


Abstract
Benzodiazepines (BZs) are prescribed for a variety of disorders, including those involving anxiety and sleep, but have unwanted side effects that limit their use. Elucidating the GABAA receptor mechanisms underlying the behavioral effects of BZs will help develop new drugs having both maximum clinical benefit and minimum adverse side effects. A recently developed compound is SL651498 [6-fluoro-9-methyl-2-phenyl-4-(pyrrolidin-1-yl-carbonyl)-2,9-dihydro-1H-pyridol[3,4-b]indol-1-one], which is a full agonist at GABAA receptors containing α2and α3 subunits and a partial agonist at GABAA receptors containing α1 and α5 subunits. We assessed the ability of SL651498 to engender anxiolytic-like, motor, and subjective effects characteristic of BZ-type drugs in nonhuman primates. Anxiolytic-like activity was assessed with a conflict procedure in rhesus monkeys. Motor effects were evaluated in squirrel monkeys using observational techniques, and the subjective effects of SL651498 were assessed in squirrel monkeys trained to discriminate the nonselective BZ triazolam from saline. SL651498 engendered anxiolytic-like effects similar to conventional BZs. In addition, SL651498 fully induced muscle relaxation, but unlike conventional BZs, engendered minimal ataxia. In drug discrimination studies, SL651498 partially substituted for triazolam. This effect was blocked with the α1 GABAA subtype-preferring antagonist β-CCT (β-carboline-3-carboxylate-t-butyl ester), implicating α1 GABAA effects receptors in the subjective of SL651498. Together, these studies suggest that compounds such as SL651498 that have high intrinsic efficacy at α2GABAA and/or α3GABAA receptors may have clinical potential as anxiolytics and muscle relaxants. Moreover, a compound with reduced efficacy at α1 GABAA and/or α5 GABAA receptors may lack some of the motor and subjective effects associated with conventional BZs.
Benzodiazepine (BZ) receptor agonists are commonly prescribed for the treatment of anxiety, sleep, and seizure disorders. Their clinical utility is limited, however, due to undesirable side effects, which include sedation, motor incoordination, memory impairment, abuse, and dependence (Griffiths and Wolf, 1990; Korpi et al., 1997; Belzung et al., 2000; Verster et al., 2002). Research efforts currently are aimed at developing novel BZ agonists that retain therapeutically beneficial properties without the unwanted side effects.
BZ agonists produce their behavioral effects by positive allosteric modulation of GABA binding at the GABAA receptor complex. The GABAA receptor is a pentameric ionophore formed by the assembly of subunits from at least five different families (Sanger et al., 1994; Lüddens et al., 1995; McKernan and Whiting, 1996). The presence of α, β, and γ subunits are necessary to confer sensitivity to BZ ligands (Pritchett et al., 1989; McKernan and Whiting, 1996; Rudolph et al., 2001), and conventional BZs exert their pharmacological effects via binding nonselectively to GABAA receptors containing α1, α2, α3, and α5 subunits.
Recent research has been aimed at elucidating the receptor mechanisms underlying the specific behavioral effects of BZs, with the goal of developing BZ-type drugs having both maximum clinical benefit and minimum adverse side effects. One such compound that has been developed recently is 6-fluoro-9-methyl-2-phenyl-4-(pyrrolidin-1-yl-carbonyl)-2,9-dihydro-1H-pyridol[3,4-b]indol-1-one (SL651498). SL651498 is a novel pyridoindole derivative that has high affinity for rat native GABAA receptors containing α1 and α2 subunits and weaker affinity for α5GABAA receptors, although having intermediate affinity for recombinant rat GABAA receptors containing the α3 subunit (Griebel et al., 2001). However, SL651498 is “functionally selective” in that it acts as a full agonist at α2GABAA and α3GABAA receptors and as a partial agonist at α1GABAA and α5GABAA receptors, as determined in vitro by modulation of GABA-mediated Cl– flux (Griebel et al., 2001). Behavioral studies in rodents demonstrated that SL651498 elicited anxiolytic-like activity similar to that of diazepam (Griebel et al., 2001, 2003). SL651498 also induced muscle weakness, ataxia, and sedation as measured by motor activity; nevertheless, the doses that engendered these effects were much higher than those producing anxiolytic-like activity. In addition, SL651498 produced neither tolerance to its anticonvulsant effects nor physical dependence after repeated administration (Griebel et al., 2001, 2003). Taken together, these results suggest that the anxiolytic and anti-convulsant activity of SL651498 may make this compound a suitable alternative to currently prescribed drugs for those indications. Furthermore, the observed behavioral effects may be a result of selective actions at specific GABAA receptor subtypes.
