


Abstract
Cyclic nucleotide phosphodiesterases (PDEs) are a superfamily of enzymes whose physiological role is the attenuation of the signaling mediated by the ubiquitous second messengers cAMP and cGMP. Given the myriad of physiological processes regulated by cAMP and cGMP, PDEs have long been studied as potential therapeutic targets. Although phosphodiesterase 3 (PDE3) activity is abundant in human cardiovascular tissues, and acute PDE3 inhibition, with agents such as milrinone, was beneficial in heart failure patients, prolonged treatments were associated with time-dependent reductions in hemodynamic effects and increased mortality. The molecular basis of this time-dependent reduction in efficacy has not been elucidated. In this context, we used a combination of approaches to determine PDE3 expression in human cardiovascular tissues and to elucidate the effects of prolonged elevations of cellular cAMP, as would occur with PDE3 inhibition, on this activity. Although our data confirms the expression of PDE3A in human blood vessel smooth muscle cells (HASMCs), we identify a previously unrecognized role for PDE3B in cAMP hydrolysis in human cardiovascular tissues. Specifically, although both PDE3A and PDE3B were expressed in HASMCs, their subcellular expression pattern and regulated expression by cAMP were distinct, with only expression of PDE3B being subject to cAMP-regulated expression. Thus, a paradigm emerges that allows for dual expression, with distinctive regulation, of both PDE3A and PDE3B proteins in cardiovascular tissues that may have profound significance for the rational design of molecules regulating this PDE activity.
Cyclic nucleotide phosphodiesterases (PDEs) are a superfamily of enzymes that, in concert with the adenylyl and guanylyl cyclases, govern the levels of the second messengers cAMP and cGMP (Beavo, 1995). Comprised of at least eleven different families, this superfamily has greater than fifty individual isoenzymes (Beavo, 1995; Fisher et al., 1998;Soderling et al., 1998, 1999; Fawcett et al., 2000). Isoenzymes are designated within a given PDE family based on molecular sequence, as well as kinetic and regulatory properties (Beavo, 1995). PDEs have long been viewed as drug targets based on the wide array of cyclic nucleotide regulated physiological processes, coupled with the differential tissue distribution and modalities of regulation intrinsic to each PDE family. One of the first PDE families envisioned to have therapeutic value was the cGMP-inhibited cAMP-hydrolyzing phosphodiesterase 3 (PDE3) family (Degerman et al., 1997). The PDE3 family includes two genes, PDE3A and PDE3B, whose products possess similar kinetic and regulatory properties (Degerman et al., 1997). Originally cloned from human myocardial tissue, PDE3A has historically been referred to as the cardiovascular PDE3 (Meacci et al., 1992; Smith et al., 1997). Consistent with this designation, a 110-kDa PDE3A protein is highly expressed in platelets as well as cardiac and vascular myocytes (Macphee et al., 1986; Rascon et al., 1992; Smith et al., 1993). Whereas most cellular PDE3A protein is soluble, particulate PDE3A has been detected in some tissues (Smith et al., 1993; Degerman et al., 1997). The PDE3B gene was originally cloned from rat adipocytes (Taira et al., 1993), and has, thus, often been referred to as the adipocyte PDE3 (Degerman et al., 1997). PDE3B has localized, as a 135-kDa protein, to the particulate fraction in all cell types examined (Miki et al., 1996; Degerman et al., 1997). PDE3B has been thought to have a markedly different profile of expression, compared with PDE3A, such that tissue selective expression is thought to be a defining characteristic of individual PDE3 genes (Reinhardt et al., 1995).
