


Abstract
The effects of nicotine on the tail-flick and hot-plate tests were determined to identify nicotinic receptor subtypes responsible for spinally and supraspinally mediated nicotine analgesia in knockin mice expressing hypersensitive α4 nicotinic receptors (L9′S), in seven inbred mouse strains (C57BL/6, DBA/2, A/2, CBA/2, BALB/cByJ, C3H/HeJ, and 129/SvEv), and in two F1 hybrids (B6CBAF1 and B6D2F1). L9′S heterozygotes were ∼6-fold more sensitive to the antinociceptive effects of nicotine than the wild-type controls in the hot-plate test but not in the tail-flick assay. Large differences in the effects of nicotine were also observed with both tests for the seven mouse strains. A/J and 129 mice were 6- to 8-fold more sensitive than CBA and BALB mice. In addition, B6CBAF1 hybrid mice were even less sensitive than CBA mice. Nicotinic binding sites were measured in three spinal cord regions and the hindbrain of the inbred strains. Significant differences in cytisine-sensitive, high affinity [125I]epibatidine binding site levels (α4β2* subtypes), but not in 125I-α-bungarotoxin binding (α7* subtypes), were observed. Significant negative correlations between cytisine-sensitive [125I]epibatidine binding and nicotine ED50 for both tests were noted. Our results indicate that α4β2* acetylcholine nicotinic receptors (nAChR) are important in mediating nicotine analgesia in supraspinal responses, while also showing that α4β2*-nAChR and at least one other nAChR subtype appear to modulate spinal actions.
Findings that nicotine is an antinociceptive agent in animals (Aceto et al., 1986) and that cigarette smoking and nicotine reduce pain in humans (Lane et al., 1995; Jammer et al., 1998) suggest that nicotinic compounds may be useful in the treatment of pain. Furthermore, the observation that the nicotinic agonist epibatidine is 100 times more potent than morphine as an analgesic in the tail-flick and hot-plate tests (Badio and Daly, 1994) has spurred renewed interest in the development of nicotinic agents as analgesics. Analgesic drugs currently used produce serious side effects on long-term use; safer and more efficacious analgesic agents are needed. It is generally agreed that central pathways modulate the analgesic actions of nicotine (Sahley and Berntson, 1979); in particular, the spinal cord is a major site of action for tail-flick nociception given that i.t. administration of nicotinic agonists produces antinociception in this test (Aceto et al., 1986; Christensen and Smith, 1990). In addition, Caggiula et al. (1995) suggested that nicotine's effects on the tail-flick and hot-plate tests involve at least partially separate pathways. Unfortunately, only limited progress has been made toward identifying the neuronal nicotinic acetylcholine receptor (nAChR) subtypes expressed in spinal cord that modulate pain, perhaps because mRNA for virtually all the nAChR subtypes have been identified in dorsal root ganglion neurons of adult (Haberberger et al., 2004) and neonatal mice (Cordero-Erausquin et al., 2004). This complexity may explain why studies using pharmacological approaches to identify the nAChR subtypes that modulate the antinociceptive effects of nicotinic agonists (Damaj et al., 1998) and antagonists (Iwamoto and Marion, 1993) are not in complete agreement.
Studies using nAChR subunit null mutant (gene knockout) mice have suggested roles for specific nAChR subtypes in nicotine-induced antinociception. Mice lacking either the α4 or β2 nicotinic receptor subunit showed reduced sensitivity to nicotine-induced antinociception in the hot-plate and tail-flick acute pain tests (Marubio et al., 1999). Both α4 and β2 null mutant mice failed to respond to nicotine on the hotplate test at doses that were fully efficacious in wild-type (WT) mice, suggesting a prominent role for α4β2-nAChR. In contrast, both α4 and β2 gene deletion resulted in only modest (2–3-fold) reductions in agonist potency with the tail-flick test.
The present study used two genetic approaches to investigate which nAChR subtypes modulate the actions of nicotine on the hot-plate and tail-flick tests. The first approach took advantage of knockin mice that express mutated α4*-nAChR (Labarca et al., 2001). These mice express α4 subunits with a leucine to serine substitution in the 9′ position (L9′S) of the pore-lining M2 region, which renders mice hypersensitive to nicotine in functional and behavioral tests (Labarca et al., 2001; Fonck et al., 2003). The second approach took advantage of the findings that outbred and inbred mouse strains differ in sensitivity to the effects of nicotine (Seale et al., 1998) and epibatidine on thermal reactivity tests (Flores et al., 1999), locomotor activity (Collins et al., 1988; Marks et al., 1989a), hypothermia (Tritto et al., 2001), and seizures (Miner and Collins, 1989). Mouse strains also differ in brain [3H]nicotine (α4β2*-nAChR) (Marks et al., 1989a) and 125I-α-bungarotoxin (BTX) (α7-nAChR) binding (Stitzel et al., 1998). Moreover, strain variability in [3H]nicotine binding shows a strong correlation with variations in nicotine-induced locomotor activity and changes in body temperature, whereas variability in 125I-α-BTX binding correlates with the severity of nicotine-induced seizures. Further nAChR subtype specificity can be achieved by measuring high affinity (KD ≈ 20 pM) radiolabeled epibatidine binding, which can be divided into two major components based on the sensitivity to inhibition by cytisine. The fraction sensitive to cytisine inhibition is primarily composed of the α4β2 subtype, whereas cytisine-resistant sites are thought to represent α3β2*, α3β4*, and α6β2* receptors (Marks et al., 2006). The observation that strain variability in responses to nicotine is significantly correlated with differences in binding of radioactive nicotinic ligands prompted us to study the effects of nicotine on the hot-plate and tail-flick tests of seven inbred mouse strains known to be differentially sensitive to nicotine for other measures (Marks et al., 1989b), as well as for two F1 hybrids. We then measured cytisine-sensitive and cytisine-resistant [125I]epibatidine binding and 125I-α-BTX binding in hindbrain and cervical, thoracic, and lumbar spinal cord of these mouse strains.