Our current knowledge of the role of GABAA receptor subtypes in the behavioral effects of BZs is predominantly based on studies with transgenic and wild-type rodents. At present, relatively little information is available regarding the roles of particular GABAA receptor subtypes in the characteristic effects of BZ-type drugs in either human or nonhuman primates. Therefore, in the present studies, SL651498 was used to investigate the role of specific GABAA receptor subtypes in models predictive of the anxiolytic, motor, and subjective effects of BZ-type drugs in nonhuman primates. Anxiolytic-like activity was assessed with a conflict procedure in monkeys in which operant responding maintained by food delivery was suppressed by response-contingent shock presentations. Motor effects characteristic of BZs were evaluated in monkeys using observational techniques based upon naturalistic and drug-induced behavior. Finally, the subjective effects of SL651498 were evaluated in a drug discrimination model, in which monkeys were trained to discriminate the representative nonselective BZ triazolam from saline. Together, these studies aimed to provide insights into the role of GABAA receptor subtypes in the therapeutic versus unwanted side effects of BZ-type drugs.
Materials and Methods
Subjects. Four adult rhesus monkeys (Macaca mulatta), two male and two female, were studied in the conflict study. Six adult male squirrel monkeys (Saimiri sciureus) were used for the observational study, and four adult male squirrel monkeys were used in the drug discrimination study. Monkeys used in the conflict and discrimination studies were maintained at 90 to 95% of their free-feeding weight. Monkeys used in the observational study were maintained under free-feeding conditions. All monkeys were housed individually and maintained under a 12-h light/dark cycle in a temperature- and humidity-controlled room. All procedures were conducted with the approval and under the supervision of the Harvard University Institutional Animal Care and Use Committees. Animals in this study were maintained in accordance with the guidelines of the Committee on Animals of the Harvard Medical School and the “Guide for Care and Use of Laboratory Animals” National Research Council, Department of Health, Education and Welfare Publication No. (NIH 85-23), revised 1996.
Monkeys in the conflict and discrimination studies were prepared with chronic indwelling venous catheters using the general surgical procedures described by Carey and Spealman (1998). Under isoflurane anesthesia and aseptic conditions, one end of a polyvinyl chloride catheter (rhesus monkey = i.d., 0.64 mm; o.d., 1.35 mm; squirrel monkey = i.d., 0.38 mm; o.d., 0.76 mm) was passed to the level of the right atrium by way of a brachial, femoral, or jugular vein. The distal end of the catheter was passed subcutaneously and exited in the mid-scapular region. Rhesus monkey catheters were flushed daily with heparinized saline (150–200 U/ml), whereas squirrel monkey catheters were flushed daily with saline that did not contain heparin. All catheters were sealed with stainless steel obturators when not in use. Monkeys wore custom-made nylon mesh jackets (Lomir Biomedical, Toronto, ON, Canada) at all times to protect the catheter.
Conflict Study. Rhesus monkeys were trained to sit in a custom-designed restraint chair (Crist Instruments, Hagerstown, MD) located in a sound-attenuating chamber. A response lever, stimulus lights (Med Associates, Georgia, VT), and a receptacle into which food pellets (1 g of marshmallow-flavored pellets; BioServ, French-town, NJ) were delivered were positioned in front of the monkey. Monkeys were trained on a multiple schedule of food reinforcement consisting of two components: 1) a schedule of food pellet delivery, and 2) a schedule of food pellet delivery plus a schedule of foot shock delivery (0.25-s duration, 1–3 mA depending on the individual monkey). Four components were available in a session, separated by 10-min timeout periods in which responding had no programmed consequences. Responding was maintained in each component under an 18 response, fixed-ratio (FR) schedule of food pellet delivery. Each component consisted of the schedule of food pellet delivery signaled by red stimulus lights, followed immediately by the same schedule of food delivery combined with a 20 response FR schedule of foot shock delivery signaled by green stimulus lights. Each response requirement was followed by a 10-s timeout. Drugs were administered during the 5th min of the 10-min timeout that preceded each component.
Training sessions were conducted 5 days per week until performance (measured as rates of responding, see below) in both “food only” and “food + shock” components was stable (i.e., no upward or downward trends for 3 consecutive days). In addition, if rates of responding in a component during a training session varied by more than 20% of the corresponding response rates in the previous training session, additional training sessions were conducted until responding was again stable. Once training criteria were met, test sessions were conducted once or twice a week, separated by at least 2 days. Dose-response functions were determined for test drugs using a cumulative dosing procedure similar to the one described by Rowlett et al. (2001). Four-point cumulative dose-response functions were determined within a single test session as a result of incremental increases in drug (one-half log units) administered at the 5th min of the 10-min timeout period. Dose-response functions were determined for SL651498 (0.1–3 mg/kg), the classical nonselective BZ agonist chlordiazepoxide (1–30 mg/kg), as well as triazolam (0.001–0.03 mg/kg).
The number of responses in a component, minus responding during pellet delivery and the 10-s timeouts, was divided by the total component time minus the 10-s timeouts to obtain rates of responding (responses per second). Data for multiple determinations were averaged for an individual monkey, and then these response rates were averaged across monkeys (N = 4, unless otherwise noted). The effects of the individual doses of drug were compared with the control rates of responding using Dunnett's test comparing the average response rate engendered by each dose to the average response rate after vehicle administration (α level = p < 0.05).
