

Abstract
Thrombin activity is a factor in acute CNS trauma and may contribute to such chronic neurodegenerative diseases as Alzheimer’s disease. Thrombin is a multifunctional serine protease that catalyses the final steps in blood coagulation. However, increasing evidence indicates that thrombin also elicits a variety of cellular and inflammatory responses, including responses from neural cells. Most recently, high concentrations of thrombin were shown to cause cell death in both astrocyte and hippocampal neuron cultures. The purpose of this study was to determine the mechanisms underlying thrombin-induced cell death. Our data show that thrombin appears to cause apoptosis as evidenced by cleavage of DNA into oligonucleosomal-sized fragments, fragmentation of nuclei, and prevention of death by inhibition of protein synthesis. Synthetic peptides that directly activate the thrombin receptor also induced apoptosis, indicating that thrombin-induced cell death occurred via activation of the thrombin receptor. The signal transduction cascade involves tyrosine and serine/threonine kinases and an intact actin cytoskeleton. Additional study revealed the involvement of the small GTP-binding protein RhoA. Thrombin induced RhoA activity in both astrocytes and hippocampal neurons, and inhibition of RhoA activity with exoenzyme C3 attenuated cell death, indicating that thrombin activation of RhoA was necessary for thrombin-induced cell death. Tyrosine kinase inhibitors blocked thrombin induction of RhoA, indicating that tyrosine kinase activity was required upstream of RhoA. These data suggest a sequential linkage of cellular events from which we propose a model for the second messenger cascade induced by thrombin in neural cells that can lead to apoptosis.
- thrombin
- apoptosis
- neurons
- astrocytes
- RhoA
- receptor
- Alzheimer’s disease
- cerebrovascular injury
- tyrosine kinase
- serine/threonine.
Thrombin is a multifunctional serine protease that has important roles in hemostasis and wound-healing. The biological activities of thrombin include a key role in blood coagulation, the cleavage of fibrinogen to fibrin and the activation of platelets (Berndt and Phillips, 1981), and the stimulation of cellular responses involved in inflammation and repair processes. Thrombin is chemotactic for macrophages (Bar-Shavit et al., 1983) and mitogenic for smooth muscle cells and fibroblasts (Carney and Cunningham, 1978; McNamara et al., 1993). Thrombin can also induce secretion of cytokines and growth factors from macrophages, smooth muscle cells, and fibroblasts (Harlan et al., 1986; Jones and Geczy, 1990; Okazaki et al., 1992). Thrombin is produced immediately from prothrombin at sites of injury and thus can act at the earliest stages of wound repair. The recent finding that elevated levels of thrombin activity are present for days after peripheral nerve injury implies that thrombin is involved throughout the healing process (Smirnova et al., 1996).
Evidence is building for an important role for thrombin in CNS injury. Studies using cultured neural cells reveal a variety of cellular responses to thrombin. Thrombin induces process retraction in neuroblastoma cells (Gurwitz and Cunningham, 1988), astrocytes (Cavanaugh et al., 1990; Nelson and Simon, 1990), and human fetal neurons (Grand et al., 1989). Thrombin is mitogenic for astrocytes (Perraud et al., 1987; Loret et al., 1989; Cavanaugh et al., 1990) and induces the synthesis and secretion of both nerve growth factor (Neveu et al., 1993) and endothelin-1 (Ehrenreich et al., 1993). Recently, moderate concentrations of thrombin were shown to protect hippocampal neurons and astrocytes from a variety of cellular insults, such as hypoglycemia, growth supplement deprivation, oxidative stress, and β-amyloid toxicity, that mimic conditions found in injury (Vaughan et al., 1995; Pike et al., 1996). High concentrations of thrombin induced cell death in these same cells when cultured under nonstressed conditions (Smith-Swintosky et al., 1995b; Vaughan et al., 1995; Debeir et al., 1996). These actions of thrombin suggest both beneficial and potentially harmful roles for thrombin in response to CNS injury. The action of thrombin on neural cells may yield increased plasticity and protection from insults; however, uncontrolled thrombin activity could result in extensive retraction of neurites and processes on astrocytes, uncontrolled astrocyte proliferation, and even neural cell death. Recently, an attempt to elucidate the consequences of thrombin exposurein vivo was performed by directly infusing thrombin into the rat caudate nucleus (Nishino et al., 1993). Infused thrombin caused increased reactive gliosis, infiltration of inflammatory cells, proliferation of mesenchymal cells, and induction of angiogenesis, effects that resemble the inflammation, reactive gliosis, and scar formation that occur after injury to the CNS. Taken together, these data underscore the importance of thrombin in regulating pathophysiological processes in the CNS.
Cellular actions induced by thrombin are in most cases attributable to activation of the thrombin receptor (Rasmussen et al., 1991; Vu et al., 1991), a proteolytically activated, seven-transmembrane spanning receptor that has been linked to a variety of cellular pathways including hydrolysis of phospho-inositides; calcium mobilization; and activation of heterotrimeric G-proteins, tyrosine kinases, and monomeric G-proteins (for review, see Grand et al., 1996). Cellular mechanisms underlying thrombin effects on neural cells are not known. In light of the increasing evidence that thrombin has a role in CNS injury, this study was undertaken to investigate the cellular pathways mediating the effects of thrombin on neural cells, specifically thrombin-induced cell death.
MATERIALS AND METHODS
Materials. Highly purified, high-specific-activity human α-thrombin was obtained from Calbiochem (San Diego, CA) and Sigma (St. Louis, MO). Because of variation in specific activity between thrombin supplies, all thrombin treatments were performed using units per milliliter, with 200 U/ml approximately equivalent to 1 μm thrombin. Peptides were obtained from Chiron Mimotopes Peptide Systems (San Diego, CA) and American Peptide Company (Sunnyvale, CA). Herbimycin A, cytochal-asin D, genistein, pertussis toxin, HA 1004, H-7, wortmannin, and exoenzyme C3 were obtained from Calbiochem (San Diego, CA). 1,2-Bis(2-aminophenoxy) ethane-N,N,N′,N′-tetra-acetic acid (BAPTA), leupeptin, aprotinin, phenylmethylsulfonyl fluoride and soybean trypsin inhibitor were obtained from Sigma (St. Louis). Monoclonal antibody to glial fibrillary acidic protein was obtained from Boehringer Mannheim (Indianapolis, IN). Monoclonal antibodies (SMI-311) to neuron-specific neurofilaments were obtained from Sternberger Monoclonals (Baltimore, MD). SYTO 11 and calcein AM were obtained from Molecular Probes (Eugene, OR).
