


Abstract
We studied the mechanism of action of methylene blue (Mblue), a putative guanylyl cyclase inhibitor, on the L-type calcium current (ICa) and the muscarinic activated K+ current (IK,ACh) in rat ventricular and atrial myocytes, respectively, and on the binding of [3H]quinuclidinyl benzylate in rat ventricular membranes. Superfusion, but not internal dialysis, with 30 μm Mblue antagonized the inhibitory effect of acetylcholine (ACh, 1 μm) on β-adrenergic stimulation of ICa with isoprenaline (Iso, 10 nm or 1 μm). However, Mblue had no effect on the basal ICa or on the stimulation of ICa by Iso in the absence of ACh. The activation of IK,ACh by 3 μm ACh was also antagonized by Mblue in a dose-dependent manner. In contrast, Mblue had no effect on the activation of IK,ACh by either guanosine-5′-O-(3-thio)triphosphate or guanosine-5′-(β,γ-imido)triphosphate. Chlorpromazine (CPZ), a piperazine derivative like Mblue, also inhibited the muscarinic activation of IK,ACh in a dose-dependent manner. The specific binding of [3H]QNB, a muscarinic ligand, to rat ventricular membranes was displaced in a dose-dependent manner by Mblue and CPZ. The piperazine derivatives behaved like competitive antagonists of [3H]QNB binding, exhibiting equilibrium dissociation constant (Ki ) values of 187 nm for Mblue and 366 nm for CPZ. In conclusion, Mblue exerts antimuscarinic effects on ICaand IK,ACh in rat cardiac myocytes that are best explained by the binding of Mblue to the M2 subtype of muscarinic receptors. This property probably contributes to the antimuscarinic effect of the putative guanylyl cyclase inhibitor reported in previous studies.
Mblue has been shown to antagonize the production of cGMP in various tissues and is generally accepted as a selective antagonist of the NO-sensitive isoform of guanylyl cyclase (1-5). As such, Mblue is often used to study the roles of NO and cGMP in the effects of neurotransmitters and hormones. For instance, Mblue antagonizes the effect of ACh on the relaxation of coronary vessels (6, 7).
In the heart, Mblue was soon recognized as an inhibitor of the response to ACh (Ref. 8 and references therein). Because ACh, like NO donors, can increase cGMP levels in cardiac myocytes (for reviews, see Refs. 9and 10), a likely mechanism for the cardiac effect of Mblue was the inhibition of guanylyl cyclase. Consistent with this hypothesis are the recent findings that Mblue (i) inhibits cGMP production induced by ACh in isolated guinea pig cardiomyocytes (11), (ii) prevents the inhibitory effect of ACh on the L-type Ca2+channel current (ICa) (12-15), (iii) prevents the inhibitory effect of ACh on the cAMP-stimulated Cl− current (16), and (iv) antagonizes the effects of NO donors on cardiac contractility (17, 18) and ICa (12, 14, 19). However, other cardiac effects of Mblue cannot be easily explained by its inhibitory effect on guanylyl cyclase. For instance, Mblue abolishes the negative chronotropic effect of ACh in rat cardiomyocytes (20), a phenomenon that is clearly unrelated to cGMP accumulation. Moreover, even in the absence of ACh, Mblue modifies the β-adrenergic stimulation of ICa (inhibition in frog myocytes: Refs. 19 and21), of the Cl− current (potentiation in guinea pig myocytes: Ref. 16), and of cardiac inotropism (potentiation in rat myocytes: Ref. 20).
Previous studies have questioned the mechanism of action of Mblue and suggested that Mblue is not a direct guanylyl cyclase inhibitor. Indeed, Mblue is a superoxide anion generator, and this anion, which is a potent NO scavenger, may account for the lowering effect of Mblue on cGMP levels in several preparations (22–24). However, Mblue has antimuscarinic properties even in the presence of SOD, which eliminates the superoxide anion (6). This SOD-resistant effect is likely to involve the core of the Mblue molecule. Like the neuroleptic compound CPZ, Mblue is a piperazine derivative, and both CPZ and Mblue inhibit NOS activity (25, 26). This inhibition of NOS, rather than guanylyl cyclase inhibition, may explain the lowering effect of Mblue on cGMP levels (25).
CPZ is not only an antagonist of dopaminergic receptors, but it was also shown to inhibit Na+, K+, and Cl− channel activity (Refs. 27 and 28 and references therein) in isolated cardiac myocytes. Due to its structural analogy with CPZ, there is the possibility that Mblue also binds to receptors and channels. Interestingly, it was recently reported that Mblue can inhibit the muscarinic activation of a potassium current (IK,ACh) that is not regulated by the NO/cGMP pathway (14, 15, 21). This side effect of Mblue was observed during external superfusion of the myocytes and not during internal dialysis of the cells (14). This finding led Han et al. (14) to hypothesize that Mblue can act as a muscarinic antagonist or interact with the coupling of the G protein with its effector. However, a direct action of Mblue on the channel itself cannot be ruled out. For instance, on illumination, Mblue was shown to modify functional histidine residues on neuronal Ca2+ channels (29).
To get further insights into the mechanism of action of Mblue in cardiac myocytes, we investigated in the current study the effects of Mblue in isolated myocytes and membrane preparations of adult rat hearts. More specifically, we examined the effects of Mblue on the muscarinic regulation of ICa and IK,ACh in rat ventricular and atrial myocytes using the whole-cell, patch-clamp technique. Moreover, we investigated the effect of Mblue on the binding of [3H]QNB to muscarinic receptors in rat ventricular membranes. Altogether, our data demonstrate that Mblue acts as a M2 receptor antagonist in cardiac myocytes. A preliminary account of some of these results has appeared in abstract form (30).
Experimental Procedures
The investigation conforms with the European Community guiding principles in the care and use of animals3 and the French decree 87/748 of October 19, 1987.4 Authorizations to perform animal experiments according to this decree were obtained from the French Ministère de l’Agriculture et de la Forêt.5
Electrophysiology.
Male Wistar rats (180–250 g) were anesthetized by intraperitoneal injection of urethane (2 g/kg) and heparin (2.5 mg/kg). Ventricular cardiomyocytes were dispersed using collagenase A (0.255 mg/ml; Boehringer-Mannheim Biochemica, Mannheim, Germany) as previously described (31). At the end of the retrograde perfusion, atria and ventricles were separated, and the isolated ventricular myocytes were resuspended, step by step, in a 1 mm Ca2+-containing solution. The atria were transferred, minced, and slowly agitated for 15–30 min at 37° in a nominally Ca2+-free solution containing 0.5 mg/ml protease (Sigma Chemical, St. Louis, MO), 0.255 mg/ml collagenase A (Boehringer-Mannheim), and 1 mg/ml bovine serum albumin. Isolated atrial myocytes were then collected and resuspended in 1 mm Ca2+-containing solution. Ventricular cardiomyocytes (stored at 37°) and atrial myocytes (kept at 4°) were used within 1–12 hr after isolation.