Observation Study. The observable behavioral effects of BZ ligands were assessed in squirrel monkeys according to the procedures described by Platt et al. (2002). Each monkey was habituated to a ventilated, transparent Plexiglas arena (114 cm × 122 cm × 213 cm). This observation arena was equipped with perches, suspended plastic chains, and a wood chip foraging substrate to allow the monkeys to express a range of species-typical behaviors. A video camera was positioned 1 m in front of the chamber and operated throughout the 30-min session.
Drug testing was conducted once or twice per week with control sessions preceded by saline injections on intervening days. Doses of SL651498 (0.3–10 mg/kg i.m., administered 10 min prior to the start of the session) or chlordiazepoxide (3.0–56 mg/kg i.m., administered 30 min prior to start of the session) were administered on separate test days. Trained observers, unaware of the drug being studied, scored the videotapes by recording the presence or absence of each of eight behaviors (Table 1) at 15-s intervals during three 5-min observation periods across the session (0–5, 12–17, and 24–29 min). For each subject, frequency scores (defined as the total number of 15-s intervals in which a particular behavior was observed; maximum score = 20) for each behavior were averaged across the three observation periods of a session because no reliable differences in scores were identified by separated repeated measures analysis of variance. Scores were then averaged across subjects to obtain group means. To determine statistical reliability of treatment effects on each behavior, the effect of dose was determined for each drug by separate repeated measures analyses of variance. Treatment effects were assessed further using Bonferroni t tests, in which the effects of different doses of each drug were compared with vehicle.
Behavioral definitions
Muscle relaxation and ataxia were measured in the same animals during the 6th, 18th, and 30th min of each 30-min observation session. The monkeys were removed briefly from the observation arena by a trained handler and evaluated for degree of muscle relaxation, which was defined as decreased resistance to extension of a hind limb. A score of 0 indicated a normal resistance to extension, a score of (–1) indicated a decreased resistance to extension, and a score of (–2) indicated no resistance to extension (i.e., the monkey was flaccid and completely relaxed). Ataxia was defined as the inability to balance on the 1-cm diameter stainless steel transport pole held in the horizontal plane. A score of 0 indicated that the monkey was able to balance normally on the pole, a score of (+1) indicated that the monkey was able to hold onto the pole but unable to maintain balance (e.g., suspended by limbs below pole), and a score of (+2) indicated that the monkey could neither balance on nor hold on to the pole.
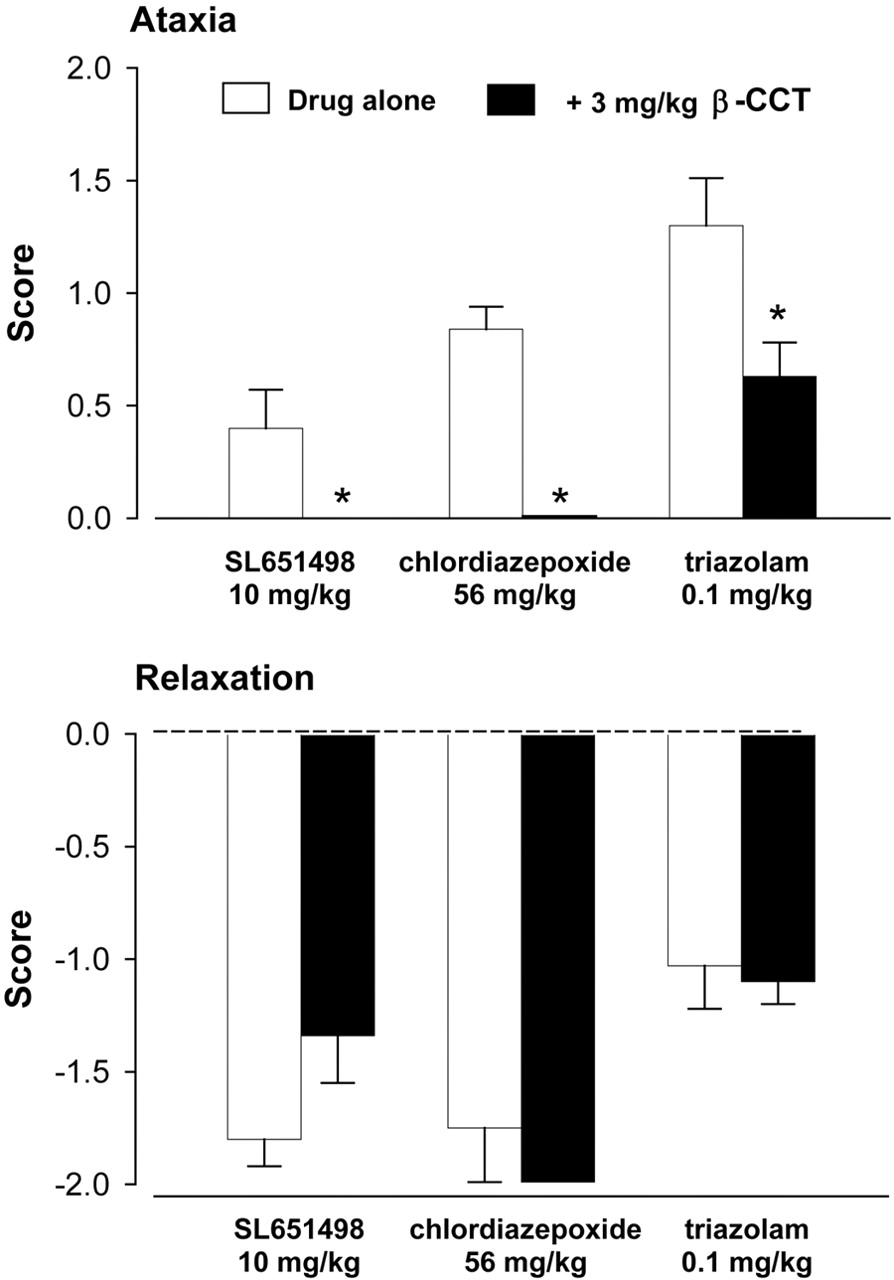
In an additional study, SL651498 (10 mg/kg i.m.), chlordiazepoxide (56 mg/kg i.m.), or triazolam (0.1 mg/kg i.m., administered 30 min prior to the start of the session) were evaluated alone and combined with the α1GABAA-preferring antagonist β-carboline-3-carboxylate-t-butyl ester (β-CCT) (Platt et al., 2002; 3.0 mg/kg i.m., administered 10 min prior to the start of the session). Monkeys were evaluated for muscle relaxation and ataxia.