A decreased cAMP-dependent contractility of the heart, and alterations in peripheral circulation, contribute to heart failure (Movsesian, 1999). As PDE3 inhibition gives rise to positive inotropic and vasodilatory responses (Beavo, 1995), PDE3 inhibitors, such as milrinone, were initially thought to hold great promise for the treatment of congestive heart failure (CHF). Whereas clinical evidence clearly indicated that short term usage of PDE3 inhibitors improved cardiac function in CHF patients (Andrews and Cowley, 1993; Movsesian, 1999), increases in patient mortality over a longer term oral administration (Packer et al., 1991) led to a restriction in their usage in CHF management. Consistent with the development of tachyphylaxis to PDE3 inhibitors, a time-dependent reduction in the hemodynamic effects of these agents was noted (Movsesian, 1999). Although, reports have demonstrated reduced PDE3A expression in a canine model of CHF (Smith et al., 1997; Sato et al., 1999), no such effect was observed in human heart (Movsesian et al., 1991). Whereas alterations in the blood vessel response to these agents could have contributed to a reduction in hemodynamic efficacy, the importance of this has yet to be assessed. In this context, increases in PDE3 activity and expression in rat aortic smooth muscle cells (RASMCs), following prolonged cAMP increases, have been documented and shown to contribute to a heterologous desensitization to the cAMP-elevating effects of β-adrenergic agonists in these cells (Rose et al. 1997). When this effect was examined at a molecular level, contrary to previous reports concerning PDE3 expression, both PDE3A and PDE3B were shown to be expressed in RASMCs. Of further note, it was observed that PDE3B, not PDE3A, was markedly up-regulated following the cAMP increases (Liu and Maurice, 1998). Given the present limitations associated with prolonged use of PDE3 inhibitors in heart failure, the renewed investigation into the merits of PDE3 inhibitors in recent clinical trials (Cuffe et al., 2000), and the potential significance of alterations to the peripheral circulation in this disease, a detailed study to determine which PDE3 isoforms were expressed in human aortic smooth muscle cells (HASMCs) was carried out. Moreover, since sustained increases in vascular cAMP would necessarily occur during prolonged treatment with PDE3 inhibitors, the effects of cAMP on PDE3 expression in these cells was assessed. Our results demonstrate clearly that HASMCs express both PDE3A and PDE3B. Furthermore, PDE3B, not PDE3A, was increased following a prolonged cAMP challenge in HASMCs. Thus, a novel paradigm emerges that allows for dual expression, with distinctive regulation, of both PDE3A and PDE3B proteins in cardiovascular tissues, which may have profound significance for the rational design of molecules regulating this PDE activity.
Materials and Methods
Cell Culture.
Primary cultures of HASMCs derived from isolated explants from thoracic aorta were provided by Dr. C. Graham (Queen's University, Kingston, Ontario, Canada). Except for growth media supplementation with fetal bovine serum (10% final concentration; GibcoBRL, Burlington, Ontario), culture of the HASMCs was carried out as previously described for RASMCs (Palmer et al., 1998). HASMCs between passages 6 and 12 were used in all experiments. Similar to protocols described previously (Rose et al., 1997; Liu and Maurice, 1998), HASMCs were treated with forskolin (10–100 μM; Calbiochem; LaJolla, CA),N 6,O2′-dibutyryl cAMP (DbcAMP, 0.1–1.0 mM; Calbiochem), actinomycin D (4 μM; Sigma-Aldrich, Oakville, Ontario, Canada) and cycloheximide (100 μM; Sigma-Aldrich) or vehicle (dimethyl sulfoxide or water). Following treatment, HASMCs were homogenized as previously described (Palmer et al., 1998).
Subcellular fractionation was performed by differential centrifugation of homogenates from DbcAMP- or vehicle-treated cells. Soluble and particulate fractions were resolved by centrifuging of a 1,000g supernatant at approximately 162,600g for 2 h at 4°C. All homogenates were stored at 4°C before their use.
cAMP Phosphodiesterase (PDE) Assay.
The cAMP PDE activity in homogenates of HASMCs was assayed using 1 μM cAMP as substrate as previously described (Palmer et al., 1998). Protein amounts were determined using the bicinchoninic acid protein assay (Pierce; Rockford, IL), with bovine serum albumin as the standard. Activities are representative of at least three determinations from a minimum of two independent experiments.
Measurement of Steady State mRNA Levels by Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR).
