


Abstract
The chronic use of opioids in humans, accompanied by the development of tolerance, is a dangerous phenomenon in its own right. However, chronic opioid use is often made more dangerous by the coconsumption of other substances. It has been observed that the blood level of opioids in postmortem analyses of addicts, who consumed ethanol along with the opioid, was much less than that observed in individuals who died from opioids alone. This relationship between ethanol and opioids led us to investigate the hypothesis that ethanol alters tolerance to opioids. In the present study, we report that ethanol significantly and dose-dependently reduced the antinociceptive tolerance produced by morphine and the cross-tolerance between [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) and morphine in the mouse tail-flick test. The reversal of morphine tolerance was partially blocked by both the gamma receptor blocker bicuculline and by the γ-aminobutyric acid (GABA)B receptor blocker phaclofen and the administration of both inhibitors completely reversed the effects of ethanol on morphine tolerance. Diazepam, like ethanol, decreased morphine tolerance. However, this inhibition was reversed by the GABAA antagonist bicuculline but not by the GABAB antagonist phaclofen. These findings have important implications for individuals who abuse opioids and ethanol as well as suggest a mechanism to reduce the amount of opioid needed in chronic pain treatment.
Introduction
Opioid overdose is a tragedy which is often assumed to be the result of excessive opioid administration alone. However opioid abusers are often polydrug abusers with ethanol being the most commonly detected substance in addition to an opioid. Examination of postmortem toxicological screens makes polydrug use apparent. While there is a very low percentage of opioid-related deaths that result from opioid use alone, the percentage that involves ethanol is much higher. In almost half of heroin-related deaths and about a third of methadone-related deaths ethanol was found to be coabused with the opioid (Ruttenber et al., 1990; Darke and Zador, 1996; Darke and Hall, 2003; Hickman et al., 2007; Oliver et al., 2007).
This analysis points to a role for ethanol in these fatalities; however, the mechanism by which ethanol is potentiating opioid overdose is not clear. An inverse relationship has been documented between the blood ethanol and blood morphine concentrations in postmortem analysis of heroin overdose victims (Darke and Hall, 2003; Hickman et al., 2007; Oliver et al., 2007). Studies in both the United States and Australia have shown that in heroin overdoses where ethanol was present at >100 mg/dl, the morphine concentration in the blood was 60% lower than in subjects where ethanol was present at <100 mg/dl (Ruttenber et al., 1990; Darke et al., 2000).
In addition to the trend for ethanol concentration to have an inverse relationship to the morphine concentration, the morphine concentrations themselves were often found to be lower than would have typically been predicted to be lethal given the level of opioid consumption in these highly tolerant individuals. The levels of opioid detected postmortem were found to be equal to or lower than the levels found in living active heroin users (Tagliaro et al., 1998; Darke et al., 2002; Hickman et al., 2007). These findings indicate that there may be a drug interaction responsible for the increased risk of overdose at lower opioid levels, and that the interaction between ethanol and opioids may be the main drug interaction responsible for these cases of polydrug overdose (Darke and Zador, 1996; Darke and Hall, 2003).
The ability of high doses of ethanol to cause respiratory depression has long been understood; however, the levels found in these postmortem studies were not high enough to cause lethal respiratory depression. This indicates that it may be the interaction of ethanol and opioids that results in fatal respiratory depression (Ruttenber et al., 1990; Darke and Hall, 2003).
Ethanol is not the only substance that has this effect when combined with opioids; benzodiazepines have also been found to pose a significant risk for opioid overdose particularly in methadone-related deaths (Oliver et al., 2007). However, these two drug classes are not identical in their interactions with opioids. The amount of opioid present in fatal overdoses when benzodiazepines are also present is not lower than the amount typically found in overdoses where opioids are the only drug found postmortem. Therefore the ability of ethanol to reduce the amount of opioids required for overdose indicates a unique mechanism that warrants further investigation to better understand how such deaths can be prevented. Establishing the difference in the mechanisms by which ethanol and benzodiazepines, such as diazepam, affect opioid tolerance is crucial to helping to prevent accidental overdoses in opioid-tolerant populations.
