

Abstract
Repeated peripheral administration of the μ-opioid agonist [d-Ala2,N-Me-Phe4,gly5-ol] enkephalin (DAMGO) produces acute tolerance and dependence on its peripheral antinociceptive effect against prostaglandin E2(PGE2)-induced mechanical hyperalgesia. In this study we evaluated the roles of protein kinase C (PKC) and nitric oxide (NO) in the development of this tolerance and dependence. Repeated administration of PKC inhibitors chelerythrine and 1-(5-isoquinolinesulfonyl)-2-methylpiperazine dihydrochloride with DAMGO did not alter the tolerance to DAMGO; however, dependence (defined as naloxone-induced withdrawal hyperalgesia) was blocked. Repeated administration ofN-(n-heptyl)-5-chloro-1-naphthalenesulfonamide, a PKC activator, which alone did not produce tolerance, mimicked the dependence produced by DAMGO. Repeated administration of the NO synthase inhibitorNG-methyl-l-arginine with DAMGO blocked the development of tolerance to DAMGO but had no effect on the development of dependence. Repeated administration of l-arginine, a NO precursor, mimicked tolerance produced by repeated administration of DAMGO (i.e., the antinociceptive effect of DAMGO was lost); however, l-arginine did not mimic dependence. These findings suggest that the development of acute tolerance and dependence on the peripheral antinociceptive effects of DAMGO have different, dissociable mechanisms. Specifically, PKC is involved in development of μ-opioid dependence, whereas the NO signaling system is involved in the development of μ-opioid tolerance.
The repeated or sustained administration of opioids produces tolerance, or loss of effect, and also dependence, manifested by abstinence withdrawal after removal of opioid or precipitated withdrawal after administration of opioid antagonist. Receptor phosphorylation and uncoupling of G-proteins from cell-surface receptors have been implicated in opioid tolerance and dependence (Fukagawa et al., 1992; Escriba et al., 1994; Sim et al., 1996). G-proteins can be coupled from opioid receptors by increased activity of the second messenger protein kinase C (PKC) (Fukushiama et al., 1994; Lin et al., 1994), and translocation and activation of PKC in spinal cord dorsal horn neurons have been implicated in the development of tolerance and dependence on the antinociceptive effects of morphine (Mayer et al., 1995a,b). The second messenger nitric oxide (NO) has also been suggested to be involved in morphine tolerance (Kolesnikov et al., 1993; Elliott et al., 1995; Herman et al., 1995; Pasternak et al., 1995; Vaupel et al., 1995; Dunbar and Yaksh, 1996); it was demonstrated that the NO synthase (NOS) inhibitor NG-methyl-l-arginine (NMLA) prevents the development of antinociceptive tolerance and/or dependence on systemic morphine in the mouse (Kolesnikov et al., 1993; Majeed et al., 1994).
In addition to their actions in CNS to produce analgesia and other effects, opioids also act in the periphery to block inflammatory mediator-induced hyperalgesia (Levine and Taiwo, 1989; Stein, 1991,1995; Khasar et al., 1995) and also inhibit primary afferent sensitization in vitro (Gold and Levine, 1996). It has been shown recently that repeated administration of the μ-opioid agonist [d-Ala2,N-Me-Phe4,gly5-ol] enkephalin (DAMGO) results in tolerance for its ability to produce peripheral antinociception, i.e., to inhibit prostaglandin E2 (PGE2)-opioid hyperalgesia. Opioid dependence manifested by withdrawal hyperalgesia precipitated by naloxone (Aley et al., 1995) is also produced. In the present study, we evaluated the roles of PKC and NO as second messengers for the development of tolerance and dependence on the peripheral antinociceptive action of the μ-opioid DAMGO to inhibit PGE2-induced hyperalgesia.
MATERIALS AND METHODS
Animals. Experiments were performed on male Sprague Dawley rats (220–300 gm; Bantin and Kingman, Fremont, CA). Animals were housed in pairs under a 12 hr light/dark cycle, lights on at 6 A.M. Food and water were available ad libitum. All experiments were performed between 10 A.M. and 4 P.M. Experiments were performed under approval of the Institutional Animal Care Committee of the University of California, San Francisco.