The whole-cell configuration of the patch-clamp technique was used to record L-type ICa and K+currents on Ca2+-tolerant cells. For ICa measurements in ventricular myocytes, a routine protocol consisted in a pulse at −50 mV (50-msec duration) followed by a pulse at 0 mV (400-msec duration) elicited every 8 sec. The holding potential was adjusted (to −60 or −50 mV) to achieve the inhibition of the fast Na+ current in the presence of 0.6 μm tetrodotoxin. When this combination failed to fully abolish the Na+ current, the tetrodotoxin concentration was raised to 32 μm (31). K+ currents were blocked by replacing K+ ions with intracellular and extracellular Cs+. Under these conditions, the time-dependent current measured during a depolarization to 0 mV was attributed to the L-type calcium channel (31). For IK,AChmeasurements, 200-msec pulses to 0 and −120 mV were separately and successively applied every 5 sec from a holding potential of −50 mV (32). The experiments were performed at room temperature (19–34°), and in a given experiment, the temperature did not change by >2°.
Solutions for patch-clamp recordings.
The external Cs+-containing solution contained 107 mm NaCl, 10 mm HEPES, 20 mm CsCl, 4 mm NaHCO3, 0.8 mmNaH2PO4, 1.8 mmMgCl2, 1.8 mmCaCl2, 5 mm d-glucose, 5 mm sodium pyruvate, and 6 × 10−7 tetrodotoxin, pH 7.4 adjusted with CsOH. In the external K+-containing solution, CsCl was substituted for 2.5 mm KCl and pH adjusted with NaOH. Control or drug-containing solutions were applied to the exterior of the cell by placing the cell at the opening of 250-μm i.d. capillary tubings flowing at a rate of ∼10 μl/min (33).
The patch pipettes (0.5–1.0 MΩ for ventricular cells and 1–2.5 MΩ for atrial cells) used to record ICa were filled with an internal Cs+-containing solution of 119.8 mm CsCl, 5 mm EGTA (acid form), 4 mm MgCl2, 5 mmNa2-phosphocreatine, 3.1 mmNa2ATP, 0.42 mmNa2GTP, 0.062 mmCaCl2 (pCa 8.5), and 10 mm HEPES, pH 7.3 adjusted with CsOH. In the internal K+-containing solution, CsCl was substituted for 102 mm KCl and pH adjusted with KOH. The internal GTPγS- or Gpp(NH)p-containing mediums were obtained by substituting GTP for GTPγS or Gpp(NH)p, respectively. Another internal Cs+-containing solution was used to study ICa and contained 90 mm aspartic acid, 30 mm CsCl, 5 mm HEPES, 10 mmEGTA, 1 mm CaCl2, 3 mmMgATP, and 3 mm Na2-phosphocreatine, pH 7.2 adjusted with CsOH (13). The results obtained with both internal Cs+-containing media were identical; therefore, all results were pooled. Intracellular solution changes were performed as previously described (33).
Data analysis.
During patch-clamp experiments, the maximal amplitude of whole-cell ICa was measured as previously described (33). IK,ACh was measured as the change of the steady state current at the end of the pulse at 0 mV after the application of ACh (32). Currents were not compensated for capacitive and leak currents. On-line analysis of the recordings was made possible by programming a PC-compatible 486/50 in Assembly language (Borland, Buffalo, NY) to determine, for each membrane depolarization, peak and steady state current values (33).
Results are expressed as mean ± standard error. Differences between mean values were tested for statistical significance by Student’s t test. In the text, the “basal” condition for ICa refers to the absence of Iso (33).
Drugs.
Tetrodotoxin was from Latoxan (Rosans, France). All other drugs were from Sigma Chemical (St Louis, MO). All solutions were prepared by dilution to the desired concentration in the physiological solution at the beginning of each experiment.
Membrane preparation.
Rats (340–400 g) were killed after chloroform anesthesia by decapitation and bleeding through the carotid arteries. The hearts were rapidly removed, washed in ice-cold 0.9% NaCl, and freed of the pericardium, atria, and great vessels. The ventricles were minced with scissors and homogenized in eight volumes of ice-cold homogenization buffer (50 mm Tris, 5 mm MgCl2, 5 mm EDTA, 1 mm EGTA, aprotinin 2 μg/ml, pH adjusted to 7.5) with an Ultraturrax homogenizer three times for 15 sec at maximal speed. The homogenate was filtered through 200-μm gauze and centrifuged for 30 min at 4° and 500 × g. The supernatant was adjusted to 107 mm KCl and 20 mm MOPS, pH 7.4; mixed; incubated for 10 min on ice; and centrifuged for 60 min at 160,000 × g at 4°. The pellet was resuspended in 160 mm KCl/20 mm Tris, pH 7.4, with a short burst of an Ultraturrax at medium speed and respun at 160,000 ×g for 45 min at 4°. The final pellet was resuspended in homogenization buffer and stored in aliquots at −80°. Protein concentration was determined according to Bradford (34) using bovine serum albumin as a standard. The protein yield was 3.2–3.7 μg/mg of wet weight.
Radioligand binding.
The density of muscarinic receptors in rat ventricular membranes was determined by saturation binding experiments with [3H]QNB (43.5 Ci/mmol; DuPont-New England Nuclear (Boston, MA) at room temperature (22–28°) using 40–50 μg of membrane protein in an assay buffer of 20 mm Tris, 100 mm NaCl, and 0.5 mmEDTA, pH 7.4, and in a total volume of 250 μl. The reaction was terminated by rapid filtration through glass-fiber filters (MAFB NOB; Millipore, Bedford, MA). The radioactivity bound to the filters was determined by scintillation counting after an overnight incubation. Association/dissociation kinetics were tested over 120 min of association and 120 min of dissociation after the addition of 1 μm atropine at 90 min. Under these conditions, binding reached an equilibrium after 60 min and remained stable between 60 and 120 min and was almost nondisplaceable (not shown). All further reactions were performed after a 90-min incubation in triplicate. Nonspecific binding was defined as bound radioligand in the presence of 1 μm atropine and was subtracted from the total binding to calculate specific binding; it amounted to <2% of total binding at the KD (see Fig. 5A).

