


Abstract
The existence of two rather than one estrogen receptor, today characterized as estrogen receptor α (ERα) and estrogen receptor β (ERβ), indicates that the mechanism of action of 17β-estradiol and related synthetic drugs is more complex than previously thought. Because the homology of amino acid residues in the ligand-binding domain (LBD) of ERβ is high compared with those amino acid residues in ERα LBD, previously shown to line the ligand binding cavity or to make direct contacts with ligands, it is not surprising that many ligands have a similar affinity for both receptor subtypes. We report that 17α-ethynyl,17β-estradiol, for example, has an ERα-selective agonist potency and that 16β,17α-epiestriol has an ERβ-selective agonist potency. We also report that genistein has an ERβ-selective affinity and potency but an ERα-selective efficacy. Furthermore, we show that tamoxifen, 4-OH-tamoxifen, raloxifene, and ICI 164,384 have an ERα-selective partial agonist/antagonist function but a pure antagonist effect through ERβ. In addition, raloxifene displayed an ERα-selective antagonist potency, in agreement with its ERα-selective affinity. However, although ICI 164,384 showed an ERβ-selective affinity, it had a similar potency to antagonize the effect of 17β-estradiol in the ERα- and ERβ-specific reporter cell lines, respectively. In conclusion, our data indicate that the ligand binding cavity of ERβ is probably more different from that of ERα than can be anticipated from the primary sequences of the two ER subtypes and that it will be possible to develop receptor-specific ligands that may form the basis of novel pharmaceuticals with betterin vivo efficacy and side effect profile than current available drugs.
Nuclear steroid/thyroid hormone receptors and their cognate hormonal ligands constitute a group of key mediators in the endocrine signaling pathways, playing an important role in the control of differentiation, growth, and metabolic homeostasis (Evans, 1988; Gronemeyer and Laudet, 1995). The receptors exercise control of these events by their function as hormone-activated transcription factors and modulators of gene expression in target cells (Beato, 1989; Gronemeyer, 1992). Classic signals to which this family of receptors respond are the steroid hormones, the thyroid hormones, and vitamins A and D, each interacting with a specific receptor of the family (Green and Chambon, 1988).
All receptors of the nuclear receptor superfamily have a similar architecture (Tsai and O′Malley, 1994). The amino-terminal A/B domain is involved in transactivation of gene expression. The C-domain contains a two-zinc finger structure, which plays an important role in receptor specific DNA-binding and receptor dimerization. The carboxyl-terminal ligand-binding domain (or E/F domain) is crucial for binding of receptor specific ligands, nuclear translocation, receptor dimerization, and modulation of target gene expression in association with corepressors and coactivators (Horwitz et al., 1996;Shibata et al., 1997).
Members of the nuclear steroid/thyroid hormone receptor family have been associated with various disorders and disease conditions such as cancer, osteoporosis, cardiovascular disease, inflammation, and metabolic disorders. A number of natural and synthetic hormonal drugs that modulate the function and activity of nuclear receptors are used for the treatment of major clinical indications, such as thiazolidinediones for treatment of type II diabetes (Lehman et al., 1995; Berger et al., 1996), glucocorticoids as anti-inflammatory drugs, estrogen antagonists for treatment of breast cancer, and androgen antagonists in prostate cancer therapy (Hayneset al., 1990).
A significant deal of attention has recently been focused on women’s health issues and hormone replacement therapy. At the onset of perimenopause/menopause, women may have symptoms like hot flushes and urogenital tract complications (Lichtman, 1996). Furthermore, women may experience serious health risks such as the development of osteoporosis or cardiovascular disease during their postmenopausal life (Lichtman, 1996). The symptoms and health risks affecting many menopausal/postmenopausal women have been attributed to the loss ofde novo production of the natural female sex hormone E2 (i.e., to an E2-deficient state).
To alleviate or prevent symptoms of the nature described above and the development of serious health risks, women are given estrogen replacement therapy in combination with a gestagen (Lichtman, 1996). However, current hormone replacement therapy is associated with certain serious concerns like fear for increased risk of breast or uterine cancer, indicating the need for development of safer therapy.
The effects of estrogen were long believed to be mediated by the ER cloned >10 years ago (Green et al., 1986; Greene et al., 1986). However, recently, a second ER, ERβ, was cloned from rat (Kuiper et al., 1996), mouse (Tremblay et al., 1997), and human (Mosselman et al., 1996; Enmarket al., 1997). The presence of two ERs, the “old” ER (renamed ERα) and ERβ, reveals that the mechanism of biological action of estrogen and related synthetic drugs is more complex than previously thought. It also provides a unique opportunity for the development of improved modulators of estrogen action and the identification of new targets for estrogens.