Behavioral testing. The nociceptive flexion reflex was quantified with a Basile Analgesymeter (Stoelting, Chicago, IL), which applies a linearly increasing mechanical force to the dorsum of the rat’s hindpaw. Before the experiments, rats were exposed to the procedure for 3 d (1 hr daily at 5 min intervals). On the day of the experiment, rats were exposed to the same procedure, and the mean of the last six readings was defined as the baseline threshold (Taiwo et al., 1989; Aley et al., 1995). The mean baseline threshold for the rats used in these experiments was 110.4 ± 0.4 (SEM) gm (n = 394). Mechanical threshold was redetermined at three time points (15, 20, and 25 min) after treatments. The mean of these three readings was used to calculate the drug-induced change from the baseline threshold.
Drugs used in this study were PGE2 (a direct-acting hyperalgesic inflammatory mediator) (Pitchford and Levine, 1991; Gold and Levine, 1996), l-arginine hydrochloride (NO precursor), NMLA (NOS inhibitor), and dimethyl sulfoxide (DMSO) (all from Sigma, St. Louis, MO); DAMGO (μ-opioid receptor agonist), naloxone methyliodide (Nal; a quaternary salt of naloxone, an opioid receptor antagonist), 1-(5-isoquinolinesulfonyl)-2-methylpiperazine dihydrochloride, (H-7; PKC inhibitor),N-(n-heptyl)-5-chloro-1-naphthalenesulfonamide (SC-10; PKC activator) (all from RBI, Natick, MA); and chelerythrine chloride (PKC inhibitor) (L. C. Laboratories, Woburn, MA). The selection of the drug doses used in this study was based on the dose–response curves determined during this study or on previous work performed in this laboratory (Aley et al., 1995). A stock solution of PGE2 (10 μg/2.5 μl) was prepared in 100% ethanol, and further dilutions were made in saline, to a final concentration of ethanol ≤1%. DAMGO was dissolved in saline, chelerythrine and H-7 were dissolved in deionized water, and SC-10 was dissolved in DMSO. All drugs administered intradermally were in a volume of 2.5 μl. When drug combinations were used, they were administered from the same syringe in such a way that the drug mentioned first reached the intradermal site first. The two drugs were separated in the syringe by a small air bubble, to prevent drugs mixing in the syringe.
Statistical analysis. Data are presented as mean ± SEM of six or more observations in each experimental group. Statistical significance was determined by ANOVA followed by Scheffe’s post hoc test, if ANOVA showed a significant difference.p < 0.05 was considered statistically significant.
RESULTS
Development of tolerance to DAMGO does not depend on PKC signaling
Intradermal injection of PGE2 (100 ng) into the hairy skin of the hindpaw of the rat significantly decreased paw-withdrawal threshold (p < 0.05 (Fig. 1). DAMGO (1 μg) attenuated PGE2 hyperalgesia (p < 0.05). Three repeated injections of DAMGO given at intervals of 1 hr, when tested 1 hr later, produced tolerance measured as a decrease of antinociception by DAMGO on PGE2-induced hyperalgesia (p < 0.05).

Effect of PGE2 (100 ng,PGE2; n = 24), DAMGO (1 μg) plus PGE2 (DAMGO+PGE2; n = 24), chelerythrine (1 μg) hourly × 3 and at the fourth hour PGE2 (Chx3,PGE2; n = 12) or DAMGO plus PGE2 (Ch×3,DAMGO+PGE2;n = 12), H-7 hourly × 3 and at the fourth hour DAMGO plus PGE2 (H7×3,DAMGO+PGE2;n = 12), DAMGO hourly × 3 and at the fourth hour DAMGO plus PGE2 (DAMGO×3,DAMGO+PGE2;n = 18), chelerythrine plus DAMGO hourly × 3 and at the fourth hour DAMGO plus PGE2[(Ch+DAMGO)×3,DAMGO+PGE2; n = 12], H-7 plus DAMGO hourly × 3 and at the fourth hour DAMGO plus PGE2 [(H7+DAMGO)×3,DAMGO+PGE2;n = 12] on mechanical paw-withdrawal threshold. In this and subsequent figures, * p < 0.05;NS, not statistically significant.