Saturation isotherms of [3H]QNB binding to washed membranes from rat cardiac ventricles. A, [3H]QNB binding under control conditions. ▵, Total binding; ⋄, nonspecific binding in the presence of 1 μmatropine; □, specific binding. B, Scatchard plot of the data depicted in A. C, Specific [3H]QNB binding in the absence (□) and presence (▪) of 1 μm Mblue. Data are mean values taken from a representative experiment carried out in triplicate and are typical for three independent experiments.
To test whether Mblue competes with [3H]QNB for binding to rat ventricular membranes, two sets of experiments were performed. First, saturation experiments with [3H]QNB were repeated in the absence and presence of 1 μm Mblue. Second, binding of a fixed concentration of [3H]QNB (200–500 pm) was displaced by different muscarinic antagonists over a wide range of concentrations. All reactions were performed at least three times in duplicate or triplicate. The computer program GraphPAD (GraphPAD Software, San Diego, CA) was used to fit displacement curves and calculate dissociation constants.
The same experiments were performed under identical conditions with the hydrophilic nonspecific β1/β2 antagonist CGP 12177 [(+)-[3H]-4-(3-t-butylamino-2-hydroxy-propoxy)benzimidazol-2-one, 45.4 Ci/mmol; Dupont-New England Nuclear] to test whether the effect of Mblue also applied to binding to other G protein-coupled receptors.
Results
Mblue has been shown to antagonize the muscarinic regulation of ICa in the presence of the β-adrenergic agonist Iso in isolated ventricular myocytes (13). The mechanism of action of Mblue on ICa was first studied under the same conditions in rat ventricular myocytes, perfused and dialyzed with Cs+-containing solutions. In a typical experiment (Fig. 1A), superfusion with 1 μm Iso elicited a 2-fold increase in ICa over its basal amplitude. In the continuing presence of the β-adrenergic agonist, the addition of 1 μm ACh reduced the stimulatory effect of Iso on ICa by ∼60%. External perfusion with Mblue (30 μm) abolished this antiadrenergic effect of ACh on ICa in a reversible manner. Mblue had no apparent effect on the kinetics of ICa (as shown by the individual traces on top of Fig. 1A) and did not modify the shape of the macroscopic current-voltage relationship (data not shown). In several similar experiments, ICa was enhanced ∼2-fold by Iso (0.01–1 μm), to 196.6 ± 9.7% of the initial basal level (16 experiments). On average, 1 μm ACh inhibited ∼20% of the Iso-stimulated ICa (Fig. 1B, left) (i.e., it reduced the Iso response by ∼40%). Superfusion with 30 μmMblue antagonized the inhibitory effect of ACh on the Iso-stimulated ICa. When Mblue (30 μm) was added to the cell before the application of ACh, it had no significant effect on Iso (10 nm)-stimulated ICa (Fig.1B, left). However, the presence of Mblue completely abolished the inhibitory effect of a subsequent application of 1 μm ACh on ICa (data included in the summary results of Fig. 1B, left). In these experiments, the effect of Mblue seemed to be selective for the muscarinic inhibition of ICa. This hypothesis was strengthened by the following results. First, in four experiments in which a low concentration (1 nm) of Iso had enhanced ICa to 142.0 ± 12.6% over basal level, external Mblue (30 μm) had no effect (8.3 ± 2.4% change over Iso level). Second, external perfusion with 30 μm Mblue did not modify the basal ICa (5.7 ± 2.8% change over basal, eight experiments).

External Mblue antagonizes the muscarinic inhibition of ICa. A, A rat ventricular cardiomyocyte was first superfused with control external Cs+ solution and internally dialyzed with intracellular Cs+ solution. □, the maximal peak amplitude of ICa obtained by depolarizing the cell every 8 sec to 0 mV over a period of 400 msec from a holding potential of −60 mV. During the periods indicated (horizontal lines), the cell was successively exposed to Iso (1 μm), Iso plus ACh (1 μm), and Iso plus ACh plus Mblue (30 μm). Top current traces, recorded at the times indicated (corresponding letters on the main graph). B, Summary of the results of several similar experiments like in A with 10 nm or 1 μm Iso. Left, Mblue (30 μm) was added to the external solution either before or after application of ACh (1 μm). Right, Mblue (30 μm) was dialyzed into the cell. The effects of ACh and Mblue were normalized to the amplitude of the Iso-stimulated ICa(100%, dotted line). Continuous line, mean basal ICa level (i.e., in the absence of Iso). Bars, mean values. Line, mean ± standard error of the number of experiments indicated near the bars. Significant statistical differences from the Iso (#) and the Iso-plus-ACh levels (*) are indicated as *#, p < 0.01; ##, p < 0.005.
The sidedness of action of Mblue was investigated by means of an intracellular perfusion system (see Experimental Procedures). In five cells in which 10 nm Iso enhanced ICato 280.2 ± 24.5% of the basal level, internal dialysis with 30 μm Mblue had no significant effect on the current (Fig.1B, right). In similar experiments, the inhibition of the β-adrenergic stimulated ICa by 1 μm ACh was neither antagonized nor prevented by internal dialysis with 30 μm Mblue (Fig. 1B, right). In four other experiments, a higher concentration of ACh (10 μm) inhibited 27.3 ± 7.5% of the Iso-stimulated ICa, and this effect was unchanged in the presence of 30 μm internal Mblue (−33.2 ± 7.67% of the Iso-stimulated ICa). In these cells, Iso (0.01 and 1 μm) had enhanced ICa to 238.4 ± 34.3% of the basal level. The lack of effect of internal Mblue is in contrast with a previous study in rat ventricular myocytes (see Discussion) (13). We took advantage of these results to characterize the mechanism of action of external Mblue.
The fact that Mblue had antimuscarinic properties when applied extracellularly but not intracellularly could be the result of an activation of acetylcholinesterases, resulting in an enhanced hydrolysis of ACh. This hypothesis was tested by using CCh, a hydrolysis-resistant agonist of muscarinic receptors. In a series of 13 experiments in which Iso (10 nm to 1 μm) enhanced ICa to 233.1 ± 12.8% of the basal level, the addition of 1 μm CCh lowered the Iso-stimulated ICa by 24.7 ± 2.8% (i.e., the Iso response was inhibited by 44.1 ± 4.8%, 10 experiments). The addition of Mblue (30 μm) in the external solution, before or after the application of CCh, prevented or antagonized the inhibitory effect of CCh, to 101.9 ± 2.2% (seven experiments) of the Iso-stimulated ICa. In these experiments, we also tested whether an oxidation of CCh by Mblue could explain its antimuscarinic effect. When added together with the reducing agent DTT (1 mm), Mblue (30 μm) still fully antagonized the inhibitory effect of CCh (1 μm) to 99.2 ± 5.5% of the Iso-stimulated ICa (four experiments). Thus, the antimuscarinic effect of Mblue on ICawas not the consequence of a stimulation of acetylcholinesterase activity or a direct oxidation of the muscarinic agonists.
