

Abstract
Diffuse axonal injury (DAI) is one of the most common and important pathologies resulting from the mechanical deformation of the brain during trauma. It has been hypothesized that calcium influx into axons plays a major role in the pathophysiology of DAI. However, there is little direct evidence to support this hypothesis, and mechanisms of potential calcium entry have not been explored. In the present study, we used an in vitro model of axonal stretch injury to evaluate the extent and modulation of calcium entry after trauma. Using a calcium-sensitive dye, we observed a dramatic increase in intra-axonal calcium levels immediately after injury. Axonal injury in a calcium-free extracellular solution resulted in no change in calcium concentration, suggesting an extracellular source for the increased post-traumatic calcium levels. We also found that the post-traumatic change in intra-axonal calcium was completely abolished by the application of the sodium channel blocker tetrodotoxin or by replacement of sodium withN-methyl-d-glucamine. In addition, application of the voltage-gated calcium channel (VGCC) blocker ω-conotoxin MVIIC attenuated the post-traumatic increase in calcium. Furthermore, blockade of the Na+–Ca2+ exchanger with bepridil modestly reduced the calcium influx after injury. In contrast to previously proposed mechanisms of calcium entry after axonal trauma, we found no evidence of calcium entry through mechanically produced pores (mechanoporation). Rather, our results suggest that traumatic deformation of axons induces abnormal sodium influx through mechanically sensitive Na+ channels, which subsequently triggers an increase in intra-axonal calcium via the opening of VGCCs and reversal of the Na+–Ca2+ exchanger.
Damage to axons is thought to be the most common pathology associated with traumatic brain injury (Maxwell et al., 1997; Smith and Meaney, 2000). Widely distributed damage to axons in the brain, termed diffuse axonal injury (DAI), results from the inertial forces exerted on the white matter tracts in the brain during traumatic incidents such as automobile accidents and, in some cases, falls and assaults (Gennarelli et al., 1982; Gennarelli, 1993;Graham et al., 1995). DAI has been postulated as the major cause of coma and death as a result of these types of injury (Gennarelli et al., 1982). Although severe inertial brain injury may induce tissue tears in the white matter resulting in immediate disconnection of axons (primary axotomy), most damaged axons undergo secondary disconnection over an extended time course (secondary axotomy) (Povlishock et al., 1983;Povlishock, 1992). Accordingly, much attention has been placed on mechanisms of this delayed response of axonal trauma to elucidate potential therapeutic strategies.
Although it has been suggested that elevated intra-axonal calcium ([Ca2+]i) levels play a pivotal role in the secondary damage to axons after mechanical deformation (Banik et al., 1987; Young, 1992; Maxwell et al., 1995;Lopachin and Lehning, 1997), this has yet to be directly demonstrated. Accordingly, in the present study we used a new in vitromodel of dynamic stretch injury of axons to evaluate changes in [Ca2+]i. Thisin vitro model induces strains to axons that replicate the deformation experienced by axons in the white matter during inertial brain injury. In previous experiments, continuous monitoring of cultured axons during and after high strain revealed severe undulations of the axons and ultrastructural changes after injury (Smith et al., 1999). In the present study, we used a Ca2+-sensitive dye to investigate changes in intra-axonal Ca2+ levels after trauma, and we explored potential mechanisms of Ca2+ entry using pharmacological manipulation.
MATERIALS AND METHODS
Cell culture. We chose the N-Tera2 cl/D1 (NT2) cell line as our neuronal substrate because of the well characterized ability of these cells to differentiate into neuron-like cells of human origin (Pleasure et al., 1992; Pleasure and Lee, 1993). In addition, this cell line has been shown to respond to excitatory injury in a manner similar to that of primary neuronal cell cultures (Munir et al., 1995). Furthermore, we have found previously that NT2 cells grow long axons with a diameter similar to that of human CNS axons (1 μm) in a sustained culture system (Smith et al., 1999). The NT2 cells were maintained in culture with OptiMEM (Life Technologies, Gaithersburg, MD) media supplemented with 5% fetal bovine serum (HyClone, Logan, UT) and 1% penicillin–streptomycin (Life Technologies). To differentiate the NT2 cells into neurons (NT2/N), NT2 cells were cultured for 5 weeks in DMEM supplemented with 10% FBS (HyClone), 1% penicillin–streptomycin (Life Technologies), and 10 μmretinoic acid (Sigma, St. Louis, MO). To isolate neurons in the culture, the cells were trypsinized, triturated with a fire-polished Pasteur pipette, and replated in DMEM supplemented with 5% FBS and mitotic inhibitors (10 μm 5-flouro-2′-deoxyuridine, 10 μm uridine, and 1 μm cytosine arabinofuranoside; Sigma) for 9 d. A final trypsinization step is used to separate the neurons from the rest of the culture. Approximately 99% of the cells remaining after this procedure have differentiated into neurons. These NT2/N cells were seeded on a treated (poly-d-lysine, fibronectin, and Matrigel) deformable substrate (Specialty Manufacturing, Saginaw, MI) in custom-designed culture wells (Smith et al., 1999). A 2 × 16 mm clear silicon barrier was placed on the membrane in the center of the well before plating of the NT2/N cells to create a 2 mm “gap” through the center of the membrane. Cells were allowed to attach for 24 hr before the barrier was removed. The temporary barrier prevents neurons from seeding in the gap region, creating a cell-free area for growth of isolated axons. After barrier removal, axons begin traversing the gap, ultimately integrating with neurons on the other side (Fig.1). The diameter of the axons crossing the gap ranged from 0.5 to 1.5 μm (typical widths of human axons in the brain). These cultures were maintained in conditioned media (50% media from the first replate and 50% DMEM with 5% FBS) for 3 weeks before experimentation, because it has been demonstrated previously that NT2/N cells express a mature neuronal phenotype similar to that ofin vivo human neurons with regard to receptor function by 2–2.5 weeks after plating (Munir et al., 1995).

