


Abstract
The constitutive androstane receptor (CAR) regulates mouse and humanCYP2B genes through binding to the direct repeat-4 (DR4) motifs present in the phenobarbital-responsive enhancer module (PBREM). The preference of PBREM elements for nuclear receptors and the extent of cross-talk between CAR and other nuclear receptors are currently unknown. Our transient transfection and DNA binding experiments indicate that binding to DR4 motifs does not correlate with the activation response and that mouse and human PBREM are efficiently ‘insulated’ from the effects of other nuclear receptors despite their substantial affinity for DR4 motifs. Certain nuclear receptors that do not bind to DR4 motifs, such as peroxisome proliferator-activated receptor-α and farnesoid X receptor, can suppress PBREM function via a coactivator-dependent process that may have relevance in vivo. In competition experiments, mouse PBREM is clearly more selective for CAR than human PBREM. Pregnane X, vitamin D, and thyroid hormone receptors can potentially compete with human CAR on human PBREM. In contrast to the selective nature of PBREM, CYP3A enhancers are highly and comparably responsive to CAR, pregnane X receptor, and vitamin D receptor. In addition, the ligand specificities of human and mouse CAR were defined by mammalian cotransfection and yeast two-hybrid techniques. Our results provide new mechanistic explanations to several previously unresolved aspects of CYP2B andCYP3A gene regulation.
Phenobarbital (PB) and many structurally unrelated xenobiotics induce same drug- and carcinogen-metabolizing cytochrome P450 and other genes as a protective response directed toward elimination of these xenobiotics from the body. Among tens of PB-inducible genes, CYP2B genes are the most efficiently activated (reviewed by Waxman, 1999;Honkakoski and Negishi, 2000). Recent studies have established that the constitutive androstane receptor (CAR, NR1I3) is crucial for induction of CYP2B genes by PB and 1,4-bis[2-(3,5-dichloropyridoxy)]benzene (TCPOBOP) because CYP2B mRNA inducibility is lost in CAR null mice (Wei et al., 2000). After forming a heterodimer with retinoid X receptor (RXR, NR2B), the xenobiotic-activated CAR binds to the phenobarbital-responsive enhancer module (PBREM) or unit located in the upstream regions of the mouse Cyp2b10, humanCYP2B6, and rat CYP2B1 and CYP2B2genes (reviewed by Honkakoski and Negishi, 2000). The PBREM contains two CAR/RXR heterodimer binding sites, NR1 and NR2, that conform to the direct repeat-4 (DR4) motif. Successive mutations of DR4 motifs result in gradual loss and, finally, abolition of trans-activation by CAR in HEK293 cells and induction in primary hepatocytes. It is known that NR1 sites alone are sufficient for CAR responsiveness (Sueyoshi et al., 1999), whereas the nuclear factor 1 (NFI) binding site between NR1 and NR2 may contribute to the full inducibility (Honkakoski et al., 1998; Kim et al., 2001).
Several nuclear receptors (NRs), such as vitamin D receptor (VDR, NR1I1), thyroid hormone receptors α/β (TR, NR1A1/2), retinoic acid receptors α/β/γ (RAR, NR1B1/2/3), liver X receptors α/β (LXR, NR1H3/2), pregnane X receptor (PXR, NR1I2) and farnesoid X receptor (FXR, NR1H4) display considerable in vitro binding and activation of DR4-type sites (Mangelsdorf and Evans, 1995; Laffitte et al., 2000;Quack and Carlberg, 2000; Xie et al., 2000b). Therefore, it has been proposed that additional NRs could bind to PBREM and modulate its activity (Waxman, 1999). This idea is in line with evidence that CYP2B mRNA induction is influenced by sex, steroid and thyroid hormones, sterol metabolites, and retinoids, all of which are known NR ligands (Honkakoski and Negishi, 2000). Several inducers such as PB and pesticides activate not only CAR (Sueyoshi et al., 1999) but also PXR, a receptor important for CYP3A gene regulation (reviewed byQuattrochi and Guzelian, 2001). CAR and PXR recognize similar DNA motifs that range from DR2 to DR5 and everted repeat-6 (ER6), and both receptors are expressed in the liver and intestine (Honkakoski and Negishi, 2000; Quattrochi and Guzelian, 2001). Collectively, these data suggest that other NRs might well affect the PBREM enhancer and influence CYP2B gene regulation through cross-talk with CAR.
Surprisingly, there is very little information or systematic studies on PBREM binding or cross-talk with CAR by other NRs. Such studies are much needed for detailed understanding of CYP2B gene regulation, modulating factors, and species differences. So far, we know that CAR can activate the PXR-responsive ER6 and DR3 motifs inCYP3A genes (Sueyoshi et al., 1999; Moore et al., 2000; Xie et al., 2000b; Smirlis et al., 2001). This is consistent with the report that PB can induce CYP3A mRNA in PXR null mice (Xie et al., 2000a). Recently, hPXR and mPXR were shown to bind to DR4 motifs and activate PBREM elements from various species by 3- to 6-fold. This effect is roughly comparable with that of CAR-mediated activation (Xie et al., 2000b; Goodwin et al., 2001; Smirlis et al., 2001). None of these studies, however, could address the preference of PBREM for CAR and PXR. The data are also mostly based on in vitro DNA binding assays with simple DR4 motifs (Sueyoshi et al., 1999; Xie et al., 2000b; Smirlis et al., 2001) instead of functional assays with receptors and PBREM elements. Finally, there is practically no data on the modulation of PBREM by other NRs. Therefore, our aim was to gain more insight to the mouse and human PBREM function and its specificity for CAR by evaluating the effects of several NRs on PBREM activity by functional and DNA binding assays. To help in this effort, the ligand specificities of human and mouse CAR were also defined.
Materials and Methods
Chemicals.
TCPOBOP was synthesized and purified according to Honkakoski et al. (1996) to more than 98% purity as assessed by 1H-NMR spectra and elemental analysis (observed: N, 6.7%; C, 46.9%; H, 2.0%; expected: N, 6.8%; C, 46.8%; H, 2.2%). Steroids were from Steraloids, Inc. (Newport, RI) or Sigma-Aldrich Chemical Co. (St. Louis, MO). WY-14,643 was bought from ChemSyn, Inc. (Lenexa, KS). Other chemicals were at least analytical grade from Sigma, Fluka (Ronkonkoma, NY), or Calbiochem (La Jolla, CA).
Reporter Plasmids.
pCMVβ was purchased from BD Clontech Inc. (Palo Alto, CA). The mPBREM-tk-luc reporter was constructed by insertion of the PBREM element plus the thymidine kinase promoter (tk) from the mouse PBREM-tk-CAT (Honkakoski et al., 1998) intoBglII site of pGL3-Basic luciferase plasmid (Promega, Madison, WI). The rat (rER6)3-tk-luc reporter was constructed similarly from (ER6)3-tk-CAT plasmid (Lehmann et al., 1998) donated by Dr. Steven Kliewer (GlaxoSmithKline, Research Triangle Park, NC). The mouse (mNR1)3-tk-luc, human PBREM-tk-luc, and (hNR1)5-tk-luc plasmids have been described previously (Sueyoshi et al., 1999). The human XREM-3A4-luc reporter containing the proximal 362 base pairs of CYP3A4 gene promoter and the distal enhancer (Goodwin et al., 1999) was a kind gift from Dr. Chris Liddle (University of Sydney at Westmead Hospital, Westmead, Australia). The UAS4-tk-luc (Janowski et al., 1996) and rat CYP3A23[−1360/+82] reporters (Xie et al., 2000b) were donated by Dr. Ronald Evans (Salk Institute for Biological Studies, La Jolla, CA). Other luciferase reporter plasmids for nuclear receptors were generated by inserting multiple copies of their cognate DNA sites intoBglII site of pGL3-Basic plasmid. All plasmids were purified with QIAGEN columns (Hilden, Germany) and verified by restriction mapping, functional testing and, when necessary, by sequencing.
Expression Plasmids.
