and the superficial gray layer of the superior colliculus. Whereas unlabeled A-585539 fully displaced [3H]A-585539 binding, 1 mM nicotine did not completely displace the [3H]A-585539 label in the CA3 region of the hippocampus, an area that was intensely labeled by [3H]A-585539. It is possible that [3H]A-585539 labeled a small population of nAChRs in this discrete area that are resistant to displacement by nicotine. However, because the concentration of [3H]A-585539 used was 25 μM, which was ∼400-fold above the Kd, it is also possible that small amounts of a localized non-nAChR receptor were labeled. Absolute comparison of the two radioligands is difficult because of the difference in attainable resolution between tritium and 125I. Phosphorimaging of tritium is inferior to film imaging of 125I, both in terms of visualization of structures and duration of exposure. Nonetheless, the obtained images show clearly that the highest levels of [3H]A-585539 binding are observed in regions known to have high density of α7 nAChRs.
In summary, [3H]A-585539 demonstrates high-affinity binding consistent with α7 nAChR pharmacology, a rapid association rate, and a relatively slow dissociation rate—the latter attributes, coupled with low nonspecific binding, make it ideal for filtration assays. It provides an advantage to be able to study agonist-agonist interactions at the α7 nAChR without having to interpret agonist displacement of tenacious antagonists. [3H]A-585539 binding also avoids the pitfalls of working with the pseudo-irreversible [125I]α-Bgt that requires 37°C incubation and the reversible nonspecific binding of [3H]MLA. A key advantage of [3H]A-585539 is that it enables the measurement of α7 nAChRs in a variety of tissues, especially human brain, unlike MLA, and should aid in the further study of ligand interactions with the α7 nAChR subtype under physiological and pathological conditions.
Acknowledgments
We thank Steven Cassar for generating the chimera construct and Kirsten Thorin-Hagene for generating the HEK-293 cell line.
Footnotes
-
This work was supported by Abbott Laboratories. The autoradiography work was supported, in part, by the Swiss National Science Foundation (to D.B.).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.130062.
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; α-Bgt, α-bungarotoxin; MLA, methyllycaconitine; A-585539, (1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azoniabicyclo[2.2.1]heptane; PNU-282987, N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride; PHA-543613, N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide; (±)-AR-R17779, (±)-spiro[1-azabicyclo[2.2.2]octane-3,5′-oxazolidin-2′-one]; A-582941, 2-methyl-5-(6-phenyl-pyridazin-3-yl)-octahydro-pyrrolo[3,4-c]pyrrole; SSR180711, 1,4-diazabicyclo[3.2.2]-noname-4-carboxylic acid, 4-bromophenyl ester; BSS, balanced salt solution; BSA, bovine serum albumen; PEI, polyethylenimine; A-85380, 3-(2(S)-azetidinylmethoxy)pyridine; PNU-120596, 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea; MDL 72222, 3-tropanyl-3,5-dichlorobenzoate; TC-5280, 2(R)-(3-pyridyl)-1-azabicyclo[3.2.2]nonane; varenicline, 7,8,9,10-tetrahydro-6,10-methano-6H-pyrazino(2,3-h)(3)-benzazepine; HPLC, high-performance liquid chromatography; BRL 43694, granisetron; 5-HT3, 5-hydroxytryptamine type-3; [3H]GR65630, 3-(5-methyl-1H-imidazol-4-yl)-1-(1-methyl-3H3-1H-indol-3-yl)-1-propanone.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}