Drug Discrimination Study. Squirrel monkeys were trained to discriminate the conventional BZ agonist triazolam from saline under an FR 10 schedule of food delivery using the procedure described by Lelas et al. (2001, 2002). The monkeys sat in a restraint chair located in a sound-attenuating chamber (Med Associates). In front of the monkey, response levers and stimulus lights were available as well as a receptacle into which food pellets (190 mg of sucrose pellets; BioServ) were delivered. Training sessions consisted of one to four components. A 10-min timeout period preceded each FR component, and either saline or triazolam (0.03 mg/kg) was administered intravenously at the 5th min. Each FR component, during which the stimulus lights were illuminated and the FR 10 schedule of food delivery was in effect, lasted 5 min or until 10 pellets were delivered, whichever occurred first. Ten consecutive responses on the correct lever resulted in food delivery and extinguished the stimulus lights for a 10-s timeout period. Responses on the incorrect lever did not result in pellet delivery and reset the response requirement on the correct lever.
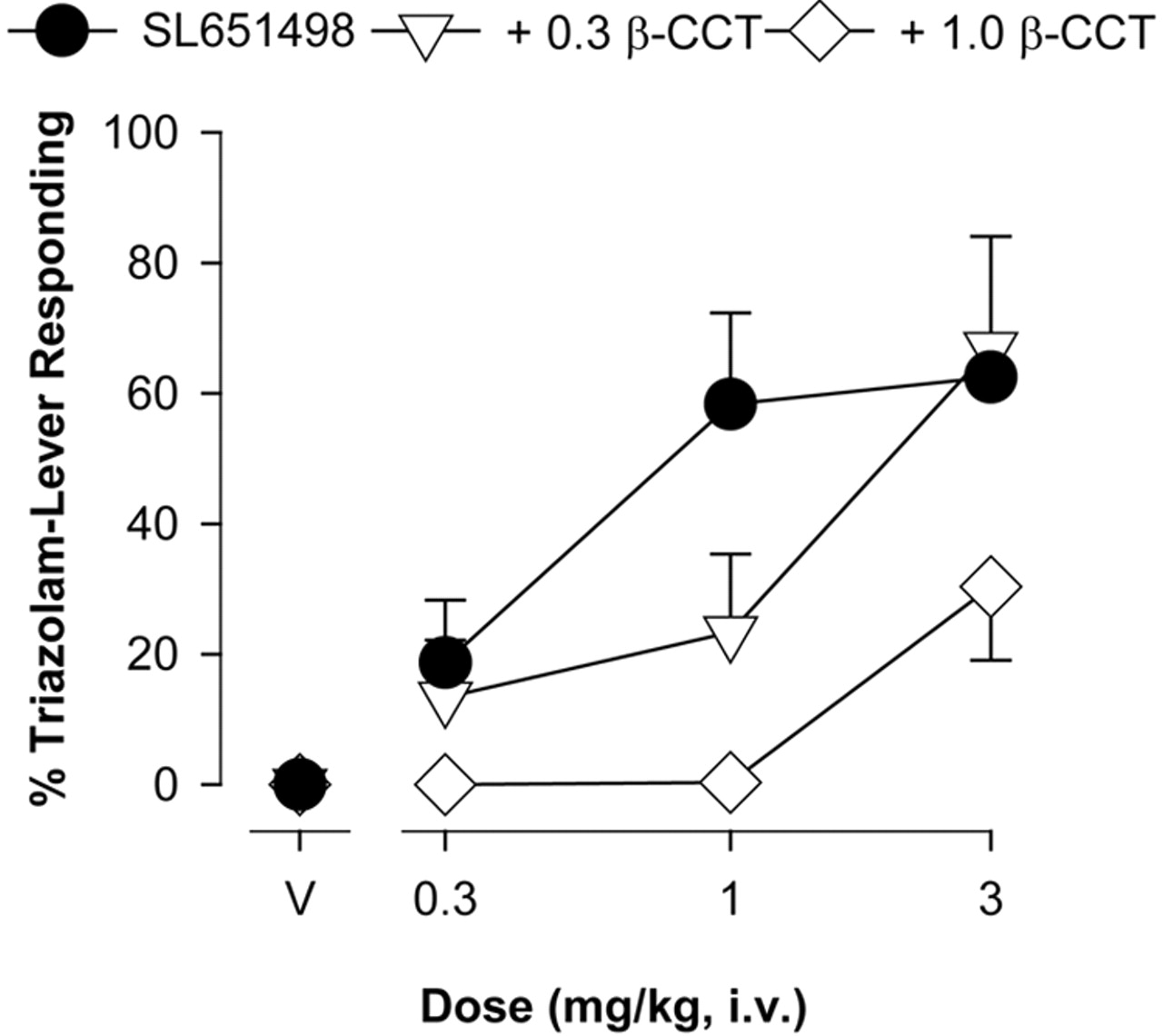
Drug testing sessions were conducted once or twice per week with training sessions scheduled on intervening days. Test sessions were conducted if 80% or more of the total responses occurred on the lever designated correct for that component for at least four of the five preceding training sessions. Test sessions consisted of i.v. injections of saline, triazolam (0.003–0.1 mg/kg), or SL651498 (0.1–1 mg/kg). Overlapping cumulative dose-response functions were administered on different test sessions until at least one dose of drug produced ≥80% drug-lever responding, decreased response rates to ≤25% of control, or resulted in two or more animals not completing a response requirement. Antagonism studies also were conducted in which selected doses of β-CCT (0.3 and 1.0 mg/kg i.v.) were studied in combination with SL651498. β-CCT and its vehicle control were administered i.v. in the first component similar to previous studies (Lelas et al., 2002).
The percentage of drug-lever responding was computed for individual subjects by dividing the number of responses on the drug lever by the total number of responses on both levers and multiplying by 100. The percentage of drug-lever responses for each dose was then averaged across monkeys and the standard error of the mean computed. A drug was considered to substitute for triazolam if the average maximum percentage of drug-lever responses reached 80%. Average maximum percentages of less than 80% but greater than 20% triazolam-lever responding were considered partial substitution.