There are a number of ways in which ethanol may be increasing the effects of opioids (Hickman et al., 2008). It is possible that an additive or synergistic relationship between the two depressant drugs is responsible for the negative consequences of their concomitant use. However it is also possible that ethanol may be altering the mechanism of opioid tolerance at the cellular level. We hypothesize that ethanol is altering opioid tolerance at the cellular level as a direct result of the effects of ethanol on the γ-aminobutyric acid (GABA)ergic system either at the level of GABA release or GABA receptors. Enhancement of GABA receptor signaling by ethanol leading to downstream inhibition may be the cause of ethanol’s effects on opioid tolerance.
The purpose of this study was to determine the receptors involved in the mechanism by which ethanol and the benzodiazepine diazepam reverse tolerance to opioids. Antinociception, as measured by the rodent tail-flick assay, was chosen for these studies because it has been shown to be a good predictor of antinociception and tolerance for a wide range of compounds in humans. Further, our group has used this assay to investigate extensively the mechanism of opioid tolerance (Smith et al., 2007; Hull et al., 2010).
Materials and Methods
Drugs and Chemicals.
Morphine sulfate, 75-mg morphine pellets and placebo pellets were obtained from the National Institutes of Health National Institute on Drug Abuse (Bethesda, MD). The morphine sulfate was dissolved in pyrogen-free isotonic saline (Hospira, Lake Forest, IL). Ethanol was obtained from AAPER Ethanol and Chemical Co. (Shelbyville, KY) and was diluted with pyrogen-free isotonic saline. [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) was obtained from Tocris Bioscience (Ellisville, MI). Bicuculline and phaclofen were obtained from Biomol Research Laboratories Inc. (Plymouth Meeting, PA) Phaclofen and DAMGO were dissolved in ddH2O; bicuculline was dissolved in dimethylsulfoxide. The final concentration of dimethylsulfoxide was 10%. Diazepam was obtained from Hoffmann-La Roche, Inc. (Nutley, NJ) and dissolved in ddH2O. Solutions made in ddH2O were made for i.c.v. injections and so were not pH balanced to limit additional ions in the solutions.
Animals.
Male Swiss Webster mice (Harlan Laboratories, Indianapolis, IN) weighing 25–30 g were housed six to a cage in animal care quarters and maintained at 22 ± 2°C on a 12-hour light-dark cycle. Food and water were available ad libitum. The mice were brought to the test room (22 ± 2°C, 12-hour light-dark cycle), marked for identification, and allowed 18 hours to recover from transport and handling. For all experiments at each dose/experimental group n = 8 animals. Protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Commonwealth University Medical Center and comply with the recommendations of the International Association for the Study of Pain (IASP).
Tail Immersion Test.
The warm-water tail immersion test was performed according to Coderre and Rollman (1983) using a water bath with the temperature maintained at 56 ± 0.1°C. Before injecting the mice, a baseline (control) latency was determined. Only mice with a control reaction time from 2 to 4 seconds were used. The average baseline latency for these experiments was 3.0 ± 0.1 seconds. The test latency after morphine treatment was assessed at 30 minutes with a 10-second maximum cut-off time imposed to prevent tissue damage. Antinociception was quantified according to the method of Harris and Pierson (1964) as the percentage of maximum possible effect (%MPE), which was calculated as: %MPE = [(test latency – control latency) / (10 – control latency)] × 100. Percent MPE was calculated for each mouse using at least eight mice per dose of drug.
Intracerebroventricular Injections.
Intracerebroventricular injections were performed as described by Pedigo et al. (1975). Mice were anesthetized with 2.5% isoflurane and a horizontal incision was made in the scalp. A needle was inserted to a depth of 3 mm into the lateral ventrical (2 mm rostral and 2 mm lateral at a 45° angle from the bregma). At intervals, 5-μl injections of drug or vehicle were made using this guide insertion to the same depth using a needle with a guard in nonanesthetized animals. The extensive experience of this laboratory has led to a method in training for this procedure that results in injections by this route of administration having greater than 95% accuracy (Smith et al., 1999; Gabra et al., 2008; Hull et al., 2010). This training method involves 5-μl i.c.v. injections of methylene blue until the handler is able to meet the accuracy requirements. In control experiments the accuracy of the injections is determined through macroscopic examinations of the brains of the animals as described in Pedigo et al. (1975). A marker cannot be administered with the drug injection due to potential chemical interactions, so proper training in this technique is required. Animals undergo the anesthetized surgery in the morning of the experiment and are then injected with drug at intervals indicated in the text without additional anesthesia. Immediately after testing, the animals were euthanized to minimize any type of distress, according to IACUC guidelines.