Three injections of the PKC inhibitor chelerythrine (1 μg), given at intervals of 1 hr, did not significantly affect PGE2-induced hyperalgesia (p > 0.05) or the antinociceptive effect of DAMGO on PGE2-hyperalgesia (p > 0.05). Similarly, three injections of H-7 (1 μg), at intervals of 1 hr each, did not attenuate the antinociceptive effect of DAMGO on PGE2-hyperalgesia (p > 0.05). Three injections of chelerythrine or H-7 plus DAMGO, at intervals of 1 hr each, did not affect DAMGO-induced tolerance (both p > 0.05); i.e., neither DAMGO antinociception nor development of tolerance to DAMGO antinociception was affected by PKC inhibitors.
The development of dependence on DAMGO requires PKC signaling
The opioid antagonist naloxone methyliodide (at 200 ng, the ID80 to inhibit the antinociceptive effect of DAMGO against PGE2-induced hyperalgesia) (Aley et al., 1995), given alone in DAMGO-tolerant paws (i.e., 1 hr after three hourly injections of DAMGO), produced hyperalgesia when compared with vehicle-treated paws (p < 0.05) (Fig. 2). Chelerythrine (1 μg) or H-7 (1 μg) coinjected with the three hourly DAMGO injections blocked the development of this naloxone-induced hyperalgesia (both p < 0.05); i.e., the development of DAMGO dependence was prevented by PKC inhibitors.

Effect of DAMGO hourly × 3 and at the fourth hour naloxone (DAMGO×3,Nal;n = 16), vehicle (saline) hourly × 3 and at the fourth hour naloxone methyliodide (Veh×3,Nal;n = 6), chelerythrine plus DAMGO hourly × 3 and at the fourth hour naloxone methyliodide [(Ch+DAMGO)×3,Nal; n = 12], H-7 plus DAMGO hourly × 3 and at the fourth hour naloxone methyliodide [(H7+DAMGO)×3,Nal; n= 12] on mechanical paw-withdrawal threshold.
Activation of PKC in the absence of DAMGO produces a state similar to opioid dependence but not tolerance
Three injections of SC-10 (PKC activator, 1 μg) alone did not induce significant hyperalgesia (p > 0.05) (Fig. 3) or affect PGE2-induced hyperalgesia (p > 0.05) or the antinociceptive effect of DAMGO on PGE2-induced hyperalgesia (p > 0.05); however, after three hourly injections of SC-10, naloxone given as the fourth hourly injection produced hyperalgesia (p < 0.05) (Fig.3B); i.e., a DAMGO dependent-like state was present. The hyperalgesia induced by naloxone after SC-10 administration was not as great as that induced by naloxone after DAMGO (p< 0.05). Naloxone administered without previous SC-10 administration did not induce significant hyperalgesia (p > 0.05). Therefore, a PKC activator produced a state similar to opioid dependence but without the characteristics of opioid tolerance.

A, Effect of SC-10 hourly × 3 (SC10×3; n = 16), SC-10 hourly × 3 and at the fourth hour PGE2(SC10×3,PGE2; n = 6) or DAMGO plus PGE2 (SC10×3,DAMGO+PGE2;n = 10), saline hourly × 3 and at the fourth hour DAMGO plus PGE2 (Veh×3,DAMGO+PGE2;n = 8) on mechanical paw-withdrawal threshold.B, Effect of DAMGO hourly × 3 and at the fourth hour naloxone methyliodide (DAMGO×3,Nal;n = 16), DAMGO hourly × 3 and at the fourth hour vehicle (DAMGO×3,Veh; n = 6), SC-10 hourly × 3 and at the fourth hour naloxone methyliodide (SC10×3,Nal; n = 16), SC-10 hourly × 3 and at the fourth hour vehicle (SC10×3,Veh; n = 8), vehicle hourly × 3 and at the fourth hour naloxone methyliodide (Veh×3,Nal; n = 6) on mechanical paw-withdrawal threshold.