In the current study, we compare the ligand binding and transcriptional responses of the human ERα and the human ERβ, respectively, to various synthetic estrogen agonists and antagonists. The results of this study indicated that there are similarities between hERα and hERβ in their responses to ligands but also that there are receptor-selective differences, which are of possible importance for design and synthesis of receptor-selective ligands for the development of drugs for hormone replacement therapy with improved in vivo efficacy and side effect profile.
Experimental Procedures
Materials.
E2, 17α-Estradiol, tamoxifen, 4-OH-tamoxifen, genistein, bisphenol A, and DES were from Sigma-Aldrich Sweden AB. Raloxifene and ICI 164,384 were synthesized according to published procedures (Jones et al., 1984; Bowler et al., 1989). [3H]E2 was purchased from New England Nuclear Research Products (Boston, MA). MEM culture media, FCS,l-glutamine, OptiMEM, lipofectamin, G418, and gentamicin were purchased from Gibco Life Technologies (Stockholm, Sweden). Hygromycin B was from Calbiochem (LabKemi AB, Sweden). Phenol red-free Coon′s/F12 medium was from SVA (Uppsala, Sweden). The SRC 3000 serum substitute was purchased from Tissue Culture Services (Botolph Claydon, England). Rabbit reticulocyte lysate was from Promega (Scandinavian Diagnostic Services, Falkenberg, Sweden). The chemiluminescence substrate CSPD was purchased from Tropix (Boston, MA).
Ligand competition binding assay.
Full-length recombinant hERα and hERβ were produced at high levels using the baculovirus expression system (Luckow and Summers, 1989). Receptor protein was prepared from the nuclear fraction as described previously (Barkhemet al., 1991) and extracted with buffer containing 17 mm K2HPO4/3 mm KH2PO4, pH 7.9, 400 mm KCl, 1 mmMgCl2, 0.5 mm EDTA, 6 mmmonothioglycerol, and 8.7% glycerol. The concentration of extracted hERα was 400 pmol/ml nuclear extract and 800 pmol/ml extract for hERβ (determined by specific binding of [3H]E2).
In all ligand binding experiments, the hERα and hERβ extracts were diluted in buffer B (20 mm HEPES, pH 7.5, 150 mm KCl, 1 mm EDTA, 6 mmmonothioglycerol, and 8.7% glycerol) supplemented with unprogrammed rabbit reticulocyte lysate (diluted 1:200) and 2% dimethylsulfoxide, to a final concentration of receptor protein of 0.2 nm. The concentration of [3H]E2 used for determination of the apparent equilibrium binding constant (Kd ) of E2 to hERα and hERβ, respectively, in the presence or absence of raloxifene or ICI 164,384 was in the range of 20–2000 pm. For all binding experiments, the incubation time was 20 hr, run at 22°. Receptor-bound [3H]E2 was separated from free ligand by G25 filtration (Salomonsson et al., 1994), and the amount of [3H]E2 bound to the receptor protein was determined by liquid scintillation counting (Rackbeta 1217; Wallac Oy, Turku, Finland).
Specifically bound [3H]E2 (obtained by subtracting total [3H]E2 bound from nonspecifically bound [3H]E2) was plotted against the amount of free [3H]E2 (obtained by subtracting the amount of [3H]E2 added from the total bound [3H]E2). The apparent equilibrium binding constant of [3H]E2 for hERα and hERβ, respectively, was then determined by applying the Hill equation [bound = (B max × [L]n)/([L]n +K d n)] (Salomonssonet al., 1994).
Vector constructs.
The cDNAs encoding the full-length human ERα (Greene et al., 1986) and the human ERβ (Enmarket al., 1997) were cloned into the BamHI andXbaI sites in the mammalian expression vector pMT-hGH (Friedman et al., 1989; Alksnis et al., 1991) after excision of human growth hormone coding sequences. The human ERα cDNA encodes the wild-type 66-kDa receptor protein, and the human ERβ cDNA used in all experiments in this study encodes the 485-amino acid residues long form with a molecular mass of ∼55 kDa.