Schematic illustration of axonal stretch injury.Top, Cutaway of the injury device reveals the culture well placed in a sealed chamber on a microscope stage. Axons grow between two populations of neurons plated on a flexible substrate.Bottom, A pressure pulse deforms only the region of axons, inducing tensile elongation.
Stretch injury. The culture wells were placed in the stretch-injury device that consists of an aluminum cover block, a stainless steel plate with a machined 2 × 18 mm slit, and an air pulse-generating system (Fig. 1). The culture well was inserted into the cover block and then placed on the slit plate so that the area of the deformable substrate contained the cultured axons. The cover plate was attached to the microscope stage, creating a sealed chamber. The top plate had a quartz viewing window in the center, an air inlet for compressed air, and a dynamic pressure transducer (Entran model EPX-V01-25P-/l6F-RF, Fairfield, NJ) to monitor internal chamber pressure. The introduction of compressed air into the chamber was gated by a solenoid (Parker General Valve, Elyria, OH). The solenoid and the pressure transducer were controlled and monitored by an analog-to-digital board (Keithley Metrabyte, Cleveland, OH) integrated with a computer data acquisition system (Capital Equipment Corporation, Bellerica, MA). The device was mounted on the stage of a Nikon inverted microscope (Optical Apparatus, Ardmore, PA), allowing for continuous observation of the axons throughout the experiments.
A controlled air pulse was used to induce stretch to only the cultured axons traversing the gap in the well (Fig. 1). A rapid change in chamber pressure (rise time, 20 msec; duration, 50 msec) deflects downward only the portion of the substrate that contains the cultured axons; as a result, only these axons are stretched transiently to mimic the in vivo conditions of traumatic brain injury. Strain on the axons was calculated geometrically as described previously (Smith et al., 1999). For the experiments presented here, internal chamber pressure was 13 psi, correlating to a transient uniaxial strain on the axons of 1.75 or 75% beyond its initial length.
Fluorescence microscopy and analysis. Cells were loaded with 2 μm fluo-4 AM ester (Molecular Probes, Eugene, OR) solubilized in DMSO (0.05% final) with pluronic F-127 [0.004% (w/v) final] in a control saline solution (CSS; 120 mm NaCl, 5.4 mm KCl, 0.8 mm MgCl2, 1.8 mm CaCl2, 15 mm glucose, and 25 mm HEPES, pH 7.4, adjusted to 330 mOsm with sorbitol) in which all experiments were run (Takahashi et al., 1999). Because of the small diameter (1 μm) and volume of the axons, fluo-4 AM, an analog of the widely used fluo-3 AM, was used to achieve the maximum fluorescence after binding calcium, because it has an increased fluorescence excitation at 488 nm compared with that of fluo-3 AM. The ion dissociation constantKd (Ca2+) is reported to be similar for the two dyes under identical conditions (fluo-3 = 325 nm; fluo-4 = 345 nm; manufacturer's specifications). Pluronic F-127 (Molecular Probes), a nonionic detergent, is used to further disperse the dye in the CSS and allow greater access to the cytoplasm by altering membrane fluidity. The dye solution was loaded at 37°C for 30 min; the cells were rinsed, then allowed to sit for another 30 min to allow for further deesterification of the dye, and then rinsed once more before injury.
The ionophore 4-bromo-A23187 at 50 μm (Molecular Probes) served as a positive control for dye response to Ca2+ influx and was used under all treatment conditions to ensure that proper dye loading had occurred and that the treatments had not altered the dye's calcium affinity. Because of the brightness of the dye at high calcium concentrations and the sensitivity setting of the camera necessary to image axons, the ionophore-treated axons uniformly reached the maximum detectable fluorescence in our system.
Fluorescence microscopy was performed on a Nikon Diaphot inverted microscope with a Hamamatsu “Orca” CCD camera attached (Optical Apparatus). A xenon light source excited the dye at 488 nm, and the emitted fluorescence was collected at 515 nm. Fluorescence images (1024 × 768 pixels) were collected and analyzed using the MetaFluor software package on a personal computer to which the camera was attached (Universal Imaging Corporation, West Chester, PA). Images were taken at 1 sec intervals for 20 sec, at which time the injury was induced (see above). For the 3 min experiments, sampling continued at 1 sec intervals after injury. The 20 min duration experiments were sampled 5 times per minute after the first minute had elapsed.