Antagonist potencies were obtained by calculating in vivo apparent pKB values using the methods described by Lelas et al. (2002). For each monkey, the dose of agonist that engendered 50% triazolam-lever responding (ED50) was calculated. The ED50 values were used to calculate in vivo apparent pKB values according to the equation described by Negus et al. (1993): pKB =–log [B/(DR – 1)]. In the equation, “B” is the dose of β-CCT in moles per kilogram and “DR” is the ED50 of agonist combined with antagonist divided by the ED50 of agonist alone. Mean apparent pKB values and 95% confidence intervals were calculated to make comparisons with previous findings (e.g., Lelas et al., 2002; Rowlett et al., 2003).
Drugs. The base forms of triazolam (Research Biochemicals Inc., Natick, MA), β-CCT (Department of Chemistry, University of Wisconsin-Milwaukee, Milwaukee, WI), and SL651498 (Sanofi-Aventis, Bagneux, France) were prepared in a vehicle of 50% propylene glycol, 40% saline, and 10% ethanol. Chlordiazepoxide (Research Biochemicals Inc.) was dissolved in 0.9% saline. The drugs were injected in a volume of 0.1 to 1.0 ml/kg, depending on the dose and solubility. All drugs were prepared the day of a test session.
Results
Conflict Study. During the course of the experiments, mean rates of responding during training sessions for nonsuppressed (food only) responding were between 2.0 and 3.0 responses/second and were at or near zero for suppressed (food + shock) responding. During tests with drug vehicle, rates of responding in both components showed a similar pattern (i.e., relatively high rates in the absence of shock, little or no responding when shock was present; Fig. 1, points above “V”). SL651498 produced a dose-dependent increase in suppressed responding at 1.0 and 3.0 mg/kg with no change in nonsuppressed responding (Fig. 1, left panel). Chlordiazepoxide increased rates of suppressed responding at 3.0 and 10 mg/kg (Fig. 1, middle panel), but decreased rates of both nonsuppressed and suppressed responding at the highest dose of 30 mg/kg. Similarly, triazolam increased rates of suppressed responding at 0.003 and 0.01 mg/kg, but decreased rates of suppressed responding at 0.03 mg/kg (Fig. 1, right panel). Rates of nonsuppressed responding also were decreased by 0.03 mg/kg triazolam.