The pΔERE2-ALP reporter vector contains one copy of the vitellogenin ERE (Klein-Hitpaβ et al., 1986) fused to the mouse mammary tumor virus core promoter sequences including the NF1 site (Brüggemeier et al., 1990) and cloned 5′ of the cDNA encoding human placental ALP (Berger et al., 1988) and the 3′ noncoding sequences from the human growth hormone gene (Friedmanet al., 1989).
DNA transfections.
All transient or stable transfections were done by using the OptiMEM/lipofectamin procedure according to the supplier′s recommendations (Gibco Life Technologies).
Generation of stable ERα and ERβ reporter cell lines.
The 293 cells (CRL-1573; American Type Culture Collection, Rockville, MD) routinely cultured in MEM supplemented with 10% FCS and 2 mm l-glutamine were first transfected with 2.5 μg of the pΔERE2-ALP reporter vector and 0.2 μg of the drug resistance vector pSV2-Neo (Southern and Berg, 1982) using the lipofectamin procedure according to the supplier′s recommendations. A G418-resistant clone mix (293/ΔERE2-ALP) was isolated and used in a second round stable transfection with 0.5 μg of pMT-hERα and pMT-hERβ, respectively, together with 0.1 μg of the drug resistance vector pKSV-Hyg (Gritz and Davies, 1983). Individual hygromycin B-resistant clones were isolated. One stable clone each of 293/hERα and 293/hERβ reporter cells was chosen for further study in response to various estrogen agonists and antagonists.
The stable cell reporter/vector clone mix (293/ΔERE2-ALP) and the 293/hERα and 293/hERβ reporter cells were cultured routinely at 37° in humidified chambers at 5% CO2 in MEM supplemented with 10% FCS and 2 mm l-glutamine.
Assay procedure for hormonal effects on 293/hERα and 293/hERβ reporter cells.
Approximately 25 × 103cells per well were seeded onto 96-well culture plates in 100 μl of Coon′s/F12 supplemented with 10% FCS (stripped twice using dextran-coated charcoal) and 2 mm l-glutamine. At 24 hr later, conditioned medium was replaced with 100 μl Coon′s/F12 supplemented with 5% serum substitute, 2 mm l-glutamine, gentamicin (50 μg/ml), and hormonal substances as indicated in the figure legends. In all experiments, cells have been exposed to hormones for 72 hr before harvest and analysis for effect on reporter gene expression. Triplicate determinations of reporter protein levels in the conditioned media for each concentration of compound have been performed in all experiments.
Assay for human placental ALP.
The level of ALP expressed from the ΔERE2-ALP reporter vector in the stably transformed 293/hERα and 293/hERβ reporter cells was determined by a chemiluminescent assay in which a 10-μl aliquot of heat-treated (at 65° for 40 min) conditioned cell culture medium was mixed with 200 μl of assay buffer (10 mm diethanolamine, pH 10.0; 1 mm MgCl2, and 0.5 mmdisodium 3-[4-methoxyspiro(1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.1.3,7]decan)-4-yl]phenyl phosphate) (Alksnis et al., 1991; Nilsson et al., 1993) in white microtiter plates (Dynatech Laboratories In Vitro AB, Stockholm, Sweden) and incubated at 37° for 20 min before being transferred to a microplate format luminometer (Luminoskan; Labsystem, Helsinki, Finland). The setting of the Luminoskan was integral measurement with 1-sec reading of each well. The ALP activity is expressed in LU, which is directly proportional to the level of ALP expressed from the cells.
Calculation of degree of agonism in percent of E2.
The degree of agonism in percent of E2 for each concentration of ligand was calculated as {[response of ligand (LU) − background (LU)]: [maximum response to E2 (LU) − background (LU)]} × 100.
Results
Stable subtype-specific ER reporter cell lines.
The human 293 kidney epithelial cell line was chosen as the host cell line for the generation of stable reporter cells for screening of compounds that act via the human ERs α (hERα) and β (hERβ) because as 293 cells did not respond to E2 in transient transfections with an ERE-driven reporter vector (Nilsson S, unpublished observations). The generation of the ERα and ERβ reporter cell lines, respectively, was done in two steps: first, the establishment of a G418-resistant reporter vector clone mix (293/ΔERE2-ALP), and, second, a second transfection with the expression vectors encoding the human ERα and human ERβ proteins, respectively, and the drug-resistance vector pKSV-Hyg. Of several hygromycin B-resistant cell clones, one clone of each receptor subtype, 293/hERα and 293/hERβ, showing the best signal-to-noise ratio and sensitivity to increasing concentration of E2, was chosen for further studies. Both the 293/hERα cells and 293/hERβ cells showed a dose-dependent transactivation of ALP reporter gene expression by E2, whereas the reporter clone mix 293/ΔERE2-ALP did not show any response to E2, as expected for a cell that does not express any endogenous or exogenous ER (Fig. 1). The level of receptor protein, hERα and hERβ, respectively, expressed by the stable cells is <4000 receptors/cell as determined by ligand equilibrium binding assay (data not shown).

