


Abstract
Drugs that antagonize D2-like receptors are effective antipsychotics, but the debilitating movement disorder side effects associated with these drugs cannot be dissociated from dopamine receptor blockade. The “atypical” antipsychotics have a lower propensity to cause extrapyramidal symptoms (EPS), but the molecular basis for this is not fully understood nor is the impact of inverse agonism upon their clinical properties. Using a cell-based functional assay, we demonstrate that overexpression of Gαo induces constitutive activity in the human D2-like receptors (D2, D3, and D4). A large collection of typical and atypical antipsychotics was profiled for activity at these receptors. Virtually all were D2 and D3 inverse agonists, whereas none was D4 inverse agonist, although many were potent D4 antagonists. The inverse agonist activity of haloperidol at D2 and D3 receptors could be reversed by mesoridazine demonstrating that there were significant differences in the degrees of inverse agonism among the compounds tested. Aripiprazole and the principle active metabolite of clozapine NDMC [8-chloro-11-(1-piperazinyl)-5H-dibenzo [b,e] [1,4] diazepine] were identified as partial agonists at D2 and D3 receptors, although clozapine itself was an inverse agonist at these receptors. NDMC-induced functional responses could be reversed by clozapine. It is proposed that the low incidence of EPS associated with clozapine and aripiprazole used may be due, in part, to these partial agonist properties of NDMC and aripiprazole and that bypassing clozapine blockade through direct administration of NDMC to patients may provide superior antipsychotic efficacy.
Blockade of dopamine receptors is the key mechanistic feature of antipsychotic medications believed to mediate many of their therapeutic benefits, but it is also responsible for many of the debilitating side effects associated with these drugs, particularly the extrapyramidal side effects (EPS) and hyperprolactinemia (Strange, 2001). The antipsychotics are divided into two major classes, the first-generation D2 blockers (henceforth typicals) and the second-generation serotonin/dopamine antagonists (henceforth atypicals). The typical antipsychotics, exemplified by drugs such as chlorpromazine and haloperidol, were the first generation of compounds used to treat schizophrenia and tend to have uniformly high affinity for D2 dopamine receptors and produce a high incidence of EPS. There is a strong correlation among D2 affinity, clinical dose, clinical efficacy, and incidence of EPS for these agents (Creese et al., 1976; Seeman et al., 1976).
The atypical antipsychotics are distinguished by their lower incidence of EPS compared with the typical antipsychotics. As a group, the atypical drugs are much more heterogenous than the typical antipsychotics; thus, it has been difficult to find a common mechanism of action explaining their distinct clinical profiles. The atypical drugs have varied affinities for D2 receptors, and they produce a variety of side effects, including metabolic disorders, weight gain, and cardiovascular effects. Many explanations have been proposed to explain the molecular basis of atypicality, including affinity differences for D2 receptors (Kapur and Seeman, 2001), 5HT1A agonism (Serretti et al., 2004), and selectivity for D3 (Schwartz et al., 2000) and D4 receptors (Jardemark et al., 2002). Furthermore, all of the atypical antipsychotics are potent 5HT2A inverse agonists (Meltzer et al., 1989; Weiner et al., 2001).
Dopamine supersensitivity caused by chronic blockade of dopamine receptors has been proposed to explain the propensity of antipsychotics to cause EPS or tardive dyskinesia (TD) (Casey 2000). Although this hypothesis has concentrated on D2 receptor occupancy, several antipsychotics are inverse agonists at D2 receptors (Akam and Strange, 2004) and it has been demonstrated that inverse agonists cause recruitment and up-regulation of G-protein-coupled receptors (GPCRs) to the cell surface (Daeffler and Landry, 2000). Therefore, D2 partial agonists may be particularly useful for treating schizophrenia because they would not up-regulate dopamine receptor tone but would still block the actions of dopamine at D2 receptors (Tamminga and Carlsson, 2002). Consistent with this concept, aripiprazole, an atypical agent with partial agonist activity at D2 receptors (Burris et al., 2002) and D3 receptors (Shapiro et al., 2003), in contrast to haloperidol, seems to have a low liability for inducing EPS/TD, does not elevate serum prolactin levels (Harrison and Perry, 2004), and does not up-regulate either D2 binding sites or D2 mRNA (Inoue et al., 1997).
These findings emphasize the importance of defining efficacy, as well as the affinity of compounds for individual receptor subtypes to fully understand their molecular basis of clinical action. However, a comprehensive efficacy profile of compounds that target D2-like receptors is lacking. The impact of inverse agonism upon the clinical properties of these compounds is not fully understood, in part because the low sensitivity of many functional assays prevents reliable measurements of constitutive activity, and precludes functional assessment of receptors that signal poorly in heterologous systems such as D3 (see Newman-Tancredi et al., 1999) and D4 receptors (see Oldenhof et al., 1998; Gazi et al., 2000). Previously, we have shown that overexpression of G-proteins increases assay sensitivity and induces constitutive activity of GPCRs (Burstein et al., 1997). We now report that overexpression of Gαo dramatically improves the dynamic range of D3- and D4-mediated functional responses and induces constitutive activity in D2, D3, and D4 receptors. A comprehensive pharmacological profile of compounds that target D2-like receptors is described, indicating that efficacy at D2 is another factor that may affect certain clinical aspects of antipsychotic drug action.
