


Abstract
Endocannabinoids may serve as retrograde messengers to inhibit neurotransmitter release during depolarization-induced suppression of inhibition (DSI) or excitation (DSE). We therefore tested whether endocannabinoids inhibit N-type voltage-dependent Ca2+ channels by activating Gi/o-protein-coupled CB1 cannabinoid receptors (CB1R)—a possible mechanism underlying DSI/DSE. Three putative endocannabinoids [2-arachidonylglycerol (2-AG), 2-arachidonyl glycerol ether (2-AGE), and anandamide (AEA)] and the cannabimimetic aminoalkylindole WIN 55,212-2 (WIN) inhibited whole-cell Ca2+ currents in rat sympathetic neurons previously injected with cDNA encoding a human CB1R. Agonist-mediated Ca2+ current inhibition was blocked by a selective CB1R antagonist [SR141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride] and pertussis toxin (PTX) pretreatment. The rank order of potency was WIN (IC50 = 2 nM) > 2-AGE (350 nM) ∼ 2-AG (480 nM) > AEA (∼3 μM), with each agonist displaying similar efficacy (approximately 50% maximal inhibition). Increasing CB1R expression level significantly enhanced AEA potency. AEA (10 μM) also inhibited Ca2+ channels in a voltage-independent, CB1R-independent, and PTX-insensitive manner, whereas 2-AG and 2-AGE were devoid of this activity. All three endocannabinoids activated G-protein-coupled inwardly rectifying potassium (GIRK) channels, GIRK1/4, heterologously expressed in sympathetic neurons. These results suggest a mechanism by which endocannibinoids might influence presynaptic function.
Δ9-Tetrahydrocannabinol, the psychoactive principle of marijuana, influences various neural functions by activating central and peripheral cannabinoid receptors. Two types of cannabinoid receptors, CB1R and CB2R, have been identified, and both couple to G-proteins of the Gi/o class. Whereas CB2Rs are limited to the immune system, CB1Rs are found in both the central and peripheral nervous system as well as in a variety of non-neuronal tissues. The molecular identification of the CB1R prompted the search for endogenous ligands (endocannabinoids) that bind to and activate cannabinoid receptors. Thus far, at least three putative endogenous ligands for cannabinoid receptors, anandamide (AEA) (Devane et al., 1992), 2-arachidonyl glycerol (2-AG) (Mechoulam et al., 1995), and noladin ether (2-arachidonyl glycerol ether, 2-AGE) (Fezza et al., 2002), have been isolated.
Depolarization-induced suppression of inhibition (DSI) or excitation (DSE) in central neurons involves presynaptic inhibition of neurotransmitter release after depolarization of the postsynaptic neuron and release of an unknown retrograde messenger. Recently, evidence supporting a role for endocannabinoids as the elusive messenger has emerged. For example, SR141716A (SR), a CB1R antagonist, prevents hippocampal and cerebellar DSI as well as cerebellar DSE (Wilson and Nicoll, 2001; Wilson et al., 2001; Ohno-Shosaku et al., 2002; Kreitzer and Regehr, 2001a,b). Furthermore, hippocampal and cerebellar DSI as well as striatal long-term depression were absent in mice genetically devoid of CB1R (Wilson et al., 2001; Gerdeman et al., 2002; Yoshida et al., 2002). Thus, a proposed sequence of events underlying DSI/DSE is: 1) depolarization of a postsynaptic neuron stimulates the synthesis of endocannabinoids via Ca2+ influx through voltage-gated Ca2+ channels; 2) the newly synthesized endocannabinoid escapes the postsynaptic neuron through as yet unknown mechanisms and diffuses across the synaptic cleft to inhibitory or excitatory presynatpic terminals containing CB1Rs; and 3) activation of heterotrimeric G-proteins results in inhibition of voltage-gated Ca2+ channels and subsequently the release of GABA or glutamate.