The sources of expression vectors for mRARα and hRARα (Zelent et al., 1989), cTRα (Harbers et al., 1996), hLXRβ (Teboul et al., 1995), mCOUP-TFI (NR2F1; Cooney et al., 1993), hPPARα and mPPARα (NR1C1; Sher et al., 1993), hFXR (Forman et al., 1995), hCAR and mCAR (Honkakoski et al., 1998; Sueyoshi et al., 1999), hVDR (Quack and Carlberg, 2000) and hPXR and mPXR (Lehmann et al., 1998) have been described previously. The expression plasmid for coactivator hTIF2 (Voegel et al., 1996) was donated by Dr. Hinrich Gronemeyer (IGBMC, Illkirch, France).
GAL4-LBD Fusion Plasmids.
The ligand binding domains (LBD) of mCAR (residues 118–358), hCAR (residues 108–348), mPXR (residues 104–431), and hPXR (residues 107–434) were amplified withPfu DNA polymerase from mouse and human liver RNAs and cloned into 5′ EcoRI and 3′ BamHI orKpnI sites of CMX-GAL4 plasmid (Janowski et al., 1996) donated by Dr. Ronald Evans. GAL4-mCARΔ8 plasmid coding for a truncated mCAR lacking eight amino acids at the C terminus (Choi et al., 1997) was donated by Dr. David Moore (Baylor College of Medicine, Houston, TX).
Ligand Specificities of mCAR and hCAR.
Ligand specificities of mCAR and hCAR were assessed for PBREM preference studies (Figs. 6-8) because CAR and PXR are reported to share some ligands (Moore et al., 2000) and to assess the effect of other NR ligands on mCAR and hCAR activity. The ligand specificities were measured first by chemical-dependent modulation of GAL4 fusion protein-driven reporter activity in HEK293 cells (Table1) according to Honkakoski et al. (2001)and then by yeast two-hybrid assays as described below.
Ligand specificities of CAR and PXR with selected xenobiotics
Human and mouse CAR LBDs were inserted between EcoRI andBamHI sites in pGBKT7 plasmid. The NR interaction domains from mouse (residues 1988–2304) and human (residues 1972–2290) corepressor NCoR (Hu and Lazar, 1999) were cloned from liver RNAs and inserted between EcoRI and BamHI sites in pGADT7 plasmid (Matchmaker GAL4 System 3, BD Clontech). All the manipulations were done essentially according to manufacturer's instructions. Random yeast colonies selected on SD/Leu−/Trp− plates were picked, amplified, and aliquots of cells were then treated with vehicle or test chemicals for 3.5 h before measurement of β-galactosidase activities and cell densities according to Nishikawa et al. (1999).
In Vitro Translation and Gel Shift Assays.
NRs were produced in vitro by first transcribing linearized expression vectors with T7 RNA polymerase and then translating these RNAs in vitro using rabbit reticulocyte lysate as recommended by the supplier (Promega). Nuclear receptor heterodimers with RXR (approximately 10 ng of specific protein; equal protein amounts verified by a parallel translation in the presence of [35S]methionine) were incubated with ligand for 15 min at room temperature in a total volume of 20 μl of binding buffer [10 mM HEPES, pH 7.9, 1 mM dithiothreitol, 0.2 μg/μl poly(dI-dC), and 5% glycerol], which was adjusted to 150 mM KCl. Approximately 1 ng of the32P-labeled human CYP2B6 or mouseCyp2b10 DR4-type NR1 motif (50,000 cpm) was then added, and incubation was continued for 20 min. Protein-DNA complexes were resolved through 8% nondenaturing poly-acrylamide gels in 0.5× Tris/borate/EDTA (45 mM Tris, 45 mM boric acid, 1 mM EDTA, pH 8.3) and were quantified on a FLA3000 reader (Fuji, Tokyo, Japan) using Image Gauge software (Fuji).
Cell Culture and Nuclear Receptor Cotransfection.
Mouse primary hepatocytes were isolated, transfected, and assayed as described previously (Honkakoski and Negishi, 1998; Honkakoski et al., 1998). HEK293 cells (American Type Culture Collection, Manassas, VA) were grown in phenol red-free Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin-100 μg/ml streptomycin (Invitrogen, Gaithersburg, MD). One day before transfection, the cells were seeded on 48-well plates in medium containing delipidated serum (Sigma) to remove potential NR-activating substances. After an overnight incubation, the medium was changed and the cells were transfected using a calcium phosphate method with pCMVβ (50 ng), various luciferase reporter plasmids (25 ng; 100 ng for XREM-3A4-luc and CYP3A23-luc), and variable amounts of expression vectors for NRs (varied from zero to 250 ng). In activation and suppression experiments, the amount of CAR expression plasmid that produced maximal activity from the reporter plasmid was 12.5 ng, and other NRs were titrated from zero to 20-fold excess (250 ng) over CAR so as to reach the effect plateau. In preference experiments, the total amount of NR expression vector was only 50 ng, much below levels that produced any unspecific squelching (≥200 ng). The balance of DNA was kept constant by addition of empty expression vector.
After a 4-h transfection period, the medium was changed. The fresh medium additionally contained an established NR ligand/activator at concentration sufficient for maximal or near-maximal NR response: 20 μM WY-14643 for h/mPPARα, 0.1 μM 1α,25-dihydroxycholecalciferol (VD3) for hVDR, 10 μM rifampicin (RIF) for hPXR, 10 μM mifepristone (RU486) for mPXR, 10 μM 3α-androstenol (ANDR) or 0.5 μM TCPOBOP for mCAR, 10 μM 5β-pregnanedione, 2 μM clotrimazole (CLOTR), or 10 μM 17α-ethynyl-3,17β-estradiol (EE2) for hCAR (see Table 1), 10 μM arotinoid acid for h/mRARα, 50 μM chenodeoxycholic acid for hFXR, 10 μM 25OH-cholesterol for hLXRβ, and 0.1 μM tri-iodothyronine (T3) for cTRα.
Reporter Assays.
Transfected HEK293 cells were cultured for 40 h, washed with PBS, and lysed. Luciferase and β-galactosidase activities (Honkakoski et al., 2001) were determined from 20 μl of lysates in 96-well plates using the Victor2 multiplate reader (PerkinElmer Wallac, Turku, Finland). All luciferase activities were normalized to β-galactosidase expression and expressed as mean ± standard deviation from three to four independent experiments.
Results
Ligand Specificities of mCAR and hCAR.
With GAL4 fusion proteins in HEK293 cells (Table 1), we found that GAL4-mCAR activity was suppressed by ANDR, as expected, and that ANDR-suppressed activity could be reactivated by 0.5 μM TCPOBOP, 10 μM EE2, and 2 μM CLOTR to varying degrees. With GAL4-hCAR, a reproducible partial deactivation (50–60%) by EE2 and about 2-fold activation by CLOTR was seen. Furthermore, the partial deactivation by EE2 could be overcome by addition of CLOTR and, to a lesser extent, by 5β-pregnanedione (Table1). The known ligand profiles of mPXR and hPXR (Moore et al., 2000) were also reproduced: both receptors were activated by CLOTR, RU486, and 5β-pregnanedione, but RIF activated only hPXR.
Yeast two-hybrid assays supported the finding that ANDR and EE2 can deactivate mCAR and hCAR, respectively, because they could dose-dependently increase association between the CAR LBD and the NR interaction domain of the corepressor NCoR by 25- to 50-fold (Fig.1, left, ■, ░). This association could be reversed by TCPOBOP and CLOTR (Fig. 1, right, ▨), whereas these activators themselves had little if any effect on the CAR LBD-NCoR interaction. These results obtained from two independent systems strongly suggest that the EE2 and CLOTR are true, reciprocally acting hCAR ligands.