Quantification of steady state levels of PDE3 mRNA was achieved using RT-PCR as previously described (Liu and Maurice, 1998). Gene-specific primer sets (Cortec DNA Service Laboratories, Inc., Kingston, Ontario) were used for the amplification of human PDE3A (5′-GAACAGGGTGATGAAGAGGC-3′ [sense], 5′-CACTGGTCTGGCTTTTGGGTTG-3′ [antisense]), PDE3B (5′-GCGGATCCGTTCTTCTCCTCAACTAGC-3′ [sense], 5′-CGCTCGAGTTCCTCTTCATCTGCCTC- TTC-3′ [antisense]), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 5′-GTTGCCATCAATGACCCCTTCATTG-3′ [sense], 5′-GCTTCACCACCTTCTTGATGTCATC-3′ [antisense]) specific mRNA.
Immunoblot Analysis of PDE3 Expression.
For analysis of the HASMC PDE3 proteins, a monoclonal antibody and a polyclonal antiserum were used as previously described (Liu and Maurice, 1998). The polyclonal antiserum, provided by Dr. J. Beavo (University of Washington, Seattle, WA) and raised against a glutathioneS-transferase-fusion protein of amino acids 887-1108 from mouse brain PDE3 (Zhao et al., 1997), was previously shown to react with human platelet PDE3A (Liu and Maurice, 1998) and was used to detect PDE3A protein in this study (1:5,000 dilution). Characterization of PDE3B expression was achieved using the supernatant from a hybridoma (281P) derived from a mouse immunized against a glutathioneS-transferase-fusion with amino acids 517–878 of human PDE3B (1:10 dilution). This monoclonal antibody was obtained from Drs. S. Wolda and V. Florio (ICOS Corporation, Bothell, WA).
Statistical Analysis.
Data are presented as means ± S.E. from multiple determinations for at least two independent experiments. Statistical differences were assessed using unpaired ANOVA with a Tukey post hoc test, or unpaired Student's t test as appropriate, with a value of P < .05 considered statistically significant.
Results and Discussion
Characterization of cAMP PDE Activity in HASMCs.
Because HASMCs could have a distinctive PDE complement compared with other species (Polson and Strada, 1996; Rybalkin et al., 1997), we first characterized the cAMP PDE activities present in these cells using family-selective modulators of PDE activity. Collectively, our data demonstrate that the most abundant cAMP PDE activities in HASMCs were from the calmodulin-stimulated PDE1 and the cGMP-inhibited PDE3 families. Consistent with demonstrated expression of PDE1C in HASMCs (Rybalkin et al., 1997), cAMP PDE activity in these cells was markedly enhanced (∼12-fold) by the addition of calcium (800 μM) and calmodulin (4 μg/ml) (Table 1). Cilostamide (1 μM), a PDE3-selective inhibitor, reduced cAMP PDE activity by approximately 25% (Table 1); an extent comparable with that seen in human coronary artery smooth muscle cells (Johnson-Mills et al., 1998). Consistent with the modest contribution of the cAMP-specific PDE4 to the total cAMP PDE activity, Ro 20-1724 (10 μM), a PDE4-selective inhibitor, had a marginal effect on cAMP hydrolysis, whether incubated alone or in the presence of cilostamide (Table 1). Furthermore, a mixed PDE3/PDE4 inhibitor, zardaverine, inhibited cAMP PDE activity to the same extent as cilostamide (not shown). Although a PDE2 (cGMP-stimulated PDE)-selective inhibitor, erythro-9-(2-hydroxy-3-nonyl)adenine (20 μM), had no effect on HASMC cAMP PDE activity (not shown), a nonselective PDE inhibitor (3-isobutyl-1-methylxanthine, 500 μM) reduced HASMC homogenate cAMP PDE activity by greater than 90% (Table 1).
Characterization of cAMP PDE Activities in HASMCs
Prolonged Incubations of HASMC with DbcAMP Increases PDE3 Activity.