72-Hour Morphine Tolerance Model.
A 75-mg morphine or placebo pellet was implanted according to Way et al. (1969). Mice were anesthetized with 2.5% isoflurane before shaving the hair around the base of the neck. The skin was cleansed with 10% providone iodine (General Medical Corp., Prichard, WV) and rinsed with alcohol before making a 1-cm horizontal incision at the base of the neck. The underlying subcutaneous space toward the dorsal flanks was opened using a sterile glass rod. Maintenance of a stringent aseptic surgical field minimized any potential contamination of the pellet, incision, and subcutaneous space. A placebo pellet or 75-mg morphine pellet was inserted in the space before closing the site with Clay Adams Brand, MikRon AutoClip 9-mm Wound Clips (Becton Dickinson, Sparks, MD) and again applying iodine to the surface. The animals were allowed to recover in their home cages where they remained throughout the experiment.
7-Hour DAMGO/Morphine Cross-Tolerance Model.
A 7-hour antinociceptive tolerance to DAMGO was developed as follows. Mice were injected intracerebroventricularly once every hour (total of seven injections) with the minimal dose of DAMGO that results in maximal analgesia in naïve mice in the tail immersion test. One hour after the final dose, mice were administered the ethanol or vehicle by intraperitoneal injection and 30 minutes later were challenged with various subcutaneous doses of morphine to construct dose-response curves for calculation of ED50 values and potency ratios.
Statistical Analysis.
Opioid dose-response curves were generated for calculation of ED50 values using least-squares linear regression analysis followed by calculation of 95% confidence limits (95% CL) by the method of Bliss (1967). Tests for parallelism were conducted before calculating the potency-ratio values with 95% CL by the method of Colquhoun (1971), who notes that a potency-ratio value of greater than one, with the lower 95% CL greater than one, is considered a significant difference in potency between groups.
Results
Effects of Acute Administration of Ethanol in Naïve Mice.
Ethanol was administered at doses of 0.1, 1, and 2 g/kg i.p., and mice were monitored over a 3-hour period for behavioral changes and assessed in the warm-water tail immersion test at 30-minute intervals over 2 hours. No behavioral changes were observed with all three doses. A slight analgesic effect (%MPE = 30%) was observed in mice treated with 2 g/kg ethanol at 60 and 90 minutes postinjection and the effect disappeared at 120 minutes. The 0.1- and 1-g/kg doses were inert (unpublished data).
Reversal of Morphine Antinociceptive Tolerance with Ethanol.
Experiments were conducted 72 hours following morphine or placebo pellet implantation. Baseline latencies were obtained in the tail immersion test before i.p. administration of ethanol (0.01, 0.1, or 1 g/kg). Thirty minutes later, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios (Fig. 1; Supplemental Table 1). Ethanol dose dependently reversed morphine tolerance.

Ethanol reversal of morphine tolerance. Ethanol (Alc) dose dependently reversed 72-hour morphine tolerance. Each data point represents eight mice. Animals were pelleted with placebo or morphine pellets for 72 hours, then injected with ethanol or vehicle i.p., and then 30 minutes later with morphine s.c. After that tolerance was determined by the tail immersion test. Various doses of morphine were used for construction of dose-response curves for calculation of ED50 values and potency ratios. MP, morphine pellet; PP, placebo pellet.
Reversal of Morphine/DAMGO Cross-Tolerance with Ethanol.