Observation Study. The highest dose of SL651498 (10 mg/kg) engendered mild ataxia (score of 0.4 ± 0.1) [F(4,12) = 7.3, p = 0.003; Bonferroni t test, p < 0.05], unlike chlordiazepoxide which reliably engendered ataxia at three of four doses tested [F(6,30) = 24.7, p < 0.001; Bonferroni t test, p < 0.05] (Fig. 2, top). Similar to chlordiazepoxide, SL651498 produced a dose-dependent decrease in muscle resistance (Fig. 2, bottom). All doses of SL651498 differed from vehicle [F(4,12) = 41.0, p < 0.001; Bonferroni t test, p < 0.05], whereas only the two highest doses of chlordiazepoxide differed reliably from vehicle [F(6,30) = 59.2, p < 0.001; Bonferroni t test, p < 0.05]. Figure 3 shows the effects of SL651498 and chlordiazepoxide on measures of sedative-like behaviors. SL651498 had no effect on locomotion, rest posture, or procumbent posture at any of the doses tested. In contrast, chlordiazepoxide reliably decreased locomotion at the highest dose (56 mg/kg) [F(6,30) = 8.2, p < 0.001; Bonferroni t test, p < 0.05], increased rest posture at an intermediate dose [F(6,30) = 5.0, p = 0.001; Bonferroni t test, p < 0.05], and increased procumbent posture at the two highest doses [F(6,30) = 55.5, p < 0.001; Bonferroni t test, p < 0.05]. No other behaviors were significantly altered by SL651498 (Table 2). In contrast, chlordiazepoxide reliably suppressed environment-directed behavior [F(6,30) = 6.9, p < 0.001; Bonferroni t test, p < 0.05].
Behavioral effects of the highest dose of SL651498, in comparison with chlordiazepoxide, in squirrel monkeys (N = 6)
Data represent the mean frequency scores (±S.E.M.).

Anticonflict effects of SL651498 compared with chlordiazepoxide and triazolam in rhesus monkeys trained under a multiple schedule of food delivery (nonsuppressed responding) and food + shock delivery (suppressed responding). Data are mean ± S.E.M. from N = 4 monkeys. *, p < 0.05 versus vehicle, suppressed responding (filled symbols); **, p < 0.05 versus vehicle, nonsuppressed responding (open symbols).
Table 3 summarizes the ability of SL651498 to engender typical BZ-like side effects. The maximum score (MS) and minimally effective dose (MD) of SL651498 were compared directly with those of chlordiazepoxide, zolpidem, and triazolam. SL651498 was distinguishable from chlordiazepoxide, zolpidem, and triazolam by its relative lack of ataxia and procumbent posture. In addition, muscle relaxant effects of SL651498 occurred at doses at least 33-fold lower than those that engendered ataxia, in contrast to chlordiazepoxide, zolpidem, and triazolam, for which muscle relaxation occurred at higher or similar doses than ataxia.
Summary of observed behavioral effects induced by BZ-type drugs in squirrel monkeys

Ataxic and myorelaxant effects of SL651498 compared with chlordiazepoxide in squirrel monkeys. Data are mean ± S.E.M. from N = 6 monkeys. Points above V represent data from vehicle test sessions. Asterisks represent significant differences relative to vehicle (p < 0.05).

Sedative-like effects of SL651498 compared with chlordiazepoxide in squirrel monkeys. Data are mean ± S.E.M. from N = 6 monkeys. Points above V represent data from vehicle test sessions. Asterisks represent significant differences relative to vehicle (p < 0.05).
Antagonism studies revealed that pretreatment with 3.0 mg/kg α1GABAA-preferring antagonist β-CCT did not alter reliably the muscle resistance induced by high doses of SL651498, chlordiazepoxide, or triazolam (Fig. 4). In contrast, the ataxia engendered by these doses of SL651498, chlordiazepoxide, and triazolam were reliably attenuated by 3.0 mg/kg β-CCT pretreatment.
Drug Discrimination Study. In monkeys trained to discriminate triazolam from saline, triazolam increased responding on the drug-appropriate lever in a dose-dependent manner (Fig. 5). As the dose of triazolam increased, rates of responding decreased, with the 0.1 mg/kg dose reliably decreasing the rate of responding compared with vehicle (Dunnett's t test, p < 0.05). SL651498 also engendered an increase in triazolam-appropriate responding; however, the maximum percentage of drug-lever responding did not reach 80% up to a dose of 10 mg/kg (Fig. 5). At 10 mg/kg SL651498, two of four monkeys did not respond, whereas the remaining two monkeys responded primarily on the saline lever at rates of responding that were approximately 25% of vehicle control levels (Fig. 5, bottom panel). This resulted in an inverted u-shaped dose-response function for the triazolam-like effects of SL6514898.
As shown in Fig. 6, the α1GABAA-preferring antagonist β-CCT inhibited the discriminative stimulus effects of SL651498 in a dose-dependent manner. Over the dose range evaluated in this study, the 0.3 mg/kg dose of β-CCT resulted in antagonism that was fully surmountable (i.e., the maximum percentage triazolam-lever responding returned to the levels observed without β-CCT treatment). It is unknown whether the effects of the 1.0 mg/kg dose of β-CCT are surmountable because higher concentrations of these drug combinations could not be evaluated without compromising catheter patency. Antagonism by the 0.3 mg/kg dose of β-CCT was analyzed by in vivo apparent pKB analysis, and an average apparent pKB of 6.3 was obtained. The 95% confidence intervals associated with the apparent pKB for 0.3 mg/kg β-CCT and SL651498 overlapped with apparent pKB values for antagonism by this dose of β-CCT of the discriminative stimulus effects of zolpidem in zolpidem-trained monkeys, as well as β-CCT antagonism of zolpidem and triazolam in triazolam-trained monkeys (Table 4). Neither dose of β-CCT had any effect on rate of responding when administered alone (data not shown).