Materials and Methods
Materials. NIH-3T3 cells were from ATCC CRL 1658 (American Type Culture Collection, Manassas, VA). O-Nitrophenyl-β-d-galactopyranoside and Nonidet P-40 were from Sigma-Aldrich (St. Louis, MO). Tissue culture media used were Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 4500 mg/l glucose, 4 nM l-glutamine, 50 U/ml penicillin G, 50 U/ml streptomycin, and 10% calf serum. Ninety-six-well tissue culture dishes were from Falcon: BD Biosciences Discovery Labware (Bedford, MA). Hanks' balanced salt solution and trypsin EDTA were from Invitrogen.
Drugs. All compounds for receptor selection and amplification technology (R-SAT) studies were solubilized as 10 mM stock solutions in either water or dimethyl sulfoxide. Working dilutions were made from 50 μM solutions in DMEM with 25% Ultraculture (Cambrex Bio Science Walkersville, Walkersville, MD) and 1% penicillin-streptomycin-glutamine. All of the compounds were obtained from Sigma/RBI (Natick, MA), with the exception of the following: spiperone and remoxipride (Tocris Cookson Inc., Ellisville, MO); sultopride (IC Rom, Milan, Italy); moperone and bromperidol (Janssen Pharmaceuticals, Antwerp, Belgium); sertindole, trans-flupenthixol, and molindone (Lundbeck A/S, Copenhagen, Denmark); and melperone (Cilag, Schaffhausen, Switzerland), whereas NDMC, sulforidazine, and 9-hydroxy-risperidone were synthesized by ACADIA Pharmaceuticals.
Cell Culture. NIH-3T3 cells (ATCC number CRL 1658) were incubated at 37°C in a humidified atmosphere (5% CO2) in Dulbecco's modified Eagle's tissue culture medium.
Constructs. The D2 (short form) and D3 receptors have been described previously (Weiner et al., 2001). The D3 receptor contained a mutation (E275V), which was repaired by QuikChange mutagenesis (Stratagene, La Jolla, CA). The D4 receptor used in this study (variant 4.2) was cloned by polymerase chain reaction using Pfu Turbo (Stratagene) using primers derived from the GenBank accession entry L12398. The ras/rap chimera used in these studies [ras/rap1B(AA)] has been described previously (Ma et al., 2004). Human regulator of G-protein signaling 1 (RGS1) was cloned by polymerase chain reaction using oligonucleotides derived from the GenBank accession entry XM_042967. The adenylyl cyclase type II (AC2) construct used in these studies has been described previously by Ma et al. (2004). The Gαo construct was described previously (Jones and Reed, 1987). cDNAs encoding bovine β1 and γ2 were generous gifts from B. Simonds (Metabolic Diseases Branch/National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD). All of the clones were subcloned into the pSI vector (Promega, Madison, WI) and sequence-verified before use.
Functional Assays. R-SAT assays were performed as described previously (Ma et al., 2004) with the following modifications. In brief, cells were plated one day before transfection using 7 × 103 cells in 0.1 ml of media/well of a 96-well plate. Cells were transiently transfected with 5 ng/well receptor DNA, 20 ng/well ras/rap1B(AA), 2 ng/well AC2, and 30 ng/well pSI-β-galactosidase (Promega) per well of a 96-well plate using Polyfect (QIAGEN, Valencia, CA) according to the manufacturer's instructions. The use of ras/rap1B(AA) and AC2 was found to enable responses of Gs and Gi-coupled GPCRs in this functional assay (Ma et al., 2004). Where indicated, 5 ng/well each of Gαo, Gβ1, Gγ2, and RGS1 were cotransfected. The β-γ subunits Gβ1 and Gγ2 were cotransfected to enhance the effects of Gαo (Fishburn et al., 1999). One day after transfection, medium was changed and cells were combined with ligands in DMEM supplemented with 25% Ultraculture synthetic supplement instead of calf serum to a final volume of 200 μl/well. After 5 days in culture, β-galactosidase activity was measured. The media were aspirated from the wells, and the cells were rinsed with phosphate-buffered saline, pH 7.4. 200 μl of phosphate-buffered saline with 3.5 mM ONPG and 0.5% Nonidet P-40 were added to each well, and the 96-well plates were incubated at room temperature. After 3 h, the plates were read at 420 nm on a plate reader (Bio-Tek EL 310 or Molecular Devices, Sunnyvale, CA). All of the data were analyzed using the computer programs Excel Fit and Prism software (GraphPad Software Inc., San Diego, CA). Data for inverse agonism are reported as negative log values (pIC50). Functional antagonist IC50 data were adjusted for agonist occupancy using the Cheng-Prusoff equation Ki = IC50/{1+[agonist]/EC50agonist} to derive Ki values.