Mice were injected i.c.v. with ddH2O or DAMGO (0.05 nmol, a dose that results in 100% MPE in the tail immersion test) at times 0, 1, 2, 3, 4, 5, and 6 hours (total of seven injections). One hour after the last injection, mice were injected with isotonic saline or ethanol (1 g/kg i.p.). Thirty minutes later, mice were challenged with various doses of morphine s.c. and their tail immersion latencies were determined for construction of dose-response curves as well as calculation of ED50 values and potency ratios. DAMGO-treated animals generated the morphine ED50 value of 21.2 mg/kg (19.3, 23.4 95% CL) compared with 4.7 mg/kg (4.3, 5.2 95% CL) in the vehicle-treated animals. The animals treated with DAMGO showed 4.5-fold (3.9, 5.1 95% CL) tolerance compared with the control group. The administration of ethanol in DAMGO-treated animals before the morphine challenge generated an ED50 of 5.0 mg/kg (4.0, 6.3 95% CL). Animals administered ddH20 then ethanol before being challenged with morphine generated an ED50 of 5.1 mg/kg (4.9, 5.5 95% CL). When compared with the DAMGO-treated animals who also received ethanol, the potency ratio was 1.0 (0.9, 1.1 95% CL) indicating a full reversal of tolerance (Fig. 2).

Ethanol reversal of morphine/DAMGO cross-tolerance. Ethanol dose dependently reversed 7-hour morphine/DAMGO cross-tolerance. Each data point represents eight mice. Animals were treated with DAMGO i.c.v. for 7 hours, then injected with ethanol or vehicle i.p., and then 30 minutes later with morphine s.c.; 30 minutes after that tolerance was determined by the tail immersion test. Various doses of the morphine were used for construction of dose-response curves for calculation of ED50 values and potency ratios.
Acute Effects of Bicuculline in Naïve Mice.
Bicuculline was administered at doses of 1, 5, 10, 20, and 40 mg/kg i.p. and mice were monitored over a 3-hour period for behavioral changes and assessed in the warm-water tail immersion test at 30-minute intervals over 2 hours. No behavioral changes were observed with all three doses. No analgesic or hyperalgesic effects were observed. All doses tested were inert. Bicuculline did not potentiate or decrease acute morphine antinociception (unpublished data).
Acute Effects of Phaclofen in Naïve Mice.
Phaclofen was administered at doses of 0.1, 1, 10, and 30 mg/kg i.p. and mice were monitored over a 3-hour period for behavioral changes and assessed in the warm-water tail immersion test at 30-minute intervals over 2 hours. No behavioral changes were observed with all three doses. No analgesic or hyperalgesic effects were observed. All doses tested were inert. Phaclofen did not potentiate or decrease morphine antinociception (unpublished data).
Experiments Following the 72-Hour Morphine Tolerance Model.
For both ethanol- and diazepam-induced reversal-of-tolerance experiments, tolerance was established using the 72-hour morphine tolerance model and in all groups n = 8. Animals were surgically implanted with either placebo pellets or morphine pellets for 72 hours and then baseline latencies were obtained in the tail immersion test. Following the baseline testing the experiments continued as described in the following sections.
Effects of Bicuculline on Ethanol-Induced Reversal of Morphine Antinociceptive Tolerance in Mice.
In mice chronically treated with morphine, bicuculline was administered i.p. (1, 5, or 20 mg/kg), followed 5 minutes later by ethanol (1 g/kg i.p.). Thirty minutes later, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios (Fig. 3A; Supplemental Table 1). Ethanol reversal of morphine tolerance was dose dependently inhibited by bicuculline, but full reversal was not reached.

Effects of bicuculline or phaclofen on ethanol reversal of morphine tolerance. Bicuculline (Bic) (A) and phaclofen (Phac) (B) were able to inhibit only partially the ethanol (Alc) reversal of 72-hour morphine tolerance in a dose-dependent manner, but when combined (C) were able to fully inhibit ethanol’s reversal of 72-hour morphine tolerance. Each data point represents eight mice. Animals were injected with bicuculline i.p. and/or phaclofen i.p. 5 minutes before ethanol i.p., then 30 minutes later various doses of morphine s.c. were used for construction of dose-response curves for calculation of ED50 values and potency ratios. MP, morphine pellet; PP, placebo pellet.