In vivo apparent pKB analyses for β-CCT antagonism (0.3 mg/kg) of the triazolam-like discriminative stimulus effects of SL651498, in comparison with zolpidem and triazolam under different training conditions
Discussion
The data presented here describe the behavioral effects engendered by SL651498, a novel BZ agonist that acts as a full agonist at α2GABAA and α3GABAA receptors and as a partial agonist at α1GABAA and α5GABAA receptors in nonhuman primates. The distribution of the subtypes of the GABAA receptors throughout the brain varies such that the α1GABAA receptor is the most abundant and ubiquitous; α2GABAA and α3GABAA receptors are located primarily in limbic areas and in the spinal cord; α5GABAA receptors are the least abundant, residing mainly in hippocampal pyramidal cells (McKernan and Whiting, 1996). The intrinsic efficacy of SL651498 at the GABAA receptors and the regional distribution of those receptors within the central nervous system together likely play an important role in the behavioral effects produced by SL651498.

Myorelaxant and ataxic effects of SL651498, chlordiazepoxide, and triazolam alone and combined with the α1GABAA-preferring antagonist β-CCT in squirrel monkeys (N = 6). Asterisks represent significant differences relative to drug alone (p < 0.05).
Punishment-based conflict procedures are a reliable and clinically relevant method for demonstrating the efficacy of anxiolytic agents (Geller and Seifter, 1962; Kleven and Koek, 1999; Millan and Brocco, 2003). SL651498 increased responding suppressed by contingent shock in a conflict procedure developed for rhesus monkeys. This outcome, moreover, was observed at doses that did not decrease responding in the absence of shock. These results are in agreement with previous studies showing the potent anxiolytic action of SL651498 in the absence of behavioral impairment in various rodent conflict tests (Griebel at al., 2001). In contrast, both chlordiazepoxide and triazolam increased suppressed responding, but decreased rates of nonsuppressed responding at the highest doses tested (cf. Hascoet and Bourin, 1997; Pattij et al., 2000).
Although there are fewer available data attributing the anxiolytic effects of BZ agonists to specific GABAA receptor subtypes in nonhuman primates, studies in rodents have provided valuable insights about the receptor mechanisms underlying these effects. Point mutations in mice rendering the α1, α2, or α3 subunits insensitive to diazepam suggested that BZ-induced anxiolytic-like effects are mediated in part by α2GABAA receptors (Rudolph et al., 1999; Löw et al., 2000; McKernan et al., 2000). Additionally, administration of a α3GABAA receptor inverse agonist had anxiogenic effects in rats tested in the elevated plus maze (Collins et al., 2002). Together, these data implicate the α2 and/or α3GABAA receptors in anxiolysis. The present results support a similarly critical role for α2 and/or α3GABAA receptors in mediating anxiolytic-like effects in primates.

Percentage of drug-lever responding (top panel) and rates of responding (bottom panel) engendered by triazolam and SL651498 in squirrel monkeys trained to discriminate triazolam from saline. Data are mean ± S.E.M. from N = 4 monkeys. In parentheses is the number of monkeys responding/total sample size.
Observational techniques were used to characterize systematically the effects of SL651498 on a range of motor behaviors in squirrel monkeys. Across the range of doses tested, SL651498 decreased muscle resistance, with only a mild ataxic effect. To evaluate the role of α1GABAA receptors in these effects, we examined the ability of the α1GABAA receptor antagonist β-CCT to block the SL651498-induced muscle relaxant and ataxic effects. Neither the relaxation engendered by SL651498 nor that engendered by chlordiazepoxide or triazolam could be blocked by β-CCT, suggesting that α1GABAA receptors play a minimal role in mediating this behavior. In contrast, the same dose of β-CCT attenuated the ataxic effects of SL651498, triazolam, and chlordiazepoxide, suggesting that ataxia engendered by BZ agonists may be mediated at least in part by α1GABAA receptors. Putative measures of sedative-like effects such as changes in locomotion, procumbent, and rest postures (cf. Platt et al., 2002) were not altered by SL651498. These results differ markedly with findings from a similar study in which triazolam and the α1GABAA agonist zolpidem engendered rest and/or procumbent postures and virtually eliminated locomotor activity (Platt et al., 2002; Table 3). The lack of sedative-like effects of SL651498 support a role for α1GABAA receptors in these effects (see Platt et al., 2002).

Antagonism of the triazolam-like discriminative stimulus effects of SL651498 by the α1GABAA-preferring antagonist β-CCT in squirrel monkeys trained to discriminate triazolam from saline. Data are mean ± S.E.M. from N = 4 monkeys.