Binding Studies. Binding studies were carried out with [3H]raclopride (D2 and D3) (87 Ci/mmol; GE Healthcare, Little Chalfont, Buckinghamshire, UK) and [3H]spiperone (D4) (98 Ci/mmol; GE Healthcare) using membranes of NIH-3T3 cells transiently transfected as described above with D2, D3, or D4 (10 μg ea/15-cm dish) either with or without Gαo (10 μg) and prepared as described previously (Ma et al., 2004) using increasing concentrations of radiolabeled ligand for saturation binding experiments. Binding reactions were terminated by filtration through type B glass fiber filters (MultiScreen Harvest plates; Millipore Corporation, Billerica, MA) presoaked for 30 min in 0.1% polyethyleneimine. Nonspecific binding was determined using 1 μM haloperidol (D2 and D3) or 1 μM spiperone (D4).
Results
Using a functional assay based on cellular proliferation (R-SAT) (see Ma et al., 2004), we have shown that overexpression of G-proteins augments both agonist-induced and constitutive responses of GPCRs and enables detection of inverse agonism (Burstein et al., 1997). In the presence of the D2/D3/D4-agonist pergolide, the D2 dopamine receptor mediates robust agonist responses; however, only weak responses of D3 and no significant response of D4 to pergolide were observed (Fig. 1 and Table 1), consistent with earlier findings that D3 and D4 receptors signal poorly in heterologous systems (discussed above). Through reconstitution of receptor/G-protein interactions, it has been shown that D2-like receptors preferentially couple to Gαo (Nickolls and Strange, 2004). When Gαo was coexpressed, there was a dramatic increase in the potency and efficacy of pergolide at D2 and in the constitutive activity of D2. The constitutive response of D2 induced by Gαo could be reversed by haloperidol demonstrating that haloperidol acts as an inverse agonist at D2 (Fig. 1B). A small component of the constitutive response could not be reversed by haloperidol and probably represents nonreceptor-regulated interactions of G-protein with endogenous cellular components, as observed previously (Burstein et al., 1997). The haloperidol-sensitive constitutive activity was not due to endogenous ligands present in the media, because it was readily observed using synthetic media or in the presence of heterologous coexpression of the human dopamine transporter (data not shown). Similarly, overexpression of Gαo induced a dramatic increase in the potency and efficacy of pergolide at D3 and significant responses to D4 could now be detected (see Fig. 1, C and E, respectively; Table 1). Gαo also induced robust increases in constitutive activity of D3 and D4; however, only constitutive activity of D3 and not D4 was reversed by haloperidol (Fig. 1, D and F).
Effect of Gαo and RGS1 on functional responses to D2, D3, and D4 receptors
Plasmids Gαo and RGS1 encoding the respective receptors and where indicated were transfected into NIH-3T3 cells and functionally analyzed in the presence of the serial dilutions of pergolide (agonist) and haloperidol (inverse agonist) using the R-SAT functional assay as described under Materials and Methods. Data are reported in nanomolar units as EC50 and IC50 for agonist and inverse agonist, respectively, and represent the means ± S.D. of four or more independent experiments in each case. To obtain expression levels, radioligand binding using [3H]raclopride (D2 and D3) and [3H]spiperone (D4) was carried out as described under Materials and Methods. Data represent the mean ± S.D. of two to three independent experiments in each case.

Effects of Gαo and RGS1 on functional responses of D2, D3, and D4 receptors. Plasmids encoding receptors (R), Gαo (G), and RGS1 (RGS) were transfected into NIH-3T3 cells and functionally analyzed in the presence of the indicated concentrations of pergolide and haloperidol using the R-SAT functional assay as described under Materials and Methods. A and B, D2. C and D, D3. E and F, D4. Data points represent the mean ± S.D. of two separate determinations in each case.
RGS1 is a protein that selectively accelerates the GTPase activity of Gαo, terminating its actions. To confirm that the observed increases in constitutive activity were the result of increased receptor activation of G-proteins, we coexpressed RGS1 and Gαo with D2, D3, or D4. RGS1 reduced the potency of pergolide and completely reversed the constitutive activity induced by Gαo in all cases (Fig. 1; Table 1).
Although increases in the level of expressed receptors could result in increased constitutive activity, coexpression of Gαo had little or no effect on the expression levels of the receptors (Table 1). Thus, the observed constitutive activity is due to the increased levels of Gαo and not because of changes in receptor levels.