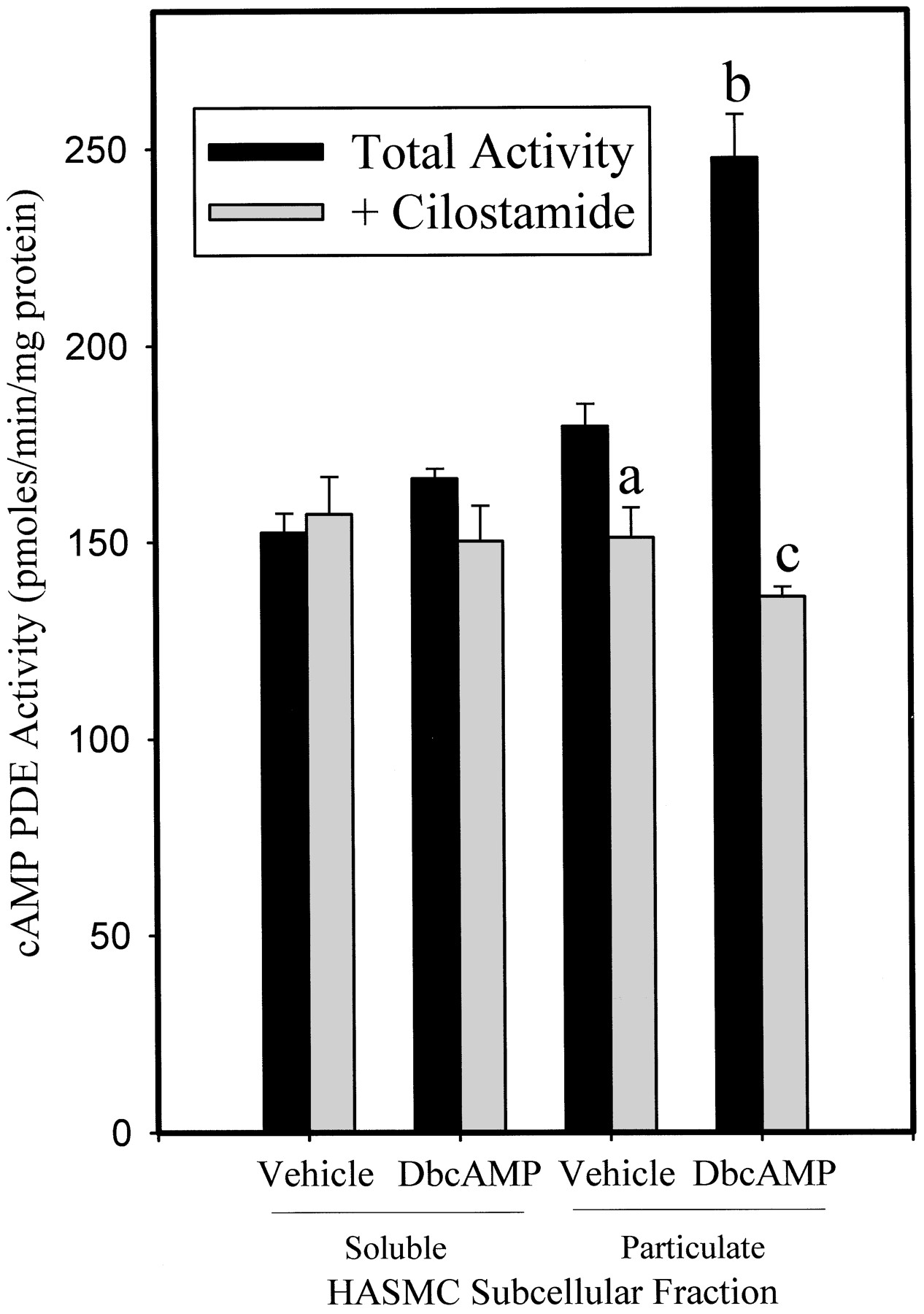
In previous work, our laboratory (Rose et al., 1997; Liu and Maurice, 1998), and others (Erdogan and Houslay, 1997; Seybold et al., 1998), have used a strategy involving prolonged incubations of cells with cAMP-elevating agents (e.g., forskolin) or structural analogs of cAMP (e.g., 8-bromo-cAMP) to study PDE3 expression. For this study, cAMP PDE expression in HASMCs was analyzed using either an activator of adenylyl cyclase (forskolin) or a structural analog of cAMP (DbcAMP). Collectively our data show that incubation of HASMCs with these agents markedly enhanced both the total cAMP PDE activity as well as the fraction attributable to PDE3 (Table2, Fig. 1). Consistent with our previous findings, both forskolin (10–100 μM) and DbcAMP (0.1–1.0 mM) caused concentration- and time-dependent increases in total HASMC cAMP PDE, which were maximal at the longest time point studied (16 h). In all subsequent experiments, DbcAMP was used as a representative regulatory molecule (1 mM, 16 h; Fig. 1).