Isolation of primary cultures of rat astrocytes. Primary cultures of type 1 astrocytes were prepared from the brains of 1- to 2-d-old rat pups (Sprague Dawley, Indianapolis, IN) using a modification of procedures described previously (McCarthy and de Vellis, 1980). Briefly, the frontoparietal cortex from eight pups was isolated, stripped of meninges, and dissociated by a combination of trypsin digestion and mechanical trituration. A single cell suspension was prepared from this dissociated tissue by passage through Nitex nylon screens. The cells were plated into 75 cm2plastic flasks and grown in DMEM containing (in mm): glucose 25, sodium bicarbonate 7.5, HEPES 20, glutamine 2, and sodium pyruvate 1, and 100 units/ml penicillin, 100 mg/ml streptomycin sulfate, and 10% fetal bovine serum. Once the cultures reached confluence (∼10–12 d), the flasks were shaken at 260 rpm for 24 hr at 37°C to remove nonadherent cells. The remaining type 1 astrocytes were trypsinized and reseeded into 75 cm2 flasks. Once these cells had reached confluence, they were replated for experiments, as detailed below. The purity of the cultures was confirmed by immunofluorescent staining with a monoclonal antibody to the type 1 astroglial-specific marker glial fibrillary acidic protein; > 95% of the cells were immunoreactive for this marker.
Experimental treatment of astrocytes. Astrocytes were removed from flasks by trypsinization and plated at a density of 1 × 104 cells/cm2. After growth to 70–80% confluence (2–3 d after plating), the cells were used in experiments. In all experiments, cells were rinsed three times with serum-free DMEM and then incubated in this medium for 16–18 hr before experimental treatments. For experiments measuring the effects of thrombin on DNA fragmentation, cells were plated into six well plates, and medium containing the indicated concentrations of thrombin was added to the cells for 48 hr. The supernatant was then removed, and and all DNA present in the culture medium was isolated using QIAamp reagents and protocols (QIAamp Blood kit, Qiagen, Hilden, Germany). This DNA was then end-labeled and analyzed as described below. For experiments measuring the effect of thrombin receptor activating peptides (TRAP) on astrocyte viability, cells were plated in 48 well plates, medium containing 15 mm TRAP was added, and cell viability was determined 72 hr after TRAP addition. For experiments measuring the effect of TRAP on nuclear morphology, astrocytes were treated as described, except that after a 48 hr exposure to TRAP, the fluorescent nucleic acid dye SYTO-11 was added to the medium and the nuclei photographed. For experiments measuring the effect of pharmacological agents on thrombin-induced astrocyte death, cells were rinsed in serum-free DMEM and then incubated in this medium alone or medium containing the indicated concentrations of each pharmacological agent. Thirty minutes or 3 hr after addition of the pharmacological agents, thrombin was added directly to the culture medium and cell viability determined 72 hr later. In all experiments, cell viability was calculated relative to its control wells, which were maintained either in medium alone or in medium containing the pharmacological agent throughout the experiment. In all experiments, cell viability was determined by assaying the medium from each well for lactate dehydrogenase (LDH) activity using a diagnostic kit according to the manufacturer’s instructions (Sigma). Released LDH is a stable enzymatic marker that correlates linearly with cell death. To determine total LDH activity, after removal of the medium, the cells were lysed in 0.5% Triton X-100, centrifuged at 16,000 × g for 1 min and the supernatants were assayed for LDH activity. This cell-associated LDH activity was then added to the LDH activity in the removed culture medium, and the total activity was considered to represent 100% cell death. The amount of LDH present in the medium was then calculated as a percentage of the total, which determines the percent cell death in that sample. For clarity, results were then presented as the reciprocal, the percent cell viability. In some experiments, SYTO 11 (Molecular Probes) was added to the control and thrombin-treated cultures to monitor viability microscopically. All results are expressed as the mean ± SEM of triplicate samples. Data were statistically examined by one-way ANOVA followed by pairwise comparisons using the Bonferroni/Dunn procedure. All studies were repeated in at least three independent experiments.
Isolation of rat hippocampal neurons. Hippocampal neuronal cultures were prepared as described previously (Pike et al., 1993). Briefly, hippocampi were dissected from embryonic day 18 Sprague Dawley rat pups and mechanically dissociated in a Ca2+/Mg++-free balanced salt buffer. The cell suspension was pelleted, then resuspended in serum-free DMEM containing (in mm): glucose 25, sodium bicarbonate 26, HEPES 20, and pyruvate 1, and supplemented with N2 components (30 nm selenium, 20 nm progesterone, 100 mm putrescine, 100 mg/ml transferrin, and 5 mg/ml bovine insulin) (Bottenstein and Sato, 1979). The purity of the cultures was confirmed using monoclonal antibodies against neuronal-specific neurofilaments (Sternberger Monoclonals, Baltimore, MD); >95% of the cells were positive for this marker (Pike et al., 1993).
Experimental treatment of rat hippocampal neurons. Neurons were plated into poly-l-lysine coated multiwell dishes at a density of 2.5 × 104cells/cm2. To analyze the effects of thrombin on DNA fragmentation, medium containing the indicated concentrations of thrombin was added to the cells for 24 hr. DNA was isolated and analyzed as described for the astrocytes. To analyze the effects of TRAP on cell viability, TRAP was added to neurons that had been cultured in DMEM with N2 supplements for 72 hr after isolation. Cell viability was determined 24 hr later. For experiments measuring the effect of TRAP on nuclear morphology, neurons were treated as described, except that after a 24 hr exposure to TRAP, the fluorescent nucleic acid dye SYTO-11 was added to the medium and the nuclei photographed. Neuronal viability was determined based on the number of cells exhibiting positive staining for the viability dye calcein AM (Molecular Probes), as described previously (Pike et al., 1996). The total number of cells in untreated wells was considered to represent 100% cell viability. All results are expressed as the mean ± SEM of triplicate samples. Data were statistically examined by one-way ANOVA followed by pairwise comparisons using the Bonferroni/Dunn procedure. All studies were repeated in at least three independent experiments.