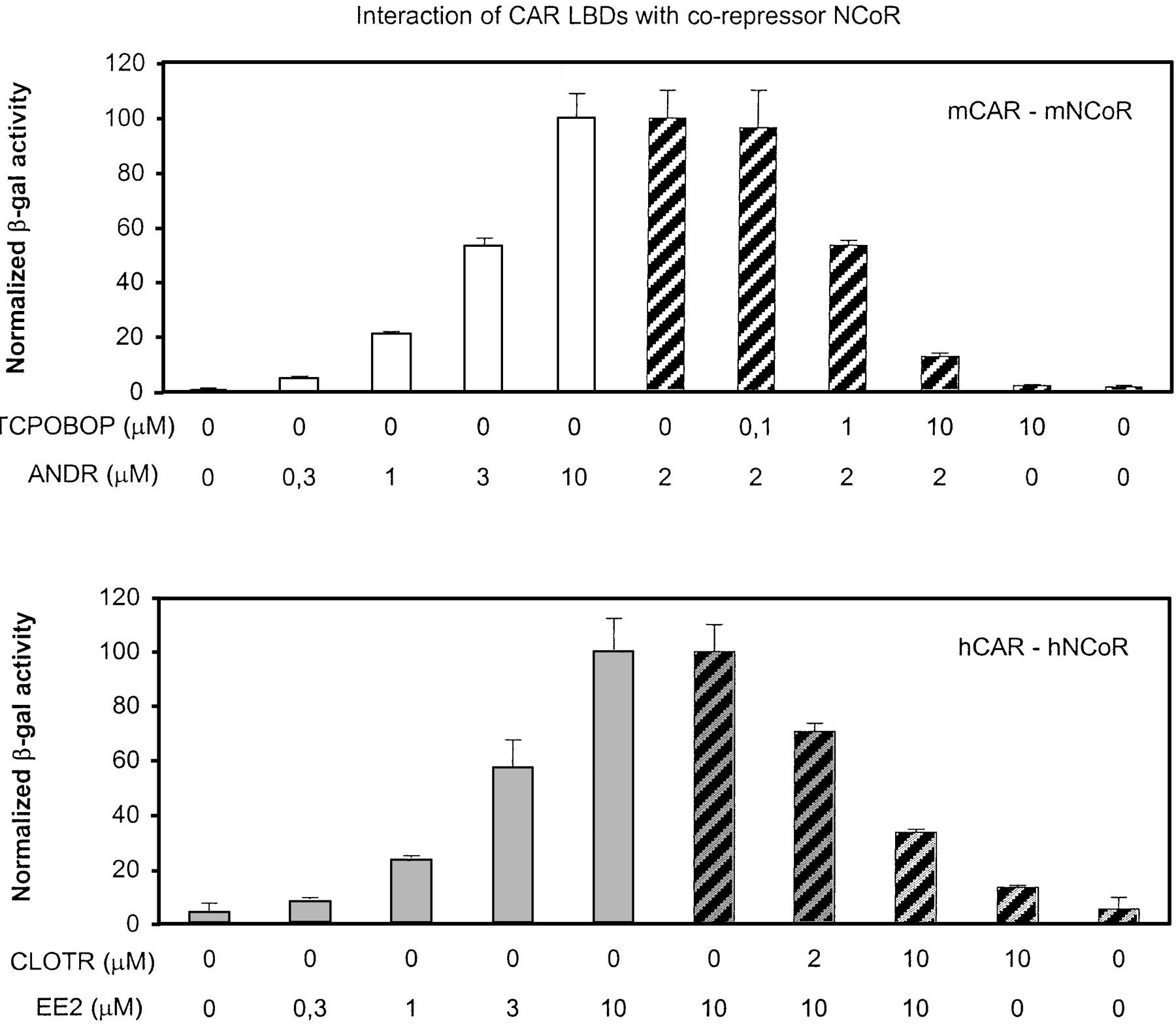
Ligand-dependent association of mouse and human CAR LBD with NR corepressor. Aliquots of yeast cells transformed with GAL4-mCAR LBD (top) or GAL4-hCAR LBD (bottom) plus NR interaction domain from NCoR plasmids were treated for 3.5 h with vehicle or test chemicals at indicated concentrations (micromolar) before cell lysis and β-galactosidase assays as described under Materials and Methods. For mCAR-mNCoR association, the reporter activity with 10 μM ANDR was set to 100 (top, ■). In TCPOBOP displacement experiment (top, ▨), the activity with 2 μM ANDR was set to 100 (same concentration as in mammalian GAL4 assays in Table 1). For hCAR-hNCoR association, the reporter activity with 10 μM EE2 was set to 100 (bottom, ░, ▨). The data shown are mean ± standard deviation from triplicate samples. The experiments were repeated independently two (mCAR) or three times (hCAR) with similar results. Activities with either GAL4-CAR or NCoR plasmid alone were below detection limit.
DNA Binding to NR1 Sites by NRs.
PBREM elements are known to confer about 10-fold activation by CAR in HEK293 cells and about 10-fold induction by TCPOBOP in primary hepatocytes (Honkakoski et al., 1998; Sueyoshi et al., 1999). When organization of the mouse PBREM (5′ NR1-NFI-NR2 3′) was changed to NR1-NFI-NR1 or to NR2-NFI-NR2, the original and NR1-containing PBREM elements retained >10-fold activation. PBREM containing NR2 motifs only conferred much lower ≈3-fold activation by mCAR and TCPOBOP inducibility (Fig.2). This indicates that NR1 is the stronger site for PBREM function.

Activation of mutated mouse PBREM elements. Reporter plasmids containing the wild-type mouse PBREM sequence, NR1 sites only, NR2 sites only, or no enhancer (tk only) was cotransfected in the presence of either empty or CAR expression vector into HEK293 cells (■). Mouse hepatocytes were electroporated with the same reporters and treated with DMSO or 0.5 μM TCPOBOP (▪). Reporter activities from 3–4 independent experiments were measured as described underMaterials and Methods.
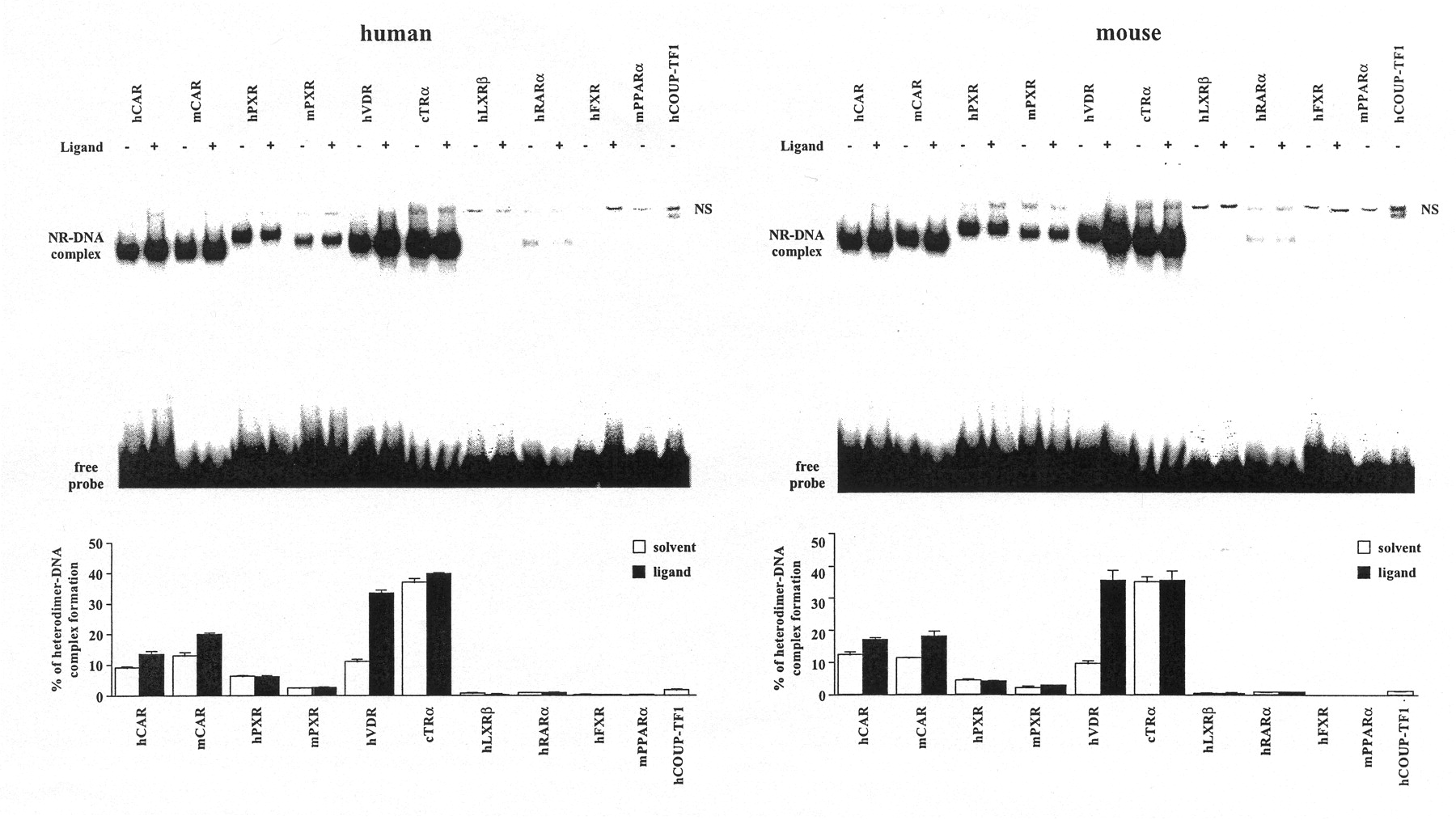
Gel shift assays were performed to compare the ability of NR/RXRα heterodimers to form complexes with NR1 sites (Fig.3). The human and mouse NR1 sites were similar in their binding patterns. In the absence of specific activating ligands, the ranking of complex formation was found to be cTRα ≫ mCAR > hCAR ≈ hVDR > hPXR > mPXR ≈ hCOUP-TFI > hRARα > hLXRβ. Heterodimers of hFXR and hPPARα showed no binding to NR1 sites but were demonstrated to bind to consensus DR1-type motifs (data not shown). Addition of specific ligands enhanced complex formation only for mCAR, hCAR, and hVDR. However, the VD3-induced complex formation of hVDR was so strong that it practically equalled that of cTRα. Thus, cTRα and hVDR surpass both CAR and PXR isoforms in their ability to bind to mouse and human NR1 sites.