NO signaling is required for the development of acute tolerance to DAMGO
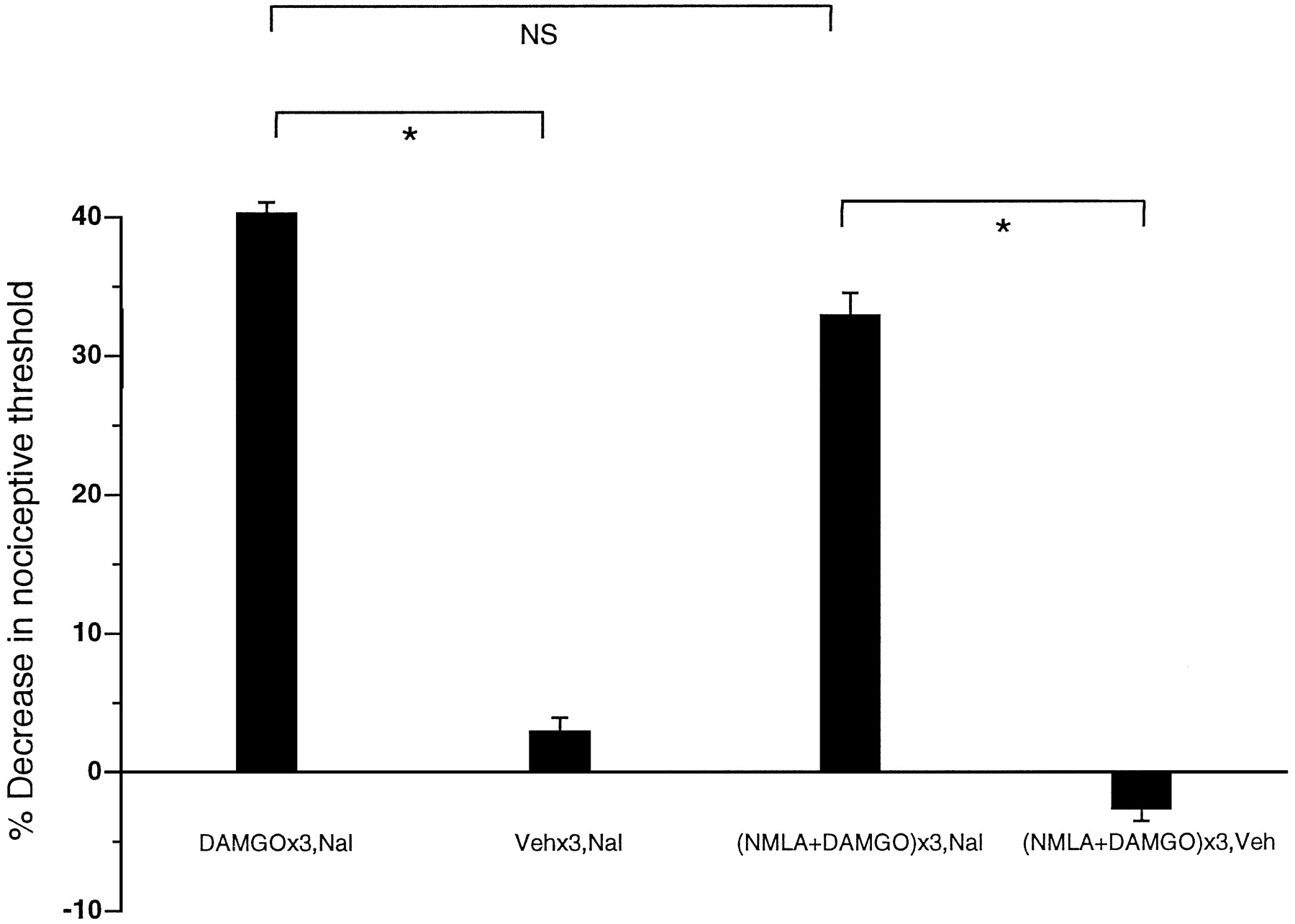
The NOS inhibitor NMLA (1 μg) had no effect on paw-withdrawal threshold after single (data not shown) or repeated injection; however, NMLA inhibited PGE2-induced hyperalgesia (p < 0.05) (Fig. 4). Therefore, it was not possible to evaluate for an effect of NMLA on tolerance using the previous protocol, because the tolerance assay, PGE2 hyperalgesia, was affected independently; however, PGE2 hyperalgesia had recovered almost completely 72 hr after the previous three injections of NMLA (1 μg). Therefore, we assessed for persisting tolerance to DAMGO at 72 hr after three hourly injections of DAMGO and found it present (p < 0.05); i.e., there was no effect of DAMGO on PGE2hyperalgesia. Therefore, an effect of NMLA on tolerance could be evaluated at the 72 hr time point. When three hourly injections of NMLA plus DAMGO were followed by DAMGO plus PGE2 72 hr later, DAMGO produced an antinociceptive effect (p < 0.05), indicating that administration of the NOS inhibitor NMLA with DAMGO prevented the development of tolerance. As a control, the effect of NMLA plus DAMGO for three hourly injections, on PGE2hyperalgesia at 72 hr, was tested; PGE2 hyperalgesia was not affected (p > 0.05).