Effects of Phaclofen on Ethanol-Induced Reversal of Morphine Antinociceptive Tolerance in Mice.
Following the 72-hour morphine-pellet implantation, phaclofen was administered i.p. (1, 10, or 30 mg/kg) followed 5 minutes later by ethanol (1 g/kg i.p.). Thirty minutes later, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios (Fig. 3B; Supplemental Table 1). Ethanol reversal of morphine tolerance was dose dependently inhibited by phaclofen, but full reversal was not reached.
Effects of Combined Administration of Bicuculline and Phaclofen on Ethanol-Induced Reversal of Morphine Antinociceptive Tolerance in Mice.
Finally in the tolerant animals, both bicuculline (40 mg/kg) and phaclofen (30 mg/kg) were administered i.p. followed 5 minutes later by ethanol (1 g/kg i.p.). Thirty minutes later, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios (Fig. 3 C; Supplemental Table 1). Bicuculline and phaclofen in combination were able to fully inhibit the reversal of morphine tolerance by ethanol.
Effects of Acute Administration of Diazepam in Drug-Naïve Mice.
Diazepam was administered at doses of 0.1, 0.25, 0.5, 1, and 5 mg/kg i.p. and mice were monitored over a 3-hour period for behavioral changes and assessed in the warm-water tail immersion test at 30-minute intervals over 2 hours. No behavioral changes were observed at any of these doses. No antinociceptive effects were observed at any of these doses. The same doses (0.1, 0.25, 0.5, 1, or 5 mg/kg i.p.) were administered in a different group of naïve mice and thirty minutes later the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios. Diazepam at 1 mg/kg and 5 mg/kg significantly enhanced the antinociceptive effect of morphine, altering the morphine ED50 to 3.2 mg/mouse (2.9–3.5 95% CL) for the 1-mg/kg diazepam dose, and altering the morphine ED50 to 1.8 mg/mouse (1.3–2.6 95% CL) for the 5-mg/kg diazepam dose (unpublished data).
Reversal of Morphine Antinociceptive Tolerance with Diazepam.
