


Abstract
Kinins, members of a family of peptides released from kininogens by the action of kallikreins, exhibit a variety of biological activities including vasodilation, increased vascular permeability, contraction of smooth muscle cells, and activation of sensory neurons. However, investigation of the physiological actions of kinins has been greatly hampered because its effects are curtailed by rapid proteolysis in blood, lung, and liver. We describe the pharmacological characteristics of a novel nonpeptide bradykinin receptor agonist FR190997 (8-[2,6-dichloro-3-[N-[(E)-4-(N-methylcarbamoyl)cinnamidoacetyl]-N-methylamino]benzyloxy]-2-methyl-4-(2-pyridylmethoxy)quinoline). FR190997 markedly stimulated phosphatidylinositol hydrolysis in Chinese hamster ovary cells permanently expressing the human bradykinin B2 receptor. The response of phosphatidylinositol hydrolysis was antagonized by the B2 receptor selective antagonist Hoe 140 (d-Arg-[hydroxyproline3,β-thienylalanine4,d-Tic7,Oic8]bradykinin). In competitive experiments using membranes prepared from Chinese hamster ovary cells expressing the human bradykinin receptor subtypes, FR190997 showed a high affinity binding to the B2 receptor with IC50 value of 5.3 nm and no binding affinity for the B1 receptor. In vivo, FR190997 mimics the biological action of bradykinin and induces hypotensive responses in rats with prolonged duration. Therefore, FR190997 is a highly potent and subtype-selective nonpeptide agonist which displays high intrinsic activity. This compound should represent a powerful tool for further investigation of the physiology and pathophysiology of bradykinin receptors.
Kinins are members of a family of peptides that have been implicated in a variety of biological activities including vasodilation, increased vascular permeability, contraction of smooth muscle cells, and activation of sensory neurons (1-3). Bradykinin and kallidin (Lys-bradykinin) are released from high- and low-molecular weight kininogens by the proteolytic action of kallikreins. Removal of the carboxyl terminus of these peptides by a carboxypeptidase generates des-Arg9-bradykinin and des-Arg10-kallidin, respectively (1, 2). The biological effects elicited by kinins are mediated through the activation of two bradykinin receptor subtypes, B1 and B2 (1, 3,4). The two bradykinin receptors have seven hydrophobic segments and share significant sequence similarity with other G protein-coupled receptors (5-7). The B2 receptor has higher affinity for bradykinin and kallidin and is prevalent in normal tissues, whereas the B1 receptor binds to the carboxyl-terminal des-Arg metabolites and is up-regulated by inflammation (1, 3-7).
The involvement of kinins in the pathology of human diseases has been suggested from studies with animal models and humans (1-3). However, investigation of the physiological actions of kinins has been greatly hampered because their effects are curtailed by rapid proteolysis in blood, lung and liver. Although we (8, 9) and Sawutz et al.(10) have reported nonpeptide compounds that behave as bradykinin receptor antagonists, nonpeptide mimics that activate bradykinin receptors and are resistant to proteolytic degradation have not yet been generated. In our previous experiments, stable expression of the human B2 receptor in CHO cells was used to demonstrate that, in response to bradykinin, the B2 receptor couples efficiently to PI hydrolysis and thus provides a useful system for the accurate characterization of ligand-receptor interactions (8). Transfection and functional expression of cDNA clones for single receptor subtypes in the same cell type also allow the quantitative measurement of the potency, subtype selectivity, and efficacy of the potential agonist, because uncertainties arising from the presence of multiple receptor subtypes and from the nonspecific activation of signaling pathways can be readily eliminated. We report here the pharmacological characteristics of a novel nonpeptide bradykinin receptor agonist FR190997 (8-[2,6-dichloro-3-[N-[(E)-4-(N-methylcarbamoyl)cinnamidoacetyl]-N-methylamino]benzyloxy]-2-methyl-4-(2-pyridylmethoxy)quinoline) that was selected from the Fujisawa chemical file for the ability to mediate PI hydrolysis in CHO cells expressing the human B2receptor. We investigated the potency, selectivity, and efficacy of FR190997 for the human bradykinin receptors in transfected CHO cells. In addition, we examined the in vivo effect of this compound on blood pressure of anesthetized rats.
Experimental Procedures
Materials.