Cell fractionation and [32P] ADP ribosylation. Cell fractionation and in vitroribosylation were performed as described previously (Jalink et al., 1994). Briefly, cells were grown in 10 cm culture dishes for 3–4 d, serum-starved as described, then exposed to the indicated concentrations of thrombin for 20 min. For experiments using pharmacological agents, cells were pretreated for 3 hr in the presence of each agent as described above; thrombin was added directly to this medium for 20 min. Once treated, cells were washed twice with PBS, scraped into ice-cold 20 mm Tris/HCl, pH 8.0, in the presence of a protease inhibitor cocktail (0.4 mmphenylmethylsulfonyl fluoride, 20 μm leupeptin, 0.05 U/ml aprotinin, 20 μg/ml soybean trypsin inhibitor), and homogenized using a tight pestle Dounce homogenizer (30 strokes). Lysates were centrifuged at 500 × g for 10 min at 4°C. The supernatants were centrifuged at 25,000 × g for 30 min to separate the cytosolic from the crude membrane fraction. Protein concentration was determined using the Bio-Rad (Hercules, CA) Bradford protein assay. Ribosylation assays were performed using 30 μg of protein from each fraction, which was heat-inactivated at 65°C for 5 min and then taken up in reaction buffer containing (in mm): Tris/HCl 90, pH 8.0, MgCl2 2.6, EDTA 1, thymidine 10, dithiothreitol 10, and ATP 1, and 100 μmGTP and 10 μCi/ml [32P]NAD. Reaction was started by addition of exoenzyme C3 (5 μg/ml) and allowed to precede at 37°C for 1 hr. Proteins were acid-precipitated (10% TCA wt/vol), centrifuged (15,000 × g for 15 min), and washed with ether. Samples were then subject to SDS-PAGE (12.5%) and ribosylated proteins visualized by autoradiography.
End-labeling DNA. Cells were grown and DNA-isolated as detailed above. As described previously in Rosl (1992), DNA was treated with 1 U/ml Klenow polymerase (5000 U/ml, Promega, Madison, WI) and 0.5 μCi/μg [32P]CTP (3000 Ci/mmol, Amersham, Arlington Heights, IL) in a reaction buffer consisting of 50 μm Tris/HCl, pH 7.2, 10 mmMgSO4, and 1 mm DTT. The reaction was allowed to proceed for 10 min at room temperature and terminated was with 10 mm EDTA. The presence of only a single nucleotide prevents the 3–5′ exonuclease activity of Klenow polymerase from producing a nick translation effect; thus, these results reflect only the labeling of 3′ DNA ends. Labeled DNA was subjected to two rounds of precipitation (1 hr each) with 2.5 m ammonium acetate and 2.5 vol ethanol in the presence of 50 μm tRNA carrier, followed by centrifugation for 30 min at 4°C. DNA was washed with 70% ethanol, resuspended in Tris–EDTA, and electrophoresed through a 1.8% agarose gel. Gels were stained with ethidium bromide, dried under vacuum in a Bio-Rad slab gel drier, and exposed to autoradiography film.
RESULTS
Thrombin-induced astrocyte death involves apoptosis
In agreement with our previous data (Vaughan et al., 1995), we observed that thrombin levels >100 U/ml or ∼500 nminduce cell death in cultured astrocytes (data not shown). We first determined whether this thrombin-induced cell death in cultured astrocytes is a passive, necrotic process, or whether it is attributable to activation of apoptosis. Experimentally, apoptosis is evidenced by the cleavage of cellular DNA into oligonucleosomal-sized DNA fragments; this is visualized as “DNA laddering” upon agarose gel electrophoresis. To determine whether thrombin-induced cell death involves this hallmark of apoptosis, we examined DNA released into the culture medium both from astrocytes treated with thrombin for 24 hr and from untreated cells. DNA recovered from culture medium was end-labeled using only [32P]dCTP, electrophoresed through a 1.8% agarose gel, and then visualized by autoradiography. As shown in Figure 1A, the DNA isolated from the supernatant of untreated cells did not give rise to a DNA ladder, whereas the DNA from cells treated with 150 U/ml and 200 U/ml thrombin showed a ladder of oligonucleosomal-sized DNA fragments. These data indicate that astrocytes treated with thrombin concentrations that induce cell death exhibited DNA fragmentation characteristic of apoptosis.

Thrombin induces apoptosis in astrocytes; this is mediated by activation of the thrombin receptor. A, Astrocytes treated with thrombin release oligonucleosomal DNA fragments indicative of apoptosis. Astrocytes were treated for 24 hr with 0 (lane 1), 150 U/ml (lane 2), and 200 U/ml thrombin (lane 3). The culture medium was harvested and all DNA present extracted. DNA was end-labeled with [32P]dCTP, electrophoresed through a 1.8% agarose gel, and subjected to radiography. B, TRAP induces cell death in cultured astrocytes. Cells were treated with 15 mmTRAP or 200 U/ml thrombin. Seventy-two hours after addition, cell viability was determined using the LDH assay. C,D, Astrocytes treated with TRAP show altered nuclear morphology. Control cultures or cultures treated with 15 mmTRAP for 48 hr were incubated with SYTO 11 to stain the nuclei. Samples were analyzed under fluorescent microscopy and photographs taken; control cultures (C) and cultures treated with 15 mm TRAP for 48 hr (D).
Thrombin-induced astrocyte death involves thrombin receptor activation
Next we examined whether thrombin-induced cell death was attributable to activation of the thrombin receptor or to some other enzymatic effect of thrombin on the cells such as degradation of extracellular matrix, cell surface, or adhesion molecules. These possibilities can be distinguished by using TRAP in place of thrombin (Vu et al., 1991; Chen et al., 1994; Gerszten et al., 1994). This six amino acid peptide, SFLLRN, is an active ligand for the receptor, but has no proteolytic activity. Accordingly, the toxicity of TRAP was evaluated in cultured astrocytes. Cells treated with 15 mmTRAP for 72 hr exhibited marked cell death as determined by the LDH assay (Fig. 1B), demonstrating that activation of the thrombin receptor by TRAP can induce cell death in astrocytes. To determine if this cell death is apoptotic, we evaluated TRAP treated cells for the presence of apoptotic nuclei. As cells undergo apoptosis, the chromatin condenses and the nuclei break down into small pyknotic spheres. The untreated cells had oval, evenly stained nuclei indicative of healthy cells (Fig. 1C). In contrast, astrocytes treated with TRAP for 48 hr showed numerous fragmented and pyknotic nuclei (Fig. 1D). These results suggest that thrombin-induced cell death in cultured astrocytes is most likely mediated by the thrombin receptor and proceeds via apoptosis.
Pharmacological characterization of the signal transduction cascade underlying thrombin-induced cell death: involvement of tyrosine kinases, serine/threonine kinases, and the actin cytoskeleton
Thrombin receptor activation can result in induction of a variety of different second messengers including hydrolysis of phosphoinositides, calcium, mobilization, and activation of heterotrimeric G-proteins, tyrosine kinases and monomeric G-proteins. To identify the second messenger pathways activated by thrombin in neural cells under conditions of thrombin-induced cell death, we tested several pharmacological agents for their ability to block thrombin-induced cell death in cultured astrocytes. Cells were preincubated with each agent, followed by 72 hr exposure to 200 U/ml thrombin. None of these pharmacological agents decreased astrocyte viability, with the exception of genistein, which caused a mild but significant injury (p < 0.05 relative to control condition). Cultures treated with thrombin alone showed a significant (∼50%) decrease in viability by the LDH assay. Two tyrosine kinase inhibitors, herbimycin A and genistein, blocked thrombin-induced cell death with thrombin-treated cells retaining ∼95% viability (Fig. 2A). Next we examined the involvement of serine/threonine kinases using the broad spectrum serine/threonine kinase inhibitors HA-1004 and H-7. H-7, at 10 μm, fully blocked thrombin-induced cell death (Fig.2A). HA-1004, at concentrations up to 100 μm, did not decrease thrombin-induced cell death (Fig.2A). The primary difference between these inhibitors is that H-7 is a more potent inhibitor of protein kinase C (PKC). This finding suggests a role for PKC activity in thrombin-induced cell death, but does not rule out the possibility that other serine/threonine kinases may be involved.