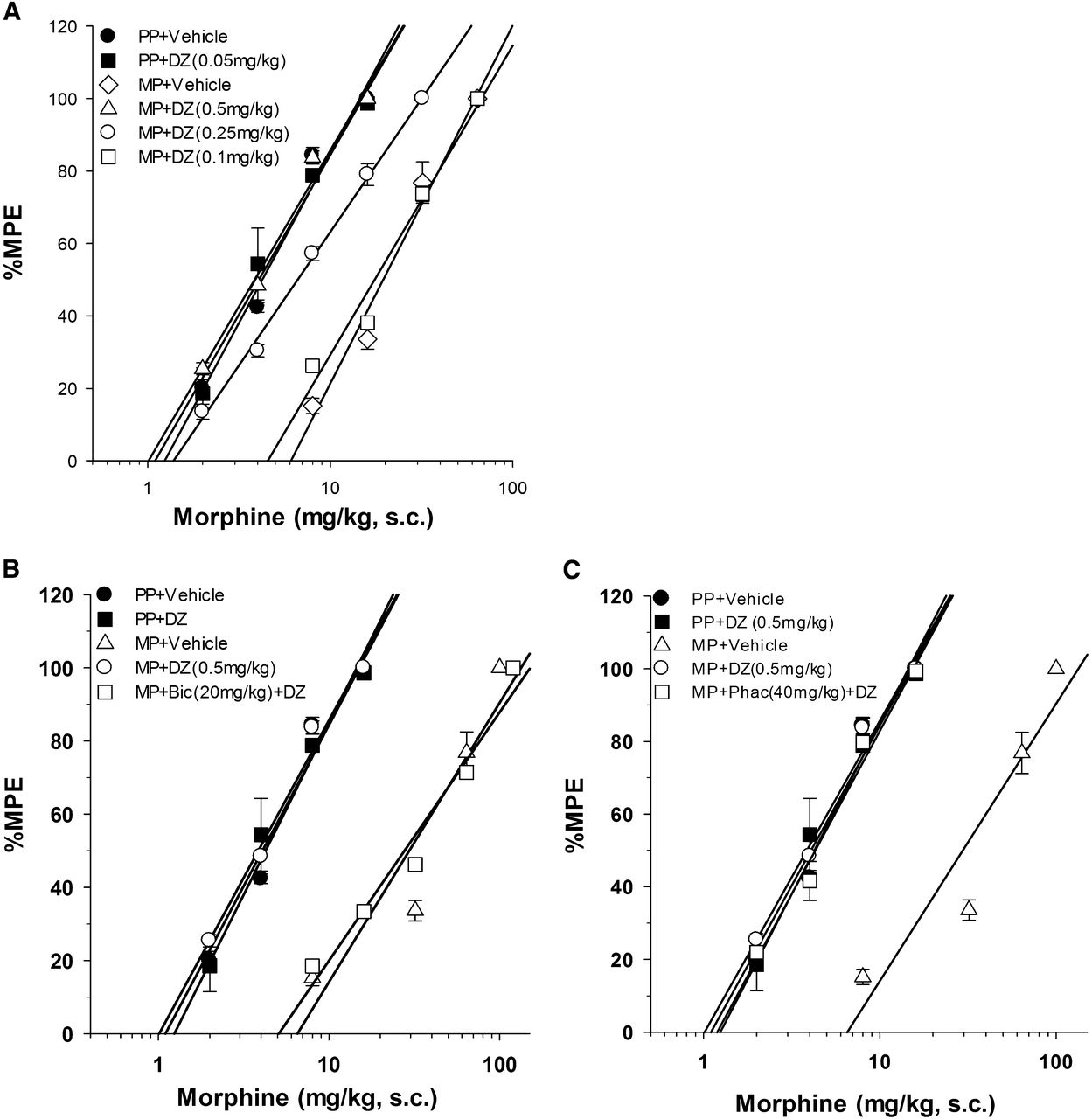
Following the development of tolerance, diazepam was administered i.p. (0.1, 0.25, or 0.5 mg/kg). Thirty minutes later, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios (Fig. 4A; Supplemental Table 2). Diazepam demonstrated a dose-dependent ability to reverse morphine tolerance, with full reversal achieved with the highest diazepam dose.

Effects of diazepam (DZ) on 72-hour morphine tolerance (A) and the effects of bicuculline (Bic) (B) and phaclofen (Phac) (C) on ethanol reversal of 72-hour morphine tolerance. Bicuculline (B) was able to fully inhibit ethanol’s reversal of 72-hour morphine tolerance while phaclofen (C) was not. Each data point represents eight mice. Animals were injected with bicuculline i.p. or phaclofen i.p. 5 minutes before ethanol i.p., then 30 minutes later various doses of morphine s.c. were used for construction of dose-response curves for calculation of ED50 values and potency ratios. MP, morphine pellet; PP, placebo pellet.
Effects of Bicuculline on Diazepam-Induced Reversal of Morphine Antinociceptive Tolerance in Mice.
After morphine tolerance was developed, bicuculline was administered i.p. (20 mg/kg) followed, 5 minutes later, by diazepam i.p. (0.5 mg/kg). Thirty minutes later, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios (Fig. 4B; Supplemental Table 2). Bicuculline fully inhibited the reversal of morphine tolerance by diazepam.
Effects of Phaclofen on Diazepam-Induced Reversal of Morphine Antinociceptive Tolerance in Mice.
Again following the development of morphine tolerance, phaclofen was administered i.p. (40 mg/kg), followed 5 minutes later by diazepam i.p. (0.5 mg/kg). Thirty minutes later, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the ED50 values and potency ratios (Fig. 4C; Supplemental Table 2). Phaclofen was unable to fully inhibit the reversal of morphine tolerance by diazepam.