Within the context of this model, it is important to establish whether voltage-gated Ca2+ channels involved in presynaptic inhibition are modulated by endocannabinoids. Although N- and P/Q-type Ca2+ channels have been shown to be modulated by synthetic CB1R agonists such as WIN 55212-2 (WIN) and CP 55,940 (Caulfield and Brown, 1992; Mackie and Hille, 1992; Mackie et al., 1993; Pan et al., 1996), evidence supporting a similar function for endocannabinoids is quite limited. Mackie et al. (1993) showed that AEA acts as a partial agonist to inhibit N-type Ca2+ channels in differentiated N18 neuroblastoma cells. In contrast, AEA had little effect on N-type Ca2+ channels in the majority of rat sympathetic neurons microinjected with CB1R cRNA (Pan et al., 1996). In terms of 2-AG and 2-AGE, very little is known about potential actions on ion channels. Finally, extrapolation of endocannabinoid action from the effects of synthetic agonists is potentially challenging because the two classes of compounds do not always produce the same effect. For example, it was reported that WIN blocked the potent emetogenic effects of 2-AG (Darmani, 2002).
In the present study, we sought to examine whether three putative endocannabinoids could modulate N-type Ca2+ channels by activating CB1Rs. We also tested whether endocannabinoids could activate G-protein-coupled inwardly rectifying potassium (GIRK) channel, another type of ion channels that has been reported be activated by WIN via CB1Rs in AtT20 cells and Xenopus laevis oocytes (Henry and Chavkin, 1995; Mackie et al., 1995). Because native CB1R expression is largely restricted to presynaptic terminals, we took advantage of a neuronal expression system in which CB1Rs are heterologously expressed in rat sympathetic neurons and couple to natively expressed G-proteins and N-type Ca2+ channels.
Materials and Methods
Preparation of Dissociated Superior Cervical Ganglion. Superior cervical ganglion (SCG) neurons from adult male Wistar rats (150–300 g) were isolated as described previously (Ikeda, 1997). Animals were anesthetized by CO2 inhalation as approved by the Institutional Animal Care and Use Committee. After decapitation, SCG were removed bilaterally, cleaned of connective tissue, minced into small pieces, and transferred into 6 ml of modified Earles' balanced salt solution containing 0.6 mg/ml collagenase D (Roche Diagnostics, Indianapolis, IN), 0.3 mg/ml trypsin (Worthington Biochemicals, Freehold, NJ) and 0.05 mg/ml DNase I (Sigma Chemical, St. Louis, MO). After incubation in a shaking water bath at 36°C under an atmosphere of 5% CO2/95% O2 for 1 h, ganglion fragments were shaken vigorously to release the neuronal somata. The dissociated cells were washed twice, transferred to minimum essential medium containing 10% fetal calf sera, plated on poly-l-lysine–coated tissue culture dishes (35 mm), and placed in an incubator (95% air and 5% CO2; 100% humidity) at 37°C. After cDNA injection, the cells were incubated overnight at 37°C, and patch-clamp experiments were performed the following day. Where indicated, neurons were incubated overnight with 500 ng/ml PTX (List Biological Laboratories Inc., Campbell, CA).
Microinjection of cDNA. The procedure for intranuclear micro-injection of cDNA has been described in detail previously (Ikeda, 1997). Briefly, injection of cDNA was performed with an Eppendorf FemtoJet microjector and 5171 micromanipulator (Eppendorf, Madison, WI) using custom-designed software. Plasmids (pcDNA3.1; Invitrogen, Carlsbad, CA) containing inserts coding for human CB1R, GIRK1, and GIRK4 were stored at -20°C as 0.6 to 1 μg/μl stock solution in 10 mM Tris and 1 mM EDTA, pH 8. Except where indicated, cDNA was injected at a pipette concentration of 10 ng/μl. Neurons were coinjected with “enhanced” green fluorescent protein cDNA (5 ng/μl; pEGFP-N1; BD Biosciences Clontech, Palo Alto, CA) to facilitate later identification of neurons receiving a successful nuclear injection.