We screened 40 typical and atypical antipsychotics at the D2, D3, and D4 receptors coexpressed with Gαo and measured the functional responses, assigning a value of 100% to the basal response. Nearly all of the ligands tested were inverse agonists at D2 and D3, reducing the response to between 50 and 70% of the basal response in most cases (Fig. 2, A and B; Table 2). Several compounds, including olanzapine, quetiapine, mesoridazine, and thioridazine, displayed substantially lower degrees of inverse agonist activity compared with haloperidol. Although virtually all of the antipsychotics tested were inverse agonists, only the primary active metabolite of clozapine, NDMC, and aripiprazole were found to have agonist activity at D2 and D3, increasing the response 2- to 3-fold over baseline at both receptors. None of the tested antipsychotics displayed significant agonist or inverse agonist activity at D4 (Fig. 2C; Table 2), although many were potent antagonists (see below). In addition, none of these compounds displayed significant agonist or inverse agonist activity at cells transfected with G-proteins alone or at cells transfected with unrelated receptors (unpublished observations).
Antagonist and inverse agonist profiles of dopaminergic ligands
For determination of intrinsic efficacy (%), D2, D3, and D4 receptors were cotransfected with Gαo and functionally analyzed using R-SAT as described above in the presence of 1 μM concentrations of the indicated drugs, with the exception of olanzapine, thioridazine, and N-desmethylolanzapine, which were tested at 300 nM to avoid nonreceptor-mediated effects. Response is defined as [(drug response/basal response) × 100%] - 100%. Thus, a neutral antagonist would have a response equal to 0%. Values shown represent the mean of two or more independent experiments with eight individual determinations per experiment. To determine inverse agonist activity (reported in nanomolar units as IC50), D2 and D3 receptors were cotransfected with Gαo and functionally analyzed in the presence of serial dilutions of the indicated ligands. To determine antagonist activity (reported in nanomolar units as Ki), D2 receptors were transfected without Gαo and D3 and D4 receptors were transfected with both Gαo and RGS1 to increase the dynamic range of the assay. Cells were incubated with a fixed concentration of pergolide (5 × the previously determined EC50 for each receptor) and serial dilutions of the indicated ligands. Ki values were calculated as described under Materials and Methods. Data represent the means ± S.D. of at least two or more independent experiments.
To confirm that the differences in efficacy noted among partial agonists, partial inverse agonists, and full (with respect to haloperidol) inverse agonists were significant, D2 and D3 receptors coexpressed with Gαo were challenged with NDMC, haloperidol, mesoridazine, NDMC and mesoridazine, or NDMC and haloperidol. As shown in Fig. 3, mesoridazine was able to block the inverse agonist activity of haloperidol and the partial agonist activity of NDMC.