Total cAMP PDE Activities in the Presence and Absence of Cilostamide from HASMCs after prolonged exposure to DbcAMP: The effect of inhibitors of mRNA and protein synthesis

Effects of DbcAMP treatment on total and cilostamide-inhibited cAMP PDE activity from soluble and particulate fractions of human aortic smooth muscle cell homogenates. cAMP-PDE activity was determined using 1 μM as substrate for quadruplicates from two independent experiments. Depicted is a representative experiment. Results are expressed as mean ± S.E.P < .05 (a) versus vehicle-treated total particulate cAMP PDE activity; P < .05 (b) versus vehicle-treated total particulate cAMP PDE activity;P < .05 (c) versus DbcAMP-treated total particulate cAMP PDE activity (by one-way ANOVA).
When determined in isolated subcellular fractions, a striking increase in PDE3 activity was observed in the particulate fraction of cells treated with DbcAMP with no increase in cytosolic PDE3 activity noted (Fig. 1). This increase in PDE activity was dependent on a de novo synthetic mechanism, as inclusion of either actinomycin D or cycloheximide, inhibitors of mRNA and protein synthesis, respectively, abolished this increase (Table 2). Although cAMP-dependent protein kinase-mediated phosphorylation and activation of PDE3 isoenzymes has been demonstrated (Degerman et al., 1997), the complete abrogation of the increase in HASMC cAMP PDE activity by the de novo synthetic inhibitors is more consistent with a DbcAMP-mediated up-regulation of enzyme levels. In addition to the increases in PDE3 activity, PDE4 activity also exhibited a significant up-regulation such that all of the increase in cAMP PDE activity subsequent to the incubation with DbcAMP was largely accounted for by the increase in PDE3 and PDE4 (our unpublished data). PDE1 activity, despite being a substantial component of the cAMP PDE activity in HASMCs, was not altered by the prolonged DbcAMP treatment (not shown).
Prolonged Elevations in cAMP in HASMC Selectively Increases PDE3B, but Not PDE3A.
Earlier work (Liu and Maurice, 1998) in RASMCs had shown the particulate PDE3B to be the major cAMP-regulated PDE3 in these cells. Because our results in HASMCs demonstrated that only particulate PDE3 activity in these cells was increased by DbcAMP, we proposed that the PDE3 variant regulated by cAMP in HASMCs could be PDE3B. However, because some tissues exhibit particulate PDE3A expression (Smith et al., 1993), the expression of PDE3A and PDE3B, the composition of the particulate PDE3 activity in HASMCs, and the effect of cAMP on the PDE3 isoforms expressed in this fraction, were each examined.
Expression of PDE3A and PDE3B was examined using a combination of molecular biological (RT-PCR) and immunological (Western blotting) approaches. PCR with oligo(dT) reverse-transcribed RNA generated from HASMCs, using PDE3A or PDE3B selective primer sets, confirmed the presence of mRNA for both PDE3A [506 base pairs (bp)] and PDE3B (416 bp) in HASMCs. The identity of the PCR-derived DNA products was confirmed by restriction digests (Fig.2A) and sequencing (Fig. 2B) and were determined to be identical with those previously published for the PDE3 genes. Although mRNA for both PDE3A and PDE3B was detected, significantly more PDE3B mRNA was detected when RNA isolated from DbcAMP-treated HASMCs was used (3.7- ± 0.5-fold increase; Fig. 2C). In contrast, a prolonged DbcAMP treatment had no effect on the amount of HASMC PDE3A mRNA (Fig. 2C). DbcAMP did not alter the mRNA level of expression of the housekeeping gene GAPDH (not shown), which was used to normalize PDE3A and PDE3B levels (Fig. 2C). Thus, whereas mRNA for both PDE3A and PDE3B was readily detected in HASMCs, prolonged increases in cAMP increased only the steady state level of PDE3B mRNA, leaving the PDE3A mRNA level unaltered (Fig. 2C).