Inhibition of thrombin-induced cell death by tyrosine and serine/threonine kinase inhibitors and cytochalasin D.A, Astrocytes were pretreated for 3 hr with each of the following agents: herbimycin A (0.5 μm), genistein (100 μm), H-7 (10 μm), and HA-1004 (100 μm). Thrombin was added to the culture media (200 U/ml) and cell viability assayed 72 hr after addition using the LDH assay.B, Astrocytes were pretreated for 30 min with pertussis toxin (1 μg/ml) and for 3 hr with each of the following agents: wortmannin (1 μm), BAPTA (100 μm), and cytochalasin D (0.5 μg/ml). Thrombin was added to the culture medium (200 U/ml) and cell viability assayed 72 hr after addition.Asterisk denotes p < 0.05 in thrombin plus pharmacological agent conditions relative to thrombin alone conditions.
Because the thrombin receptor is a G-protein-linked receptor, we tested the role of the Gi subfamily of heterotrimeric G-proteins in thrombin-induced cell death. Accordingly, we treated cells with pertussis toxin (0.1 and 1 μg/ml) for 30 min followed by addition of thrombin. Pretreatment with either concentration of pertussis toxin did not inhibit thrombin-induced cell death (Fig. 2B). To test the possible involvement of phospholipase D (PLD) and phosphatidylinositide 3-kinase (PI 3-K), cells were treated with wortmannin at 10 nm and 1 μm. Wortmannin at either concentration did not affect thrombin-induced cell death (Fig.2B). The calcium chelator BAPTA used at 100 μm also did not block thrombin-induced cell death in astrocytes (Fig. 2B), suggesting that large influxes of calcium into the cell are not required. Use of calcium-free medium or the calcium ionophore A23187 for the time course of this experiment was toxic to the cells (data not shown). Finally, we tested cytochalasin D, an inhibitor of actin filament assembly and a regulator of the actin cytoskeleton shown previously to block other thrombin activities (Li et al., 1994; Ezumi et al., 1995; Banno et al., 1996) and also reported to protect neurons from excitotoxic insults and β-amyloid toxicity (Furukawa and Mattson, 1995; Furukawa et al., 1995). Cytochalasin D at 0.5 μg/ml relaxed stress fibers in the astrocytes (visually observed) and blocked thrombin-induced cell death (Fig. 2B). This finding indicates strongly that an intact actin cytoskeleton is necessary for thrombin-induced cell death.
Thrombin induces RhoA activity, and this overactivation of RhoA leads to cell death
The small GTP-binding protein RhoA has been shown previously to regulate the actin cytoskeleton (Ridley and Hall, 1992; Narumiya and Morii, 1993), and its deregulation has been linked to both apoptosis and oncogenesis (Perona et al., 1993; Esteve et al., 1995; Jimenez et al., 1995). A previous report examining thrombin-signaling in a neuroblastoma cell line implicates RhoA in thrombin-induced neurite retraction. Using NEI-115 cells, Jalink et al. (1994) showed that a specific inhibitor of RhoA, exoenzyme C3, blocked thrombin-induced neurite retraction. This series of observations suggests that RhoA activation may participate in the cellular pathway of thrombin-induced cell death. To evaluate this possibility, we first examined whether thrombin induced RhoA activity. Astrocyte cultures were treated with increasing concentrations of thrombin for 20 min and then lysed, and membrane and cytosolic fractions were prepared. These cell fractions were assayed for the level of available RhoA activity byin vitro ribosylation assay. This assay labels any RhoA that has not been inactivated by ribosylation from cellular ADP ribosyl transferases and thus measures the level of available RhoA. It should be noted that intracellularly, many factors contribute to RhoA activity. In this assay, proteins were ADP-ribosylated with32P-NAD, precipitated, and electrophoresed through 12.5% SDS-polyacrylamide gels. Ribosylated proteins were visualized by autoradiography (Fig. 3A). Thrombin increased RhoA activity beginning at 100 U/ml thrombin (Fig. 3A,lane 4), but only at concentrations that lead to cell death was there a marked increase in RhoA activity (Fig. 3A,lane 5 vs lanes 1–3). This observation is consistent with the possibility that RhoA activity is responsible for transducing thrombin-induced cell death.