All of the compounds identified as having substantial inverse agonist activity (defined as less than –30%) were analyzed in full-concentration response experiments at D2 and D3 (Fig. 4), and all of the compounds, regardless of their activities as inverse agonists, were tested as antagonists of pergolide-induced activity at D2, D3, and D4 (Table 2). The IC50 values for reversal of G-protein induced constitutive activity at D2 and D3, and the Ki values for reversal of agonist-induced activity at these receptors were, in general, very similar. Many of the tested ligands were potent functional antagonists at D4, despite the fact that they lack inverse agonist activity at D4. The vast majority of compounds displayed limited selectivity between D2 and D3 receptors but relatively strong selectivity for D2 and D3 over D4. A notable exception was clozapine, which displayed 3- to 10-fold selectivity for D4 over D2 and D3, although NDMC was not selective for D4.
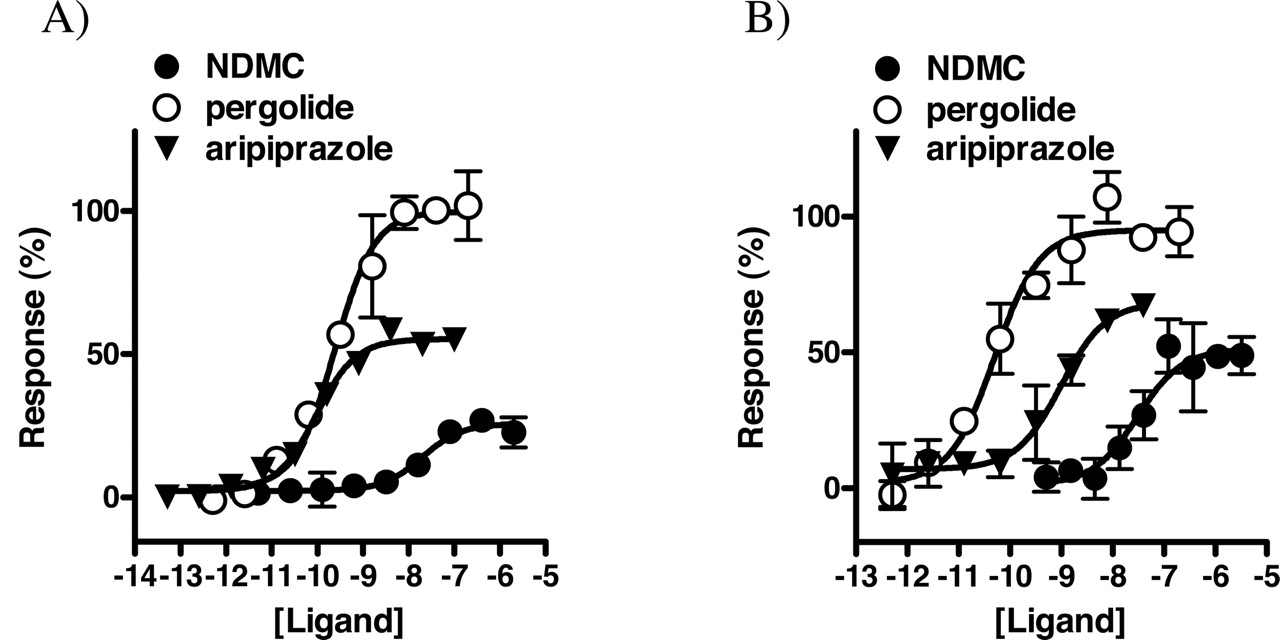
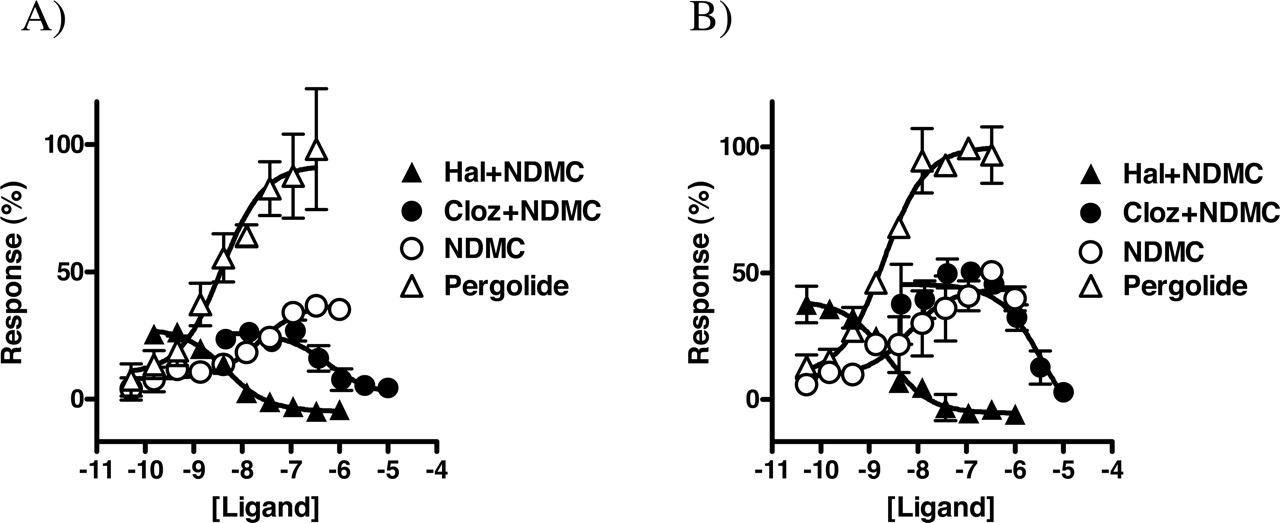
Aripiprazole and NDMC, which were the only compounds with agonist activity identified in the screen (see Fig. 2), were further analyzed in full-concentration response experiments. Aripiprazole was a potent partial agonist at both D2 and D3 (EC50 = 0.1 and 1 nM, respectively), with ∼60% efficacy relative to pergolide in each case (see Fig. 5). Compared with aripiprazole, NDMC was found to be a less potent, less efficacious agonist at both D2 (20 nM, 35%) and D3 (30 nM, 50%). Both aripiprazole and NDMC also functionally antagonized pergolide-induced activation of D2 and D3 (see Table 2). In comparison, the partial agonist S-(–)-PPP had significantly higher efficacy than aripiprazole at both D2 and D3 (unpublished observations). Neither aripiprazole nor NDMC displayed appreciable agonist or inverse agonist activity at D4 (not shown), although NDMC was a moderately potent antagonist at D4 (Table 2). Haloperidol could block D2 and D3 responses to both NDMC (Fig. 6) and aripiprazole (not shown). Similarly, clozapine blocked D2 and D3 responses to NDMC, demonstrating that these compounds have opposing actions at these receptors (Fig. 6).
Discussion
These studies have provided a comprehensive pharmacological profile of an array of antipsychotics and related compounds at the human D2, D3, and D4 dopamine receptors. Overexpression of Gαo induced constitutive activation of these receptors, facilitating detection of both agonism and inverse agonism. Virtually all of the antipsychotics were D2 and D3 inverse agonists, whereas none was D4 inverse agonist, although many were potent D4 antagonists. A wide range of inverse agonist activities was observed from high inverse agonist activity (e.g., haloperidol, pimozide, etc.) to very low inverse agonist activity (e.g., mesoridazine and quetiapine). Surprisingly, the major active metabolite of clozapine, NDMC, like aripiprazole, was a partial agonist at D2 and D3 receptors.