Recent work in transgenic mice attributes myorelaxation to the α2 and α3GABAA receptors (Crestani et al., 2001), and our results are consistent with these GABAA receptors mediating muscle relaxation. The muscle relaxant effects of SL651498 reported here likely were not mediated by α1GABAA receptors, as β-CCT did not inhibit this behavior. That β-CCT did attenuate ataxia engendered by SL651498, as well as chlordiazepoxide and triazolam, suggests that ataxia may be mediated by α1GABAA receptors, a result consistent with SL651498's low efficacy at α1GABAA receptors. Recent studies have demonstrated that α1GABAA receptors are critically involved in motor effects of BZs, suggesting that SL651498's low efficacy at α1GABAA receptors may explain the absence of ataxia/sedation in this study (Rudolph et al., 1999; McKernan et al., 2000; Crestani et al., 2001).
Drug discrimination procedures provide a method for studying receptor mechanisms underlying the subjective effects of drugs. Typically drugs that occasion the same behavioral response as the training drug (i.e., substitute for the training drug) share the same receptor mechanism of action, converge upon the same neurotransmitter system, or both (Lelas et al., 2000). Using this paradigm, the receptor mechanisms mediating the discriminative stimulus effects of SL651498 were assessed in squirrel monkeys trained to discriminate triazolam from saline. The conventional BZ agonist triazolam increased responding on the drug-appropriate lever in a dose-dependent manner, an effect that may be mediated by α1GABAA receptors (Lelas et al., 2001, 2002). SL651498 partially reproduced the discriminative stimulus effects of triazolam. To evaluate the role of α1GABAA receptors in this effect, we examined the ability of the α1GABAA receptor-preferring antagonist β-CCT to block the partial triazolam-like subjective effects of SL651498. β-CCT, at doses that antagonized the discriminative stimulus effects of both triazolam and the α1GABAA-preferring agonist zolpidem (Lelas et al., 2002; Rowlett et al., 2003), dose-dependently prevented SL651498's partial substitution for triazolam. The apparent pKB value for antagonism of the triazolam-like discriminative stimulus effects of SL651498 was similar to those calculated from previous studies examining β-CCT antagonism of zolpidem- and triazolam-like discriminative effects (Lelas et al., 2002; Rowlett et al., 2003). This analysis suggests that β-CCT blocked the discriminative stimulus effects of SL651498, zolpidem, and triazolam via a similar population of receptors, presumably α1GABAA receptors. Consistent with the involvement of α1GABAA receptors, SL651498 previously was shown to partially substitute for the α1GABAA-preferring BZ agonist zolpidem in rats (Griebel et al., 2001). Altogether, these findings suggest that SL651498 shares some subjective effects with triazolam that may be mediated by α1GABAA receptor subtypes.
Drug discrimination studies also provide information about a drug's intrinsic efficacy, a factor that contributes to its subjective effects. An agonist with low intrinsic efficacy will engender less than full substitution or substitute in a subset of animals trained to discriminate an agonist with high intrinsic efficacy (Ator and Griffiths, 1999). In the present studies, SL651498 only partially substituted for the high efficacy agonist triazolam (Ducic et al., 1993), suggesting that it has lower intrinsic efficacy than triazolam. Interestingly, the partial substitution of SL651498 for triazolam (64% triazolam-lever responding) is nearly identical to the 66% triazolam-lever responding engendered by chlordiazepoxide in a previous study (Lelas et al., 2001). Thus, it has been suggested that chlordiazepoxide is a partial agonist, a hypothesis that is supported by its similarities to the known low efficacy BZ agonist bretazenil (Puia et al., 1992; Sannerud and Ator, 1995). The similar substitution profiles of SL651498 and chlordiazepoxide in triazolam-trained animals together with the blockade of the partial substitution by the α1GABAA receptor antagonist, underscore SL651498's low intrinsic efficacy at α1GABAA receptors.
Altogether, these data suggest that the receptor mechanisms underlying the behavioral effects of the BZ agonist SL651498 likely reflect its relative efficacy at the various α subunits of the GABAA receptor complex. These studies particularly implicate α1GABAA receptors in the discriminative stimulus and ataxic effects of SL651498, whereas α2GABAA and α3GABAA receptors may mediate the anxiolytic and myorelaxant effects of this drug. These data further suggest that SL651498, a drug with functional selectivity, may have therapeutic potential as a nonsedating anxiolytic and muscle relaxant.
Acknowledgments
We thank Shana Elkind and Walter Tornazky for expert technical assistance.
Footnotes
-
This work was supported by U.S. Public Health Services Grants DA11792, DA00499, and RR00168, as well as the National Institute of Mental Health Grant MH-46851. A preliminary report of these data was made at the 2004 Annual Meeting of the College on Problems of Drug Dependence.
-
doi:10.1124/jpet.104.081612.
-
ABBREVIATIONS: BZ, benzodiazepine; SL651498, 6-fluoro-9-methyl-2-phenyl-4-(pyrrolidin-1-yl-carbonyl)-2,9-dihydro-1H-pyridol[3,4-b]indol-1-one; FR, fixed ratio; β-CCT, β-carboline-3-carboxylate-t-butyl ester; MS, maximum score; MD, minimally effective dose.
- Received December 3, 2004.
- Accepted January 31, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}