Analysis of changes in fluorescence of the fluo-4 dye was performed on six representative axons from each culture. Each axon was ∼1 μm in diameter and oriented along the axis of stretch. Axons were excluded from analysis if they had undergone primary axotomy (≤5% of injured fibers). Three random regions from each axon were analyzed and then averaged. The axons were continuously sampled, except for a 2–10 sec period after injury during which the microscope is refocused. To account for potential variation in dye loading among axons or experiments, we used a standard procedure for nonratioable indicators in which self ratios were taken (F/F0) between the measured fluorescence (F) and the initial fluorescence (F0). Background fluorescence subtraction was accomplished by continuously sampling three areas in the field that had no axons in them for the duration of the experiment. The mean of these values was obtained at every time point and subtracted from the raw value obtained at each analyzed region of the axon before analysis.
Modulation of calcium changes. To evaluate modulation of Ca2+ in these axons, we used agents that blocked voltage-gated Ca2+ and Na+ channels and the Na+–Ca2+exchanger, and we removed extracellular cations. All pharmacological agents and toxins were solubilized and added to the CSS and then to the culture 10 min before experimentation. Injuries with no treatment were performed using CSS with no modifications (n = 9 wells at 25°C; n = 3 at 37°C). Tetrodotoxin (Sigma), a Na+ channel blocker, was used at 1 μm (n = 5 at 25°C;n = 3 at 37°C); ω-conotoxin MVIIC (Sigma), a P/Q- and N-type voltage-gated calcium channel (VGCC) blocker, was used at 1 μm (n = 4 at 25°C) (Hillyard et al., 1992; McDonough et al., 1996), and bepridil (Sigma Research Biochemicals, St. Louis, MO), a Na+–Ca2+exchanger blocker, was used at 50 μm(n = 6 at 25°C; n = 3 at 37°C) (Garcia et al., 1988). Calcium-free CSS was made by removing the CaCl2 and adding back an equal part of MgCl2 (final 2.8 mm) and 1 mm EGTA (Sigma) (n = 3 at 25°C). In a subset of these experiments, CSS with Ca2+ was added back to the culture 2 min after injury (n = 2). Sodium-free CSS was made by removing the NaCl from the CSS and replacing it with an equimolar amount of N-methyl-d-glucamine [NMDG(+)] (n = 3 at 37°C). Both the calcium-free and sodium-free CSS were adjusted to a pH of 7.4, and their osmolarity was corrected to 330 mOsm using sorbitol (Sigma). To compare experimental groups, mean values for each axon analyzed in the culture were then averaged to obtain a mean value for each experiment. Mean values of fluorescence over initial fluorescence (F/F0) for the experimental groups were then compared, and statistical significance was calculated using the post hoc Newman–Keuls test.
RESULTS
Axonal response to stretch injury
In agreement with our previous observations, immediately after trauma the axons became severely undulated but gradually regained most of their prestretch orientation over a period of 20 min in a classic “delayed elasticity” response (Smith et al., 1999). A small subset of axons (1–3 per field of ∼50) underwent primary axotomy at the induced strain level. In these axons, increases in [Ca2+]i were most pronounced at the disconnected ends and were higher than those of the injured but still intact axons. These results were not combined with the data from the injured, nontransected axons.
Calcium response in untreated stretch-injured axons
In noninjured axons, we were able to observe a baseline fluorescence of the Ca2+ indicator fluo-4. Although there was slight variation in the brightness of the fluorescent signal between axons in the same well, the basal fluorescence was consistent between experiments (mean = 301 and SD = 109, arbitrary fluorescence units). In nondisconnected axons, a large increase in the measured fluorescence was observed immediately after injury (F/F0 = 2.61;p < 0.001), consistent with a large increase in intra-axonal calcium concentration [Ca2+]i (Figs.2, 3). This average represented a range of no influx (F/F0 ≤ 1) to a maximumF/F0 of 7.30. However, there was a relatively homogeneous response within each individual axon that was measured. After the immediate post-traumatic increase in the mean fluorescence, there was a downward trend at the 1 min time point. The fluorescence then underwent a modest but significant incremental increase over the next 20 min (Figs. 3,4) (t = 1 min,F/F0 = 1.74;t = 20 min, F/F0 = 2.49;p < 0.05). Although there was a range in these levels among injured axons, all changes followed the same progression of [Ca2+]i increase over the 20 min time period.

Changes in intra-axonal calcium fluorescence immediately after stretch injury in control saline solution (CSS; n = 12 wells, 72 axons) compared with those after injury in calcium-free CSS (0 Ca2+; n = 3 wells) or Na+-free CSS [N-methyl-d-glucamine replacement (NMDG); n = 3 wells] or with pretreatment with tetrodotoxin (TTX;n = 8 wells), ω-conotoxin MVIIC (CTX; n = 4 wells), or bepridil (BEP; n = 9 wells).F/F0 = change in calcium fluorescence over initial fluorescence (*p < 0.01; **p < 0.001).