Materials and Methods
Animals
Generation of the L9′S strain, by replacing the native α4 subunit of nAChR with a mutated form, was described previously (Labarca et al., 2001). Only heterozygous L9′S mice with an intact neomycin resistance cassette were used because L9′S homozygous mice are neonatal-lethal; therefore, the L9′S designation used throughout the article refers to these heterozygotes. Male mice from the following inbred strains were used: C57BL/6J, DBA/2JIbg, A/J, CBA/J, C3H/HeJ, BALB/cByJ, 129/SvEv, B6CBAF1, and B6D2F1. DBA/2JIbg mice were obtained from the breeding colony at the Institute for Behavioral Genetics (Boulder, CO). The 129/SvEv strain was obtained from Taconic (Germantown, NY). The remaining strains were purchased from Jackson Laboratories (Bar Harbor, ME) and housed in the mouse colony at the Institute for Behavioral Genetics for at least 2 weeks before testing. Mice were housed in groups of three to four per cage and had free access to food and water. All the animals were 60 to 90 days of age at the time of testing. The study was reviewed and approved by the Institutional Animal Care and Use Committee of the Virginia Commonwealth University and the Animal Care and Utilization Committee of the University of Colorado.
Drugs
Mecamylamine hydrochloride was a gift from Merck Sharp and Dohme (West Point, PA). Morphine sulfate and (-)-nicotine were obtained from Sigma-Aldrich (St. Louis, MO). All the drugs were dissolved in physiological saline (0.9% sodium chloride), and all the doses are expressed as the free base of the drug.
Antinociceptive Tests
Tail-Flick Test. Mice were lightly restrained by hand while a radiant heat light source was shone onto the upper portion of the tail. Latency to remove the tail from the heat source was recorded for each animal. A control response (2–4-s latency) was determined for each mouse before treatment, and test latency was determined after drug administration. To minimize tissue damage, a maximal latency of 10 s was imposed. Antinociceptive response was calculated as percentage of maximal possible effect (%MPE), where %MPE = {[test value - baseline]/[cutoff time (10 s) - control value]} × 100, where baseline represents the value before nicotine. Groups of 8 to 12 animals were used for each dose and for each treatment. Mice were tested 5 min after an s.c. injection of nicotine.
Hot-Plate Test. Mice were tested 2 h before and 5 min after an s.c. injection of nicotine. The animals were placed on a 55°C platform (Harvard Apparatus, Holliston, MA) and were observed until they started to show pain-avoidance behavior, such as jumping or licking of the paws. Animals that did not respond to the noxious heat stimulus after 40 s were removed from the plate. Latency to pain avoidance measured in seconds was used to calculate %MPE with the following equation: {[test value - baseline]/[cutoff time (40 s) - baseline]} × 100. Baseline latency that lasted 8 to 12 s was assessed with a saline injection.
To assess specificity of the nicotine-induced analgesia in L9′S, animals were injected with mecamylamine (1 mg/kg, s.c.) 5 min before receiving a single nicotine injection in the hot-plate and tail-flick tests. For comparison purposes, the specificity of nociceptive responses in L9′S mice was also evaluated by determining the potency of morphine in the hot-plate and tail-flick tests. Animals were tested 20 min after an s.c. injection of morphine.
Binding Experiments
Tissue Preparation. Mice were killed by cervical dislocation. Their brains were removed and placed on ice, and the hindbrains (pons medulla) were dissected. The spinal column was isolated and divided into thoracic, cervical, and lumbar regions. Each segment of the spinal cord was removed from the spinal column by gentle flushing with ice-cold, isotonic saline. The dissected tissue was subsequently placed in hypotonic buffer (14 mM NaCl, 0.2 mM KCl, 0.2 mM CaCl2, 0.1 mM MgSO4, 2 mM HEPES, pH = 7.5). Samples were homogenized using a glass-Teflon tissue grinder. Homogenized samples were centrifuged at 10,000g for 20 min, and the supernatant was discarded. The pellet was resuspended in hypotonic buffer and centrifuged again at 10,000g for 20 min. This procedure was repeated two more times. The final washed pellet was resuspended in dilute buffer to a concentration of 1 to 2 mg/ml protein.