Effect of PGE2 (PGE2), NMLA plus PGE2 (NMLA+PGE2; n = 10), NMLA hourly × 3 (NMLA×3; n = 6), NMLA hourly × 3 and 72 hour post-PGE2 [NMLA×3,PGE2(72 h post); n= 12], NMLA plus DAMGO hourly × 3 and PGE2 72 hour later [(NMLA+DAMGO)×3,PGE2(72 h post);n = 12], NMLA plus DAMGO hourly × 3 and 72 hr post DAMGO plus PGE2[(NMLA+DAMGO)×3,DAMGO+PGE2(72 h post);n = 12], and DAMGO hourly × 3 and DAMGO plus PGE2 72 hr post [DAMGO×3,DAMGO+PGE2(72 h post); n = 8] on mechanical paw-withdrawal threshold.
NO signaling is not required for development of dependence on DAMGO
Administration of naloxone 1 hr after three hourly injections of NMLA plus DAMGO resulted in significant hyperalgesia (p < 0.05) (Fig. 5), similar to the withdrawal hyperalgesia produced by naloxone after DAMGO alone; administration of vehicle after three similar injections had no effect. Therefore, inhibition of NOS does not affect the development of DAMGO-induced dependence.

Effect of DAMGO hourly × 3 and at the fourth hour naloxone methyliodide (DAMGO×3,Nal;n = 16), vehicle (saline) hourly × 3 and at the fourth hour naloxone methyl iodide (Veh×3,Nal;n = 6), NMLA plus DAMGO × 3 and at the fourth hour naloxone [(NMLA+DAMGO)×3, Nal;n = 10], NMLA plus DAMGO hourly × 3 and at the fourth hour saline [(NMLA+DAMGO)×3,Veh;n = 6] on mechanical paw-withdrawal threshold.
Activation of NO system produces a state similar to opioid tolerance but not dependence
Intradermal injection of the NO precursor l-arginine (100 ng) alone produced hyperalgesia (p < 0.05) (Fig. 6A), which increased with three hourly injections (p < 0.05). At 72 hr after the last injection, however, paw-withdrawal threshold had returned to the basal level, and the magnitude of hyperalgesia produced by PGE2 was similar to that in normal animals (p > 0.05). Therefore, an effect ofl-arginine to produce a tolerant-like or dependent-like state was assessed 72 hr after the last injection ofl-arginine; DAMGO inhibition of PGE2-induced hyperalgesia was reduced (p < 0.05); i.e., the antinociceptive effect of DAMGO was similar to that in DAMGO-tolerant animals (Fig. 1). After similar treatment, however, the opioid antagonist naloxone methyliodide did not produce hyperalgesia (Fig.6B), in contrast to the significant hyperalgesia produced by naloxone in tolerized animals (Fig. 2). Injection of vehicle 72 hr after the last of three hourly injections ofl-arginine did not significantly affect mechanical nociceptive threshold (p > 0.05) (Fig.6B). Therefore, administration of the NO donorl-arginine produced an opioid tolerant-like but not dependent-like state.