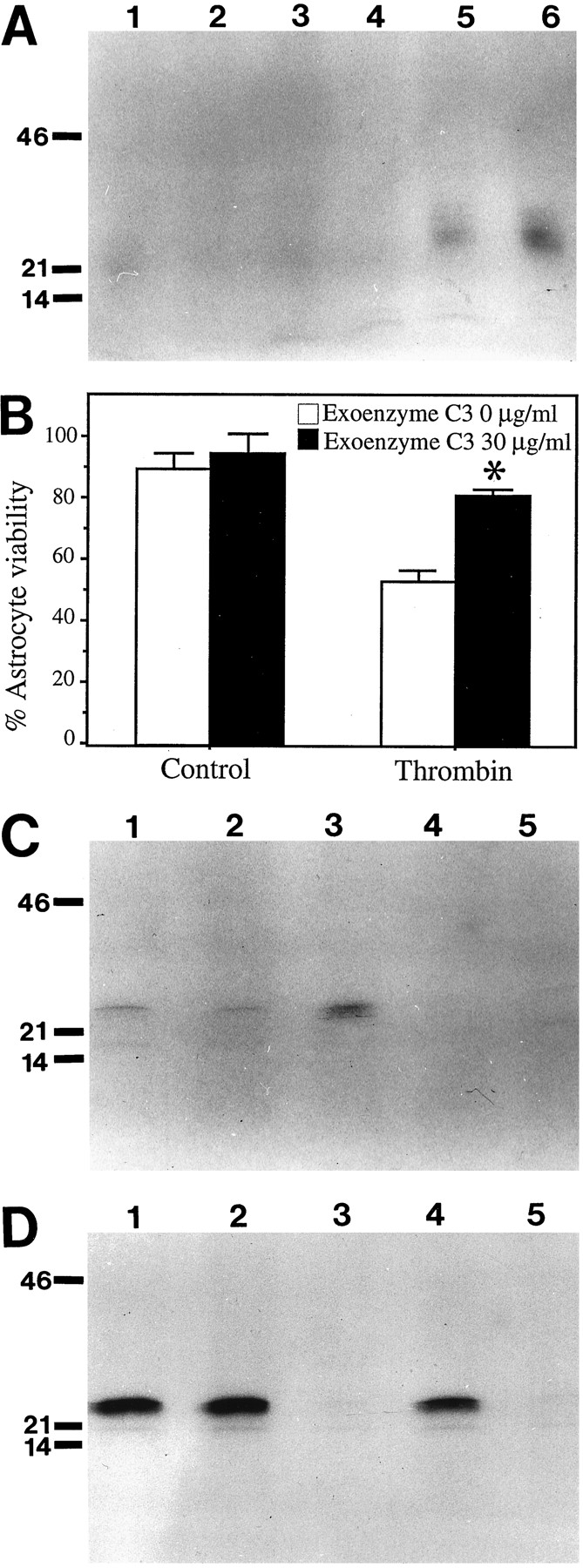
The small GTP-binding protein RhoA is induced by thrombin and is required for thrombin-induced cell death.A, Thrombin induces RhoA activity in astrocytes. Astrocytes were treated with thrombin for 20 min. Crude membrane fractions were prepared and RhoA activity assayed by ribosylation with exoenzyme C3 and [32P] NAD. Control for endogenous ribosylation activity; no exoenzyme C3 added to the reaction mixture (lane 1); untreated cells (lane 2); 20 U/ml thrombin (lane 3); 50 U/ml thrombin (lane 4); 100 U/ml thrombin (lane 5); 200 U/ml thrombin (lane 6.)B, Inactivation of RhoA with exoenzyme C3 decreases thrombin-induced cell death. Astrocytes were pretreated with exoenzyme C3 (30 μg/ml) for 3 hr, then thrombin was added to the media (200 U/ml) of pretreated and control cultures. Cell viability was assayed using LDH assay 72 hr after the addition of thrombin.Asterisk denotes p < 0.05 in thrombin plus exoenzyme C3 conditions relative to thrombin alone conditions. C, Pretreatment with tyrosine kinase inhibitors blocked thrombin induction of RhoA. Astrocytes were pretreated with genistein (100 μm) for 3 hr. Cells were then exposed to thrombin (200 U/ml) for 20 min. Membrane fractions were prepared and the activity of RhoA measured by in vitroribosylation; untreated cells (lane 1); genistein treatment only (lane 2); thrombin treatment only (lane 3); genistein pretreatment followed by thrombin treatment (lane 4); no C3 added to reaction (lane 5). D, RhoA activity moves from the cytosol fraction to the membrane fraction after thrombin treatment. Genistein blocks this translocation. Cells were treated as for those inC. Cytosolic fractions were prepared and assayed by ribosylation; untreated cells (lane 1); genistein treatment only (lane 2); thrombin treatment only (lane 3); genistein pretreatment followed by thrombin treatment (lane 4); no C3 added to reaction (lane 5).
To evaluate this result further, we tested whether inactivation of RhoA by the bacterial toxin exoenzyme C3 would block thrombin-induced cell death. Previous reports have shown that exoenzyme C3 specifically inactivates the RhoA protein (Aktories et al., 1987; Sekine et al., 1989). Cells were pretreated with exoenzyme C3 (30 μg/ml) for 3 hr, then 200 U/ml thrombin was added and the cell viability assayed 72 hr later (Fig. 3B). Pretreatment with exoenzyme C3 blocked >50% of thrombin-induced cell death in astrocytes. This indicates that not only does thrombin increase available RhoA activity, but this increase in RhoA activity appears necessary to transduce thrombin-induced cell death.
Previous studies have linked tyrosine kinase activity and RhoA activation. Some reports suggest that tyrosine kinases act upstream from RhoA (Zubiaur et al., 1995), whereas others report that tyrosine kinase activity acts downstream of RhoA (Ridley and Hall, 1994). Both tyrosine kinases and RhoA were required in the thrombin cell death pathway, and it was possible that they were linked sequentially. If tyrosine kinase activity acted upstream of RhoA, then pretreatment with tyrosine kinase inhibitors should block thrombin-induced activation of RhoA. If the tyrosine kinase activity acts downstream of RhoA, then pretreatment with tyrosine kinase inhibitors should not affect thrombin-induced activation of RhoA. Accordingly, we pretreated astrocytes with genistein as previously. Cells were then treated with thrombin (200 U/ml) for 20 min and cell lysates prepared. RhoA activity was assayed for and an autoradiograph of a typical result is pictured in Figure 3C. Astrocytes pretreated with genistein (lane 2) or herbimycin A (data not shown) had approximately the same basal level of RhoA activity as untreated cells (lane 1). Thrombin treatment produced the expected increase in activity (lane 3). In contrast, cells pretreated with genistein and then treated for 20 min with thrombin did not show an increase in RhoA activity. This result suggests that blockage of thrombin-induced cell death with the tyrosine kinase inhibitor genistein was at least partially mediated by preventing induction of RhoA activity.
A cytosol-to-membrane redistribution of RhoA is thought to be essential for its activity (Mackay et al., 1995). To determine whether thrombin induced such a redistribution, we examined the level of available RhoA in the cytosolic fraction prepared from each of the above treatments. Thrombin treatment of astrocytes resulted in greatly decreased unribosylated RhoA in the cytosol, and this was prevented by pretreatment with genistein (Fig. 3D). This data supports further our conclusion that thrombin-induced cell death involves RhoA activity and that tyrosine kinase activity is required upstream of RhoA activation. It should be noted that there was some variability in the degree of redistribution of RhoA to the membrane fraction by thrombin, and a measurable loss of RhoA activity from the cytosol may not be necessary to see increased membrane-associated RhoA activity.
Thrombin-induced neuronal death involves apoptosis
Thrombin can also induce cell death in neurons, and neuronal cell loss is arguably of greater functional significance in CNS injury. Using a strategy parallel to the one described previously for astrocytes, we examined the mechanism(s) of thrombin-induced neuron death. Although not all of the astrocyte experiments could be conducted successfully in the more inherently fragile neuronal cultures, the more significant experiments were performed.