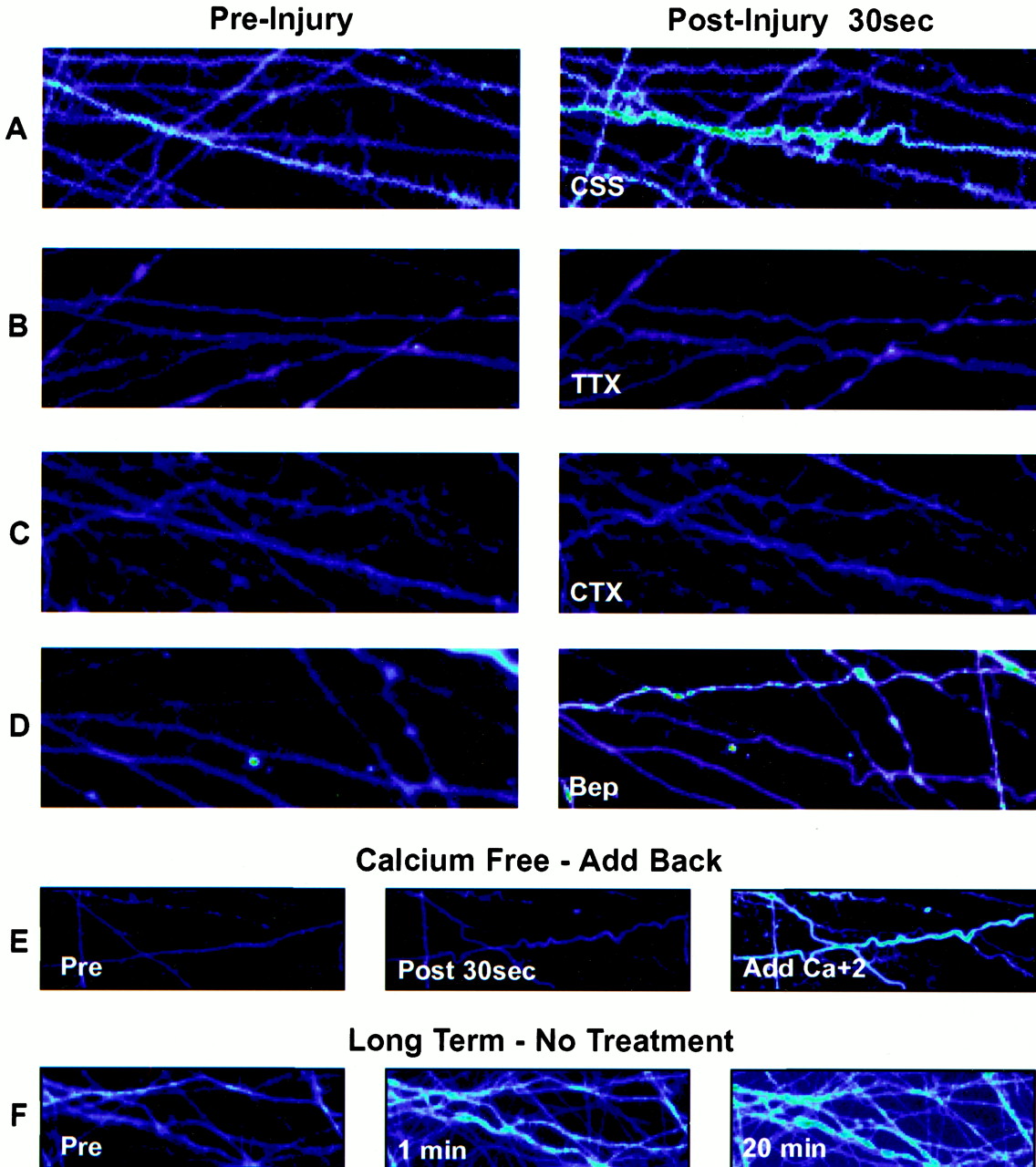
A–D, Representative photomicrographs of changes in calcium fluorescence before (left) and after (right) axonal stretch injury in control saline solution (CSS) or with pretreatment with tetrodotoxin (TTX), ω-conotoxin MVIIC (CTX), or bepridil (Bep). E, Demonstration of changes in calcium fluorescence after axonal stretch injury in calcium-free CSS with calcium added back 2 min after injury. F, Demonstration of the persistent increase in calcium fluorescence over 20 min after axonal injury.

Temporal change in calcium fluorescence over 20 min after axonal stretch injury (n = 3 wells).F/F0 = change in calcium fluorescence over initial fluorescence (*p < 0.05).
Calcium response in axons with ionic or pharmacological manipulation
For axons injured in calcium-free CSS, there was no post-traumatic increase in [Ca2+]i(F/F0 = 0.97; Figs. 2, 3). However, replacement of the calcium-free CSS with normal CSS containing Ca2+ 2 min after injury led to an immediate rise in calcium (F/F0 = 2.53 by 1 min after addition; Figs. 3, 5).