[125I]Epibatidine Binding. [125I]Epibatidine (specific activity = 2200 Ci/mmol; PerkinElmer Life and Analytical Sciences, Boston, MA) binding was conducted essentially as described previously (Marks et al., 1998). Samples were incubated for 3 h in 30 μl of buffer (135 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 0.02% bovine serum albumin at 20°C, 25 mM HEPES, pH = 7.5) using a final [125I]epibatidine concentration of 400 pM. Cytisine-resistant binding was initially measured by constructing inhibition curves for cytisine (cytisine concentrations from 1 × 10-9 to 5 × 10-5 M) for two mice of each strain. Subsequently, cytisine-resistant sites were calculated from data obtained for binding in the presence of 0, 50, and 150 nM cytisine. Blanks were established using 0.1 mM nicotine. Blanks represented less than 5% of specific binding. Following the incubation, samples were filtered using an Inotech Biosystems (Rockville, MD) harvester and two layers of glass fiber filter [top filter MFS GC, bottom filter Gelman (Ann Arbor, MI) A/E, both of which had been soaked in 0.1% polyethylenimine]. Samples were washed six times with ice-cold buffer without serum albumin. Samples were counted using a Packard Tri-Carb liquid scintillation spectrometer (PerkinElmer Life and Analytical Sciences) at 45% efficiency after the addition of 1.5 ml of Budget Solve scintillation mixture (Sigma/RBI, Natick, MA).
125I-α-BTX Binding.125I-α-BTX (specific activity = 200 Ci/mmol; NEN) binding was essentially measured as described previously (Marks et al., 1986b). Samples were incubated for 3 h in 100 μlof buffer (135 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 0.02% bovine serum albumin at 20°C, 25 mM HEPES, pH = 7.5) using a final 125I-α-BTX concentration of 1 nM. Blanks were determined by including 1 mM nicotine in the incubations. Blanks represented approximately 50% of specific binding at this concentration of [125I]α-BTX. Although maximal binding is not measured at this concentration, use of higher ligand concentrations resulted in very unfavorable signal/noise ratios. After the incubation, samples were filtered using an Inotech harvester and two layers of glass fiber filter (top filter MFS GC, bottom filter Gelman A/E, both of which had been soaked in 0.1% polyethylenimine). Samples were washed six times with ice-cold buffer without serum albumin. Samples were counted using a Packard gamma counter (PerkinElmer Life and Analytical Sciences) at 80% efficiency.
Protein. Protein was measured with the method of Lowry et al., (1951) using bovine serum albumin as the standard.
Statistical Analysis
Statistical analysis of all the behavioral studies was performed using either a t test or analysis of variance with a Dunnett's post hoc t test when appropriate. All the differences were considered significant at p < 0.05. ED50 values with a 95% confidence level (CL) for behavioral data were calculated by unweighted least-squares linear regression as described by Tallarida and Murray (1987). One-way analysis of variance followed by Dunnett's post hoc t test was used to evaluate differences in binding in each tissue for each binding site. Relationships among the parameters were assessed by linear regression.
Results
Antinociceptive Effects of Nicotine in the α4 L9′S Mice
Antinociceptive effects of nicotine on L9′S heterozygous and WT mice in the tail-flick and hot-plate tests are shown in Fig. 1, A and B. The latency response of noninjected control mice to a painful stimulus did not differ significantly between WT and L9′S animals in either test, indicating that the endogenous activation of the α4-nAChR subunit, even with a mutation conferring hypersensitivity, is not essential to the perception of acute thermal nociception.

Dose-response curves of nicotine and morphine-induced antinociception in L9′S heterozygote (dark gray circles) and WT (black circles) mice. Animals were tested in the hot-plate and the tail-flick apparatus as described under Materials and Methods. Animals were tested 5 min after an s.c. nicotine injection in the tail-flick (A) and hot-plate (B) tests. Each data point is the mean ± S.E. of n = 6 to 8 animals. A different set of animals was tested 20 min after an s.c. morphine injection in the tail-flick (C) and hot-plate (D) tests. Each data point is the mean ± S.E., n = 6 to 8.
The tail-flick test revealed no obvious hypersensitivity to nicotine in the L9′S mice (Fig. 1A) after s.c. injection of various doses of nicotine. However, it should be noted that doses higher than 0.5 mg/kg were not tested in L9′S mice because animals displayed tremors, shaking, and stereotypy when higher doses of nicotine were administered (Fonck et al., 2003). These behavioral responses in L9′S mice are thought to precede seizures, which are probably initiated in subcortical structures such as the hippocampus, thalamus, or medial habenula and are not thought to be directly involved in antinociceptive responses. WT mice exhibited a dose-dependent antinociception following nicotine administration at doses greater than 0.5 mg/kg. In the hot-plate test (Fig. 1C), WT mice showed a dose-dependent antinociceptive response with an ED50 (± CL) = 0.92 (0.7–1.1) mg/kg. In L9′S mice, the nicotine dose-response relation was shifted to the left with an ED50 value of 0.17 (0.1–0.2) mg/kg (Fig. 1C). Thus, a 6-fold shift in the dose-response relation for nicotine-induced antinociception was observed between L9′S and WT mice.
The nicotinic receptor antagonist mecamylamine (1 mg/kg) effectively blocked nicotine-induced analgesia described in these experiments. Mecamylamine blocked the nicotine-induced decrease in hot-plate nociception (data not shown) in both L9′S and WT animals. Mecamylamine also blocked the nicotine-induced delay in tail-flick responses in WT mice.