External Mblue was shown to block the muscarinic activation of the background K+current, IK,ACh, in rabbit nodal myocytes (14). In subsequent experiments, we studied the sensitivity of IK,ACh to Mblue in the rat atrial myocytes in which the density of this current allows a quantitative analysis. K+ currents were recorded in K+-containing solutions at three membrane potentials (−120, −50, and 0 mV), and the changes of the end-pulse current at 0 mV were kept for subsequent statistical analysis. The muscarinic antagonist atropine (1 μm) had no effect on the K+ currents (96.2 ± 4.2% of basal, seven experiments), demonstrating that IK,ACh was not significantly active under basal conditions. In the typical experiment of Fig. 2A, the application of 3 μm ACh increased the steady state current at all three potentials in the outward direction (at −50 and 0 mV) or the inward direction (at −120 mV). When Mblue (10 μm) was added together with ACh, it totally antagonized the muscarinic activation of IK,ACh. However, IK,ACh was quickly recovered on washout of Mblue. The effect of Mblue was similar at all three voltages with the routine protocol and was found to be independent of membrane potential in current-voltage relationships (data not shown). On average, 3 μm ACh enhanced the end-pulse current at 0 mV to 308.4 ± 32.0% of the control amplitude (24 experiments). This stimulation was reduced dose-dependently by Mblue in the micromolar range of concentrations (Fig. 2B), and the current in the presence of 3 μm ACh plus 30 μm Mblue was reduced to basal level. This complete inhibition of IK,ACh by Mblue was selective for the muscarinic-activated current because Mblue (30 μm) did not change the basal K+currents in the absence of ACh (94.4 ± 5.9% of basal, seven experiments).
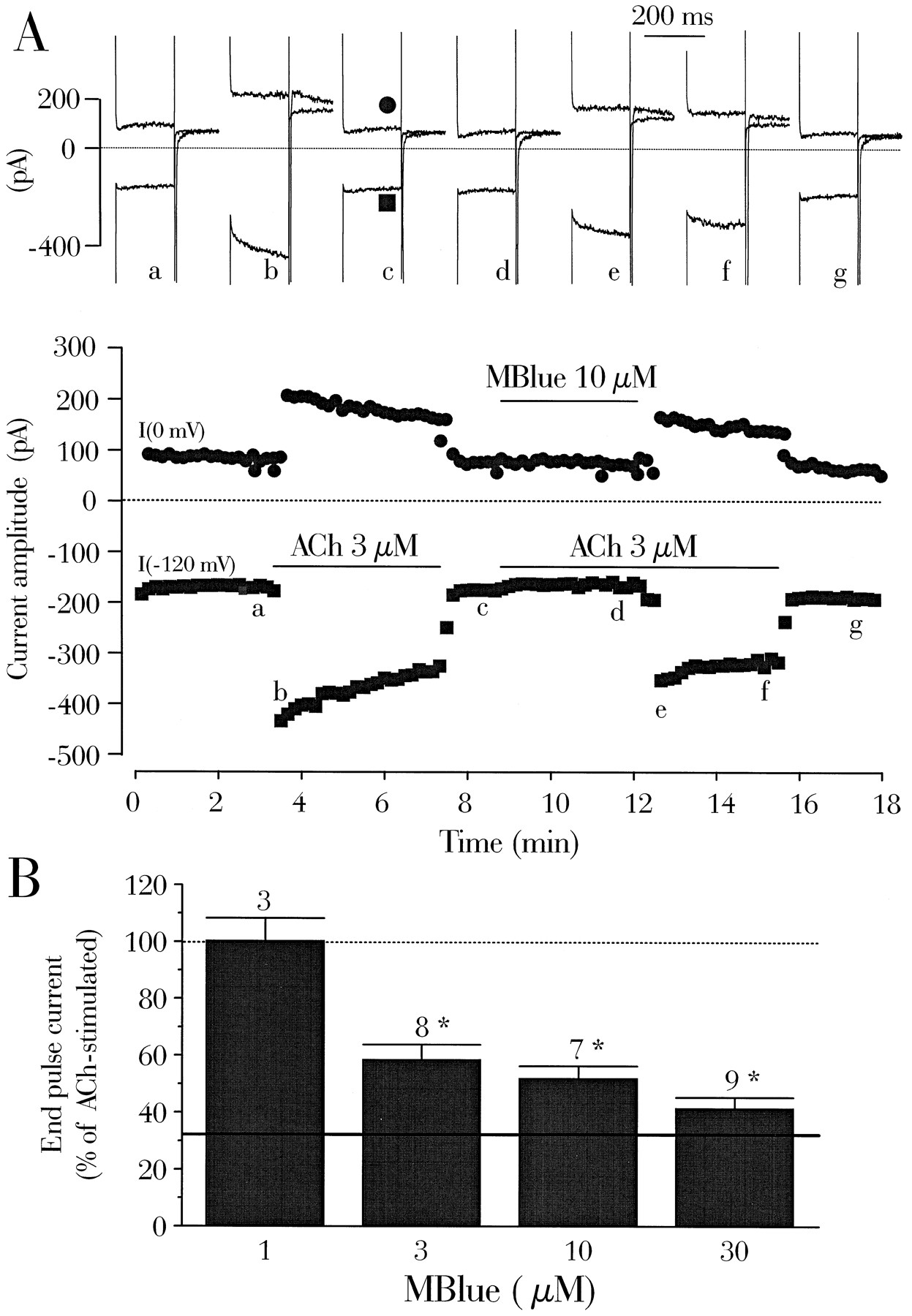
Mblue antagonizes the muscarinic activation of IK,ACh. A, A rat atrial cardiomyocyte was first exposed to external and internal K+-containing solutions.Symbols, amplitude of the current measured every 10 sec at the end of a 200-msec pulse to 0 mV (•) or −120 mV (▪), from a holding current of −50 mV. Solid lines, addition of ACh 3 μm, alone or in combination with Mblue. Top current traces, recorded at the times indicated (correspondingletters on the main graph) (see Experimental Procedures). In each group of two traces, the top trace is the current obtained at 0 mV and the bottom traceis the current obtained at −120 mV, with each followed by the current recorded during the return to the holding potential of −50 mV. B, Summary of the results of several experiments like in A, with four different concentrations of Mblue (1, 3, 10, and 30 μm), in the presence of 3 μm ACh. The amplitude of the end-pulse current at 0 mV was normalized to the amplitude of the current with ACh alone. Dotted line, 100%; continuous line, mean steady state current level at 0 mV, in the absence of ACh. Bars, mean values. Lines, mean ± standard error of the number of experiments indicated near thebars. Significant statistical differences from the ACh level are indicated (*, p < 0.0001).