Dose-response to E2 in the 293/hERα, 293/hERβ, and 293/ERE2-ALP reporter vector clone mix. The different genetically engineered clones and clone mix were exposed to the indicated concentrations of E2 for 72 hr before assay for the amount of ALP reporter protein expressed as described in the text (5 × 10−12 m indicates no added E2; solvent only). Values are the mean from triplicate determinations for each concentration of E2, with error bars (standard deviation) indicated for each value. For some concentrations, error bars are not visible because they were smaller than the symbol.
As also shown in Fig. 1, the potency of E2 to activate reporter gene expression is slightly higher for hERα than for hERβ, a finding in agreement with what was reported for mouse ERβ in comparison with the mouse ERα (Tremblay et al., 1997). Furthermore, the background ALP gene expression (in the absence of any added E2) is slightly higher in the 293/hERβ cells than in the 293/hERα cells (Fig. 1). The reason for this seemingly unprovoked reporter gene expression is under investigation but seems to be dependent on the presence of hERβ because neither the 293/ΔERE2-ALP nor the 293/hERα cells showed elevated ALP gene expression in the absence of E2.
In Fig. 2, the response of 293/hERα and 293/hERβ to a number of known estrogen agonists is shown. The response to the different ligands is expressed as percent agonism of E2 in 293/hERα and 293/hERβ cells, respectively. The environmental estrogen bisphenol A (Fig. 2C) showed a partial agonist activity in both the 293/hERα and the 293/hERβ reporter cells, whereas DES (Fig. 2B) displayed a similar degree of estrogen agonism as E2 for both receptors. 17α-E2 (Fig. 2A) only showed partial agonism in the 293/hERβ cells but full agonist activity in the 293/hERα. None of these ligands showed any clear receptor selective potency (Fig. 2, A–C).

Response in the 293/hERα and 293/hERβ reporter cell lines to increasing concentrations of different commercially available estrogens. The two stable reporter cell lines, 293/hERα and 293/hERβ were treated with (A) E2, (B) DES, (C) bisphenol A, (D) 17α-ethynyl,17β-estradiol, (E) 16β,17α-Epiestriol, and (F) genistein at the concentrations indicated. The level of ALP reporter protein expressed was assayed 72 hr after the addition of the ligands. The response is displayed as percent of the maximum response to E2 in 293/hERα and 293/hERβ. The response values for each concentration of ligand are the mean of triplicate determinations with the mean ± standard deviation for each value indicated. For some concentrations, error bars are not visible because they were smaller than the symbol.
Ligands with receptor selective potency or efficacy.
Among a number of commercially available ligands with known estrogen activity, 17α-ethynyl-17β-E2 (Fig. 2D) displayed the largest hERα selectivity with a 35-fold (range, 35–60-fold) higher potency in transactivating ALP gene expression in 293/hERα cells compared with 293/hERβ cells. 16β,17α-Epiestriol, on the other hand, displayed the largest hERβ selectivity with a 7-fold lower EC50 value for hERβ than for hERα (Fig. 2E). The phytoestrogen genistein showed receptor selective efficacy, being more efficacious than E2 in the 293/hERα cells (range, 107–130% of E2) but demonstrating only partial agonism via hERβ in the 293/hERβ cells (Fig. 2F). However, genistein was slightly more potent via hERβ compared with hERα, which is in agreement with its higher relative binding affinity to ERβ than to ERα (Kuiper et al., 1997) (in a regular ligand binding assay, the IC50 value for genistein in competition with [3H] E2 for binding to hERα and hERβ is 30 nm and 1 μm, respectively).
Effect of partial estrogens/antiestrogens.
The potency and agonist/antagonist activity of tamoxifen, 4-OH-tamoxifen, ICI 164,384, and raloxifene were tested in the 293/hERα and 293/hERβ cells, respectively (Fig. 3, A–D). All four ligands showed a partial agonist activity in the 293/hERα cells (Fig.3, A and C) but only estrogen antagonism in the 293/hERβ cells (Fig.3B). In the 293/hERβ cells, these ligands behaved as inverse agonists, suppressing the background reporter gene expression in a dose-dependent fashion. The relative agonism in percent of E2 in the 293/hERα was in the range of 9–13% for tamoxifen, 4-OH-tamoxifen, and raloxifene, respectively, and ∼3% for ICI 164,384 (Table1).