Antinociceptive effects of morphine in the hot-plate and tail-flick tests are shown in Fig. 1, B and D. L9′S and WT mice had similar dose-dependent antinociceptive responses to morphine in the hot-plate test (Fig. 1D), yielding ED50 values of 6.8 (5.8–7.9) and 5.9 (4.8–7.1) mg/kg, respectively. Similar potency for morphine was also observed for L9′S and WT mice in the tail-flick (Fig. 1B) test, with ED50 values of 0.90 (0.5–1.6) and 0.85 (0.4–1.8) mg/kg, respectively.
Antinociceptive Effects of Nicotine in Seven Inbred Mouse Strains
Tail-Flick Test. No differences among the strains in basal latencies for the tail-flick test were noted following saline injection (latencies range, 1.7–2.4 s).
The effects of nicotine administration on the tail-flick response of the seven inbred strains of mice are shown in Fig. 2, A and B, and the ED50 values calculated from these curves are shown in Fig. 2C. Antinociceptive effects of nicotine differed significantly among the inbred strains. The dose-response curves for nicotine-induced antinociception for mice of the A/J and 129SvEv strains were similar and yielded similar ED50 values [ED50 (95% CL), 0.48 (0.34–0.67) and 0.62 (0.05–0.70) mg/kg, respectively]. Dose-response curves of nicotine-induced antinociception for C57BL/6 and DBA/2 were also similar, but these mice were less sensitive to nicotine than either the A or 129 mice [ED50 (95% CL), 1.07 (0.97–1.22) for C57BL/6 and 1.20 (0.94–1.60) mg/kg for DBA]. C3H [ED50 (95% CL), 1.97 (1.4–2.6)] and BALB [ED50 (95% CL), 3.2 (2.6–3.8)] required significantly higher nicotine doses. Antinociceptive effects of nicotine in CBA mice were also small. Even at the highest dose of nicotine tested (2 mg/kg, s.c.), the effect (16% MPE) was not significantly different from saline control. Higher doses of nicotine (>2 mg/kg) were not tested because of the increased risk of triggering seizures. The rank order of the strains for sensitivity to the antinociceptive effects of nicotine measured by tail-flick was 129 ≥ A/J > C57BL/6 ≈ DBA/2 > C3H > BALB ≈ CBA. Mice of the 129 strain were at least 4-fold more sensitive than those of the CBA strain and 6-fold more sensitive than those of the BALB strain.
Antinociceptive effects were also measured with two F1 hybrids. The B6D2F1 (F1 cross of C57BL/6 and DBA/2) were more sensitive to nicotine [ED50 (95% CL), 0.68 (0.2–1.1) mg/kg] than the parental strains, whereas B6CBAF1 (F1 cross of C57BL/6 and CBA) were significantly less sensitive than either parent (1% MPE after a dose of 3.5 mg/kg).

Antinociceptive effects of nicotine in seven inbred mice strains and two F1 hybrids measured using the tail-flick and hot-plate tests. Effects of nicotine were measured in inbred A/J (open circles), C57BL/6 (black squares), DBA (light gray upright triangles), 129 (dark gray diamonds), BALB (white squares), C3H (dark gray inverted triangles), and CBA (dark gray circles) mice and in hybrid B6CBAF1 (dark gray hexagons) and B6D2F1 (light gray diamonds) mice after s.c. administration of the indicated doses of the drug. Mice were tested 5 min after injection. Each point represents the mean ± S.E., n = 8 to 12 mice. Dose-response curves for nicotine effects on tail-flick test are shown in A and B, and ED50 values calculated from these curves are shown in C. Dose-response curves for nicotine effects on hot-plate test are shown in D and E, and ED50 values calculated from these curves are shown in F.
Hot-Plate Test. Significant strain differences in baseline responsiveness in the hot-plate test were observed (latencies range, 5.5–9.7 s) [F(6,90) = 5.8, p < 0.05]. These differences were not unexpected because strain differences in baseline responsiveness have previously been noted for this test in inbred mouse strains (Flores et al., 1999).
Dose-response curves for the antinociceptive effects of nicotine measured with the hot-plate test are shown in Fig. 2, D and E, and ED50 values calculated from these curves are shown in Fig. 2F. Similar to the results of the tail-flick test, the potency of nicotine as an antinociceptive agent differed significantly among the strains. In general, ED50 values were lower for the hot-plate test than for the tail-flick test. A/J mice were the most sensitive [ED50 (95% CL), 0.2 (0.16–0.24) mg/kg]. 129SvEv mice were also quite sensitive [ED50 (95% CL), 0.45 (0.41–0.49) mg/kg]. DBA/2 and C57BL/6 mice displayed similar responsiveness to nicotine [ED50 (95% CL), 0.85 (0.5–1.2) and 0.92 (0.73–1.2) mg/kg, respectively], whereas C3H mice were slightly less sensitive [ED50 (95% CL), 1.05 (0.8–1.3) mg/kg]. The BALB [ED50 (95% CL), 1.3 (0.97–1.8) mg/kg] and CBA [ED50 (95% CL), 1.55 (1.2–1.85) mg/kg] mice required higher nicotine doses for antinociception. In contrast to the tail-flick test, nicotine-mediated antinociception was measurable in CBA mice in the hot-plate test, but the ED50 value for CBA was the highest of the seven strains tested [ED50 (95% CL), 1.55 (1.2–1.85) mg/kg]. Thus, the rank order for sensitivity to nicotine-induced antinociception for the hot-plate test was A/J > 129 > DBA/2 ≈ C57BL/6 > C3H > BALB > CBA. The ED50 value for CBA mice was approximately 7.5-fold higher than that for A/J mice.