Materials were obtained from the following sources: α-minimal essential medium lacking ribonucleosides and deoxyribonucleosides from Flow Laboratories (Irvine, Scotland); Dulbecco’s modified Eagle’s medium from Nissui (Tokyo, Japan); dialyzed fetal bovine serum from Sigma Chemical (St. Louis, MO); kallidin, [des-Arg10], [3,4-prolyl-3, 4-3H]-([3H] des-Arg10-kallidin) and bradykinin, [2,3-prolyl-3,4-3H]- ([3H]bradykinin)from Dupont-New England Nuclear (Boston, MA); myo-[2-3H]inositol (18.8 Ci/mmol) from Amersham. (Arlington Heights, IL); bradykinin from Peptide Institute (Osaka, Japan); des-Arg10-kallidin from Peninsula Laboratories (Belmont, CA). FR190997 and Hoe140 (11) were prepared by Fujisawa Pharmaceutical (Osaka, Japan).
Cell culture.
CHO (dhfr−) cells that were transfected with and stably expressed the human B1 and B2 receptors have been described previously (8). Cells were maintained in a α-minimal essential medium lacking ribonucleosides and deoxyribonucleosides and supplemented with 10% dialyzed fetal bovine serum.
Measurements of PI hydrolysis.
PI hydrolysis was measured essentially as described previously (8). CHO cells expressing the human B2 receptor were seeded in 12-well plates at a density of 1 × 10 5 cells/well and cultured for 1 day. The cells were labeled with [3H]inositol (1 μCi/ml) for 24 hr. The cells were washed twice with PBS containing 0.2% BSA and incubated with the same solution for 30 min and then with PBS containing 0.2% BSA and 10 mm LiCl for 30 min at 37°. Agonist stimulation was started by replacing the medium with fresh PBS containing 0.2% BSA, 10 mm LiCl, and test reagents. The reaction was terminated by 5% (w/v) trichloroacetic acid after incubation for 30 min at 37°. Separation of [3H]inositol phosphates was carried out by Bio-Rad AG 1-X8 chromatography essentially as described elsewhere (12). A mixture of 3H-labeled IP1, IP2, and IP3 was eluted from the column with 0.1 m formic acid/1.0 m ammonium formate. The radioactivity in the eluates was determined by a liquid scintillation spectrometer.
Ligand binding of bradykinin receptors.
Cell membranes (12.5–75 μg/ml), prepared from CHO cells permanently expressing the bradykinin receptor subtypes, were incubated with 500 pm of [3H]bradykinin (for the B2 receptor) or [3H]des-Arg10-kallidin (for the B1 receptor) as described previously (8). All experiments were carried out at least three times in duplicate. The specific binding was calculated by subtracting the nonspecific binding, determined in the presence of 1 μm unlabeled bradykinin (for the B2 receptor) or des-Arg10-kallidin (for the B1 receptor), from the total binding. The specific binding activity amounted to 90–92% of the total binding activity.
Measurements of rat blood pressure.
Male Wistar rats were anesthetized with an intraperitoneal administration of sodium pentobarbital (40 mg/kg) and sodium phenobarbital (160 mg/kg). The left femoral artery and vein of each animal were cannulated, and systemic blood pressure was measured directly in the left femoral artery by use of a pressure transducer, connected to a multichannel recorder. Bradykinin or FR190997 was administered as a bolus injection into the left femoral vein through the cannula, in increasing doses of 1, 10, and 100 μg/kg at intervals of 30 min in each animal. Hoe 140 (d-Arg-[hydroxyproline3, β-thienylalanine4, d-Tic7, Oic8]bradykinin) (100 μg/kg) was given at 10 min before the first injection of the agonists.