Amplification of PDE3A and PDE3B mRNA from human aortic smooth muscle cells. Total RNA was isolated from HASMCs treated for 16 h with vehicle (H2O) or DbcAMP. Conventional RT-PCR methodologies were used to amplify products specific to PDE3A, PDE3B and GAPDH. A, validation of PDE3A and PDE3B RT-PCR product identities via select restriction endonuclease digests. Digestion of the 506-bp PDE3A product with PvuII generates 70- and 436-bp fragments. Similarly, digestion of the 417-bp PDE3B product withEcoRI gives fragments of 67 and 350 bp, respectively.PstI digestion of the PDE3B partial cDNA generated fragments of 352 and 65 bp. The products were resolved electrophoretically using a 2% agarose gel. Bands were visualized using ethidium bromide staining under UV light for photographic documentation. B, nucleotide sequence from PCR-amplified reverse-transcribed HASMC mRNA for PDE3A and PDE3B; C, quantification of PDE3A and PDE3B mRNA levels from human aortic smooth muscle cells. The histogram displays the effect of long-term (16 h) treatment with DbcAMP on HASMC PDE3A and PDE3B mRNA. Densitometric analyses of amplified PDE3A, PDE3B, and GAPDH mRNA were carried out to allow for semiquantitative determination of mRNA levels. Determinations were carried out from several amplifications. *P < .05 versus untreated HASMCs (Student's t test).
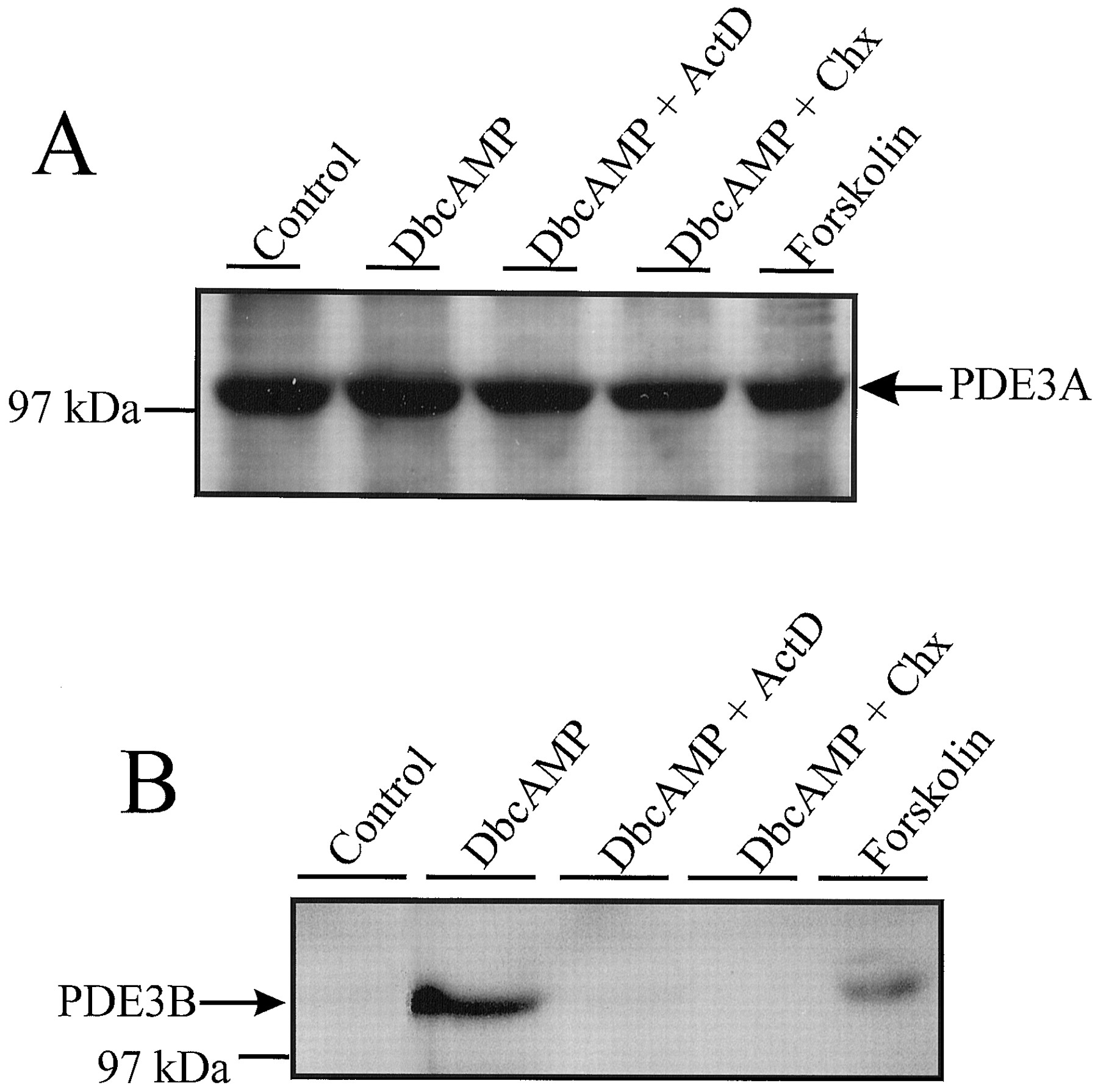
To validate the observed changes in PDE3 gene-specific mRNA species, immunoblotting methodologies to detect PDE3A and PDE3B protein were used. An approximately 110-kDa PDE3A-immunoreactive protein, which comigrated with the well characterized human platelet PDE3A, was readily detected in HASMC lysates, as well as in the resolved cytosolic and particulate fractions (Fig. 3A). Similarly, a 135-kDa PDE3B-immunoreactive protein, which comigrated with recombinant human PDE3B, was detected in HASMC lysates (Fig. 3B). In contrast to PDE3A, which was present in both the cytosolic and particulate fractions of HASMCs, PDE3B was only detected in the resolved particulate fraction of HASMCs (Fig. 3B). When DbcAMP-, or forskolin-treated HASMC lysates were examined, both treatments