RXRα-Heterodimer complex formation of various NRs on the human (left) and mouse (right) NR1 sites. Gel shift experiments were performed with in vitro translated heterodimers of equal amounts of the indicated NRs with RXRα that were preincubated at room temperature with saturating concentrations of activators (▪) [5β-pregnanedione (hCAR), TCPOBOP (mCAR), RIF (hPXR), RU486 (mPXR), VD3 (hVDR), T3 (cTRα), 25OH-cholesterol (hLXRβ), all-trans-retinoic acid (hRARα), chenodeoxycholic acid (hFXR)] or solvent (■) and the 32P-labeled hNR1 or mNR1 site. Protein-DNA complexes were separated from the free probe through 8% nondenaturing polyacrylamide gels. Representative experiments are shown. The amount of heterodimer-DNA complexes in relation to free probe was quantified by bioimaging. Columns and bars indicate mean and S.D., respectively, from three experiments. NS, nonspecific complex.
Activation of NR1 Sites and PBREM Enhancers by NRs.
Binding to NR1 may indicate a potential influence on PBREM activity. To compare between NRs, increasing amounts of NR expression vectors were cotransfected with NR1- or PBREM-driven reporter genes. The NRs were then activated by established ligands, and the reporter activities were measured. The results shown below are the maximal effects observed for each NR, usually at the same concentration as the optimal CAR concentration. The NRs themselves could ligand-dependently activate reporters driven by their consensus response elements (data not shown), demonstrating that the constructs were functional.
First, the maximal effect of NRs on simple DR4 motifs was tested (Fig.4, top, ■, ░). Mouse (NR1)3-tk-luc was activated, in descending order, by mCAR (11.2-fold) ≫ hVDR (3.4-fold) > cTRα (2.6-fold) > mPXR (2.0-fold) ≈ mRARα (1.9-fold). Addition of hLXRβ resulted in a slight 30% increase in activity, whereas COUP-TFI suppressed it by 25%. Human (NR1)3-tk-luc was activated by hCAR (8.9-fold) ≫ hPXR (3.5-fold) > hVDR (2.2-fold) ≈ cTRα (2.1-fold). Human LXRβ and mRARα increased and hFXR and COUP-TFI decreased the human NR1-driven activity slightly. Control experiments with tk-luc plasmid lacking any enhancers established the specificity of NR effects (data not shown). In addition, control experiments with activating NR ligands (Fig. 4, top, ▨, ▧) showed that NR1-elements were not activated in the absence of NR expression vectors. In summary, almost all NRs capable of NR1 binding in vitro were able to activate NR1-driven gene transcription to varying degrees, whereas COUP-TFI inhibited it.
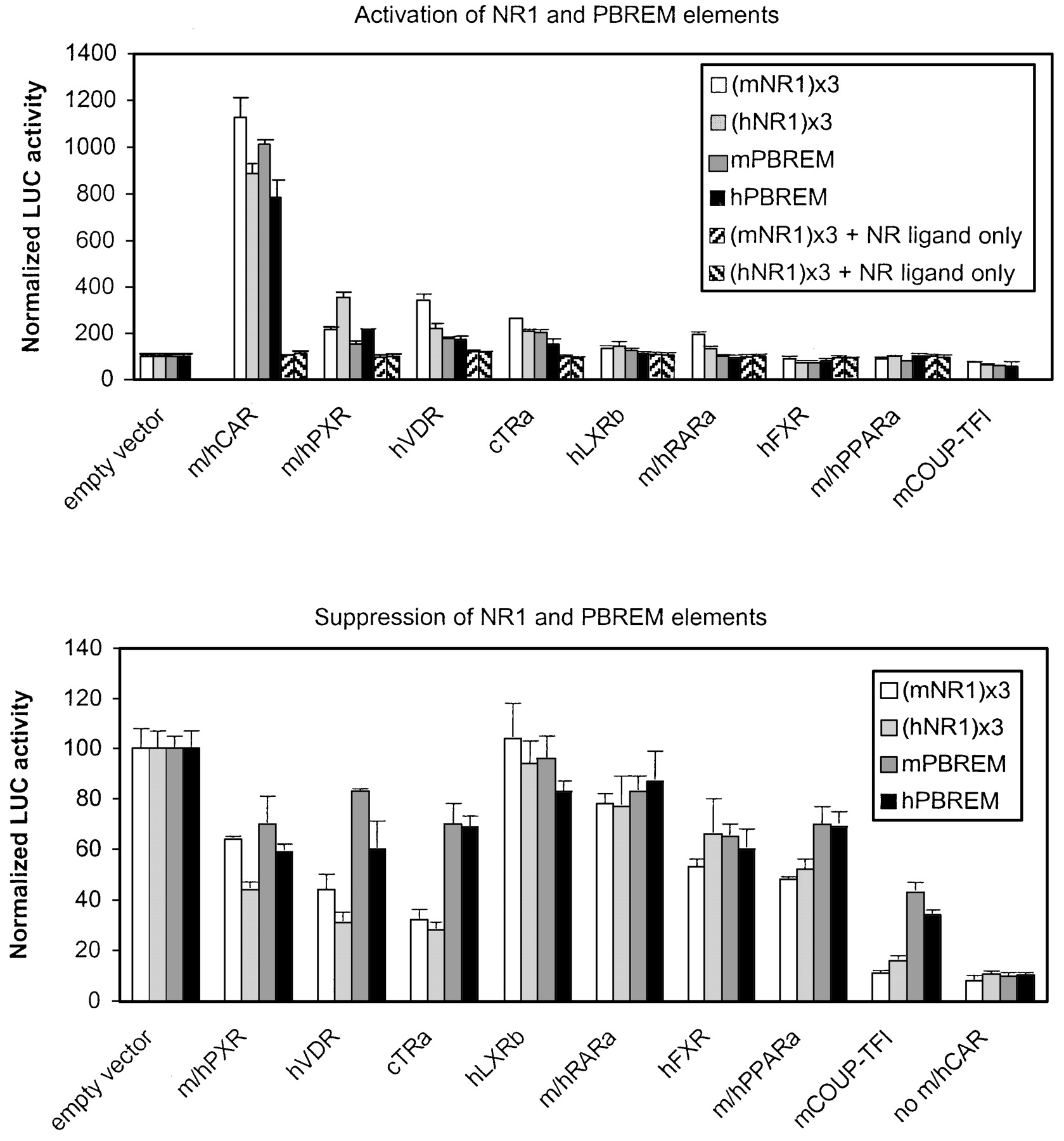
Activation and suppression of mouse and human NR1- and PBREM-driven reporter genes by various NRs. Activation by NRs (top) was assessed by cotransfection into HEK293 cells of increasing amounts of indicated NR expression vectors (0–250 ng) and reporter genes (25 ng) driven by mNR1 (■), hNR1 (░), mPBREM (
 ), or hPBREM (▪) elements and addition of NR-specific ligands. ▨, ▧, ligand controls (empty vector + activating NR ligand). Representative results at optimal 12.5 ng of NR expression vectors (37.5 ng for RARα) are shown, with columns and bars denoting mean and S.D., respectively. Suppression by NRs (bottom) was assessed by cotransfection of increasing amounts of indicated NR expression vectors (0–250 ng) together with saturating amount of mouse or human CAR expression vector (12.5 ng) and reporter genes (25 ng) driven by mNR1 (■), hNR1 (░), mPBREM (
), or hPBREM (▪) elements and addition of NR-specific ligands. ▨, ▧, ligand controls (empty vector + activating NR ligand). Representative results at optimal 12.5 ng of NR expression vectors (37.5 ng for RARα) are shown, with columns and bars denoting mean and S.D., respectively. Suppression by NRs (bottom) was assessed by cotransfection of increasing amounts of indicated NR expression vectors (0–250 ng) together with saturating amount of mouse or human CAR expression vector (12.5 ng) and reporter genes (25 ng) driven by mNR1 (■), hNR1 (░), mPBREM (
 ), or hPBREM (▪) elements. Transfected HEK293 cells were treated with NR-specific ligands. Activity with empty vector was set to 100. Representative results at maximal effect (125 ng of NR expression vectors) are shown, with columns and bars denoting mean and S.D., respectively. No CAR, basal reporter activity in the presence of empty expression vector substituted for both CAR and the competing NRs.