Results and Discussion
CHO cells permanently expressing the human B2bradykinin receptor show a marked stimulation of PI hydrolysis in response to bradykinin interaction (8), allowing the quantitative measurement of the potency of potential agonists for the cloned receptor. Accordingly, we examined the ability of FR190997 (Fig.1), a quinoline derivative, to mediate PI hydrolysis in clonal CHO cells expressing the human B2 receptor. Interestingly, we found that FR190997, although structurally similar to previously reported bradykinin receptor antagonists FR167344 and FR173657 (Fig. 1) (8), stimulated PI hydrolysis, suggesting that this compound acts as a nonpeptide agonist. PI hydrolysis was measured by incubating cells with different concentrations of bradykinin or FR190997 for 30 min and monitoring the maximal formation of total inositol phosphates (IP1, IP2, and IP3) as described previously (8) (Fig. 2A). A significant increase in inositol phosphate formation was obtained at 1 nm FR190997, and maximal stimulation was achieved at 1 μm or higher. Exposure to 1 μm FR190997 enhanced inositol phosphate formation 8.7-fold over basal and was 67% of the maximal effect elicited by bradykinin. To eliminate the possibility that the FR190997-induced stimulation of PI hydrolysis was due to nonspecific activation of signaling pathways rather than activation of the B2 receptor, we first examined the stimulatory effect of FR190997 on inositol phosphate formation in CHO cells permanently expressing the endothelin ETB receptor (13). Although ET-1 caused a marked increase in the formation of inositol phosphates as reported previously (13), FR190997 showed no stimulatory effect on PI hydrolysis in ETBreceptor-expressing cells (data not shown). Second, we examined the effect of Hoe 140 (11), a B2 receptor selective antagonist, on FR190997-induced inositol phosphate formation. B2receptor-expressing cells were preincubated in the absence and presence of 100 nm Hoe 140 for 30 min. The amount of total inositol phosphates was determined after the application of either 100 nm bradykinin (Fig. 2B) or FR190997 (Fig. 2C). Under these conditions, Hoe 140 inhibited the bradykinin-stimulated and FR190997-stimulated PI hydrolysis by 60 and 63%, respectively, indicating that the effect of FR190997 on PI hydrolysis is mediated by the B2 bradykinin receptor.
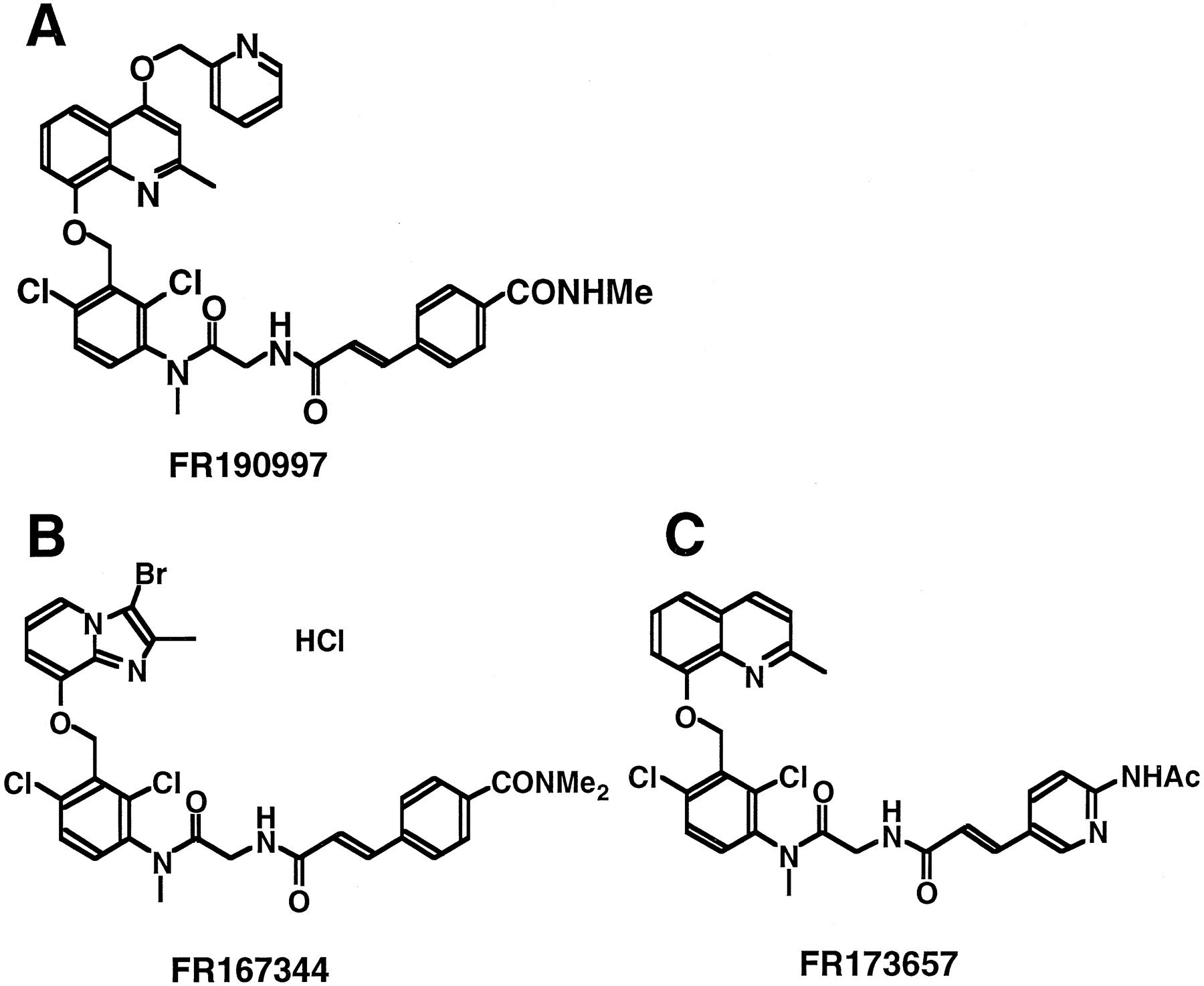
Structures of nonpeptide agonist FR190997 (A) and antagonists FR167344 (B) and FR173657 (C).

A, FR190997-induced inositol phosphate formation in CHO cells expressing the human B2 receptor. The amount of total inositol phosphates (IP1, IP2, and IP3) was determined after activation of the human B2 receptor with various concentrations of bradykinin (○) and FR190997 (•). Total inositol phosphate formation is expressed as the fold increase in inositol phosphate levels, compared with cells not treated with ligands. The data indicated were taken from a representative experiment