Discussion
Opioid tolerance is a complicated and multifaceted phenomenon that is not well understood at the cellular, tissue, or organism levels. It is also well established that tolerance develops to differing effects of opiates in the same individual at different rates and to different degrees. Further, since humans who become tolerant and then addicted to opiates usually are consuming other substances as well, it is important to investigate the effects of these other substances on opioid tolerance. In the present study we have investigated the effects of ethanol, the most widely coabused substance along with opiates, on opioid antinociceptive tolerance. Our laboratories have used studies at different levels to increase our understanding of these complex mechanisms. In the present study we evaluated the mechanism of tolerance in the whole animal.
Doses of ethanol that were inactive alone dose-dependently reversed morphine-induced antinociceptive tolerance. The generality of this effect of ethanol on opioids was demonstrated when we found that it also reversed the cross-tolerance produced in animals by the chronic administration of DAMGO challenged with doses of morphine. This observation of the ability of ethanol to reverse the cross-tolerance among opioids is very important considering the high level of human use of multiple prescription opioids as well as the coabuse of heroin and these agents. DAMGO is not an opioid used in human populations due to its inability to pass the blood brain barrier. However, because DAMGO is the most efficacious opioid agonist (McPherson et al., 2010) we feel that using this compound in cross-tolerance studies is justified. Tolerance to high efficacy opioids has been shown by our laboratory and others to be supported by different kinases compared with lower efficacy opioids (Hull et al., 2010; Williams et al., 2013). Demonstrating cross-tolerance between two opioids of very different efficacies illustrates a much fuller representation of cross-tolerance than that shown by experiments identifying cross-tolerance between two opioids of more similar efficacies.
We next determined the neurochemical mechanism of this reversal. We investigated whether ethanol reversed morphine tolerance through its interaction with one or more types of GABA receptors. We found that both bicuculline and phaclofen were able to partially inhibit ethanol’s reversal of morphine’s tolerance, but even at the highest tolerated dose neither inhibitor fully reversed ethanol’s effect. When administered in combination they fully inhibited ethanol’s reversal of morphine tolerance indicating that ethanol’s reversal of morphine tolerance was due to ethanol’s actions at both the GABAA and GABAB receptors.
We investigated whether diazepam, another drug which alters the function of GABA receptors, would also reverse morphine tolerance. We found that diazepam also reversed morphine tolerance in a dose-dependent manner. However, further studies showed that the mechanism of this reversal differed from that of ethanol. The diazepam reversal was fully inhibited by bicuculline, a GABAA antagonist, but not by phaclofen, a GABAB antagonist, indicating that, as expected, the benzodiazepine was responsible for opioid tolerance reversal through actions at the GABAA receptor.
It is our hypothesis that the actions of ethanol at inhibitory GABA receptors result in either an enhancement of GABAergic signaling or GABA release leading to a reversal of opioid tolerance. The precise mechanism and neuronal circuitry involved remain to be elucidated. This tolerance-reversal effect could be responsible in part for the high incidence of polydrug use among opioid abusers. The addition of chemicals that act on the GABA receptors could increase the rewarding effects of opioid abuse by reducing tolerance. Increasing the effectiveness of the main drug of abuse would be more of an impetus for concomitant use than an additive or synergistic depressant effect of the drugs.
The interaction between opioids, the GABAA receptor, and benzodiazepines has been reported in the literature with some disagreement. Our findings that diazepam alone had no antinociceptive activity at any dose up to 5 mg/kg is in conflict with several observations in the literature which indicate an antinociceptive effect of benzodiazepines (Sierralta and Miranda, 1992; Jimenez-Velazquez et al., 2008; Jimenez-Velazquez et al., 2010) but in agreement with others who found no antinociceptive effect of benzodiazepines (Rodgers and Randall, 1987; Rosland et al., 1987; Moreau and Pieri, 1988). In addition to the effects of benzodiazepines on antinociception alone, there are as well mixed findings on the interactions between this class of drugs and opioid actions. Intrathecal administration of midazolam was found to enhance morphine antinociception in a number of studies (Bergman et al., 1988; Yanez et al., 1990; Rattan et al., 1991). On the other hand, other studies have demonstrated that benzodiazepines attenuate the antinociceptive effect of morphine (Mantegazza et al., 1982; Rosland et al., 1990). We demonstrated that diazepam administered i.p. at doses of 1 and 5 mg/kg in mice significantly enhanced the antinociceptive effect of morphine. The possible reasons for these contradictory findings include the chemical agent (diazepam versus midazolam versus alpreazolam), the species (mouse versus rat), the analgesic test (writhing versus tail immersion), as well as the route of administration (i.t. versus i.c.v. versus i.p.). These mixed findings are of interest and may be due to further complexities in this interaction that are specific to the areas of binding on the GABAA receptor, the systematic distribution of the drug, or in species differences.