Antinociceptive effects were also measured with two F1 hybrids. As was the case with the tail-flick test, the B6D2F1 (F1 cross of C57BL/6 and DBA/2) were slightly more sensitive to nicotine [ED50 (95% CL), 0.68 (0.2–1.1) mg/kg] than the parental strains, whereas B6CBAF1 (F1 cross of C57BL/6 and CBA) were significantly less sensitive than either parent (40%MPE after a dose of 3.5 mg/kg).
Receptor Determinations in Seven Inbred Mouse Strains
The density of three nicotinic binding sites was measured to explore possible relationships between nAChR levels and sensitivity to nicotine in the mouse strains studied. Binding was measured in hindbrain and the cervical, thoracic, and lumbar regions of spinal cord. The three binding sites measured were 125I-α-BTX (measuring α7-nAChR), cytisine-sensitive [125I]epibatidine (measuring β2*-nAChR, primarily α4β2*-nAChR), and cytisine-resistant [125I]epibatidine [measuring a mixed population of β2*-nAChR (non–α4β2*-nAChR) and β4*-nAChR] (Marks et al., 2006).
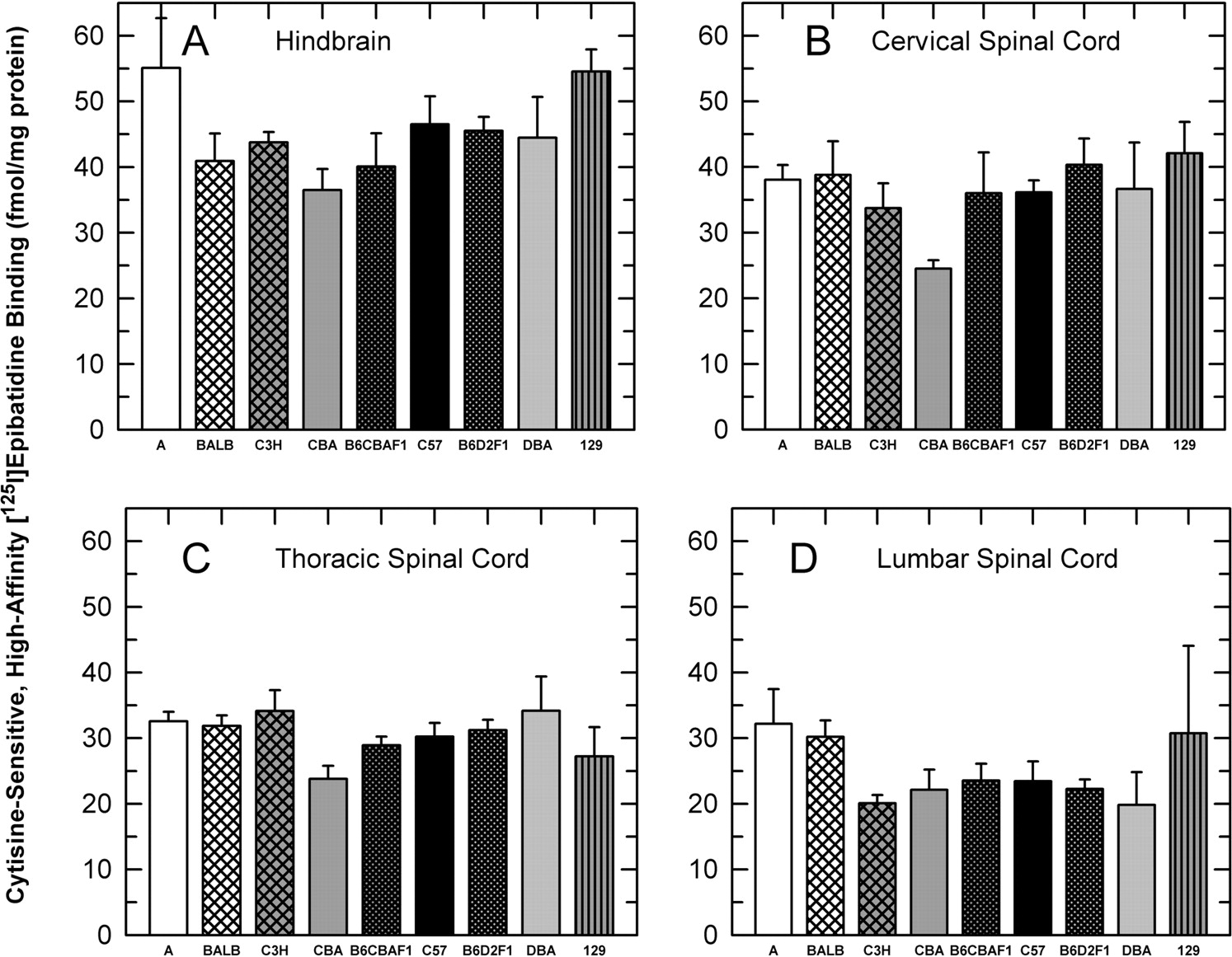
Cytisine-Sensitive [125I]Epibatidine Binding. The density of high-affinity [125I]epibatidine binding sites sensitive to inhibition by cytisine was measured using a [125I]epibatidine concentration of 400 pM. This concentration is saturating, thereby providing a measurement of maximal binding site density. Results for these measurements are shown in Fig. 3.