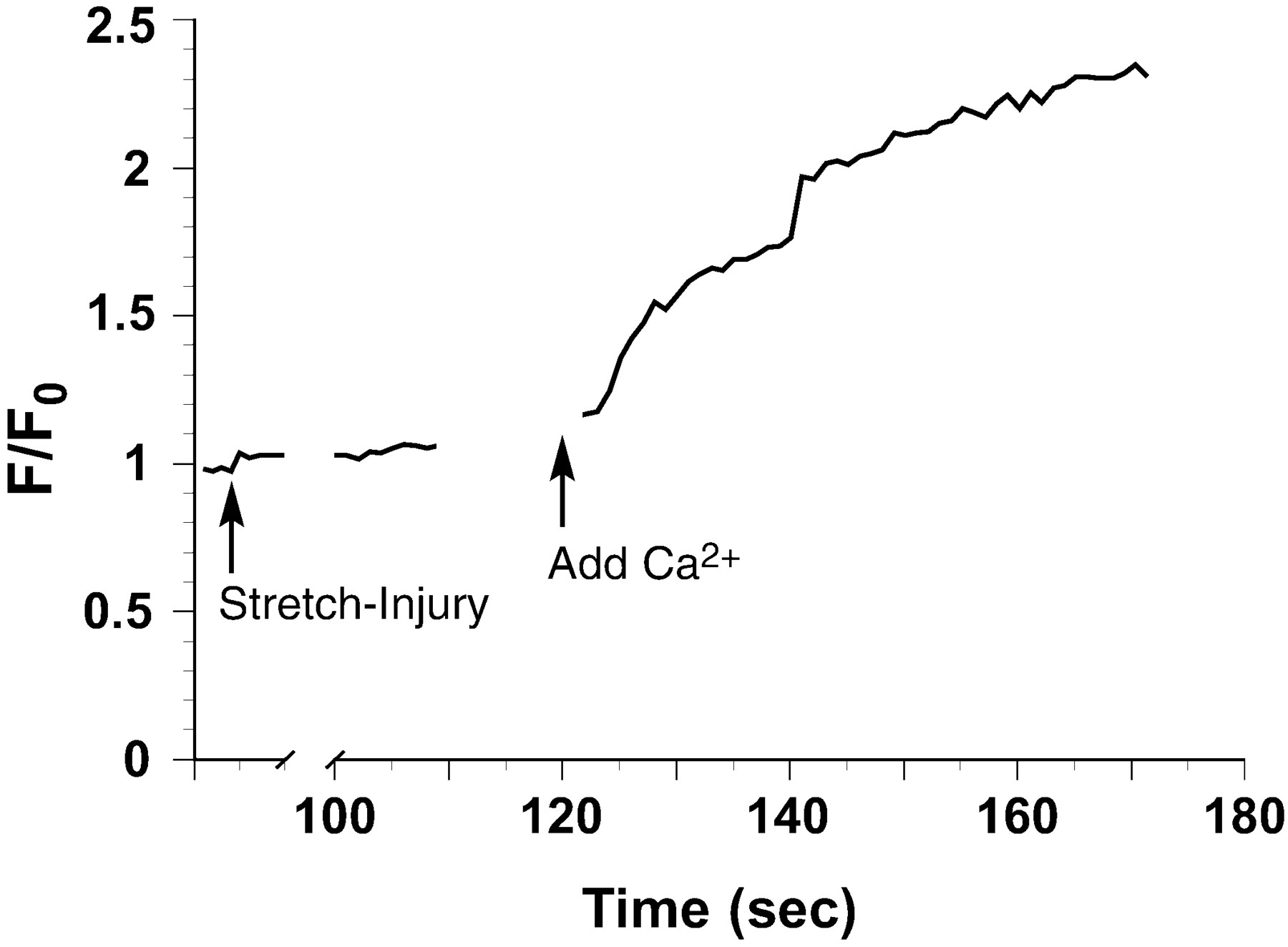
An example tracing (mean of 6 axons) from a Ca2+-free experiment in which Ca2+ is added back 2 min after injury. No data are taken during removal of the top plate after injury to gain access to the well (t, 20–100 sec) or addition of the Ca2+ back to the extracellular solution.F/F0 = change in calcium fluorescence over initial fluorescence.
In axons that were pretreated with the Na+channel blocker tetrodotoxin (TTX; 1 μm) and then injured, we found a complete attenuation of the Ca2+ influx response (F/F0 = 0.98) compared with untreated axons that underwent stretch injury (p< 0.001; Figs. 2, 3). In addition, replacement of extracellular Na+ with NMDG(+) before injury also dramatically attenuated Ca2+ influx compared with injured untreated axons (F/F0 = 0.96;p < 0.001).
Partial attenuation of the Ca2+ increase after injury was observed by modulating either VGCCs or the activity of the Na+–Ca2+exchanger. Pretreatment of axons with the P/Q- and N-type VGCC blocker ω-conotoxin MVIIC (1 μm) attenuated Ca2+ influx (F/F0 = 1.30;p < 0.001). Moreover, pretreatment with bepridil (50 μm), a Na+–Ca2+exchanger blocker, partially attenuated the Ca2+ influx after injury (F/F0 = 1.74;p < 0.01). No significant difference because of ambient temperature (25 or 37°C) was noted in these pharmacological studies.