As suggested in a previous study, the antimuscarinic effects of external Mblue on ICa and IK,ACh may share a common mechanism that may involve the G proteins (14). Substitution of internal GTP for a hydrolysis-resistant analog of GTP allowed us to examine this hypothesis. Indeed, the slow load of GTPγS or Gpp(NH)p on the G protein activates spontaneously IK,ACh even though the receptor is free of agonist. In the typical experiment shown in Fig. 3A, the GTP analog GTPγS was directly included in the patch pipette, and no attempt was made to follow the spontaneous rise of IK,ACh. After ∼10 min of dialysis of the atrial myocyte, the addition of ACh (3 μm) alone or in combination with Mblue (30 μm) had no effects on the currents. In similar experiments, the mean end-pulse current density at 0 mV in the presence of internal GTPγS (5.4 ± 0.7 pA/pF, five experiments) was close to that recorded in GTP-dialyzed myocytes in the presence of 3 μm ACh(7.4 ± 0.6 pA/pF, 24 experiments). Accordingly, superfusion with 3 μm ACh enhanced the steady state current by only 15.9 ± 4.4% in the GTPγS-dialyzed cells (six experiments, Fig. 3B), and this small increase was irreversible. Superfusion of the myocyte with Mblue (30 μm) alone or in combination with 3 μm ACh did not significantly reduce the GTPγS-stimulated current (Fig. 3B). Thus, Mblue is unlikely to interfere with G protein activation of IK,ACh. This conclusion is also supported by experiments in which we used another hydrolysis-resistant analog of GTP, Gpp(NH)p. Dialysis of the myocyte with Gpp(NH)p raised the mean end-pulse current density at 0 mV to 6.2 ± 1.0 pA/pF (four experiments). In these cells, 3 μm ACh slightly increased the Gpp(NH)p-stimulated IK,ACh, although not significantly (+10.3 ± 5.9% change, three experiments), and Mblue (30 μm) had a small but nonsignificant effect on IK,ACh in the presence of ACh (−12.5 ± 9.8%, three experiments).
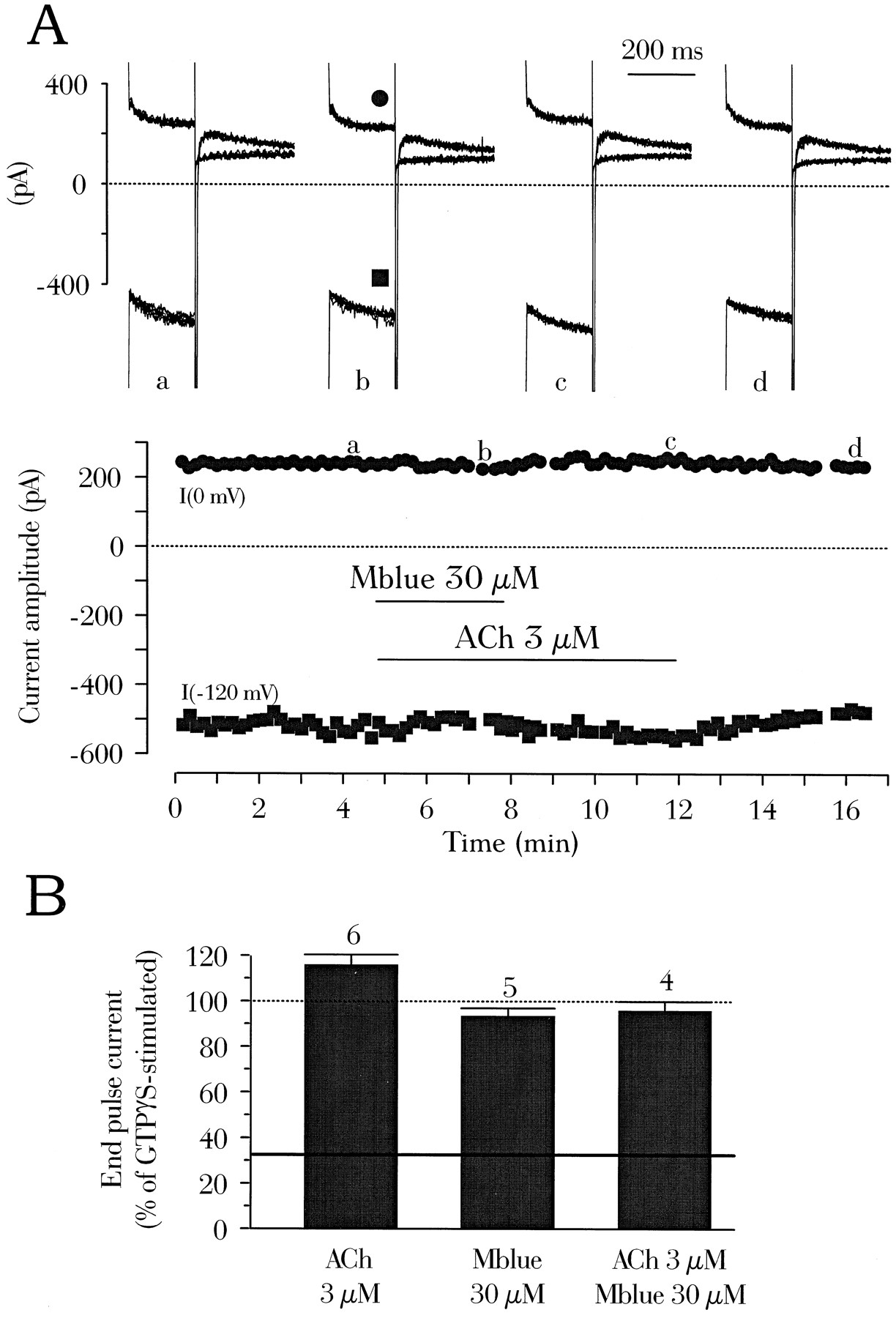
Mblue does not antagonize the GTPγS-stimulation of IK,ACh. A, A rat atrial cardiomyocyte was superfused with external K+-containing solution and dialyzed with the internal GTPγS-containing solution for 10 min before initiation of current recording. Solid lines, addition of ACh (3 μm), alone or in combination with Mblue. Each symbol, amplitude of the current measured at the end of a pulse to 0 mV (•) or −120 mV (▪). Top current traces, recorded at the times indicated (corresponding letters on the main graph) under similar conditions as indicated in the legend to Fig. 2A, except that three consecutive traces have been superimposed at each voltage (see Experimental Procedures). B, Summary of the results of several similar experiments as in A. The amplitude of the end-pulse current at 0 mV was normalized to the amplitude of the current with GTPγS alone (100%, dotted line).Continuous line, mean steady-state current level at 0 mV in the presence of GTP (same as in Fig. 2B). The effects of ACh (3 μm), Mblue (30 μm), or ACh plus Mblue on the GTPγS-stimulated IK,ACh are shown. Bars, mean values. Lines, mean ± standard error of the number of experiments indicated near the bars.