Agonist/antagonist response to tamoxifen, 4-OH-tamoxifen, raloxifene, and ICI 164,384 in the presence or absence of 0.5 nm E2 in 293/hERα and 293/hERβ. The effect on ALP gene expression to increasing concentrations of tamoxifen, 4-OH-tamoxifen, raloxifene, and ICI 164,384, respectively, in the absence (▪, ▴, ♦, •) or presence (□, ▵, ⋄, ○) of 0.5 nm E2 in (A) 293/hERα cells and (B) 293/hERβ cells. C, Agonist function of tamoxifen, 4-OH-tamoxifen, raloxifene, and ICI 164,384 is a magnification of their agonism displayed in A. D, Antagonist potency of raloxifene in the 293/hERα and 293/hERβ reporter cell lines is shown for comparison. The response values are the mean values from triplicate samples for each concentration of ligand. For some concentrations, error bars are not visible because they were smaller than the symbol.
Character of tamoxifen, 4-OH-tamoxifen, ICI 164,384, and raloxifene as estrogen agonists, antagonists, or both in the 293/hERα and 293/hERβ reporter cells, respectively
All four partial agonists/antagonists displayed antagonism to E2 in the 293/hERα and 293/hERβ cells, respectively (Fig. 3, A and B). Their potency in antagonizing E2-induced gene expression was very similar in both reporter cell lines (Table 1), except for raloxifene, which was a ∼15-fold more potent estrogen antagonist for hERα than for hERβ (Fig. 3D and Table 1).
The relative agonism in percent of E2 induced by tamoxifen, 4-OH-tamoxifen, ICI 164,384, and raloxifene in the 293/hERα and 293/hERβ reporter cells, respectively, and their potency as agonists and antagonists are summarized in Table 1.
To study further the transcriptional effects of the synthetic partial estrogens/antiestrogens after their binding to hERα and hERβ, respectively, we ran repeated dose-titrations of E2 in the presence of fixed concentrations of raloxifene (Fig.4, A and B) or ICI 164,384 (Fig. 4, C and D). Both raloxifene and ICI 164,384 acted as competitive antagonists to E2 on hERα and hERβ, but their concentration-dependent effect on the rightward shift of the EC50 values for E2 was ligand and receptor selective (Fig. 4). Raloxifene had a much greater impact on the shift of the EC50 value for E2 in the 293/hERα cells than in the 293/hERβ cells (Fig. 4, A and B), whereas the effect on the EC50 value for E2 in the presence of increasing concentrations of ICI 164,384 was similar in the 293/hERα and 293/hERβ cells, causing roughly a shift of the EC50 value for E2 in the order of 4 log-units at the highest concentration of ICI 164,384 used compared with E2 alone (Fig. 4, C and D).

Effect of increasing concentrations of raloxifene and ICI 164,384 on the agonist potency of E2 in the 293/hERα and 293/hERβ reporter cell lines. The cells were exposed for 72 hr with no or 1, 5, 50, or 500 nm raloxifene or ICI 164,384 and increasing concentrations of E2 (10−12 to 10−5 m; 10−13 stands for no E2 added, only solvent or solvent and the indicated concentrations of raloxifene or ICI 164,384). Effect on the EC50 value for E2 in the presence of increasing concentrations of raloxifene in (A) 293/hERα cells and (B) 293/hERβ cells and in the presence of increasing concentrations of ICI 164,384 in (C) 293/hERα cells and (D) 293/hERβ cells. The values represent the mean of triplicate samples for each concentration of ligand with the mean ± standard deviation for each value indicated. For some concentrations, error bars are not visible because they were smaller than the symbol.
The characteristics of raloxifene and ICI 164,384 in modulating the binding affinity of [3H]E2 to hERα and hERβ also were studied. Fig. 5 describes the resulting apparent equilibrium binding constants (Kd app) for [3H]E2 binding to hERα and hERβ, respectively, in the presence of low, fixed concentrations of raloxifene and ICI 164,384, respectively. Both ligands were found to be competitive inhibitors to [3H]E2 for both receptor subtypes, manifested as a concentration-dependent increasedKd app for [3H]E2. The concentration of [3H]E2 binding-sites (B max) was not influenced. Raloxifene was found to be a more potent inhibitor of [3H]E2 binding to hERα, showing a dramatic increase in theKd app at concentrations of raloxifene above 0.2 nm, than to hERβ. In contrast to raloxifene, ICI 164,384 affected theKd app of [3H]E2 for hERβ more than for hERα (Fig.5).