Electrophysiological Recordings. SCG neurons were voltage-clamped using the whole-cell patch-clamp technique with an Axopatch 200B amplifier (Axon Instruments Inc., Union City, CA). Patch electrodes were fabricated from 7052 borosilicate glass capillaries (Garner Glass, Claremont, CA) using a Model P-97 micropipette puller (Sutter Instrument Company, Novato, CA), coated with Sylgard (Dow Corning, Midland, MI), and fire-polished to final resistances of ∼2 MΩ when filled with internal solution. Uncompensated series resistance was <6 MΩ and generally electronically compensated ∼ 80%. Voltage protocol generation and data acquisition were performed using a custom-designed software (S5) on a Macintosh G4 computer (Apple Computer, Inc., Cupertino, CA) equipped with an ITC-18 data acquisition interface (InstruTECH Corporation, Port Washington, NY). Currents traces were filtered at 2 kHz (-3 dB) using a four-pole low-pass Bessel filter, digitized at 10 kHz with a 16-bit analog-to-digital converter in the ITC-18 data acquisition interface, and stored on the computer for later analysis. All recordings were performed at room temperature (21–24°C).
Solutions and Chemicals. For recording Ca2+ currents, the external (bath) solution consisted of 140 mM methanesulfonic acid, 145 mM tetraethylammonium hydroxide (TEA-OH), 10 mM HEPES, 10 mM glucose, 10 mM CaCl2; and 0.0003 mM tetrodotoxin, pH 7.4 with TEA-OH. The internal (pipette) solution contained 120 mM N-methyl-d-glucamine, 20 mM TEA-OH, 11 mM EGTA, 10 mM HEPES, 10 mM sucrose, 1 mM CaCl2, 4 mM MgATP, 0.3 mM Na2GTP, and 14 mM Tris creatine phosphate, pH 7.2, with methanesulfonic acid. For recording GIRK channel currents, the external solution contained 130 mM NaCl, 5.4 mM KCl, 10 mM HEPES, 10 mM CaCl2, 0.8 mM MgCl2, 15 mM glucose, 15 mM sucrose, and 0.0003 mM tetrodotoxin, pH 7.4, with NaOH. The pipette solution contained 135 mM KCl, 11 mM EGTA, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 4 mM MgATP, and 0.3 mM Na2GTP, pH 7.2, with KOH. The osmolalities of the bath and pipette solutions were adjusted with sucrose to 325 and 300 mosmol/kg, respectively.
All drugs and control solutions were applied to cells by positioning a custom-designed gravity-driven perfusion system ∼100 μm from the cell body. A fused silica gas chromatography column at the end of the perfusion system was connected to six parallel columns of the same diameter in series. The column containing normal external solution was kept open continuously to wash off drugs and to avoid flow-induced artifact until the desired test solution was applied.
2-AG, AEA, and 2-AGE were purchased from Tocris Cookson Inc. (Ellisville, MO); SR141716A was obtained from SANOFI Research Center (Montpellier, France); and WIN was purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA). 2-AG, WIN, and SR were dissolved in dimethyl sulfoxide as 10 mM stock solutions. AEA and 2-AGE were dissolved in ethanol as 14.4 and 13.7 mM stock solutions, respectively. Fatty acid-free bovine serum albumin (0.5 mg/ml) was added to all external solutions to prevent the lipid-soluble drugs from sticking to tubing. The final concentration of dimethyl sulfoxide and ethanol was <0.1%—a concentration that produced no significant effect on the Ca2+ current (data not shown). Stock solutions of norephinephrine bitartrate salt (Sigma) and PTX (List Biological Laboratories) were prepared in H2O at a stock concentration of 10 mM. All drugs were diluted in the external solutions from stock solutions to their final concentrations just before use.
Data Analysis and Statistics. Currents were analyzed using Igor Pro software (Wavemetrics, Lake Oswego, OR) on a Macintosh G4 computer. All data were expressed as mean ± S.E.M. The percentage inhibition (%) was determined using the equation (Icon - Idrug)/Icon × 100, where Icon and Idrug are the Ca2+ currents before and after agonist application, respectively. The concentration-response curves were fit to the Hill equation B = Bmax/{1 + (IC50/[agonist])nH}, where B, Bmax, IC50, [agonist], and nH are percentage inhibition, maximum inhibition, half-inhibition concentration, agonist concentration, and Hill coefficient, respectively. Statistical comparisons among groups were determined by analysis of variance followed by Student-Newman-Keuls post hoc test. The statistical significance between two groups was determined with the use of paired or unpaired Student's t tests, as appropriate. P < 0.05 was considered significant.