DISCUSSION
Calcium influx after axonal trauma has been hypothesized to be the major activator of deleterious processes after injury (Banik et al., 1987; Young, 1992; Lopachin and Lehning, 1997). It is thought that these processes lead to a delayed disconnection of axons after traumatic axonal injury (secondary axotomy) and to Wallerian degeneration of damaged axons in the white matter (Pleasure et al., 1992; George et al., 1995; Maxwell et al., 1997; Smith and Meaney, 2000). However, increased intra-axonal Ca2+ after stretch injury has yet to be directly demonstrated. In the present study, we used an in vitro axonal stretch-injury system that allows for the continuous examination of changes in intra-axonal Ca2+ levels. Using the Ca2+-sensitive dye fluo-4, we found that there is a substantial increase in axonal Ca2+ levels after stretch injury, dependent on extracellular Ca2+. In addition, we observed that this post-traumatic rise in intra-axonal Ca2+ was completely dependent on Na+ entering through TTX-sensitive Na+ channels.
It has been demonstrated that the unique loading conditions that occur during brain trauma lead to traumatic axonal injury. These conditions include dynamic deformation of axons, with the strain duration lasting <50 msec (Meaney et al., 1995). In our in vitro model, applying these parameters of dynamic injury to the axons with a high level of uniaxial strain (75%) results in severe undulations in the axons; only a small portion of the axons undergoes primary axotomy (immediate disconnection) (Smith et al., 1999).
To date, few studies have examined directly post-traumatic Ca2+ accumulation in axons. Previous studies using Ca2+-sensitive microelectrodes revealed a substantial drop in extracellular Ca2+ in the vicinity of injured spinal white matter, indicating influx of this cation into intracellular compartments (Young et al., 1982). Lopachin et al. (1999)subjected isolated strips of spinal cord white matter to compressionin vitro and measured axonal elemental content using electron probe microanalysis. These authors found a gradual increase of axoplasmic Na+ and Ca2+ developing over many minutes after injury. TTX reduced the deregulation of Na+ and Ca2+in many, but not all, fibers. Another model that examines the in vivo response of dynamic deformation of axons uses stretch injury of the guinea pig optic nerve (Gennarelli et al., 1989). This model has been used previously to provide indirect evidence of an increase in Ca2+ in axons after injury (Maxwell et al., 1995). Pyroantimonate fixation of injured optic nerves suggested a change in Ca2+ levels in mitochondria and the axoplasm of axonal swellings. However, this in vivoexperimental paradigm does not allow for the examination of mechanisms of Ca2+ influx or continuous observation in vital tissue.
Although stretch injury in our model induced a substantial rise in intra-axonal Ca2+, removal of extracellular Ca2+ and subsequently injuring axons resulted in virtually no change in the level of Ca2+. These data suggest that the source of the accumulating intra-axonal Ca2+ was predominantly extracellular. However, a possible delayed Ca2+ release from intracellular Ca2+ stores such as the endoplasmic reticulum and mitochondria cannot be excluded.
The increased Ca2+ levels observed immediately after stretch injury remained elevated and even increased slightly over time. This finding may indicate that the normal Ca2+-buffering ability of axons is overwhelmed after dynamic trauma. It has been suggested that a sustained intra-axonal Ca2+ increase may initiate a series of deleterious cascades, including activation of proteases, second messenger systems, and failure of the mitochondria (Banik et al., 1987; Gitler and Spira, 1998; Buki et al., 2000). In previous ultrastructural studies using a model of white matter anoxia, sustained elevated Ca2+ levels were linked with the disruption of neurofilaments and microtubules (Waxman et al., 1994). Therefore, the sustained Ca2+increase found in stretch-injured axons may play an important role in the observed damage of the cytoskeleton and disruption of axonal transport after dynamic stretch injury of axons (Povlishock, 1993;Maxwell et al., 1997).