Significant differences in binding site density among the seven strains were observed [hindbrain, F(6,44) = 2.34, p < 0.05; thoracic spinal cord, F(6,44) = 2.84, p < 0.05; and lumbar spinal cord, F(6,44) = 2.37, p < 0.05; however, cervical spinal cord was not significant, F(6,44) = 2.01, p > 0.05]. In every sample prepared, tissue prepared from A/J mice had the highest densities. Binding for A/J mice was significantly higher than that for CBA and C3H mice in every region. Relative binding site densities measured for tissue obtained from DBA/2 and C57BL/6 mice were similar to each other and varied among the regions. Binding for these two strains was relatively high in hindbrain and cervical and thoracic spinal cord, similar in magnitude to that of the A/J mice.

Cytisine-sensitive, high affinity [125I]epibatidine binding. The density of cytisine-sensitive high affinity [125I]epibatidine binding sites in the hindbrain and cervical, thoracic, and lumbar regions of the spinal cord was measured using 400 pM ligand in membranes prepared from each of the seven different inbred mouse strains and the two F1 hybrids as described under Materials and Methods. Results are expressed in fmol/mg protein ± S.E. for preparations from five mice of each strain.
Cytisine-sensitive [125I]epibatidine measured in hindbrain and cervical spinal cord for the B6CBAF1 mice was intermediate between that of the two parental strains, whereas that measured in thoracic spinal cord resembled that of the CBA parent (lower than midpoint), and that in the lumbar spinal cord was lower than that for either parent. A similar pattern was also observed for the B6D2F1 mice, although binding in both thoracic and lumbar spinal cord was lower than that of either parent.
Cytisine-Resistant [125I]Epibatidine Binding. The density of high affinity [125I]epibatidine binding sites was calculated from binding using a saturating concentration of 400 pM [125I]epibatidine in the presence of 0, 50, and 150 nM cytisine (Marks et al., 1998). Results for these measurements are shown in Fig. 4.
The cytisine-resistant component of the high affinity [125I]epibatidine binding represented a relatively small fraction of total high affinity binding (<20% in hindbrain, <15% in the spinal cord). Whereas no significant differences in the number of these binding sites among the strains were detected in hindbrain, cervical spinal cord, or lumbar spinal cord, significant differences among the strains were noted for thoracic spinal cord [F(6,44) = 2.82, p < 0.05] with samples prepared from A/J mice exhibiting higher binding than those prepared from DBA/2, BALB, C3H, and 129 mice.
125I-α-BTX Binding. The density of 125I-α-BTX binding sites was measured using a ligand concentration of 1 nM, which is near the KD. Therefore, these measurements do not represent maximal site densities. Results for 125I-α-BTX binding in the four regions of all the inbred strains are shown in Fig. 5. No significant differences in 125I-α-BTX binding were observed for hindbrain or any area of the spinal cord.
Relationship between Nicotinic Binding Sites and Antinociception
Because both the potency of nicotine's antinociceptive effects and the density of nicotinic binding sites have been measured in each of the seven inbred strains, linear regression analyses were conducted to compare the various measures. Scattergrams comparing the ED50 values for nicotine-induced antinociception and density of cytisine-sensitive [125I]epibatidine binding sites, cytisine-resistant [125I]epibatidine binding sites, and [125I]α-BTX binding sites in hindbrain are shown in Fig. 6. ED50 values for the two thermal pain tests for the seven inbred mouse strains were very highly correlated (Fig. 6A, r = 0.92). Highly significant (p < 0.01) correlations between ED50 for tail-flick (Fig. 6E, r =-0.89) and hot-plate (Fig. 6B, r =-0.85) tests and the density of cytisine-sensitive [125I]epibatidine binding sites were also observed. No significant correlations between the other two binding sites and the pain tests or among the three binding sites were noted. Correlations between the density of cytisine-sensitive [125I]epibatidine binding sites in cervical, thoracic, or lumbar spinal cord and the ED50 values for the hot-plate tests were not so robust (r ≈-0.62, p ≈ 0.06) as those for binding sites in hindbrain.
Discussion
Nicotinic agonists elicit antinociceptive responses in several acute pain tests, including tail-flick and hot-plate tests. Responses to the hot-plate are thought to be centrally mediated, whereas the tail-flick is considered a spinal reflex (Caggiula et al., 1995). This study takes advantage of the rare opportunity to study a behavioral test by exploiting both specific targeted mutations in a candidate molecule and inbred strains that differ in many more genes. The studies reported here further tested the postulate that activation of α4β2*-nAChR evokes the antinociceptive effects of nicotine on the hot-plate and tail-flick tests.