Results
Inhibition of Ca2+ Currents by 2-AG via CB1 Receptors. As a first step toward elucidating the potential roles of endocannabinoids in N-type Ca2+ channel modulation, we tested whether 2-AG modulates Ca2+ currents by activating CB1Rs heterologously expressed in rat SCG neurons. Figure 1 illustrates the effects 2-AG on Ca2+-channel currents elicited from uninjected and CB1R cDNA-injected SCG neurons. Ca2+-channel currents were evoked at 0.1 Hz from a holding potential of -80 mV with a double-pulse protocol (Elmslie et al., 1990) consisting of two identical 25-ms test pulses to +10 mV separated by a 50-ms depolarizing conditioning pulse to +80 mV (Fig. 1, A and B). Prepulse and postpulse currents were measured isochronally at 10 ms from the start of the test pulse. The major component of Ca2+ current evoked from rat SCG neuron under these conditions is ω-conotoxin GVIA sensitive N-type Ca2+ current (Ikeda, 1991). In uninjected cells, under control conditions (i.e., in the absence of agonist), the postpulse current amplitude usually exceeded the prepulse current amplitude—a phenomenon attributed to voltage-dependent relief of tonic G-protein–mediated Ca2+-channel inhibition (Ikeda, 1991). Basal facilitation ratio (BFR), defined as the ratio of the postpulse-to-prepulse current amplitude in the absence of agonist, was 1.22 ± 0.03 (n = 78). Application of 10 μM 2-AG did not significantly affect Ca2+ current elicited from uninjected neurons (Fig. 1A), further indicating that functional CB1 receptors are not present on the somata of rat SCG neurons (Pan et al., 1996). For CB1R cDNA-injected neurons, there was a bimodal distribution of 2-AG–induced inhibition versus the number of cells tested. In 73 (73%) of 100 cells, 2-AG (1–10 μM) produced significant inhibition (>15%) of the Ca2+ current, whereas it did not significantly inhibit Ca2+ current in the remaining cells. We thus did not include the “negative” cells in the later analysis. Figure 1B shows current traces elicited from a neuron previously microinjected with CB1R cDNA in the absence or presence of 2-AG. The 2-AG–induced Ca2+ current inhibition displayed the hallmarks of a membrane-delimited, Gβγ-mediated voltage-dependent pathway used by many G-protein-coupled neurotransmitter receptors (Herlitze et al., 1996; Ikeda, 1996) [i.e., slowed activation kinetics in the prepulse and partial relief of inhibition by a strong depolarizing conditioning pulse (Fig. 1B)]. Figure 1C shows that 2-AG (10 μM) produced a relatively rapid inhibition of Ca2+ current, reaching a steady-state level within ∼60 s, which usually completely reversed upon washing the cell with control external solution. In the presence of 2-AG (10 μM), the mean Ca2+ current inhibition was 47 ± 2% (n = 54) (Fig. 2C) and the facilitation ratio increased from 1.30 ± 0.03 to 2.13 ± 0.07 (n = 54).

2-AG induced Ca2+-current inhibition in rat SCG neurons expressing CB1Rs. A and B, superimposed Ca2+-current traces evoked with the double-pulse voltage protocol (bottom of A) in the absence and presence of 10 μM 2-AG from a control (A) and a CB1R-expressing (B) neuron. Currents were evoked every 10 s. The dashed lines indicate the zero current level. C and D, time courses of the Ca2+-current amplitudes (top) and facilitation (bottom) for CB1R-expressing neurons. Ca2+ current was measured 10 ms after initiation of the test pulse (+10 mV). Facilitation was calculated as the ratio of Ca2+-current amplitude determined from the test pulse (+10 mV) occurring after (postpulse) and before (prepulse) the +80-mV conditioning pulse. The filled bars indicate the application of 10 μM 2-AG, and the open bar indicates the application of 1 μM SR.