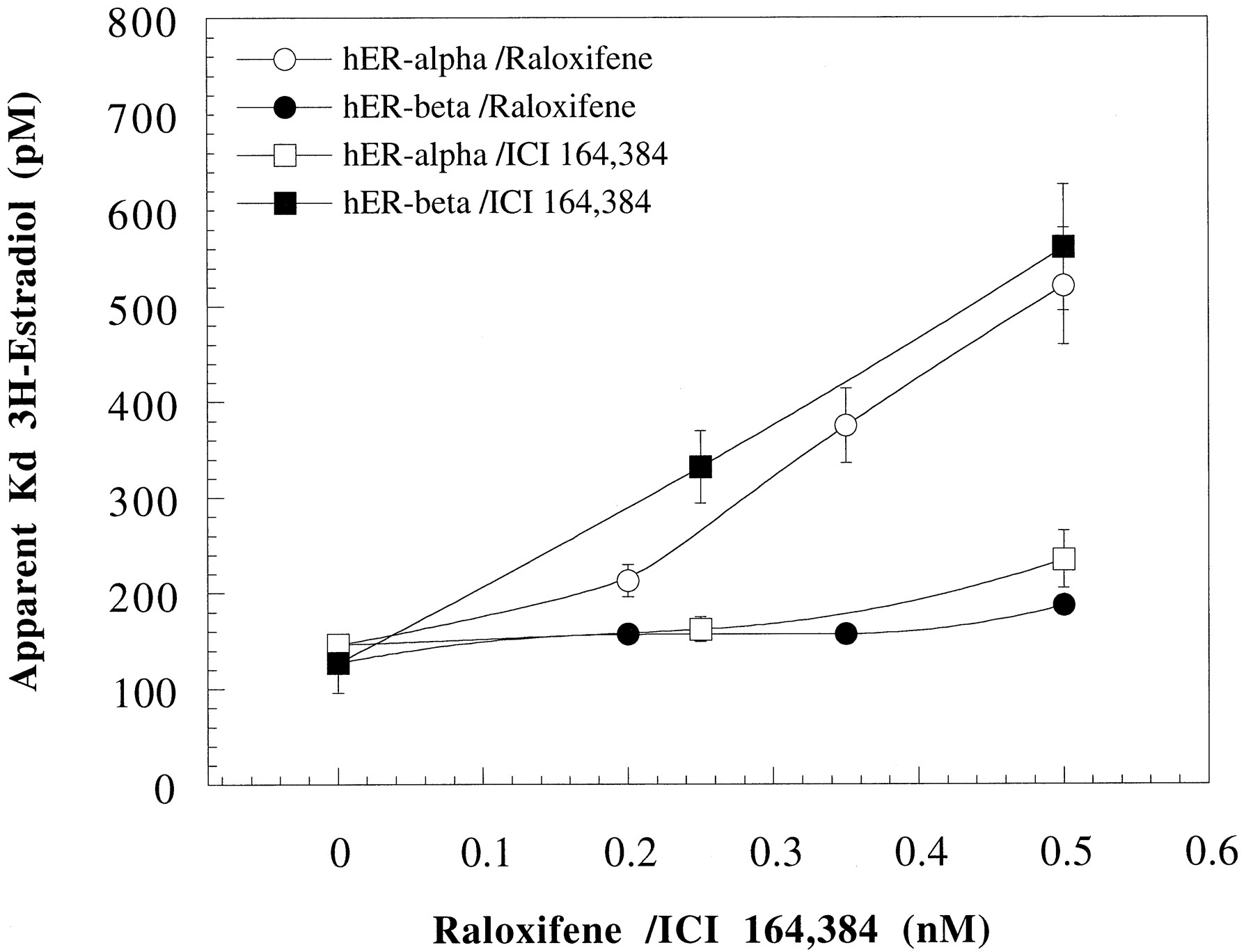
Effect of raloxifene and ICI 164,384 on the affinity of E2 for hERα and hERβ. The apparent equilibrium binding constant (Kd app) of [3H]E2 for hERα and hERβ in the presence of low, fixed concentrations of raloxifene and ICI 164,384 was determined as described in the text. Binding parameters for [3H]E2 (including the estimated error in the determination) were obtained from nonlinear curve fitting of binding data to the Hill equation using Kaleidagraph 3.08 (Synergy Software, Reading, PA). Representative data from one of three independent experiments are shown.