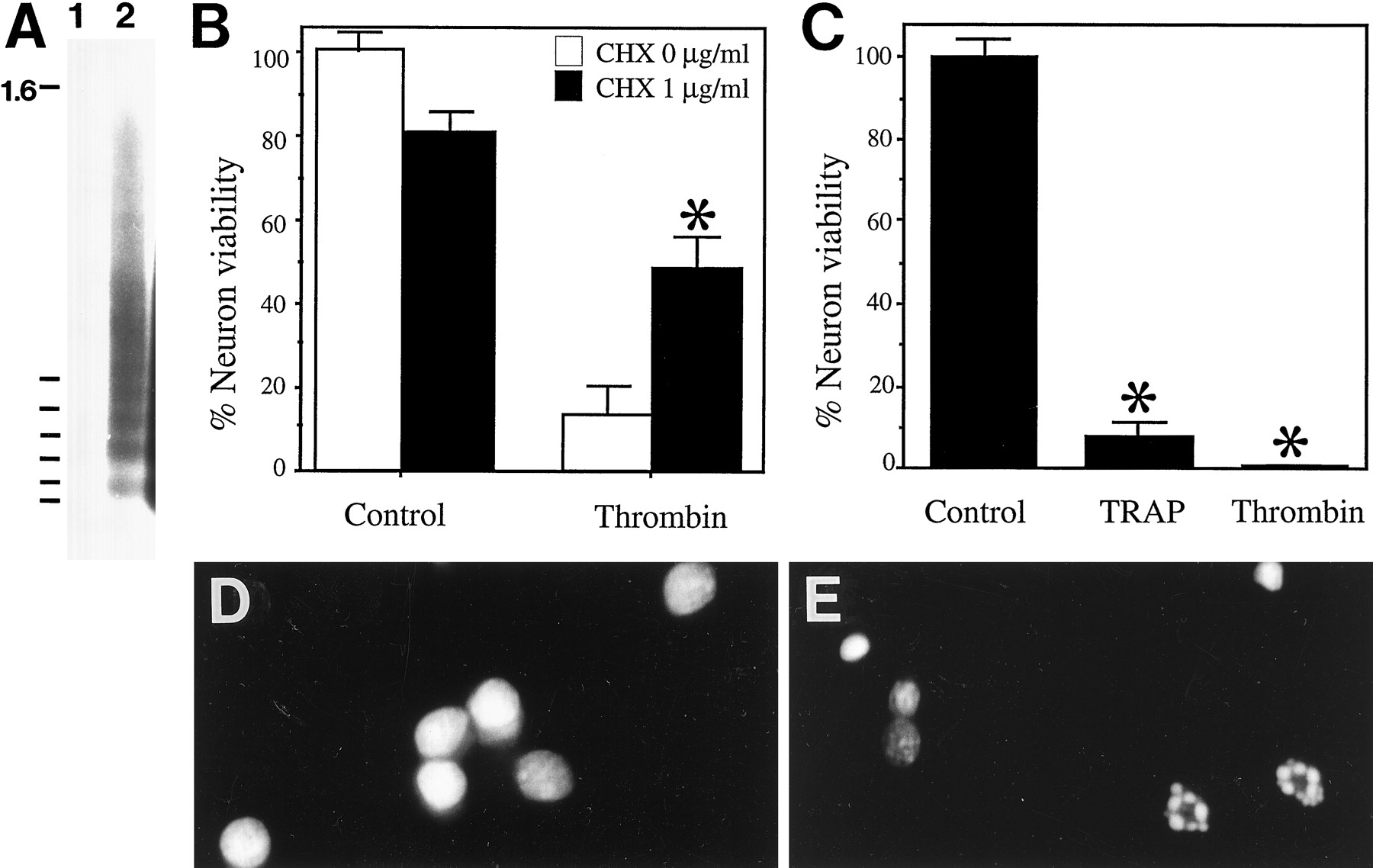
As with the astrocytes, our first tasks were to establish whether thrombin-induced neuron death involved receptor activation and occurred by an apoptotic pathway. Consistent with our previous observations (Vaughan et al., 1995), we determined that thrombin concentrations >150 U/ml (750 nm) induced neuronal death (data not shown). To determine whether thrombin induced apoptosis in neurons, we examined the DNA of thrombin-treated hippocampal neurons for DNA fragmentation, a hallmark of apoptotic cell death. Neurons were treated with 0 or 200 U/ml thrombin for 24 hr, and DNA released into the supernatant was recovered. DNA from untreated cells did not give rise to a DNA ladder (Fig. 4A, lane 1). DNA recovered from cells treated with 200 U/ml thrombin showed a pattern of bands corresponding to a DNA ladder of oligonucleosomal-sized fragments (Fig. 4A, lane 2). This indicated that thrombin-induced cell death involved an apoptotic pathway.

Thrombin induces apoptosis in hippocampal neurons; this is mediated by activation of the thrombin receptor.A, Hippocampal neurons treated with thrombin release oligonucleosomal-sized DNA fragments indicative of apoptosis. Neurons were treated with 0 (lane 1) and 200 U/ml thrombin (lane 2) for 24 hr. The culture supernatant was harvested and all DNA present extracted. DNA was end-labeled with [32P]dCTP, electrophoresed through a 1.8% agarose gel, and subjected to radiography. B, Cycloheximide pretreatment inhibits thrombin-induced cell death in hippocampal neurons. Neurons were pretreated with cycloheximide (1.3 μg/ml) for 30 min. Thrombin was added to the culture media, and the number of viable cells were counted 24 hr after the addition of thrombin. Asterisk denotes p < 0.05 in thrombin plus cycloheximide conditions relative to thrombin alone conditions. C, TRAP induces cell death in hippocampal neurons. Neurons were treated with TRAP (15 mm) or thrombin (200 U/ml), and viable cells were counted 24 hr after additions.Asterisk denotes p < 0.05 in thrombin and TRAP conditions relative to control conditions.D, E, Neurons treated with TRAP show altered nuclear morphology. Control cultures or cultures treated with 15 mm TRAP for 24 hr were incubated with SYTO 11 to stain the nuclei; control culture (D); TRAP-treated neurons (E).
In many pathways leading to apoptosis, synthesis of new proteins is required. Thus, we examined whether the protein synthesis inhibitor cycloheximide reduced thrombin-induced neuronal death. Such studies were more conducive to neuronal cultures than to astrocytes because the toxic side effects of cycloheximide are only minimally apparent during the relatively short 24 hr treatment required to observe thrombin-induced neuronal death. Neurons were pretreated for 30 min with cycloheximide (this treatment alone caused mild cell injury over 24 hr), then thrombin was added and the cell viability determined 24 hr later. We observed that a 30 min pretreatment with cycloheximide (1.3 μg/ml) yielded a robust but incomplete protection against thrombin cell death, further implicating an apoptotic pathway in thrombin-induced neuronal death (Fig. 4B). To determine that protein synthesis was inhibited by this cycloheximide treatment in our culture system, we measured the incorporation of [35S]methionine into cellular proteins at 3 hr after cycloheximide treatment. The incorporation of [35S]methionine into cellular proteins was decreased by ∼85% in these neuron cultures at this time point; thus, we concluded that protein synthesis was largely inhibited in this experimental system and that inhibition of protein synthesis by cycloheximide was most likely the cause of the inhibition of thrombin-induced cell death (data not shown).