and the values are means ± standard deviation of triplicate determinations. The radioactivity incorporated into total inositol phosphates in control cells was 908 ± 35 cpm. B and C, Effect of the bradykinin antagonist, Hoe 140, on bradykinin (BK)-induced (B) or FR190997-induced (C) inositol phosphate formation in CHO cells expressing the human B2receptor. The values are means ± standard deviation of triplicate determinations. ∗∗, p < 0.01 compared with incubation with bradykinin or FR190997 alone.
To assess the binding affinity of FR190997, we determined the potency of FR190997 for the inhibition of specific [3H]bradykinin binding to membranes prepared from CHO cells expressing the human B2 receptor. Competition curves of [3H]bradykinin binding by bradykinin and FR190997 are presented in Fig. 3A. FR190997 potently competed with the specific binding of [3H]bradykinin to the human B2 receptor. IC50 of FR190997 to inhibit specific [3H]bradykinin binding to the human B2 receptor was 5.3 ± 0.88 nm. The IC50 value obtained for FR190997 was comparable to that obtained for bradykinin (1.1 ± 0.17 nm) and was consistent with its potency to stimulate PI hydrolysis. In contrast, FR190997 did not inhibit [3H]des-Arg10-kallidin binding to the human B1 receptor expressed in CHO cells (8) (Fig. 3B). These results indicate that FR190997 selectively binds to the human B2 receptor, and this interaction leads to stimulation of inositol phosphate formation.

Competition for specific radioligand binding to the cloned bradykinin receptors, B2 (A) and B1 (B) receptors. The unlabeled ligands added in the binding assays are bradykinin (○), des-Arg10-kallidin (□) and FR190997 (•).
Next, we examined the in vivo effect of FR190997 on blood pressure of anesthetized rats. Intravenous injection of bradykinin (10 μg/kg) evoked an immediate fall in blood pressure (Fig.4A), which rapidly returned to preinjection levels after 1.9 ± 0.4 min. The immediate drop in blood pressure was followed by hypertension, most likely due to a compensatory adrenergic response as reported previously (14, 15). Likewise, intravenous injection of FR190997 (10 μg/kg) also caused a fall in blood pressure. However, the duration of the hypotensive response was significantly longer than the response to bradykinin (12.4 ± 3.8 min). The hypotensive responses to bradykinin and FR190997 were dose-dependent and were significantly inhibited by prior administration of Hoe 140 (100 μg/kg) (Fig. 4, B and C) (16), indicating that these hypotensive effects are mediated by the B2 receptor. These results demonstrate that the nonpeptide FR190997 functions as a potent agonist of the B2 receptor and mimics the biological activity of bradykinin in vivo. The most interesting feature distinguishing this nonpeptide compound from the endogenous peptide bradykinin is its markedly prolonged duration of action. This property of FR190997 is presumably a consequence of its resistance to proteolytic degradation.