markedly increased the levels of particulate PDE3B, without affecting the level of PDE3A in either subcellular fraction (Fig. 3, A and B). To further characterize the mechanism by which these isoforms were regulated by the DbcAMP treatments, PDE3 isoform expression was characterized in whole homogenates from HASMCs treated with DbcAMP in the presence or absence of de novo synthetic inhibitors (Fig.4, A and B). Consistent with PDE3B being the DbcAMP-induced cilostamide-inhibited enzymatic activity, both actinomycin D and cycloheximide abrogated the increase in PDE3B without modulating PDE3A expression. (Fig. 4, A and B). These data confirm that the increased PDE3 activity results from a cAMP-mediated increase in the expression of PDE3B mRNA and protein in these cells. The potential for gene-specific functions of PDE3 isoforms has been hinted at by an examination of the kinetic properties of baculovirally expressed PDE3A and PDE3B (Leroy et al., 1996). Specifically, it was observed that PDE3B exhibited an approximately 10-fold lesser sensitivity to cGMP-mediated inhibition, compared with PDE3A. Although this would indicate that cGMP-inhibition of the PDE3 activity in these subcellular fractions could provide a valid approach to characterize the observed changes in PDE3 activity, the presence of the dual specificity (cAMP- and cGMP-hydrolyzing) PDE1C, as well as PDE3A, in the particulate fractions of these cells makes such an approach unfeasible, speaking to the need for isoform-specific probes.

Immunoblotting analysis for the presence and regulation of PDE3A and PDE3B in HASMC. HASMC protein samples from whole homogenates, particulate fractions and soluble fractions of vehicle- and DbcAMP-treated (1 mM; 16 h) HASMCs were analyzed. A, immobilized proteins were probed with an antiserum raised against murine brain PDE3 that has been shown to recognize human PDE3A. An immunoreactive protein of approximately 110 kDa was visualized by chemiluminescence. B, samples were probed with the supernatant from a hybridoma immunized against human PDE3B. In whole homogenates and particulate fractions, a 135-kDa immunoreactive protein was detected.