Thrombin-induced neuronal death involves the thrombin receptor
To determine whether thrombin-induced neuronal apoptosis involved activation of the thrombin receptor, we evaluated the effect of TRAP on neuronal viability. Hippocampal neurons treated for 24 hr with 15 mm TRAP exhibited significant cell death (Fig.4C). As evidenced by the presence of numerous apoptotic nuclei (Fig. 4D,E), TRAP-induced neuronal death is apoptotic.
Thrombin induces RhoA activity in neurons
Next we determined whether the pathway of thrombin-induced neuronal death shared with astrocytes a requirement of RhoA activation. Hippocampal neurons were treated with increasing concentrations of thrombin for 20 min and then lysed, and membrane and cytosolic fractions were prepared. These fractions were assayed for the level of available RhoA activity by the in vitro ribosylation assay. Thrombin increased RhoA activity present in membrane fractions, but only at concentrations that led to cell death (Fig. 5,lane 5 vs lanes 1–4).

Thrombin induction of RhoA activity in hippocampal neurons. Hippocampal neurons were treated with thrombin for 20 min. Membrane fractions were prepared and RhoA activity assayed by in vitro ribosylation with exoenzyme C3 and [32P]NAD; control (lane 1); 20 U/ml thrombin (lane 2); 50 U/ml thrombin (lane 3); 100 U/ml thrombin (lane 4); 200 U/ml thrombin (lane 5); control for endogenous ribosylation activity (lane 6). Samples are as above except that no exoenzyme C3 was added to the reaction mixture.
Exoenzyme C3, a potent inhibitor of RhoA, blocks thrombin-induced neuronal death
As with the astrocytes, thrombin activation of RhoA activity led us to test whether inactivation of RhoA with exoenzyme C3 attenuated thrombin-induced cell death. Experimentally, this possibility was examined by comparing thrombin-induced neuron death in the presence and absence of a 3 hr pretreatment with 50 μg/ml exoenzyme C3. As shown in Figure 6, pretreatment with exoenzyme C3 significantly reduced neuronal vulnerability to thrombin, but did not provide complete protection (p < 0.05, thrombin plus exoenzyme C3 vs control). In the absence of thrombin, exoenzyme C3 did not significantly alter neuronal viability (data not shown).

Pretreatment with exoenzyme C3 attenuates thrombin-induced cell death in hippocampal neurons. Cells were pretreated with exoenzyme C3 (50 μg/ml) for 3 hr. Thrombin was added to the medium (200 U/ml) of pretreated and control cultures. Cell viability was determined 24 hr after the addition of thrombin. Cells were incubated with the viable dye calcein AM and images taken using fluorescent microscopy. A, Control cultures, and (B) thrombin (200 U/ml) for 24 hr.C, Cultures pretreated with exoenzyme C3 followed by thrombin (200 U/ml) for 24 hr. D, Graph of cell viabilities of each culture condition. Asterisk denotesp < 0.05 in thrombin plus exoenzyme C3 conditions relative to thrombin alone conditions.
DISCUSSION
A novel and important conclusion of this study is that thrombin induces apoptosis in astrocytes and neurons. Apoptosis is a common feature of CNS injury and contributes significantly to neuron cell loss (Bredesen, 1995; Chopp et al., 1996). Apoptosis is believed to occur after acute injury, such as cerebrovascular trauma or ischemia, and during such chronic pathological processes as Alzheimer’s disease (Cotman and Anderson, 1995). In both acute and chronic CNS pathologies, apoptotic nuclei and DNA fragmentation indicative of apoptosis have been observed (Su et al., 1994; Linnik et al., 1995; Smale et al., 1995). Thrombin is present during both acute and chronic CNS pathologies. Thrombin is produced immediately at sites of cerebrovascular trauma and may persist for days after injury (Smirnova et al., 1996). The level of prothrombin in the brain has not been quantified; however, the plasma levels of prothrombin are reportedly 1–5 μm (Walz et al., 1985). Therefore, the concentration of thrombin produced locally is potentially very high. In chronic disease states, compromised blood–brain barrier function may allow focal regions of the brain to be exposed to thrombin, perhaps over periods of years. Thrombin has been found in the plaques of Alzheimer’s disease brains (Akiyama et al., 1992; McGeer et al., 1994) and a decrease in the endogenous thrombin inhibitor, protease nexin-1, is found in the brains of Alzheimer’s patients, implying an imbalance in the protease-to-inhibitor ratio favoring thrombin activity (Wagner et al., 1989; Vaughan et al., 1994; Choi et al., 1995). The recent finding that high concentrations of thrombin induced cell death in hippocampal neuron and astrocyte cultures (Smith-Swintosky et al., 1995b; Vaughan et al., 1995) prompted us both to determine whether thrombin-induced cell death is apoptotic and to investigate the mechanism underlying thrombin-induced cell death.
In this study, we have demonstrated that thrombin induced apoptosis in astrocytes and neurons. Use of TRAP demonstrated that activation of the thrombin receptor by TRAP could induce cell death and, in parallel with thrombin, examination of the nuclei of TRAP-treated astrocytes and neurons revealed the pyknotic breakdown indicative of apoptotic cell death. Since the cloning of the thrombin receptor and the discovery that TRAP could activate the receptor, almost every action of thrombin on cells has been shown to be caused by thrombin activation of this receptor (Rasmussen et al., 1991; Vu et al., 1991). In this study, TRAP was used at 15 mm, ∼104-fold greater than the thrombin concentration needed to induce cell death. Other studies have also shown that TRAP must be present at concentrations at least 104-fold greater than the enzymatically active thrombin molecule to produce cellular effects (Grabham and Cunningham, 1995; Grand et al., 1996). The specificity of TRAP toxicity was confirmed by examining the same pharmacological agents used to block thrombin cell death. Astrocytes pretreated with genistein and neurons pretreated with cycloheximide were protected from TRAP-induced cell death (data not shown), results that support thrombin-induced cell death as a specific process mediated by activation of the thrombin receptor. It should be noted that TRAP can activate the PAR-2 receptor as well, and although it has a substantially lower affinity for this receptor, possible involvement of this receptor cannot be ruled out in TRAP-induced cell death. Thrombin does not activate the PAR-2 receptor (Blackhart et al., 1996).