), or hPBREM (▪) elements. Transfected HEK293 cells were treated with NR-specific ligands. Activity with empty vector was set to 100. Representative results at maximal effect (125 ng of NR expression vectors) are shown, with columns and bars denoting mean and S.D., respectively. No CAR, basal reporter activity in the presence of empty expression vector substituted for both CAR and the competing NRs.
When the same experiment was done with PBREM-driven reporters (Fig. 4, top,
 , ▪), a more restricted and attenuated response to NRs was noted. Mouse CAR was by far the strongest activator of the mPBREM (10.1-fold), followed by ≤2-fold activation by cTRα, hVDR, and mPXR. The human PBREM was activated, in descending order, by hCAR (7.9-fold) and ≤2.1-fold by hPXR, hVDR, and cTRα. Human RARα did not affect human PBREM, even though the NR1 element was modestly but reproducibly activated. In addition, the extent of activation by other NRs was always less on PBREM than on NR1 sites. For instance, the activation of NR1 by hVDR or PXR reached 28 and 40% of that by CAR, respectively. On PBREM, hVDR and PXR reached only 18 and 27% of CAR-dependent activity. PBREM seems to be activated preferentially by CAR and then by similar efficiency (≤2-fold) by cTRα, hVDR, and PXR. Mouse PXR was a poorer activator of both NR1 and PBREM elements than hPXR.
, ▪), a more restricted and attenuated response to NRs was noted. Mouse CAR was by far the strongest activator of the mPBREM (10.1-fold), followed by ≤2-fold activation by cTRα, hVDR, and mPXR. The human PBREM was activated, in descending order, by hCAR (7.9-fold) and ≤2.1-fold by hPXR, hVDR, and cTRα. Human RARα did not affect human PBREM, even though the NR1 element was modestly but reproducibly activated. In addition, the extent of activation by other NRs was always less on PBREM than on NR1 sites. For instance, the activation of NR1 by hVDR or PXR reached 28 and 40% of that by CAR, respectively. On PBREM, hVDR and PXR reached only 18 and 27% of CAR-dependent activity. PBREM seems to be activated preferentially by CAR and then by similar efficiency (≤2-fold) by cTRα, hVDR, and PXR. Mouse PXR was a poorer activator of both NR1 and PBREM elements than hPXR.
Suppression of CAR-Activated NR1 Sites and PBREM Enhancers by NRs.
Because NRs may influence PBREM function by competing for DNA binding sites or for common NR coregulators, NRs were cotransfected in the presence of CAR and the maximal NR-mediated suppression of CAR-dependent NR1- or PBREM-driven activities were analyzed. Increasing amounts of NR expression vectors (0- to 20-fold excess over CAR) were used and effects at plateau only (typically 10-fold excess) are shown for clarity.
Figure 4, bottom, indicates that mCAR-activated NR1-driven activity (Fig. 4, ■) was suppressed most efficiently by COUP-TFI (to 11% of control activity), followed by cTRα (32%) and hVDR (44%). Mouse PPARα, hFXR, and mPXR displayed a comparable 50 to 60% decrease, followed by mRARα and hLXRβ. Human CAR (Fig. 4, ░) was suppressed, in descending order, by COUP-TFI (to 16% of control activity) > cTRα ≈ hVDR (about 30%) > hPXR ≈ hPPARα (about 50%), followed by hFXR (65%). Again, hRARα and hLXRβ had little or no effect. A less prominent suppression by the NRs was found on PBREM-driven reporters (Fig. 4, bottom,
 , ▪). Instead of the 50 to 90% decrease in activity that was observed on NR1 sites with COUP-TFI, cTRα, hVDR, PXR, or PPARα isoforms, PBREM was inhibited by only 20 to 60% by the same receptors. There was also a tendency for human NR1 and PBREM to be inhibited more than corresponding mouse elements by NR1-binding PXR isoforms, hVDR, and cTRα. In line with activation results, mPXR was a weaker suppressor than hPXR. In the context of PBREM, hFXR, and PPARα isoforms suppressed CAR as efficiently as PXR isoforms hVDR and cTRα, which bind to and inhibit NR1 more avidly.
, ▪). Instead of the 50 to 90% decrease in activity that was observed on NR1 sites with COUP-TFI, cTRα, hVDR, PXR, or PPARα isoforms, PBREM was inhibited by only 20 to 60% by the same receptors. There was also a tendency for human NR1 and PBREM to be inhibited more than corresponding mouse elements by NR1-binding PXR isoforms, hVDR, and cTRα. In line with activation results, mPXR was a weaker suppressor than hPXR. In the context of PBREM, hFXR, and PPARα isoforms suppressed CAR as efficiently as PXR isoforms hVDR and cTRα, which bind to and inhibit NR1 more avidly.
Suppressive Effects of NRs Occurring through CAR LBD.
Expression vectors for GAL4-m/hCAR and UAS4-tk-luc reporter were used in the above suppression assay to determine whether suppression could be attributed to competition for factors associated with the CAR LBD. This approach would eliminate any competition at the level of DNA binding, which was prominent for cTRα, hVDR, and PXR isoforms. Figure5, top, indicates that with both GAL4-mCAR and GAL4-hCAR as activators, the strongest suppressors were full-length CAR and cTRα (<20% of control activity), followed by COUP-TFI ≈ PXR isoforms ≈ hFXR (25–30%), whereas hVDR, RARα, and PPARα isoforms were weaker suppressors (35–55%). No suppression was seen with GAL4-mCARΔ8 that lacks the AF2 core sequence (data not shown), indicating that NR-mediated suppression of CAR depends on the presence of intact AF2 domain. Therefore, suppression of CAR LBD probably reflects competition for NR coactivators. Indeed, cotransfection of TIF2 vector in this suppression assay (Fig. 5, bottom) resulted in partial restoration of mCAR-dependent and especially hCAR-dependent reporter activity. Differences in the extent of suppression and restoration of reporter activity further imply that NRs may have different affinities for various NR coactivators. In summary, NR1-binding cTRα and hVDR showed a remarkable difference in their ability to suppress CAR LBD. PPARα isoforms and hFXR that do not bind to NR1 sites could inhibit PBREM through an AF2- and coactivator-dependent mechanism.