Analysis for the de novo synthetic regulation of PDE3A and PDE3B in HASMC. SDS-polyacrylamide gel electrophoresis was used to resolve proteins from vehicle-, forskolin- (100 μM; 16 h), and DbcAMP- (1 mM;16 h) treated HASMC whole homogenates. In other lanes, samples from HASMCs treated with DbcAMP in the presence of actinomycin D (ActD; 4 μM) or cycloheximide (CHX; 100 μM) were resolved. A, using a polyclonal antisera that recognizes human PDE3A, a 110-kDa band was detected in all samples. No alteration in the amount of this immunoreactive band was observed following any of the described treatments. B, resolved proteins were probed using a supernatant from hybridoma 281P. A 135-kDa band that comigrated with recombinant PDE3B was detected in HASMC samples, only following treatment with forskolin or DbcAMP.
Potential Therapeutic Implications of Our Findings.
This study represents the first demonstration that both PDE3A and PDE3B can be coexpressed in human cells, and that their subcellular expression pattern and sensitivity to regulation by cAMP are distinct. These findings are significant in the context of PDE3 inhibition in cardiovascular tissues and perhaps more broadly in relation to PDE3 inhibition in other tissues. Although the acute effects associated with PDE3 inhibition in cardiovascular tissues were consistent with the use of PDE3 inhibitors such as milrinone in the treatment of CHF, their negative impact on patient survival precluded their long-term use. Emerging from our work is a hypothesis that states that the effects of prolonged increases in cAMP in cardiovascular tissues on PDE3B expression could have deleterious effects in CHF patients. Although no difference in particulate PDE3 activity was seen when tissues isolated from healthy or failing hearts were compared (Movsesian et al., 1991), the prior identification of two PDE3 proteins of molecular weights consistent with PDE3A and PDE3B in the particulate fraction of human myocardium (Smith et al., 1993) suggests that this question should be reinvestigated with PDE3 isoform-specific probes. In addition, because previous work has identified PDE3B as an important enzyme in mediating the effect of insulin on lipolysis (Degerman et al., 1997), the development of PDE3B-selective inhibitors could be considered an attractive strategy in modulating lipid metabolism. However, given our finding, one might propose that such efforts may bring about untoward cardiovascular effects of these agents, especially if used for prolonged periods. That recent efforts manifesting in the form of clinical trials (Cuffe et al., 2000) have been focused toward reassessing the therapeutic value of PDE3 inhibition in the management of CHF only reinforces the potential significance of these findings. The generation of isoform-selective inhibitory small molecules should ultimately speak to the significance of our findings.
Acknowledgments
We are indebted to Drs. Sharon Wolda and Vince Florio (ICOS Corporation, Bothell, WA) as well as Dr. Joseph Beavo (University of Washington, Seattle, WA) for generously providing immunological reagents for this study and Dr. Charles Graham (Queen's University, Kingston, Ontario, Canada) for kindly providing the human aortic smooth muscle cells.
Footnotes
- Received February 23, 2000.
- Accepted May 10, 2000.
-
Send reprint requests to: Dr. Donald H. Maurice, A221 Botterell Hall, Department of Pharmacology & Toxicology, Queen's University, Kingston, ON, Canada, K7L 3N6. E-mail:Mauriced{at}post.queensu.ca
-
Support for this study came from the Heart and Stroke Foundation of Ontario (Grant-in-aid T-3671) and the Medical Research Council of Canada (Grant-in-aid MT-15540). D.P. was the recipient of an Ontario Graduate Scholarship and a Medical Research Council of Canada Doctoral Research Award. D.H.M. is a Career Research Scientist in Health Sciences sponsored by the Pharmaceutical Manufacturer's Association of Canada-Health Research Foundation/Medical Research Council of Canada.
Abbreviations
- Abbreviations
- PDEs, cyclic nucleotide phosphodiesterases
- PDE3
- phosphodiesterase 3
- HASMCs
- human aortic smooth muscle cells
- RASMCs
- rat aortic smooth muscle cells
- N6,O2′-dibutyryl cAMP
- DbcAMP
- CHF
- congestive heart failure
- bp
- base pair(s)
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}