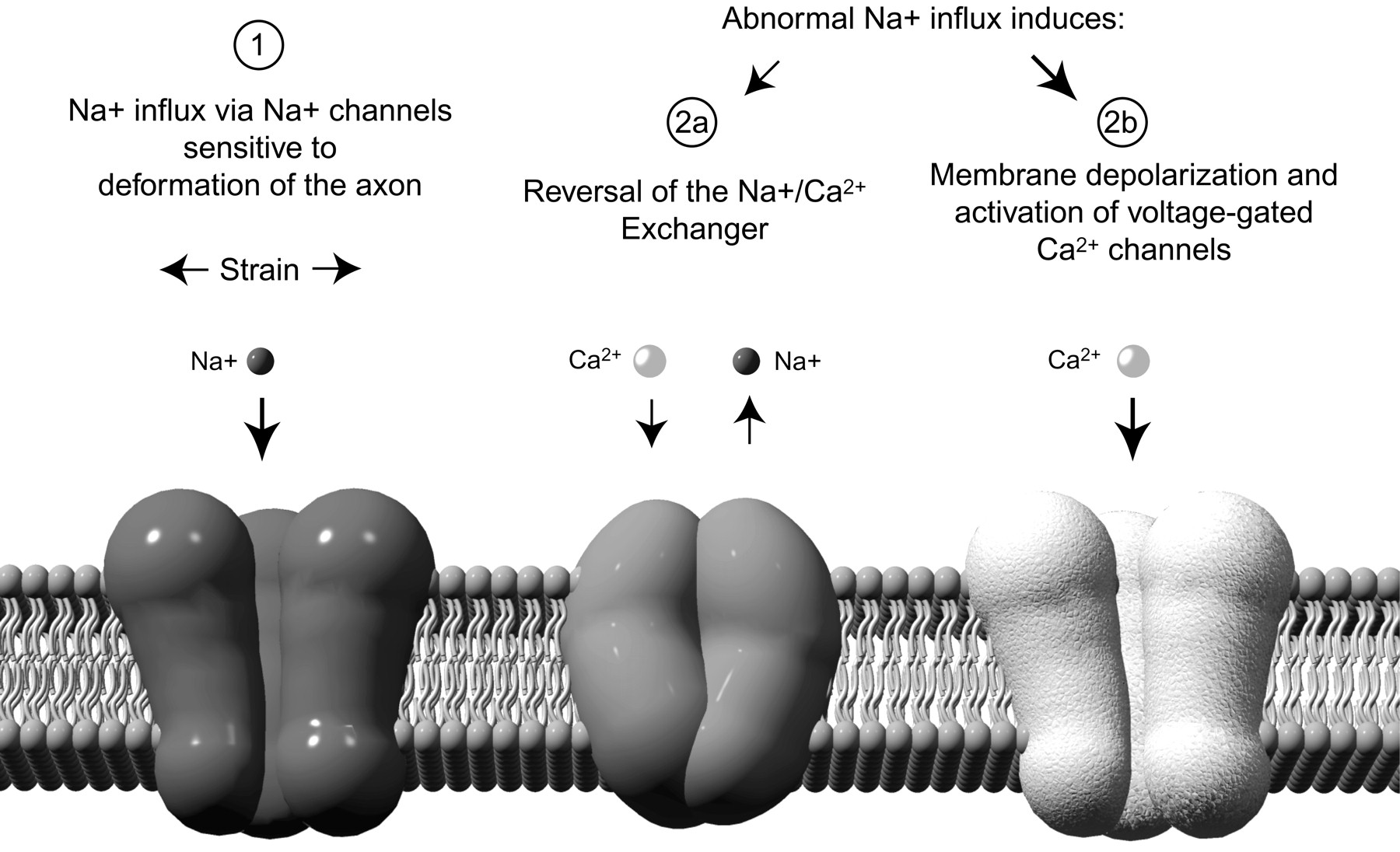
Previous investigations of white matter injury have established the prominent role of Na+ influx via voltage-sensitive Na+ channels in models of anoxia or crush injury of white matter (Stys et al., 1991; Agrawal and Fehlings, 1996; Imaizumi et al., 1997). In particular, it was shown that Na+ influx into axons through TTX-sensitive Na+ channels is an integral mediator of white matter injury in these models. In the present study, we found that treatment with TTX completely blocked Ca2+ influx after dynamic stretch injury of axons. This demonstrates a strong causal relationship between excessive Na+ influx through voltage-gated Na+ channels and an increase in intra-axonal Ca2+ after axonal trauma. Several mechanisms have been proposed that link Na+ channels and Ca2+ entry after axonal injury: (1) Ca2+ could directly enter the axon through Na+ channels, (2) increased membrane Na+ conductance could depolarize the axons opening VGCCs, or (3) Na+ influx and depolarization could induce reversal of gradient-dependent ion exchangers, such as the Na+–Ca2+exchanger (Stys, 1998).
In the present study, we addressed the potential mechanism of Ca2+ entry into injured axons by replacing Na+ with an impermeable ion, NMDG(+). This treatment, which will prevent both depolarization and Na+ influx, completely eliminated the Ca2+ influx into axons, indicating that Na+ entry is essential for Ca2+ influx after stretch injury and that Ca2+ influx through Na+ channels is insignificant.
One consequence of Na+ channel activation is the effect that the resultant depolarization of the axonal membrane may have on other voltage-gated channels, such as VGCCs. Although it was originally thought that VGCCs were not present in white matter, recent data have demonstrated significant protection from anoxic injury after blocking VGCCs (Stys et al., 1990; Fern et al., 1995; Imaizumi et al., 1999).
In addition, it has been shown that activation of VGCCs is a primary mechanism leading to Wallerian degeneration of axons after transection (George et al., 1995). In the present study, we found that pretreatment of axons with ω-conotoxin MVIIC, which blocks P/Q- and N-type VGCCs (Hillyard et al., 1992; McDonough et al., 1996), substantially decreased the influx of Ca2+ into stretch-injured axons. Therefore, one of the major routes of Ca2+ entry into axons after stretch injury appears to be through these conotoxin-sensitive Ca2+ channels.
Our current data also suggest that Ca2+influx into axons after trauma may partially occur by reversal of the Na+–Ca2+exchanger. Under normal circumstances, the Na+–Ca2+exchanger uses the high Na+ gradient to remove intracellular Ca2+. However, it has been suggested that high Na+ influx into axons may induce reversal of the Na+–Ca2+exchanger, resulting in a rise in intracellular Ca2+ concentration (Stys et al., 1992;Stys and Lopachin, 1998). In the present study we found that treatment of axons with the Na+–Ca2+exchanger blocker bepridil lowered the influx of Ca2+ after stretch injury. However, this decrease was relatively modest, suggesting that reversal of the Na+–Ca2+exchanger is not the dominant mechanism of Ca2+ entry shortly after traumatic deformation of axons. Bepridil has also been demonstrated previously to have an affinity for VGCCs, and because we show a severe reduction in Ca2+ influx with conotoxin treatment, we cannot exclude the possibility that it may be exerting its effects on VGCCs (Hosey and Lazdunski, 1988).