Suppression of GAL4-CAR-activated reporter activities by various NRs. Top, suppression by NRs was assessed by cotransfection of increasing amounts of indicated NR expression vectors (0–250 ng) together with saturating amount of mouse (■) or human (▪) GAL4-CAR LBD expression vector (12.5 ng) and UAS4-tk-luc reporter (25 ng) into HEK293 cells, followed by addition of NR-specific ligands. Reporter activity with empty vector was set to 100. Representative results at maximal effect (125 ng of NR expression vectors, 10-fold excess over CAR) are shown, with columns and bars denoting mean and S.D., respectively. GAL4 only, basal activity in the absence of any LBD in the construct. Bottom, reactivation of suppressed GAL4-mCAR- or GAL4-hCAR-dependent activity was performed as above with cotransfection (500 ng) of empty expression vector (■,
 ) or hTIF2 plasmid (
) or hTIF2 plasmid (
 , ▪).
, ▪).
Preference of NRs for PBREM Enhancers.
As shown above, DNA binding studies were not sufficient to assess the effect of NRs for PBREM enhancers. Furthermore, the activation and suppression experiments yielded information on only the maximal effect by an NR, not on the preference of PBREM for a particular NR. Therefore, detailed titrations with selected NR1-binding NRs were performed. The transfected cells were treated with CAR-deactivating chemical and an activator specific for the competing NR. We selected ANDR and EE2 for mCAR and hCAR, respectively, because ANDR can completely deactivate mCAR (Forman et al., 1998; Sueyoshi et al., 1999), and EE2 is a partial deactivator of hCAR (Table 1, Fig. 1). In contrast, ANDR and EE2 had either a slight positive effect on PXR isoforms (Table 1) or did not affect other NRs at all (see below). These “reciprocal” effects on NR activity allowed us to better assess the functional preference of PBREM.
It should be noted that in preference studies, much lower total amounts of NRs than in suppression assays (Fig. 4, bottom) were used (50 versus 250 ng), and therefore unspecific squelching effects are not likely. In activation experiment titrations (data not shown), we did not see any significant self-suppression by increasing amounts of CAR, PXR, or any other NR plasmid. This would happen if NR coregulators were a limiting factor and result in squelching. This did not seem to be the case.
Figure 6, top, shows that in the presence of saturating amounts of mPXR only, mPBREM was activated 2-fold by RU486, regardless of the presence of ANDR, as expected from data in Table 1. In the presence of mCAR only, mPBREM was activated 7-fold, which was completely abolished by ANDR. Already at 1:25 ratio of mCAR to mPXR expression vectors, the combined RU486+ANDR treatment suppressed the mPBREM-driven activity to control levels, suggesting that mCAR clearly dominates mPXR on PBREM. Mouse PBREM was activated 2-fold by 0.1 μM VD3 in the presence of hVDR (Fig. 6, middle). When the ratio of mCAR to hVDR was increased stepwise, the combined VD3+ANDR treatment gave slightly higher LUC activities than ANDR alone up to 1:25 ratio, suggesting that hVDR binds to mPBREM slightly more avidly than mPXR. However, the hVDR-mediated suppression at 5:25 ratio (about 15%) was less than for mPXR. Both these results are well in line with DNA binding and GAL4-mCAR suppression studies (Figs. 3 and 4, bottom). Because of very modest activation of mPBREM by cTRα (see Fig. 3), the experiment was done with mNR1 reporter (Fig. 6, bottom). Already at 1:25 and greater ratios of mCAR to cTRα, the combined T3+ANDR treatment decreased activities to control levels, suggesting a strong mCAR dominance over cTRα.

Dominance of mCAR over other NRs on mPBREM. Activation of mPBREM- or mNR1-driven reporter genes (25 ng) was assessed by cotransfection of indicated amounts of NR expression vectors (0–25 ng) into HEK293 cells, with balance of DNA kept constant by addition of empty expression vector. The transfected cells were treated with vehicle (■), mCAR-specific deactivator ANDR (▪), competitor NR-specific activating ligand (░), or their combination (▨). Activity with empty vector plus vehicle only was set to 100. Columns and bars denote mean and S.D., respectively, from three independent experiments.
Human PBREM was activated 1.8-fold by EE2 and 2.5-fold by RIF in the presence of hPXR only. As predicted, hPBREM activity was decreased to 40% by EE2 but not affected by RIF in the presence of hCAR only (Fig.7, top). Compared with results with mCAR-to-mPXR titration on mPBREM, hPXR predominated strikingly over hCAR at a 1:25 ratio, showed substantial activity at a 5:25 ratio; hCAR predominated only at a 25:25 ratio. Cotransfection of hVDR (Fig. 7, middle) that combined VD3+EE2 treatment reduced the activity below those elicited by VD3 alone beginning at hCAR-to-hVDR ratio of 5:25. In contrast to mPBREM, hPBREM activity was substantially reduced (to 60%) by VD3, matching the similar difference seen in suppression assay in Fig. 4. On human NR1 sites, cTRα was the dominant receptor up to 5:25 ratio of hCAR to cTRα, above which the combined T3+EE2 treatment began to decrease the activity (Fig. 7, lower). These results show that human PBREM was less selective for CAR than mouse PBREM.

Dominance of hCAR over other NRs on hPBREM. Activation of hPBREM- or hNR1-driven reporter genes (25 ng) was assessed as in Fig. 6. The transfected cells were treated with vehicle (■), hCAR-specific partial deactivator EE2 (▪), competitor NR-specific activating ligand (░) or their combination (▨).
For comparison, PXR-responsive XREM-3A4-luc and CYP3A23-luc reporters were used in similar preference studies with hCAR, mCAR, and hVDR. Figure 8, top, shows that the human XREM enhancer was activated 2- to 3-fold by EE2, RIF, and their combination when hPXR only was present. XREM activity driven by hCAR only was again decreased 50% by EE2 but not affected by RIF. Titration with increasing amounts of hPXR vector indicated that the decreasing effect of EE2 on XREM-driven activity was lost only at 25:25 ratio of hPXR to hCAR. This suggests that hCAR has considerable affinity to XREM motifs ER6 and/or DR3. Experiments with (rER6)3-tk-luc reporter proved that at least ER6-driven activity could be enhanced comparably by hCAR (6-fold) and hPXR (4-fold) (data not shown). Transfection of hVDR and VD3 treatment increased XREM-driven activity very strongly (Fig. 8, middle). Transfection of equal amounts of hPXR and hVDR resulted in more than 60 and 40% suppression of hVDR- and hPXR-dependent activities, respectively. This suggested similar competition between hVDR and hPXR for the XREM binding sites but higher activation potential by hVDR. This notion was supported by the finding that (rER6)3-tk-luc and CYP3A23-luc reporters were induced 5- and 20-fold, respectively, by ligand-activated hVDR (data not shown). Figure 8, bottom, shows that mCAR and mPXR activated the CYP3A23-luc reporter over 4- and 3-fold, respectively. Mouse CAR has substantial activity over mPXR, because the combined RU486+ANDR treatment began to increase reporter activity above ANDR levels only at 25:25 ratio of mPXR to mCAR. Similar mCAR dominance over mPXR were seen with (rER6)3-tk-luc reporter (data not shown). Our results indicated that, in contrast to PBREM elements more selective for CAR, the CYP3A enhancers are very responsive to hVDR, CAR, and PXR isoforms.