Functional screen of antipsychotics. Receptors were cotransfected with Gαo and functionally analyzed as described above in the presence of 1 μM concentrations of the indicated drugs, with the exception of olanzapine, thioridazine, and N-desmethylolanzapine, which were tested at 300 nM to avoid nonreceptor-mediated cellular effects. The basal response in the absence of added ligand was assigned a value of 100%, and all other responses were normalized to that value. A, D2. B, D3. C, D4. Values shown represent the mean ± S.D. of at least two or more independent experiments with eight individual determinations per experiment.
Inverse agonism at D2-like receptors was first noted for haloperidol, which increased prolactin production and cyclic AMP formation in GH4C1 cells transfected with D2, and from GH3 cells expressing D2 endogenously (Nilsson and Eriksson, 1993). Subsequently, other D2 antagonists, including several antipsychotic drugs, were identified as D2 inverse agonists (Akam and Strange, 2004). Haloperidol, fluphenazine, and chlorpromazine were described as D3 inverse agonists (Griffon et al., 1996). Other studies have reported mixed findings. Several D2 antagonists, including haloperidol, raclopride, and clozapine, were reported to be inverse agonists at D3 but neutral antagonists at D2 (Malmberg et al., 1998). In contrast, a series of D2 antagonists were tested at D2 and D4, and all were reported to be neutral antagonists (Gilliland and Alper, 2000). A comprehensive functional profile of dopaminergic compounds at D2, D3, and D4 subtypes addressing inverse agonism has not previously been available.
Compared with D2, the effect of expressing Gαo on functional responses was greater for D3 (see Fig. 1), possibly reflecting the low efficiency of D3 signaling previously observed (Newman-Tancredi et al., 1999). Our results show that D3 may have a greater requirement than most Gi/o-coupled receptors for Gαo, which is highly expressed in brain but is expressed at much lower levels in peripheral tissues, and in many commonly used cell lines (Asano et al., 1992).
Despite these differences in signaling, a strikingly similar pharmacology was observed at D3 compared with D2. All compounds identified as inverse agonists at D2 were also inverse agonists at D3, and the degree of inverse agonism for most of these compounds was very similar at D2 and D3. NDMC and aripiprazole were both D3 partial agonists. Finally, the potencies of all of the compounds examined were similar at D2 and D3, strongly suggesting that affinity for D3 receptors does not confer an atypical antipsychotic profile as proposed previously (Schwartz et al., 2000).
Compared with D2 and D3 receptors, the D4 receptor behaved differently. No response to D4 was observed without coexpression of Gαo, consistent with previous reports documenting poor coupling (Gazi et al., 2000). Constitutive internalization of the receptor has been proposed to explain the poor coupling observed with D4 (Oldenhof et al., 1998). The D4 receptor exists in several isoforms distinguished by the number of sequence repeat elements contained within the third intracellular (i3) loop. Only D4.2 was examined in this study; thus, the possible functional differences between isoforms were not addressed. However, no functional differences between D4 isoforms have been reported. Coexpression of Gαo induced a high degree of constitutive activity at D4, but neither haloperidol nor any of the other tested ligands was able to act as inverse agonists, although many were able to potently block pergolide-induced activity. Because no D4 inverse agonists were identified, we cannot exclude the possibility that the constitutive activity observed is not D4-mediated. However, given that the constitutive activity is greatly diminished in cells expressing Gαo without D4 (not shown) and that RGS1 suppressed the basal activity and increased the fold-response to pergolide in cells expressing D4 and Gαo suggests that the observed constitutive activity was D4-mediated.

Blockade of agonism and inverse agonism. Individual dopamine receptors were cotransfected with Gαo and functionally analyzed as described above in the presence of mesoridazine (500 nM, denoted Meso) or haloperidol (10 nM, denoted Haldol) or both (Mes+Hal). A and C, D2. B and D, D3. Responses were normalized to the response in the absence of added ligands, which was assigned a value of 100%. Shown are representative individual experiments. Data represent the means ± S.D. of 24 individual determinations. Statistical significance was assessed using the unpaired Student's t test. *, significantly different (p < 0.01) from haloperidol (A and B) or mesoridazine (C and D) alone; #, significantly different (p < 0.01) from no drug.