Potential mechanisms leading to altered intra-axonal Na+ concentration after trauma include (1) increased Na+ channel permeability, (2) a direct reduction of Na+–K+-ATPase activity, or (3) a cellular energy deficit that would secondarily result in Na+ pump failure. Direct mechanical inhibition of the Na+ pump or a rapid failure of energy supply might also result in the observed effect, although these mechanisms were not directly examined in this study. Studies on stretched cortical neurons suggest that failure of ATP supply is the first event (Tavalin et al., 1997), possibly related to mitochondrial failure (Ahmed et al., 2000), leading secondarily to pump inhibition. However, this effect took much longer to develop than did our rapid Ca2+ response.
Although we have identified a few potential sources of the increase in intracellular Ca2+ after traumatic axonal injury, our results do not support another widely accepted mechanism of Ca2+ entry, “mechanoporation.” This mechanism is postulated to involve the mechanical formation of transient membrane pores during axonal injury, acting as a primary factor in the Ca2+-induced pathophysiology in several models of brain trauma (Maxwell et al., 1993; Graham and Gennarelli, 1997; Gennarelli et al., 1998; Buki et al., 2000). However, in our in vitro model we have found previously that small molecules (∼500 Da) do not enter stretch-injured axons unless they undergo primary axotomy (Smith et al., 1999). Most important, in the present study Ca2+ influx was completely blocked by TTX or by the removal of Na+. Therefore, we found no evidence of Ca2+entry through membrane “pores” formed during trauma. It is also important to consider that Ca2+ entry in our model was not transient, as is the hypothesized mechanism of mechanoporation. We found an immediate and substantial increase in intra-axonal Ca2+ levels with the addition of Ca2+ back into the culture 1 min after injury was induced in a calcium-free medium.
Although we found no evidence of mechanoporation in the present study, physical tearing of the axonal membrane, i.e., primary axotomy, was induced in some stretch-injured axons. We observed a post-traumatic increase in intra-axonal Ca2+ at the ends of these disconnected axons that was greater than the Ca2+ changes in nondisconnected stretch-injured axons. In some animal models of brain trauma, permeability of injured axons to large molecules may be caused by primary axotomy or physical tearing of the axonal membrane across obstructions such as the foramen magnum (Pettus et al., 1994;Povlishock and Jenkins, 1995). However, it is important to note that immediate disconnection of axons during brain trauma is not thought to be the predominant form of traumatic axonal pathology. Rather, most axonal pathology has been proposed to develop over hours to even months after trauma (Erb and Povlishock, 1988).
Our current findings clearly indicate a Na+ channel-dependent Ca2+ entry into stretch-injured axons. Furthermore, these data strongly imply that axonal Na+ channels are sensitive to mechanical deformation, which may represent the watershed event leading to increased intra-axonal Ca2+ concentrations after traumatic axonal injury (Fig. 6). Voltage-gated Na+ channels are known to be sensitive to mechanical stress with irreversible changes in their kinetics (Shcherbatko et al., 1999; Tabarean et al., 1999). Interestingly, the β subunit of the Na+channel is required for normal rapidly inactivating kinetics, and abnormal β subunits have been shown to alter channel kinetics in favor of increased permeability (Wallace et al., 1998). During trauma, large mechanical forces may disturb the normal association of the Na+ channel subunits or modify the pore-forming α subunit directly.

Proposed mechanisms of calcium entry into stretch-injured axons. 1, Strain on the axonal membrane inducing an abnormal influx of Na+ through mechanosensitive sodium channels. 2, In response, reversal of axonal Na+–Ca2+exchangers (a) and activation of voltage-gated calcium channels (b), collectively contributing to a pathological influx of Ca+2 into the axons.
It has been shown recently that Na+channel blockers improve functional outcome and reduce axonal pathology in models of spinal cord crush injury (Teng and Wrathall, 1997;Rosenberg et al., 1999; Schwartz and Fehlings, 1999), suggesting, together with the present results, that this therapeutic strategy may also have application in treating diffuse axonal injury. The significant attenuation of Ca2+ influx by the VGCC blockers in our model suggests that blockade of VGCCs might also be considered as a potential therapeutic strategy for traumatic axonal injury.
In summary, these experiments are the first to demonstrate an influx of Ca2+ after stretch injury of axons in a vital system and, furthermore, to demonstrate the primary role of TTX-sensitive sodium channels in this process.
Footnotes
This work was funded in part by National Institutes of Health Grants AG12527, NS38104, NS08803 (D.H.S.), and NS35712 and by Centers for Disease Control Grant R49/CCR312712 (D.M.). We thank Alisa Plesco for her excellent technical assistance, Dr. C. E. Morris for her helpful discussions, and Jeanne Marks for her skillful preparation of this manuscript.
Correspondence should be addressed to Dr. Douglas H. Smith, Department of Neurosurgery, Room 105 Hayden Hall, 3320 Smith Walk, Philadelphia, PA 19104. E-mail: smithdou{at}mail.med.upenn.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}