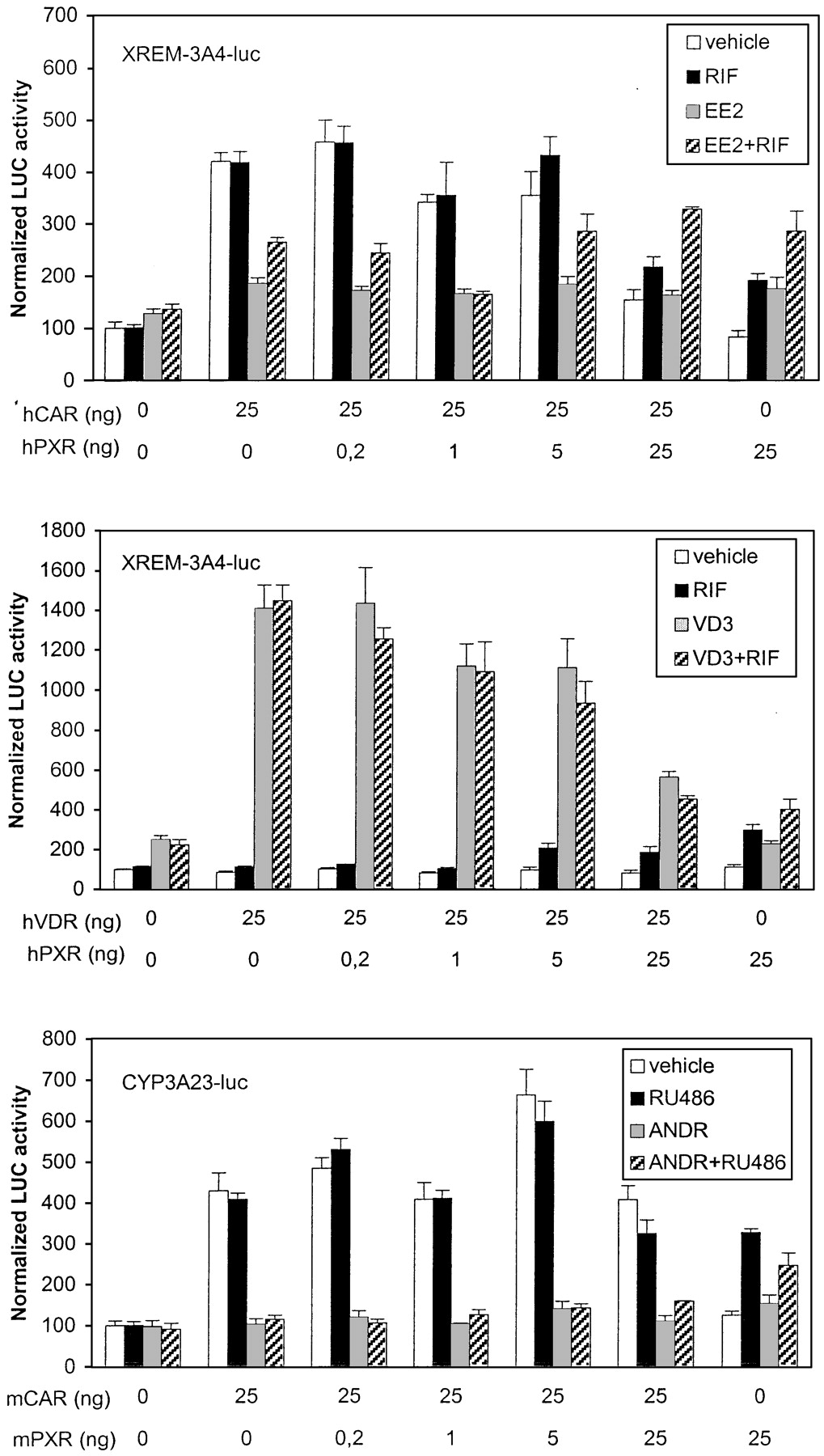
Dominance of hPXR and mPXR over other NRs on CYP3A enhancers. Activation of XREM-3A4- or CYP3A23-driven reporter genes (100 ng) was assessed as in Fig. 6. The transfected cells were treated with vehicle (■), hPXR-specific RIF or mPXR-specific RU486 (▪), competitor NR-specific ligand (░), or their combination (▨).
Discussion
The preference of PBREM for various NRs is not known although many NRs can bind to DR4-type motifs contained in PBREM. Thus, assessment of NRs with respect to their PBREM-modulating activity is important for understanding of CYP2B gene regulation, mechanisms, and species differences therein. Our studies were aimed at resolving the functional interplay between several NRs expressed in the liver and the mouse and human PBREM elements. To help in this task, CAR ligand binding specificities had to be defined in more detail as well.
Ligand Specificities of mCAR and hCAR.
The known ligand profiles of mCAR and PXR isoforms (Forman et al., 1998; Lehmann et al., 1998; Sueyoshi et al., 1999; Moore et al., 2000) were well reproduced in our GAL4 fusion protein assays. With respect to hCAR, we confirmed that 5β-pregnanedione was a modest activator, TCPOBOP had no effect, and ANDR was a weak deactivator, as shown earlier by Moore et al. (2000). Intriguingly, EE2 proved to be an activator of mCAR but a partial deactivator of hCAR. Because HEK293 cells do not express estrogen receptors (Kahlert et al., 2000), EE2 cannot inhibit hCAR activity via estrogen receptor-dependent squelching. EE2 was found to strongly promote the interaction between hCAR LBD and a NR corepressor, lending strong support for direct inhibitory action of EE2 on hCAR. In contrast to the report by Moore et al. (2000), we did not detect any suppression by CLOTR of hCAR activity. Instead, CLOTR activated GAL4-hCAR on its own and could also overcome the inhibition by EE2. This finding was also supported by our yeast two-hybrid experiments.Moore et al. (2000) reported decreases by CLOTR in hCAR activity in CV-1 cells and in in vitro association between hCAR LBD and coactivator SRC-1 with a FRET-based assay. Perhaps cell- and assay-specific differences may explain these differences. For instance, the decrease by ANDR of mCAR–SRC1 interaction that was seen in a GST pulldown assay (Forman et al., 1998) could not be reproduced by Moore et al. (2000). In our view, deactivators may be studied best with corepressor association assays.
NR1 Binding and Activation Specificity.
Although NR1 sites alone confer CAR responsiveness (Sueyoshi et al., 1999), the presence of both NR1 and NR2 sites in the natural PBREM enhancer seems crucial for optimal activation (Honkakoski et al., 1998; Goodwin et al., 2001). Despite previous observations that mouse CAR/RXRα heterodimer binds to NR1 and NR2 sites with equal efficiency in vitro (Tzameli et al., 2000), the present functional studies indicated that NR1 site is the stronger of these DR4 motifs. Paquet et al. (2000) have also suggested that NR1 and NR2 in rat CYP2B2 gene are not identical. Among many NRs capable of DR4 binding, only hPXR and mPXR have been reported to bind to the NR1 site with affinity similar to CAR (Xie et al., 2000b; Goodwin et al., 2001; Smirlis et al., 2001). Here, many other NRs were assessed through in vitro translation and NR1 probe binding under optimized conditions. Human VDR and cTRα bound to NR1 with greater efficiency than CAR, which in turn displayed better binding than PXR isoforms. If the binding efficiency to NR1 were the sole determinant of PBREM activation, then one would predict that cTRα and hVDR would be strong activators of PBREM. Clearly, this was not the case. On simple NR1 sites, activation by CAR greatly surpassed that of cTRα, VDR, or PXR, which showed a maximal 2- to 3.5-fold activation. On natural PBREM elements, these three receptors were even less efficient. This is in contrast with the results of Smirlis et al. (2001) and Xie et al. (2000b), who found similar or 40% smaller activation of rodent PBREMs by mPXR or hPXR than by mCAR, respectively. However, they found ∼2-fold activation by PCN of PBREM in hepatocytes, which is similar to the 2- to 2.5-fold activation by RU486 seen in HEK293 cells.
NR Cross-Talk Is Attenuated on PBREM Elements.
The activation potential of NRs was significantly weaker on PBREM enhancers compared with NR1 sites. This suggests that NR interaction with DR4 motifs imbedded in PBREM is restricted, resulting in increased specificity for CAR. Furthermore, PBREM enhancers are more ‘insulated’ than simple DR4 motifs from the repressive effects, as shown by diminished suppression by, for example, cTRα, hVDR, PXR, and PPARα isoforms. Only COUP-TFI, a well-known suppressor (Cooney et al., 1993), could bring the CAR-dependent PBREM activity below 50%. We observed that hPBREM is notably less selective for CAR and more prone to NR-mediated suppression than mPBREM. In hPBREM, the NFI site seems to be mutated (Sueyoshi et al., 1999); therefore, NFI might play a role in the high selectivity of mPBREM. Kim et al. (2001) have recently shown that NFI and CAR can bind simultaneously to rat PBREM in vitro and that NFI coexpression may enhance trans-activation by CAR. This attractive mechanism cannot yet explain the selectivity of PBREM for CAR because CAR and NFI bound independently of each other, at least in vitro, and other NRs could potentially substitute for CAR. It may be possible that specific cofactors, lacking from in vitro studies, mediate the interaction between NFI and CAR. Other possibilities include co-operation between CAR-bound NR1 and NR2 sites that cannot be reproduced on multimeric NR1 sites. This option is consistent with the earlier report that mutation of any NR half-site in mPBREM reduced the PB inducibility to a similar extent (Honkakoski et al., 1998). Further studies into these hypotheses are warranted.