Discussion
In this report, we have shown that the two ER subtypes, α and β, respond in a similar fashion to some ligands but that there also are receptor-specific responses. The overall homology between hERα and hERβ is 47% with only ∼18% homology in their amino-terminal domains and 59% homology in their ligand binding domains (Enmarket al., 1997). The highest homology between hERα and hERβ is in the DNA binding domain with identical amino acid sequence in the P-box, the primary determinant for sequence specific recognition of a hormone response element (Umesono and Evans, 1989).
The ligand binding cavity of hERα is delimited by amino acid residues interspersed between amino acid residue 342 and amino acid residue 547 (Brzozowski et al., 1997). The homology between hERα and hERβ in this region of their ligand binding domains is very high, especially with reference to amino acid residues indicated in the hERα structure to make direct contacts with the ligand or lining the cavity (Brzozowski et al., 1997). It therefore is not surprising that E2 and other ligands bind to hERα and hERβ with approximately similar affinity. However, ligands with higher affinity for hERα than for hERβ, and vice versa, have been described (Kuiperet al., 1997). One such example is genistein, which has been shown to have a significantly higher affinity for rat and human ERβ than for hERα (∼30-fold) (Kuiper et al., 1997) (in a regular ligand binding assay, the IC50 value for genistein in competition with [3H] E2 for binding to hERα and hERβ is 30 nm and 1 μm, respectively). However, the degree of receptor-selective difference in affinity was not mirrored by the same degree of receptor-selective potency in the cell-based gene transcription assay. The potency of genistein in the 293/hERβ reporter cells was only 4–5-fold higher than that in the 293/hERα cells, and in contrast to expectations, genistein was only a partial agonist via the β receptor but (at least) a full agonist when the transcriptional response was mediated by ERα. As reported by others, the gene modulatory effect of a receptor after binding of a ligand depends on the conformational change of the receptor induced by the ligand and the subsequent events, including receptor dimerization, receptor/DNA interaction, formation of a preinitiation complex, and recruitment of and interaction with coactivators and other transcription factors (Beekman et al., 1993; McDonnellet al., 1995; Katzenellenbogen et al., 1996;Shibata et al., 1997). Based on our current knowledge of what is required for a ligand to be recognized by the cellular transcription machinery as a full agonist, it is obvious that genistein most likely does not induce an optimal agonist conformation of the ERβ protein, thus explaining its partial agonist activity and possibly also its relatively smaller receptor-selective gene modulatory potency compared with its receptor-selective binding affinity.
As expected, the well known partial agonists/antagonists tamoxifen, 4-OH-tamoxifen, and raloxifene displayed a low but significant estrogenic activity in the 293/hERα reporter cells (Fig. 3, A and C). Even the pure antagonist ICI 164,384 induced a low degree of transcriptional activity when bound to hERα (Fig. 3C). However, none of these ligands displayed any degree of estrogen agonism in the 293/hERβ reporter cells; only antagonism was displayed (Fig. 3B). Most likely, all four ligands inactivated the ligand-dependent AF2 function of both hERα and hERβ, and therefore the most likely explanation for the observed differences in transcriptional response to these ligands is to be found in the amino-termini of hERα and hERβ, respectively. Recently, it was described that the partial agonism of tamoxifen is mediated by a slightly different part of the ERα AF1 region than required for E2 (McInerney and Katzenellenbogen, 1996). A plausible explanation for the absence of any reporter gene transactivation activity by hERβ in the presence of tamoxifen, 4-OH-tamoxifen, and raloxifene is that hERβ lacks this particular function of hERα AF1.