Cytisine-resistant, high affinity [125I]epibatidine binding. The density of cytisine-resistant high affinity [125I]epibatidine binding sites in the hindbrain and cervical, thoracic, and lumbar regions of the spinal cord was measured using 400 pM ligand in membranes prepared from the seven different inbred mouse strains and the two F1 hybrids as described under Materials and Methods. Results are expressed in femtomole/milligram protein ± S.E. for preparations from five mice of each strain.

125I-α-BTX binding. The density of 125I-α-BTX binding sites in the hindbrain and cervical, thoracic, and lumbar regions of the spinal cord was measured using 1 nM ligand in membranes prepared from the seven different inbred and two hybrid mouse strains as described under Materials and Methods. Results are expressed in fmol/mg protein ± S.E. for preparations from five mice of each strain.
Previous data based on gene knockouts (Marubio et al., 1999) and antisense-mediated knockdown (Bitner et al., 2000) indicate that α4* and β2* receptors are necessary for a major antinociceptive component as measured in the hot-plate test; however, the present finding that L9′S mice, which express a hypersensitive α4 receptor, are much more sensitive to the effects of nicotine in the hot-plate test than are the controls shows that nicotine activation of α4* receptors in mice is sufficient to produce such antinociception. Thus, the combination of necessity and sufficiency does provide compelling indications that, among the set of nAChR receptor subtypes, α4β2*-nAChR dominate nicotine's actions on the hot-plate test.
Marubio et al. (1999) observed that α4 and β2 null mutant mice were 2- to 3-fold less sensitive to nicotine than WT mice on the tail-flick test and concluded that the effects of nicotine on this test are modulated by the α4β2*-nAChR subtype plus another, lower affinity nAChR. We did not detect an increase in sensitivity to nicotine's analgesic effects on the tail-flick test in L9′S mice. However, L9′S could not be tested with the full range of nicotine doses. The highest dose that was tested (0.5 mg/kg) produced no measurable change in response in either WT or mutant mice. The higher doses (1.0 and 2.0 mg/kg), which elicited effects in WT mice, were not used in L9′S mice because doses that exceed 0.5 mg/kg elicit tremors, shaking, stereotypy, and convulsions in these mice (Fonck et al., 2003). Thus, the data obtained with the L9′S mutant mice support the assertion that α4β2*-nAChR play a more dominant role in regulating nicotine's effects on the hot-plate test than for the tail-flick response.
Studies with both knockout and knockin mice provide valuable insights into the role of α4*-nAChR in pain modulation, but adaptive changes elicited by these mutations could affect the nociceptive phenotype. For example, the number of dopamine neurons in the substantia nigra is reduced by ∼35% in adult L9′S mice (Orb et al., 2004), and α4 null mutant mice exhibit alterations in arborization of the dendrites of dopamine neurons (Parish et al., 2005). These concerns prompted us to evaluate the effects of nicotine in seven inbred mouse strains to evaluate the variation in nicotine-induced antinociception.
Inbred mouse strains also vary substantially in sensitivity to nicotine, as measured by a battery of behavioral tests, and the seven inbred strains selected for the current study represent a wide range of sensitivity to nicotine (Marks et al., 1989a). The seven strains tested also differed markedly in sensitivity to the effects of nicotine in both the hot-plate and tail-flick tests. Indeed, the 7.5-fold difference in sensitivity to nicotine's effects on the hot-plate test between the most sensitive (A/J) and the least sensitive (CBA) inbred strains was as great as the difference between the L9′S mice and their WT littermates. These results are consistent with the finding that inbred mouse strains exhibit a 4-fold variation in sensitivity to epibatidine-induced antinociception for the tail-immersion withdrawal thermal assay, with the A/J strain being the most sensitive (Flores et al., 1999).
The rank orders of ED50 values for the strains tested on the hot-plate (A/J > 129 > DBA/2 ≈ C57BL/6 > C3H > BALB > CBA) and tail-flick (129 ≥ A/J > C57BL/6 ≈ DBA/2 > C3H > BALB ≈ CBA) tests are very similar (correlation coefficient for ED50 values = 0.94) but not identical, inasmuch as the ED50 values for the hot-plate test tend to be lower than those for the tail-flick test. One possible explanation for these findings is that α4β2*-nAChR dominate the effects of nicotine on the hot-plate test, whereas an additional, lower affinity nAChR contributes to the response in the tail-flick test. Indeed, α7-selective antagonists (α-BTX and methyllycaconitine) and partial agonists (dimethoxybenzylidine anabaseine, 4-OH-DMXB) blocked the antinociceptive effects of choline (an α7 agonist) on the tail-flick test (Damaj et al., 2000). These results suggest that the α7-nAChR may be the lower affinity receptor that also modulates the tail-flick test.

Correlations among ED50 values for hot-plate and tail-flick tests and nicotinic binding sites in hindbrain. Each point represents the mean ± S.E.M. for the indicated value for the seven inbred strains. Correlation coefficients for each panel are listed in the parentheses. Regression lines are shown for correlations that are statistically significant (p < 0.05).