The pharmacology observed for D4 also differed considerably. Most compounds had significantly lower pKi values at D4 than at D2 or D3, with thiothixene and raclopride displaying the greatest selectivity for D2/D3 over D4 (Table 2). Based on their D2/D4 selectivity, most of these compounds would not be predicted to occupy D4 at clinical doses. Octoclothepin displayed the highest affinity for D4, although it had no appreciable selectivity for D4. Other than the D4 reference compound L-745,870, only tiapride, molindone, and clozapine displayed even modest selectivity for D4. D4 selectivity, particularly the relative selectivity of clozapine for D4 over D2 and D3, has been proposed to confer atypical profiles (Jardemark et al., 2002); however, the data presented above suggest that D4 does not mediate the therapeutic effects of antipsychotic medications nor does selectivity for D4 predict atypicality, in agreement with previous results (Roth et al., 1995).
Propensity to produce EPS/TD and other movement disorders, side effects that have been previously ascribed to D2 receptor occupancy (Creese et al., 1976; Seeman et al., 1976), may also depend on efficacy at D2. The dramatic clinical differences seen with aripiprazole and clozapine, which are distinguished even from other atypical antipsychotics by their much lower liability for producing EPS/TD coupled with their unique in vitro profiles, suggest that partial agonism at D2 is a desirable feature of an antipsychotic drug. D2 partial agonists offer several potential advantages for treating schizophrenia (see Tamminga and Carlsson, 2002). With the appropriate level of intrinsic activity, a partial agonist might act as an agonist at presynaptic D2 receptors (“D2 autoreceptors”), where receptor reserve is high, and act as an antagonist at postsynaptic D2 receptors, where receptor reserve is low (Meller et al., 1986). Such a drug could provide superior efficacy for negative symptoms, may display reduced propensity to cause extrapyramidal side effects, and yet still block the hyperdopaminergic activity thought to cause positive symptoms.

Profiles of dopaminergic inverse agonists. Respective dopamine receptors were cotransfected with Gαo and functionally analyzed in the presence of the indicated concentrations of ligands using the R-SAT functional assay as described above. A and B, D2. C and D, D3. Data points represent the mean ± S.D. of two separate determinations in each case. Responses were normalized to the basal response in the absence of ligand, which was assigned a value of 100%.
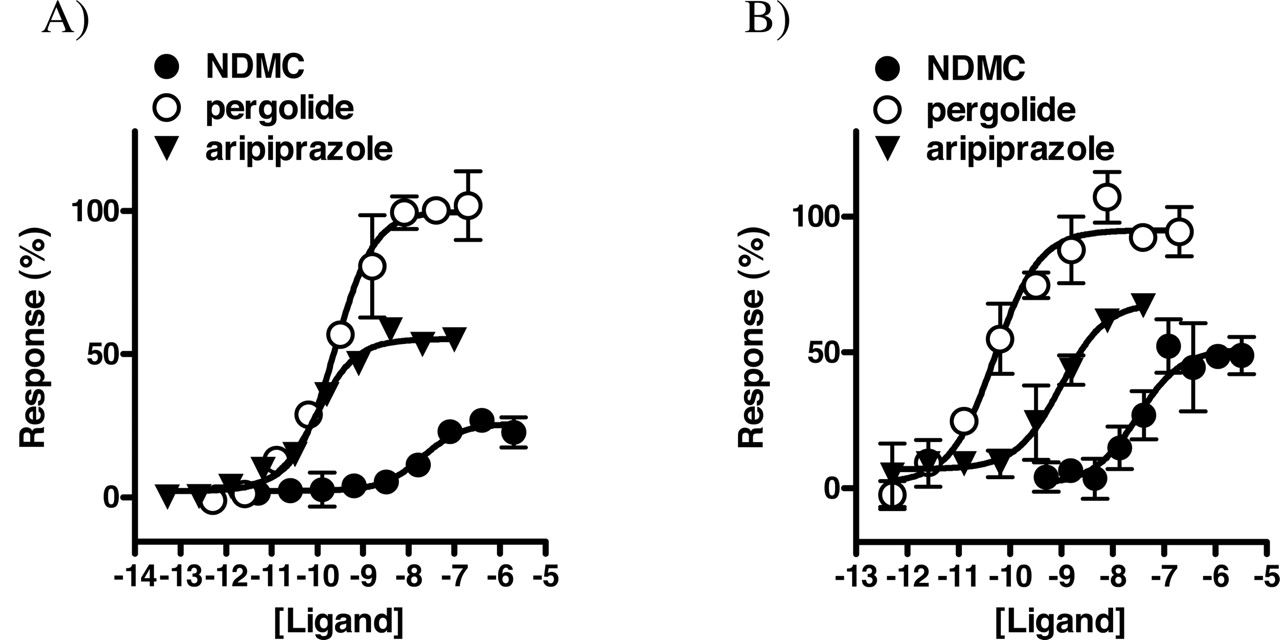
Profiles of dopaminergic agonists. Respective dopamine receptors were cotransfected with Gαo and functionally analyzed in the presence of the indicated concentrations of ligands using the R-SAT functional assay as described above. A, D2. B, D3. Data points represent the mean ± S.D. of two separate determinations in each case. Responses were normalized to the response to pergolide, which was assigned a value of 100%.