Next, we analyzed the antagonistic character of raloxifene and ICI 164,384 in the 293/hERα and 293/hERβ reporter cells, respectively, and compared this with their characteristics as modulators of theKd app of [3H]E2 for hERα and hERβ, respectively. Both ligands behaved as competitive antagonists (Fig. 4) and competitive receptor binders to E2 for both hERα and hERβ (Fig. 5). However, already low concentrations of raloxifene caused a dramatic shift in the EC50 value for E2 in the 293/hERα cells (Fig. 4A) compared with the raloxifene effect in the 293/hERβ cells (Fig. 4B) and the ICI 164,384 effect in the 293/hERα cells (Fig. 4C). The effect of raloxifene on the potency of E2 in the 293/hERα cells was confirmed by the increasedKd app of E2 for hERα, especially at concentrations of raloxifene above 0.2 nm (Fig. 5). Similar concentrations of raloxifene had only moderate effect on theKd app of E2 in binding to hERβ. Although ICI 164,384 affected theKd app of E2 for hERβ more than for hERα (Fig. 5), that effect was not reflected in the cells in which ICI 164,384 caused a ∼4 order of magnitude shift in the EC50 value for E2 in both 293/hERα and 293/hERβ cells (Fig. 4, C and D). However, ICI 164,384 was a more potent antagonist than raloxifene in the 293/hERβ cells (Table 1), shifting the EC50 value of E2 ∼4 orders of magnitude at its highest concentration compared with raloxifene, which shifted the EC50 value of E2 ∼3 orders of magnitude (Fig. 4, B and D). Thus, although raloxifene showed both an hERα-selective affinity (Fig. 5) and potency as antagonist (Figs. 3D and Fig. 4, A and B), ICI 163,384 showed hERβ-selective affinity (Fig. 5) but no receptor-selective antagonist potency (Table 1 and Fig.4, C and D). The explanations for the hERα-selective affinity and antagonist potency of raloxifene and the hERβ-selective affinity of ICI 164,384 are difficult but may be examined when the three-dimensional structure of hERβ has been determined. That ICI 164,384 showed a similar antagonist potency for both hERα and hERβ (Table 1 and Fig. 4, C and D) despite its hERβ-selective affinity (Fig. 5) may be explained by the assumption that ICI 164,384 interferes with the homodimerization function of both hERα (Fawell et al., 1990) and hERβ with equal potency.
Although some of the differences between hERα and hERβ in response to partial estrogens could be explained by the lack in hERβ of the part of AF1 that is responsible for mediating the partial agonism of, for example, tamoxifen when bound to hERα, it cannot explain all the differences between hERα and hERβ in their transcriptional responses to various agonists and antagonists described in this report. The affinity of a ligand for a particular receptor and the conformational change induced by a ligand after its binding to the receptor are two important parameters that determine the transcriptional potency and agonist/antagonist character of a ligand. Both affinity and ligand-induced conformational change of the receptor depend, in part, on the three-dimensional structure of the ligand and its hydrophobic/hydrophilic character and, in part, on the volume and shape of the ligand binding cavity and the type of amino acid residues lining the cavity. As mentioned, the differences between hERα and hERβ in the part of LBD that makes up the ligand binding cavity of hERα are small, explaining why many ligands have a similar affinity and biological character when bound to hERα or hERβ. Nevertheless, there are a number of ligands (Kuiper et al., 1997) (Nilsson S, unpublished observations) displaying receptor-selective affinity or biological character (e.g., genistein) that binds to hERβ with a ∼30-fold higher affinity than to hERα. However, the most striking receptor-selective difference with genistein perhaps is its partial agonism mediated by hERβ but slight superagonist character (range, 107–130% of E2) when bound to hERα. This receptor-selective character of genistein and, in particular, its α- versus β-selective difference in efficacy are difficult to explain by just comparing the primary sequences of the LBDs of hERα and hERβ, respectively. A possible explanation is that the ligand binding cavity of hERβ is more different from the cavity of hERα than can be expected based on comparisons between primary amino acid sequences. Another explanation might be that hERβ has somewhat different requirements for coactivators than hERα, a difference that may only become apparent with particular ligands like genistein.
In conclusion, there are good reasons to believe that it will be possible to develop selective hERα and hERβ ligands. Such ligands may form the basis of novel pharmaceutical agents, providing significant advantages over currently available drugs for hormone replacement therapy in women.
Footnotes
- Received January 23, 1998.
- Accepted March 16, 1998.
-
Send reprint requests to: Dr. Stefan Nilsson, Karo Bio AB, Novum, S-14157 Huddinge, Sweden. E-mail:stefan.nilsson{at}karobio.se
-
This work was supported in part by the MISTRA program.
Abbreviations
- E2
- 17β-estradiol
- LBD
- ligand-binding domain
- DES
- diethylstilbestrol
- MEM
- minimum essential medium
- FCS
- fetal calf serum
- ALP
- alkaline phosphatase
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- LU
- light units
- ERE
- estrogen-responsive element
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}