A, Effect of three hourly injections of vehicle (saline) (Veh×3; n = 6),l-arginine (100 ng) (L-Arg×1;n = 12), three hourly injections ofl-arginine (L-Arg×3; n= 12), 72 hr after three hourly injections of l-arginine [L-Arg×3(72hpost); n = 12], PGE2 72 hr after three hourly injections ofl-arginine [L-Arg×3,PGE2(72hpost);n = 6], PGE2 (PGE2), DAMGO plus PGE2 72 hr after three hourly injections ofl-arginine [L-Arg×3,DAMGO+PGE2(72hpost);n = 12], DAMGO plus PGE2(DAMGO+PGE2), on mechanical paw-withdrawal threshold.B, Effect of three hourly injections of DAMGO and 72 hr after naloxone methyliodide [DAMGO×3,Nal(72hpost)], naloxone methyl iodide 72 hr after three hourly injections ofl-arginine [L-Arg×3,Nal(72hpost);n = 8], vehicle 72 hr after three hourly injections of l-arginine [L-Arg×3,Veh(72hpost); n = 8], on mechanical paw-withdrawal threshold.
DISCUSSION
Our data strongly suggest that the development of μ-opioid acute tolerance for peripheral antinociception involves NO but not PKC signaling mechanisms, and that the development of dependence involves PKC but not NO signaling mechanisms. These findings support the hypothesis that there are different mechanisms for the development of tolerance and dependence on the peripheral antinociceptive effect of the μ-opioid agonist DAMGO.
The hyperalgesia produced by naloxone after repeated administration of the PKC activator SC-10 was significant but less than that produced by naloxone after similar treatment with the μ-opioid DAMGO. A tenfold increase in the dose of SC-10 produced no further increase in the naloxone-induced hyperalgesia (unpublished observation), suggesting that other mechanisms in addition to PKC contribute to the development of dependence. Because PKC antagonists completely block the development of dependence, however, our results suggest that PKC activity is necessary for dependence to develop. Of note, what we report as dependence refers to naloxone-precipitated withdrawal hyperalgesia; animals may not have been followed for a long enough period of time to observe a hyperalgesic syndrome after cessation of opioids.
The development of tolerance to an opioid agonist generally is accompanied by the development of dependence. Various clinical and experimental studies suggest that the two phenomena are closely related (Loh et al., 1969; Way et al., 1969; Klee and Nirenberg, 1974; Sharma et al., 1977). Recent evidence, however, has suggested that dependence may occur independently. A temperature-dependent functional dissociation of tolerance and dependence on morphine has been demonstrated in guinea-pig in vitro (David et al., 1993). Our results demonstrate that in the model of peripheral antinociception, tolerance and dependence can be dissociated. Some previous studies, in agreement with our findings, report reduction of opioid-induced tolerance in the CNS by NOS inhibitors (Kolesnikov et al., 1992, 1993; Elliott et al., 1994; Bhargava and Zhao, 1996) and the attenuation in the CNS of antagonist-induced hyperalgesia by PKC inhibitors (Tokuyama et al., 1995a,b). In contrast, studies of opioid tolerance in the CNS report prevention of tolerance by PKC inhibitors (Narita et al., 1994, 1995; Bilsky et al., 1996) and prevention of dependence as well as tolerance by NOS inhibitors (Majeed et al., 1994;Bhargava, 1995), different from our findings. These disparities may reflect differences in peripheral and central mechanisms of μ-opioid tolerance and dependence and/or be attributable to the complexity of neuronal interaction in the CNS.
Conclusion
The results of this study demonstrate, for the first time,in vivo dissociation of opioid acute tolerance and dependence. Pharmacological agents selectively affecting PKC and NO second messenger systems affected either opioid dependence or opioid tolerance, respectively. After administration of these agents, individual animals could demonstrate a tolerance-like but not dependence-like state and vice versa. The results have potential significance for the clinical use of opioids as well as for future study of mechanisms that underlie opioid tolerance and dependence.
Footnotes
This work was funded by National Institutes of Health Grant DE08973.
Correspondence should be addressed to Dr. Jon D. Levine, Departments of Anatomy, Medicine, and Oral Surgery, and Division of Neuroscience, Box 0452, University of California, San Francisco, CA 94143-0452.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}