Discovery of the unique clinical properties of clozapine demonstrated that it was possible to separate the therapeutic benefits of antipsychotics from their side effects. The realization of the clinically beneficial characteristics of clozapine spurred the development of second generation or “atypical” antipsychotic drugs. Clozapine is still considered distinct from other “atypicals” with respect to its use in psychotic patients suffering from EPS/TD (Charfi et al., 2004) and in treatment-induced psychosis in patients with Parkinson's disease (Parkinson Study Group, 1999). Clozapine is active in treatment-resistant patients (Kane et al., 1988) and demonstrates superior improvements over other antipsychotics in cognitive function (Hagger et al., 1993). Thus, we have sought to define other molecular properties, if any, that might be responsible for the unique clinical features of clozapine, but to date, we have not identified any obvious features that distinguish clozapine itself from other atypical agents.

Opposing actions of NDMC and clozapine. Plasmids encoding D2 or D3 and Gαo were transfected into NIH-3T3 cells and functionally analyzed in the presence of the indicated concentrations of NDMC or pergolide or in the presence of 300 nM NDMC and the indicated concentrations haloperidol and clozapine using R-SAT as described under Materials and Methods. Data points represent means ± S.D. of two separate determinations. A, D2. B, D3. Responses were normalized to the response to pergolide, which was assigned a value of 100%.
Several molecular features distinguish NDMC from clozapine and all other typical and atypical antipsychotics. Uniquely among antipsychotics, NDMC is a potent M1 muscarinic receptor agonist, whereas clozapine is a potent antagonist at M1 receptors (Sur et al., 2003; Weiner et al., 2004; Davies et al., 2005). Similarly, we now show that NDMC is a D2/D3 partial agonist, whereas clozapine and all other antipsychotics, with the exception of aripiprazole, are D2/D3 receptor inverse agonists. Activating muscarinic cholinergic neurotransmission is widely considered to be a viable approach to treating cognitive deficits in schizophrenia (Friedman 2004), and as discussed above, attenuating dopaminergic neurotransmission may actually worsen cognitive deficits. Therefore, we propose that the M1, D2, and D3 partial agonist properties of NDMC may account, in part, for the unique biochemical and clinical aspects of clozapine pharmacotherapy.
NDMC is the primary metabolite of clozapine and achieves average plasma concentrations approximately 60 to 80% of that observed for clozapine in humans (Perry et al., 1991). Therefore, during clozapine therapy, significant levels of both a D2/D3 receptor inverse agonist (clozapine) and a D2/D3 receptor partial agonist (NDMC) are present in substantial concentrations in most patients. We have observed that there is a positive correlation between superior clinical outcomes and the metabolism of clozapine to NDMC in schizophrenic subjects (Weiner et al., 2004). Specifically, higher ratios of NDMC to clozapine and not the absolute levels of either compound alone predicted better improvements in clinical response, including multiple measures of cognition and quality of life scores, suggesting that a pharmacological interaction between the two molecules may affect clinical efficacy. Given that clozapine blocks the agonist actions of NDMC at M1, D2, and D3, the observations that improved clinical outcomes are positively correlated with increased NDMC/clozapine ratios suggests that overcoming clozapine blockade of these receptors with NDMC may confer some of the unique clinical benefits ascribed to clozapine pharmacotherapy. Interindividual variation and the complex pharmacodynamic relationships between clozapine and NDMC would be bypassed if NDMC were directly administered to patients. These observations, coupled with the D2/D3 partial agonist properties of NDMC reported herein, add additional support to the hypothesis that NDMC may itself display superior antipsychotic activity. This possibility is currently being tested clinically.
Acknowledgments
We thank D. Bonhaus, D. P. van Kammen, K. Vanover, J. Lameh, and L. Iverson for critical reading of the manuscript.
Footnotes
-
doi:10.1124/jpet.105.092155.
-
ABBREVIATIONS: EPS, extrapyramidal symptoms; TD, tardive dyskinesia; DMEM, Dulbecco's modified Eagle's medium; GPCR, G-protein-coupled receptor(s); NDMC, N-desmethylclozapine [8-chloro-11-(1-piperazinyl)-5H-dibenzo [b,e] [1,4] diazepine]; PTX, pertussis toxin; 5HT, 5-hydroxytryptamine; RGS1, regulator of G-protein signaling 1; R-SAT, receptor selection and amplification technology; ONPG, O-nitrophenyl-β-d-galactopyranoside; AC2, adenylyl cyclase type II; S-(–)-PPP, (S)-(–)-3-(3-hydroxyphenyl)-N-propylpiperidine hydrochloride; L-745,870, 3-(4-[4-chlorophenyl]piperazin-1-yl)methyl-1H-pyrrolo[2,3b]pyridine.
- Received July 7, 2005.
- Accepted August 30, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}