In vivo effect of FR190997. A, Decrease in mean arterial blood pressure in anesthetized rats elicited by intravenous injections of bradykinin (10 μg/kg) (○) and FR190997 (10 μg/kg) (•). The values are changes in mean arterial pressure (percent decrease) expressed as mean ± standard error (n = 6–8 rats). B and C, Decrease in mean arterial pressure in anesthetized rats by intravenous injections of bradykinin (B) or FR190997 (C) pretreated with (•) or without (○) Hoe 140. ∗∗, p < 0.01 compared with the effect of bradykinin or FR190997 alone. The values depicted represent maximal changes in mean arterial pressure (percent decrease) expressed as mean ± standard error (n = 6–8 rats).
In the two-state model of receptor activation, G protein-coupled receptors are thought to be in an equilibrium between an inactive (R) and an active (R*) conformation (17-19). The binding of agonist to receptor stabilizes and shifts the equilibrium toward the active conformation (R*) resulting in productive receptor-G protein coupling. The present study demonstrates that a nonpeptide compound, whose structure is quite different from the natural peptide ligand, functions as a potent and subtype-selective agonist of a peptide-activated G protein-coupled receptor. In contrast to the many examples of nonpeptide antagonists that have been previously developed (20), only very few nonpeptide agonists have been reported so far. A few examples include; nonpeptide opioid receptor agonists (21), the recently characterized nonselective angiotensin II receptor partial agonist (22), and the submicromolar affinity benzodiazepine derivative cholecystokinin receptor agonists (23). The molecular mechanisms underlying the interaction of FR190997 with the human B2 receptor remains to be elucidated. However, FR190997 exhibits the unique properties that it not only activates a G protein-coupled receptor, but also shows high potency, subtype selectivity, and high efficacy. FR190997 is structurally similar to the nonpeptide antagonists that we have reported recently. Similar to our observation, Perlman et al. (24) have presented that nonpeptide ligands that differ by only a single methyl group have agonistic and antagonistic properties in angiotensin AT1receptor. This aspect should help in elucidating agonist and antagonist determinants in the B2 receptor.
In addition to being the first potent and subtype-selective nonpeptide agonist with high intrinsic activity at the human B2receptor, FR190997 demonstrated the anticipated utility of nonpeptide compounds as potential efficacious pharmacological agents by its prolonged duration of action in vivo. FR190997 should facilitate the study and elucidation of the physiological paradigms associated with the B2 receptor and help in evaluating the therapeutic potential of selective B2 receptor agonists. Studies of animal models and humans have suggested a role for kinins in several pathological conditions such as hypertension (25), renal failure (26), myocardiac ischemia (27), and male infertility (28). Kinins also have a potential beneficial value in increasing the delivery of coadministered drugs beyond the blood-brain barrier (29). The nonpeptide bradykinin receptor agonist FR190997 should represent a powerful tool for further investigation of the physiology and pathophysiology of bradykinin receptors. Further studies will be required to assess its potential as a therapeutic agent for the management or treatment of certain disease conditions.
Acknowledgments
We thank Professor Marc G. Caron and Drs. Stephen S. G. Ferguson and Kenneth J. Valenzano for reading the manuscript.
Footnotes
- Received February 25, 1997.
- Accepted March 24, 1997.
-
Send reprint requests to: Ichiro Aramori, Ph.D., Molecular Biological Research Laboratory, Fujisawa Pharmaceutical Co., Ltd., 5-2-3 Tokodai, Tsukuba 300-26, Japan. E-mail: ichiro_aramori{at}rnd.fujisawa.co.jp
Abbreviations
- CHO
- Chinese hamster ovary
- PI
- phosphatidylinositol
- IP1
- inositol monophosphate
- IP2
- inositol bisphosphate
- IP3
- inositol trisphosphate
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- BSA
- bovine serum albumin
- TES
- trimethylaminoethanesulfonic acid
- PBS
- phosphate-buffered saline
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}