Thrombin treatments that produced cell death were blocked by tyrosine kinase inhibitors, and a tyrosine kinase activity is required for RhoA activation. In our current studies, the tyrosine kinase inhibitors genistein and herbimycin A fully blocked thrombin-induced cell death. Thrombin-induced tyrosine kinase activity has been reported in various cell types including astrocytes (Grabham and Cunningham, 1995). It has been reported previously that thrombin-induced tyrosine kinase activity is induced independent of heterotrimeric G-proteins (Grand et al., 1996). This may be true in our studies also, because preincubation with pertussis toxin did not block thrombin-induced cell death. RhoA, a member of the Ras superfamily of small GTP-binding proteins, is thought to be activated by an upstream tyrosine kinase activity (Zubiaur et al., 1995) and also appears to be activated by a pertussis toxin-insensitive pathway (Katoh et al., 1996; Koyama and Baba, 1996;Post and Brown, 1996). Reports also demonstrate that downstream effectors of RhoA may include tyrosine kinases (Ridley and Hall, 1994). In our study, pretreatment with genistein blocked thrombin induction of RhoA, indicating that tyrosine kinase activity was needed to induce RhoA. The possibility remains that downstream effectors of RhoA include tyrosine kinases as well. The specificity of these inhibitors must also be considered. Genistein can effect other kinases by acting as a competitor for ATP, and herbimycin A can inhibit PLD at high concentrations; however, other kinase inhibitors used in this study, wortmannin and HA-1004, inhibitors of PI 3-K, phospholipase A2, PLD, CaM kinase II, and protein kinase A and G, respectively, were unable to block thrombin-induced cell death, making it most likely that the attenuation of cell death brought on by genistein and herbimycin A is attributable to their tyrosine kinase inhibitory properties.
We hypothesize that thrombin can overstimulate RhoA activity, and this activates a pathway that causes astrocytes and neurons to undergo apoptosis. A study by Jimenez et al. (1995) showed that high levels of expression of the Aplysia rho gene, a homolog of RhoA, stimulated apoptosis in a fibroblast cell line. In our study, thrombin increased available RhoA activity in a dose-dependent manner. Only the higher doses of thrombin that also result in cell death caused a rapid and significant increase in RhoA activity. Preincubation of astrocytes and neurons with the RhoA inhibitor exoenzyme C3 attenuated thrombin-induced cell death, indicating that RhoA activity was required for thrombin-induced apoptosis.
A serine/threonine kinase activity, possibly PKC, was involved in thrombin-induced cell death. The broad spectrum serine/threonine kinase inhibitor H-7 fully blocked thrombin-induced cell death. Another serine/threonine kinase inhibitor, HA 1004, was ineffective at blocking thrombin cell death, even at concentrations 10 fold higher than H-7. H-7 is a more effective inhibitor of PKC than HA-1004, and thus it is possible that H-7 blocked PKC activity that was necessary for thrombin-induced cell death. A model of this cascade consistent with our data are shown in Figure 7. Activation of the thrombin receptor by thrombin or TRAP results in activation of one or more tyrosine kinases. This event activates RhoA, which translocates to the membrane where it can transduce the signal to its effector proteins. Potential effector proteins of RhoA include phosphatidylinositol 4-phosphate 5-kinase (Chong et al., 1994), which in turn increases the available phosphotidylinositol-4,5-bisphosphate (PIP2). Phospholipase Cγ, also activated by tyrosine kinases, could produce diacylglycerol and inositol triphosphate from PIP2, and this would result in increased PKC activity and mobilization of calcium. It is also possible that H-7 inhibited some other serine/threonine kinase activity as well, such as ROKα and protein kinase N, two serine/threonine kinases recently shown to be effector proteins of RhoA (Leung et al., 1995,1996; Watanabe et al., 1996). Although recent reports link thrombin and RhoA to PLD and PI 3-K activity (Kuribara et al., 1995), inhibitors such as wortmannin and HA-1004 had no effect indicating that PLD, PI 3-K, protein kinase A, and myosine light chain kinase are probably not involved in this process. The final pathway to which RhoA couples and induces apoptosis is not known; however, calcium deregulation may be involved. A recent study by Smith-Swintosky et al. (1995b) showed that thrombin caused neurodegeneration and an increase in intracellular calcium; in the absence of calcium, both the rise in calcium and the cell death were blocked, implying that the thrombin-induced degeneration was due to the increased calcium load on the cells. Thrombin was also shown to potentiate β-amyloid toxicity (Smith-Swintosky et al., 1995a), toxicity that is thought to involve apoptosis and was also associated with a rise in intracellular calcium. In the current study, incubation of astrocytes with the calcium chelator BAPTA did not block thrombin-induced cell death, suggesting that large influxes of extracellular calcium are not responsible for thrombin-induced cell death; however, this finding does not rule out the possible deregulation of intracellular stores of calcium. It should also be noted that Rho proteins are implicated not only in regulation of cytoskeletal proteins, but also in gene transcription and cell cycle control (Yamamoto et al., 1993; Hill et al., 1995; Olson et al., 1995). As with many of the Ras family members, RhoA may be linked to a MAP kinase or similar cascade that ultimately regulates gene transcription and cell fate (Vojtek and Cooper, 1995).

A proposed model for a signal transduction cascade, initiated by activation of the thrombin receptor, that leads to apoptosis. See Discussion for details. Tyr Kinase, tyrosine kinases; PtdIns 4,5 kinase, phosphatidylinositol 4,5-kinase.
An overall model of thrombin effects during CNS injury must take into account the different effects of thrombin on CNS cells. Thrombin protects astrocytes and neurons from hypoglycemia, oxidative stress, and β-amyloid toxicity. Thrombin-induced cell death, while requiring higher concentrations of thrombin than those that induce neuroprotection, may also contribute to CNS repair processes. We propose the following model: at the center of an injury, cells are the most traumatized and are exposed to the highest concentrations of thrombin; under these conditions thrombin induces apoptosis, and in doing so, decreases necrotic cell death and the spread of inflammation. As thrombin diffuses out from the center of the wound, the peripheral cells are exposed to lower concentrations of thrombin as well as to nutrient deprivation and oxidative stress. The lower concentration of thrombin stimulates neuroprotective signals in these cells and thus decreases cell death and excitotoxicity in the tissue surrounding the wound. Thus thrombin-induced apoptosis and neuroprotection may serve as mechanisms to control the extent of neural cell loss after CNS trauma. Uncontrolled thrombin activity, however, could contribute to an undesirable stimulation of apoptosis. Continued elucidation of the signals underlying thrombin-induced apoptosis should facilitate therapeutic intervention against thrombin-induced cell death without compromising beneficial functions of thrombin such as neuroprotection and clot formation and lead to a better understanding of how to control the consequences of CNS exposure to thrombin.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant AG00538 and the American Paralysis Association (D.D.C., C.W.C.), and by NIH Training Grant AG00096 (F.M.D.).
Correspondence should be addressed to Dr. Dennis D. Cunningham, Department of Microbiology and Molecular Genetics, University of California, Irvine, CA 92717.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}