A noticeable characteristic of Mblue is that it is a piperazine derivative, and as such, it may interact directly with ionic channels or membrane receptors. Therefore, we next compared the effect of Mblue on IK,ACh with that of another piperazine derivative, CPZ. As shown in Fig. 4A, superfusion of a rat atrial myocyte with a combination of ACh (3 μm) plus CPZ (10 μm) prevented the development of IK,ACh. However, IK,ACh developed on removal of CPZ in the continuing presence of ACh. The inhibitory effect of CPZ on ACh-induced IK,ACh was elicited again on a second application of the drug. The experiment of Fig. 4A shows also that the inhibitory effect of Mblue (10 μm) was as fast as that of CPZ. However, the reversibility of the effect of Mblue was much faster than that of CPZ. It should be noted that unlike Mblue, CPZ at 10 μm exerted an inhibitory effect on the background current in the absence (not shown) or presence of ACh (Fig. 4A). In addition, the residual Na+ current activated on repolarization from −120 to −50 mV was diminished by CPZ (not shown). In summary (Fig. 4B), the activation of IK,ACh by 3 μm ACh was inhibited by CPZ in a dose-dependent manner, with a threshold of ∼30 nm. At high concentrations, CPZ also clearly inhibited other ion currents. The main finding of these experiments is that two piperazine derivatives tested, CPZ and Mblue, share a common inhibitory action on the muscarinic activation of IK,ACh.

CPZ antagonizes the muscarinic activation of IK,ACh. A, A rat atrial cardiomyocyte was first exposed to external K+ medium and dialyzed with internal K+-solution. Solid lines, additions of CPZ (10 μm) or Mblue (10 μm) in the presence of 3 μm ACh. Symbols, amplitude of the current measured at the end of a pulse to 0 mV (•) or −120 mV (▪).Top current traces, recorded at the times indicated (corresponding letters on the main graph) under similar conditions as indicated in the legend to Fig. 2A, except that three consecutive traces have been superimposed at each voltage (see Experimental Procedures). B, Summary of the results of several experiments like in A with four different concentrations of CPZ (0.03, 0.1, 3, and 10 μm) in the presence of 3 μmACh. The amplitude of the end-pulse current at 0 mV in the presence of CPZ plus ACh was normalized to the amplitude of the current with ACh alone. Solid line, mean basal level of the steady state current at 0 mV in the absence of the compounds. Bars, mean values. Lines, mean ± standard error of the number of experiments indicated near the bars. Significant statistical differences from the ACh level are indicated (*, p < 001; **, p < 0.001).
CPZ binds to dopaminergic receptors, which belong to the family of G protein-coupled receptors. Because this family includes muscarinic receptors, we investigated the effects of CPZ and Mblue on the specific binding of [3H]QNB, a muscarinic antagonist, in rat cardiac membranes. Binding of [3H]QNB to rat ventricular membranes was saturable between 500 and 1000 pm (Fig. 5A). Scatchard analysis indicated a homogeneous population of binding sites, aB max value of 434 ± 25 fmol/mg of protein (three experiments), and a KD value of 74 ± 8 pm (three experiments) (Fig. 5B). These values are compatible with published values for the maximal number of QNB binding sites (157–478 fmol/mg of protein) and theKD value (35–99 pm) in rat heart membranes (35-37).
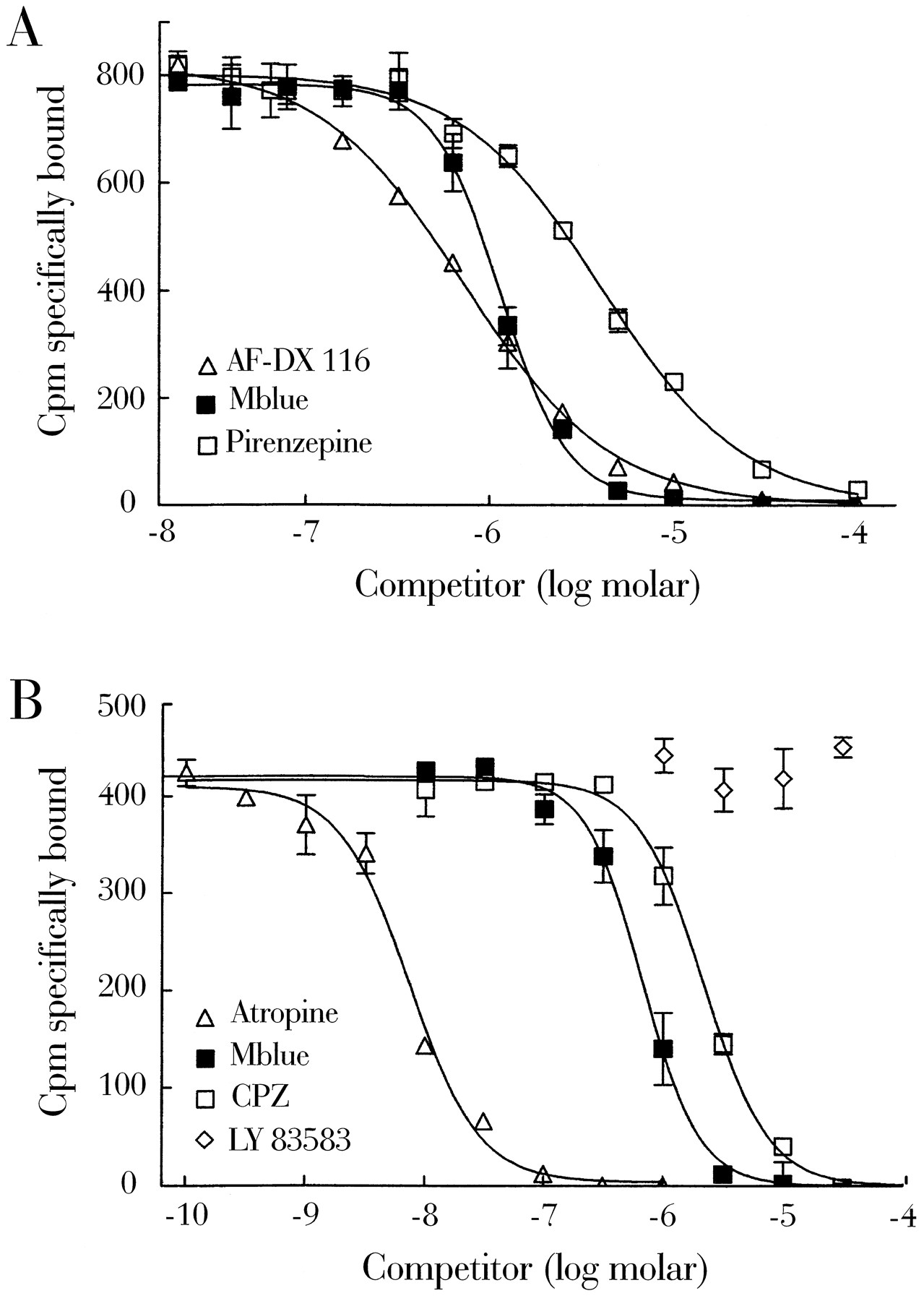
In the presence of 1 μm Mblue, the binding curve of [3H]QNB was shifted to the right by the factor of 14, but maximal specific binding (B max) was unchanged (Fig. 5C). This indicates that Mblue acts as a competitive ligand at muscarinic receptors. In accordance, Mblue displaced [3H]QNB from its binding sites with a calculated equilibrium dissociation constant (Ki ) of 187 ± 27 nm (three experiments, Fig.6). Compared with other known muscarinic antagonists, the rank order of potency (Ki ) was atropine (2 nm) > AF-DX 116 (90 nm) > Mblue (187 nm) > CPZ (366 nm) = pirenzepine (599 nm). In most experiments, the displacement by Mblue was somewhat steeper than that of all other compounds, yielding Hill slopes of 2.09 ± 0.10 (three experiments). To test whether the effect of Mblue on the muscarinic receptor is related to its guanylyl cyclase-inhibiting activity or its potential oxidative capacity, we tested the effect of the guanylyl cyclase inhibitor LY 83583 (6-anilino-5,8-quinolinedione) and the reducing agent DTT on [3H]QNB binding. LY 83583 did not interfere with binding of [3H]QNB (Fig. 6B). Similarly, 1 mm DTT alone was without effect on [3H]QNB binding and did not alter the effect of Mblue (not shown).