Preference of PBREM and XREM for NRs.
The NR preference studies indicated that mCAR predominates on mPBREM over weaker effectors such as mPXR. Therefore, only weak activation by pure NR ligands of Cyp2b10 gene might be expected in vivo. Indeed, hepatocytes transfected with a PBREM construct showed only 2-fold activation after PCN treatment (Xie et al., 2000b; Smirlis et al., 2001); hepatic CYP2B10 was induced 37-fold by PB but only 7-fold by PCN (Pellinen et al., 1994); CYP2B10 mRNA induction was not affected either by thyroid hormone or by retinoic acid in mouse hepatocytes (Honkakoski and Negishi, 1998); T3 does not seem to affect PBREM or its associated factors in rats (Ganem et al., 1999). The identity of 5′-flanking nucleotides in DR4 motifs is important for TR-mediated activation (Harbers et al., 1996; Zhang and Lazar, 2000) and this property may explain the discrepancy between the strong binding and inefficient function by TRα on PBREM. Although TR isoforms are expressed in liver (Zhang and Lazar, 2000), and TR can inhibit CAR LBD, the levels of TR relative to CAR may be too low for significant suppression via competition for NR coregulators. On the other hand, hPBREM seems to allow some hVDR and especially hPXR interactions. This probably explains why RIF, a specific hPXR ligand, can efficiently induce CYP2B6 mRNA in human hepatocytes (e.g., Goodwin et al., 2001). To our knowledge, there are no data available on the response ofCYP2B genes to VDR ligands or VDR status.
The CYP3A4 and CYP3A23 enhancers, in contrast, respond not only to PXR but also to CAR and VDR. These experiments now give, for the first time, a mechanistic explanation of the strong inducibility ofCyp3a genes by the mCAR ligand TCPOBOP (e.g., Smith et al., 1993), to the induction of CYP3A4 mRNA by VD3 in Caco-2 cells (Schmiedlin-Ren et al., 1997), and suggest why CYP3A andCYP2B genes tend to be coregulated in humans. Our results are in contrast with Moore et al. (2000), who found that hPXR predominated hCAR on the XREM enhancer. Their conclusion was based on the repressive effect of CLOTR on hCAR, a finding that we could not reproduce with either full-length hCAR, GAL4 fusion plasmids, or with yeast two-hybrid assays.
Interference of CAR Signaling without Significant NR1 Binding.
PPARα and FXR that bind poorly if at all to NR1 sites can still significantly inhibit CAR-mediated signaling. This suppression seems to be caused by reversible competition for coactivators. Recent experiments with NR null mice suggest that the interference of CAR function, detected here by cotransfection assays, may have physiological relevance in the liver where all these NRs are predominantly expressed. For example, the lack of PPARα greatly enhances the mitogenic effects of TCPOBOP (Columbano et al., 2001) that are mediated by CAR (Wei et al., 2000). Because CAR/RXRα heterodimer binding is important for both basal and inducible CYP2B gene expression (Wan et al., 2000; Wei et al., 2000), it is possible that PPARα suppresses CAR and exerts an effect on PBREM. The activation ofCYP3A and CYP2B gene expression in the absence of FXR (Schuetz et al., 2001) is difficult to interpret similarly because bile acids that accumulate in FXR null mice are also activators for PXR (e.g., Schuetz et al., 2001) and possibly weak ligands for mCAR as well (see Table 1). An FXR-specific chemical probe should help resolve this question and further elucidate interactions between NRs and P450 gene expression.
Finally, we have attempted to classify NRs based on the observed DNA binding and PBREM-suppressive effects (Table2). This data combined with approximate hepatic levels of mouse NRs reported in the literature (e.g., Wan et al., 2000; Xie et al., 2000a; Zhang and Lazar, 2000) may allow us to predict the relevance of NR cross-talk with CAR signaling. As positive signs of this predictability, the moderate or weak efficiency of mPXR, TRα, and RARα for PBREM activation is indeed reflected in some in vivo studies (Pellinen et al., 1994; Honkakoski and Negishi, 1998;Ganem et al., 1999). Similarly, PPARα seems to suppress CAR activity in vivo (Columbano et al., 2001).
Summary of NR effects on CAR signaling
Collectively, our studies generate hypotheses for further in vivo experiments that must, however, be carefully controlled to rule out any nonspecific effects occurring outside PBREM. As examples of the associated problems, CYP3A and CYP2B genes contain NR binding sites also outside of XREM and PBREM. For examples, the CYP3A promoter contains DR1 sites that are crucial for CYP3A basal activity (Quattrochi and Guzelian, 2001) but are also targets for COUP-TFI, hepatocyte nuclear factor-4, and perhaps other NRs as well. T3 suppresses CYP2B mRNA expression in rats but does not affect PBREM (Ganem et al., 1999). Moreover, these experiments would require truly monospecific NR ligands to avoid, for example, interference with glucocorticoid signaling that is essential for CYP2Bregulation (Honkakoski and Negishi, 2000) that plagues the mPXR ligands PCN and RU486, and the cross-activation of NRs such as FXR and PXR by bile acids. Given the lower selectivity of human PBREM and XREM for NRs, human hepatocytes would probably be the best system in which to run the experiments. Most importantly, tissue or hepatocytes from NR null mice would be most valuable in confirming the observed NR interferences.
In conclusion, our results indicate that binding of an NR to NR1 sites does not correlate with its functional effects in the context of PBREM. The use of simple NR motifs for binding and trans-activation assays may not reveal actual function of an NR on natural DNA elements. Mouse PBREM was found to be more selective for CAR than human PBREM, which is also activated by PXR, VDR, and TRα. In contrast to PBREM elements, CYP3A enhancers were highly responsive to VDR, CAR, and PXR. PPARα and FXR may use mechanisms dependent on coactivators to interfere with CAR signaling.
Acknowledgments
We thank Drs. Pierre Chambon (IGBMC, Illkirch, France), Ronald Evans, Frank Gonzalez (NCI, Bethesda, MD), Hinrich Gronemeyer, Steven Kliewer, David Mangelsdorf, (University of Texas Southwestern Medical Center, Dallas, TX), Masahiko Negishi and Cary Weinberger (NIEHS, Research Triangle Park, NC), Ming-Jer Tsai (Baylor College of Medicine, Houston, TX), Björn Vennström (Karolinska Institute, Stockholm, Sweden), Steven Kliewer, Chris Liddle, David Moore, for plasmids, and Kaarina Pitkänen for technical assistance.
Footnotes
- Received October 24, 2001.
- Accepted May 1, 2002.
-
This study was supported by Academy of Finland grants 44040 and 51610 (to P.H.) and 50331 (to C.C.).
Abbreviations
- PB
- phenobarbital
- CAR
- constitutive androstane receptor
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- RXR
- retinoid X receptor
- PBREM
- phenobarbital-responsive enhancer module
- DRn
- direct repeat with n base-pair spacing
- ERn
- everted repeat with nbase-pair spacing
- HEK
- human embryonic kidney
- NR
- nuclear receptor
- NFI
- nuclear factor 1
- VDR
- vitamin D receptor
- TR
- thyroid hormone receptor
- RAR
- retinoic acid receptor
- LXR
- liver X receptor
- PXR
- pregnane X receptor
- FXR
- farnesoid X receptor
- ERn
- everted repeat with n base-pair spacing
- WY-14,643
- [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid
- tk
- thymidine kinase promoter
- LBD
- ligand-binding domain
- NCoR
- nuclear receptor corepressor
- VD3
- 1α,25-dihydroxycholecalciferol
- RIF
- rifampicin
- RU486
- mifepristone
- ANDR
- 3α-androstenol
- CLOTR
- clotrimazole
- T3
- tri-iodothyronine
- COUP-TFI
- chicken ovalbumin upstream promoter-transcription factor I
- AF2
- activation function-2
- trVD3
- 1α,25-dihydroxycholecalciferol
- XREM
- xenobiotic-responsive enhancer module
- EE2
- 17α-ethynyl-3,17β-estradiol
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}