Morphine tolerance is a multifaceted mechanism which involves many different cellular and neuronal systems. The high prevalence of overdose deaths involving lower levels of heroin among polydrug users indicates that tolerance is lowered in those ingesting both ethanol and heroine concomitantly. We found that ethanol can reverse morphine tolerance in the whole animal and that this action involved both the GABAA and GABAB receptors. These results have clinical applications both for drug abusers and for chronic pain patients. Education about the risks of concomitant use of ethanol and heroin should be disseminated among the drug abusing population so that the risks are clear. Additionally, low doses of ethanol in combination with opioid pain treatment may allow a reduction of antinociceptive tolerance so that a smaller dose of opioid may be used allowing a reduction in problematic side effects for chronic pain patients.
Initially it was somewhat surprising to find that both the GABAB antagonist phaclofen and the GABAA antagonist bicuculline partially inhibited the effect of ethanol, and that the combination of the two was necessary for complete reversal of ethanol’s effects on morphine tolerance. Whether these effects are due to a direct effect of ethanol on these receptors or through a modulation of GABA release remains to be seen. The GABAA receptor has been more closely linked in vitro with ethanol’s actions (Mehta and Ticku, 1990; Frye et al., 1991); however, there is evidence in the literature that these two receptors work together in the actions of ethanol (Ogata, 1987; Kaneko and Hicks, 1988; Hahner et al., 1991). There are experiments that indicate that in vivo GABAB receptors are involved in ethanol-induced locomotor stimulation, hypothermia, motor incoordination, and narcosis (Allan and Harris, 1989; Humeniuk et al., 1993; Kruse et al., 2012). These papers suggest that some of the effects of ethanol, especially at low doses, might be mediated by agonist effects (or at least positive allosteric modulation effects) at GABAB receptors, and that there also appears to be some synergy between the effects at GABAA and GABAB receptors. There have also been investigations into how modulation of the GABAB receptor may have potential in the treatment of alcohol abuse in humans (Agabio et al., 2012; Loi et al., 2013). Taken together these findings make clear that there is a role for the GABAB receptor in the effects of ethanol, but what specific role it plays along with the GABAA receptor has yet to be fully elucidated.
These findings are an important step toward understanding the complex interactions between drugs which are often abused together with devastating results. Further work applying these findings to the respiratory depressive effects is the next step in understanding polydrug-related deaths.
Acknowledgments
The authors thank Joshua Seager and David Stevens for technical help performing the experiments needed for this study, and Matthew Hickman and Anne Lingford-Hughes for helpful discussions.
Authorship Contributions
Participated in research design: Henderson, Dewey, Gabra, Bailey.
Conducted experiments: Gabra.
Performed data analysis: Hull, Gabra.
Wrote or contributed to the writing of the manuscript: Hull, Bailey, Henderson, Dewey.
Footnotes
- Received December 6, 2012.
- Accepted March 21, 2013.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA007027 and DA020836].
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- %MPE
- percentage of maximum possible effect
- Bicuculline
- (6R)-6-[(5S)-6-methyl-5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinolin-5-yl]furo[3,4-e][1,3]benzodioxol-8(6H)-one
- CL
- confidence limit
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- diazepam
- 7-chloro-1,3-dihydro-1-methyl-5-phenyl-1,4-benzodiazepin-2(3H)-one
- GABA
- γ-aminobutyric acid
- methylene blue
- 3,7-bis(dimethylamino)-phenothiazin-5-ium chloride
- phaclofen
- [3-amino-2-(4-chlorophenyl)propyl]phosphonic acid
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}