Inbred strain differences in drug sensitivity could be attributed to pharmacodynamic or pharmacokinetic differences. Nicotine metabolism and elimination do not differ among several of these inbred strains (Petersen et al., 1984). Therefore, it is reasonable to assume that the pharmacogenetic variability observed in our studies is caused by differences in nicotinic receptors or any of the various downstream mechanisms engaged by these receptors. In particular, although the L9′S mice have a subset of α4* receptors that bear a mutation conferring a difference in acetylcholine (ACh) sensitivity, it seems possible that overall ACh sensitivity can also be governed by straightforward differences in the number of receptors. Inbred mouse strains differ in the number of brain nAChR, as measured by high affinity [3H]nicotine (α4β2*) and 125I-α-BTX (α7) binding (Marks et al., 1989a). Indeed, differences in binding site density are significantly correlated with differences in effects of nicotine on maze activities and hypothermia (Marks et al., 1989b). These results prompted us to examine the potential associations between nicotine-induced antinociception and brainstem/spinal cord nAChR numbers.
We measured nAChR binding in the spinal cord and the hindbrain using [125I]epibatidine and 125I-α-BTX as ligands. The high affinity epibatidine binding that is sensitive to cytisine inhibition measures α4β2*-nAChR (Marks et al., 2006). Strain differences in cytisine-sensitive [125I]epibatidine binding sites were seen in hindbrain and cervical, thoracic, and lumbar regions of spinal cord. Regression analysis indicated a highly significant inverse correlation between cytisine-sensitive [125I]epibatidine binding and ED[50 values for both tail-flick (r =-0.89) and hot-plate (r =-0.85) tests. This result and the fact that similar, but less robust, correlations were found between binding site densities in spinal cord and ED50 values (r ≈-0.65) indicate that mice expressing higher α4β2*-nAChR levels are more responsive to nicotine-mediated antinociception. These correlations between nicotinic analgesia and α4β2-nAChR binding sites are similar to those obtained for other nicotine-mediated behavioral responses such as hypolocomotion and hypothermia (Marks et al., 1989a). It is also likely that nicotine-evoked responses in these complex behavioral assays depend on downstream events not directly related to receptor levels. Such a possibility is reinforced by the results with the F1 hybrids, where the B6CBAF1 showed striking overdominance toward resistance to nicotine, whereas the B6D2F1 showed modest dominance toward sensitivity. Such complex inheritance patterns for nicotine-mediated behavioral responses have been observed for both nicotine-induced locomotor (Marks et al., 1986a) and hypothermic (Marks et al., 1984) responses.
nAChR are known to mediate release of several neurotransmitters and neuropeptides such as ACh, serotonin, and norepinephrine (Wonnacott et al., 1996); thus, differences in these parameters could contribute to the strain difference observed. Alternatively, activation of the endogenous opiate system may occur downstream from α4β2*-nAChR. Many studies have addressed the question of whether nicotinic agonists elicit their antinociceptive effects via interaction with endogenous opiate systems. There is substantial evidence that nicotinic agonists increase release of endogenous opiates and increase their synthesis in the brain (Houdi et al., 1998). The findings that pretreatment with opiate antagonists inhibits the antinociceptive effects of nicotine in rats (Biala et al., 2005) and mice (Berrendero et al., 2002), as well as the observation that preproenkephalin knockout mice exhibit decreased sensitivity to nicotine's effects on tail-immersion and hot-plate tests (Berrendero et al., 2005), suggest that nicotinic agonists exert some of their analgesic effects via endogenous opiate release. However, we did not find any change in sensitivity to the effects of morphine on either the hot-plate or tail-flick tests in L9′S mutant mice. Likewise, α4 knockout mice did not show a detectable change in sensitivity to the effects of morphine on the hot-plate and tail-flick tests (Marubio et al., 1999). These findings argue that opiate analgesia does not require direct activation of α4*-nicotinic receptors.
In summary, the studies reported here provide a new dimension that complements the conclusions drawn from the α4 and β2 knockout studies (Marubio et al., 1999). Taken together, studies using knockout, gain-of-function, and inbred mouse strain indicate that within the set of nAChR subtypes, activation of α4β2*-nAChR is both necessary and sufficient for nicotine-evoked antinociception in the hot-plate test. In contrast, the α4β2*-nAChR and at least one other nAChR subtype regulate the effects of nicotine in the tail-flick test. The inbred strain studies also indicate a role for α4β2-nAChR, but they also clearly illustrate that factors downstream from nAChR activation influence responses to nicotine. Future studies with inbred strains and their hybrids may provide a basis for further genetic analysis and could be useful for identifying the gene(s) that influence variability of nicotinic receptor-mediated analgesic mechanisms.
Acknowledgments
We thank Tie Han for technical assistance.
Footnotes
-
This work was supported by National Institute on Drug Abuse Grants DA-12610, DA-0527, DA-03194, and DA-17279, by the National Institute of Mental Health (MH-49176), and by the California Tobacco-Related Disease Research Program.
-
M.I.D., C.F., and M.J.M. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.112649.
-
ABBREVIATIONS: nAChR, acetylcholine nicotinic receptor(s); WT, wild-type; BTX, bungarotoxin; %MPE, percentage of maximal possible effect; CL, confidence limit(s); ACh, acetylcholine; 4-OH-DMXB, 4-OH-dimethoxybenzylidine anabaseine.
-
↵* Denotes other undetermined nicotinic subunits.
- Received August 17, 2006.
- Accepted March 16, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}