Binding isotherms of Mblue, AF-DX 116, pirenzepine, atropine, CPZ, and LY 83583 for the displacement of [3H]QNB binding to washed membranes from rat cardiac ventricles. The [3H]QNB concentration was 300 pm (A) and 200 pm (B), respectively. Specific binding was determined as that displaced by 1 μm atropine. Curves are representative of at least two independent experiments carried out in duplicate or triplicate.
β-Adrenergic receptors also belong to the family of G protein-coupled receptors. To examine whether Mblue interfered with cardiac β-adrenergic receptors, we investigated the effects of Mblue on the specific binding of [3H]CGP 12177, a β-adrenoceptor antagonist, in rat cardiac membranes. [3H]CGP 12177 bound to an apparently homogeneous population of binding sites in rat ventricular membranes with high affinity (KD = 88 pm) and low nonspecific binding (3% at theKD value as defined by 1 μm propranolol). In contrast to [3H]QNB, saturation binding of [3H]CGP 12177 was completely unaffected by 10 μm Mblue (Fig.7A). Similarly, binding of 300 pm [3H]CGP 12177 was not displaceable by Mblue, even at a 100 μmconcentration (Fig. 7B).

Saturation isotherms of [3H]CGP 12177 binding to washed membranes of rat cardiac ventricles. A, [3H]CGP 12177 binding in the absence (open symbols) and presence (filled symbols) of 10 μm Mblue. ▵, Total binding. ♦, Nonspecific binding in the presence of 1 μm propranolol. □ and ▪, Specific binding. Total and nonspecific binding in the presence of Mblue was omitted for graphical reasons and did not differ from that in the absence of Mblue. B, Binding isotherms of Mblue for the displacement of [3H]CGP 12177 binding to washed membranes from rat cardiac ventricles.
Discussion
The current results allow to characterize the mechanism of action of Mblue on the muscarinic regulation of ICa and IK,ACh in rat myocytes. In electrophysiological experiments, Mblue behaved like a muscarinic antagonist. Indeed, Mblue inhibited the modulation of ICa by ACh and CCh in rat ventricular myocytes, as well as the muscarinic activation of IK,ACh in atrial myocytes. Furthermore, intracellular dialysis with Mblue did not affect the regulation of ICa by ACh. Finally, Mblue failed to modify the stimulatory effects of GTPγS and Gpp(NH)p on IK,ACh (i.e., when the muscarinic receptors are bypassed). Binding experiments in rat ventricular membranes demonstrated that Mblue acts as a competitive antagonist of [3H]QNB binding on muscarinic receptors. Because in this preparation muscarinic receptors bound AF-DX 116 with a higher affinity than pirenzepine, the M2 subtype of muscarinic receptor is the likely target of Mblue.
Among the known properties of Mblue, its connection with the piperazine family of compounds is likely to account for the antimuscarinic effects reported in the current study. Indeed, CPZ, another piperazine derivative, also inhibited the muscarinic activation of IK,ACh in atrial myocytes, as well as [3H]QNB binding in ventricular membranes. The inhibition of IK,ACh by Mblue and CPZ developed with a similar time course. However, Mblue was found more selective for muscarinic receptors than CPZ. Mblue, like atropine, had no effect on background currents and did not inhibit IK,AChactivity when muscarinic receptor was bypassed with GTPγS or Gpp(NH)p. In contrast, CPZ inhibited background currents as well as the Na+ current, as shown in earlier studies in isolated cardiac myocytes (27, 28). The long lateral chain present in the molecule of CPZ, which is lacking in the molecule of Mblue, may favor its insertion in the membrane and account for the perturbation of various membrane proteins. This may provide an explanation for the slow recovery of IK,ACh from CPZ inhibition compared with Mblue.
Superoxide anion generation by Mblue is unlikely to participate in the antimuscarinic effects reported in the current study. Indeed, LY 83583, another superoxide anion generator, did not antagonize [3H]QNB binding in ventricular membranes. Previous studies also demonstrated that LY 83583 does not inhibit the muscarinic activation of IK,ACh in rabbit nodal myocytes (14) or frog atrial myocytes (21). In addition, the reducing agent DTT did not modify the effect of Mblue on [3H]QNB binding in ventricular membranes, nor did it modify the antimuscarinic effect of Mblue on ICa. The inhibition of IK,ACh by Mblue was also shown to be insensitive to reducing agents in frog atrial myocytes (21). Thus, a chemical modification of the muscarinic receptors by Mblue is unlikely to account for its antimuscarinic effect. This interpretation is supported by the fact that Mblue is able to antagonize the vasorelaxant effect of ACh in the presence of SOD, the enzyme responsible for the elimination of superoxide anion (6). In contrast, superoxide anion generation was shown to be a significant mechanism of action of Mblue in other tissues (21-23). Because the M2 isoform is not the major muscarinic receptor subtype expressed in these tissues, the binding of Mblue on muscarinic receptors may exhibit some subtype selectivity.
In our preparations, Mblue affected only muscarinic receptors and muscarinic responses. Mblue did not modify basal ICa or Iso-stimulated ICaand had no effect on the specific binding of [3H]CGP 12177, a nonselective β-adrenergic ligand. Unlike Mblue, the classic muscarinic antagonist atropine was demonstrated to enhance basal ICa in rat myocytes (38). Atropine is thought to shift the ratio of active (R*) to inactive (R) receptors toward R, the consequence of which is a reduction of the spontaneous turnover of the Gi protein and, subsequently, an increase in adenylyl cyclase activity (38). Thus, in the context of ICa regulation, only atropine behaves like an inverse agonist, whereas Mblue seems rather a neutral antagonist. In contrast, Zakharov et al. (16) reported a potentiating effect of both Mblue and LY 83583 on the Iso-stimulated Cl− current in guinea pig ventricular myocytes. However, the effect of Mblue, which was smaller than that of LY 83583, required higher concentrations (0.1–1 mm) than those used in the present study (16). Because we found that Mblue binds to muscarinic receptors with a Ki<200 nm, it is unlikely that muscarinic occupancy by Mblue accounts for the stimulatory effect of the drug on the Cl− current. It is possible that ICa and the cAMP-stimulated Cl− current are differentially regulated by Mblue in rat and guinea pig heart and/or that an effect of Mblue mediated through superoxide anion generation was favored in the experimental conditions used to record the Cl−current.
In contrast to superfusion of Mblue, internal dialysis with Mblue had no effects on the inhibition of Iso-stimulated ICa by ACh at 1 and 10 μm. However, inclusion of Mblue in patch pipettes has been reported to prevent the Iso-stimulated ICa from muscarinic inhibition in rabbit nodal myocytes (14, 15) and in rat ventricular myocytes (13). This antimuscarinic effect of Mblue was mimicked by inclusion of LY 83583 (14, 15, 39) orN G-monomethyl-l-arginine, a NOS inhibitor (13-15) in the patch pipette. On the basis of these results, it was concluded that the muscarinic inhibition of ICa depended on the stimulation of the NOS/cGMP pathway. However, this interpretation has been questioned (10, 40), and results of the current study suggest that the putative intracellular targets of Mblue (NOS and/or guanylyl cyclase) play a minor role in the muscarinic regulation of ICa in our preparation of rat ventricular myocytes. The possibility that superoxide anion generation by Mblue is weak under some (this study) but not all experimental conditions (13-16) provides a reasonable explanation for these discrepancies. Nevertheless, we demonstrated that Mblue was a muscarinic antagonist, and this property most likely contributes to some of the antimuscarinic effects of the putative guanylyl cyclase inhibitor reported in previous studies.
Acknowledgments
We thank Patrick Lechêne for skillful technical assistance, Florence Lefebvre for preparation of the cells, Françoise Boussac for editorial help, and Drs. Michel Chesnais and Rémy Hanf for helpful discussions. We thank Dr. E. Carmeliet for bringing to our attention the pioneer work of Dr. R. P. Cook.
Footnotes
- Received January 23, 1997.
- Accepted June 2, 1997.
-
Send reprint requests to: Dr. Pierre-François Mery, INSERM U-446, Faculté de Pharmacie, F-92296 Châtenay-Malabry, France. E-mail: u446{at}vjf.inserm.fr
-
↵1 Current affiliation: Pharmakologisches Institut, Universitäts-Krankenhaus Eppendorf, D-2000 Hamburg 20–38, Germany.
-
↵2 Current affiliation: Department of Biologia Celular, Fisiologia Animal, Fac. Ciencias, Universidad Autonoma de Barcelona, E-08193 Cerdanyola, Spain.
-
This work was supported by a grant from the Association Française contre les Myopathies. L.H.-M. was a Fellowship recipient of the Carlsberg Foundation. T.E. was supported by a grant from the Deutsche Forschungsgemeinschaft.
-
↵3 86/609/CEE, CE off J L358, December 18, 1986.
-
↵4 J Off République Française, October 20, 1987, pp. 12245–12248.
-
↵5 No. 04226, April 12, 1991.
Abbreviations
- NO
- nitric oxide
- ACh
- acetylcholine
- EGTA
- ethylene glycol bis(α-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- CCh
- carbachol
- Iso
- isoprenaline
- Mblue
- methylene blue
- CPZ
- chlorpromazine
- ICa
- L-type calcium current
- IK,ACh
- muscarinic activated potassium current
- SOD
- superoxide dismutase
- NOS
- nitric oxide synthase
- GTPγS
- guanosine-5′-O-3-triphosphate
- Gpp(NH)p
- guanosine-5′-(β,γ-imido)triphosphate
- DTT
- dl-dithiotreitol
- QNB
- quinuclidinyl benzylate
- AF-DX 116
- 11-[((2-diethylamino)methyl-1-piperidinyl)acetyl]5,11-dihydro-6H-pyrido-2,3-b)(1,4)-benzodiazepine-6-one
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}