Facilitation and NE-mediated inhibition of Ca2+ currents in the CB1R-expressing neurons. A, superimposed Ca2+-current traces evoked from a neuron expressing CB1Rs in the absence (bottom) and presence of either 10 μM NE (top) or 10 μM 2-AG (middle). The dashed lines indicate the zero current level. B, time course of the effect of 2-AG and NE in the same cell as that shown in A. The top shows the change of Ca2+-current amplitude, and the bottom shows the change of facilitation. The measurement of Ca2+ current and facilitation is illustrated in Fig. 1, C and D. The filled bars indicate application of either 10 μM 2-AG or NE. C, summary graph of mean ± S.E.M. Ca2+-current inhibition by 10 μM NE, WIN, and 2-AG from control neurons or neurons expressing CB1Rs. Ca2+-current inhibition was measured isochronally 10 ms after initiation of the test pulse (+10 mV) in the absence or presence of drugs. D, summary box plots of basal facilitation for control neurons and neurons injected with CB1R cDNA at concentrations of 10, 100, and 500 ng/μl. The horizontal line represents the median, and the box and bars represent the middle 50% and 80% of data, respectively. •, outliers. Numbers in parentheses indicate the number of neurons tested. *, P < 0.05 compared with uninjected neurons.
To confirm that 2-AG modulates Ca2+ current via CB1Rs, a selective CB1R antagonist, SR, was applied before 2-AG application. In all cells tested, pretreatment with SR (1 μM) nearly abolished Ca2+ current inhibition in response to 10 μM 2-AG (5 ± 2%, n = 5), strongly suggesting that the 2-AG–induced Ca2+-current inhibition was mediated by CB1Rs. As reported previously (Pan et al., 1998), the application of 1 μM SR alone slightly increased the Ca2+ current amplitude in CB1R-expressing neurons (Fig. 1D). Overnight pretreatment of PTX (500 ng/ml) attenuated the 2-AG–induced Ca2+-current inhibition (4 ± 3%, n = 8), suggesting a PTX-sensitive G-protein; i.e., Gi/o, was involved in 2-AG–induced Ca2+-current inhibition.
Previously, it was shown that overexpression of CB1Rs (in a concentration-dependent fashion) abolished the ability of norepinephrine (NE) and somatostatin to inhibit Ca2+ currents in SCG neurons. It was proposed that the sequestration of G-proteins from a common pool prevented coupling to natively expressed receptors (Vasquez and Lewis, 1999). In addition, Ruiz-Velasco and Ikeda (1998) reported that heterologous expression of GIRK channels in rat SCG neurons could decrease NE-mediated Ca2+-channel inhibition by altering the receptor-G-protein-effector stoichiometry. These studies suggest that overexpression of a heterologous protein might alter native signaling pathways. We therefore sought to optimize the expression of CB1R such that an appreciable CB1R response was obtained while preserving native signaling pathways. Microinjection of CB1R cDNA at a concentration of 10 ng/μl resulted in expression levels that largely preserved NE-mediated Ca2+ current inhibition. Figure 2A illustrates superimposed current traces elicited from a CB1R cDNA injected neuron in the absence or presence of either 10 μM 2-AG or NE. The time course of the effects is shown in Fig. 2B. Under these conditions, NE (10 μM) produced mean inhibitions of 46 ± 3% (n = 32) and 57 ± 2% (n = 30) in CB1R-expressing and uninjected neurons, respectively (Fig. 2C).
As described above, BFR is an indicator of tonic G-protein activation. Pan et al. (1998) reported that expression of CB1Rs significantly increased BFR. The enhanced BFR was attributed to a population of constitutively active CB1Rs coupled to PTX-sensitive G-proteins. However, in the present study, expression of CB1R did not significantly increase mean BFR, which was 1.22 ± 0.03 (n = 78) for uninjected neurons compared with 1.33 ± 0.02 (n = 185) for CB1R cDNA-injected (10 ng/μl) neurons (Fig. 2D). It should be noted that a few (14 of 200) CB1R-expressing neurons displayed a BFR of 1.8 to 2.5. To test whether BFR was dependent on the injected CB1R cDNA concentration, SCG neurons were injected with 100 and 500 ng/μl of CB1R cDNA. Under these conditions, the mean BFR (Fig. 2D) was 1.20 ± 0.03 (n = 27) and 1.22 ± 0.03 (n = 25), respectively. Thus, BFR in CB1R-injected neurons was not correlated with the injected cDNA concentration over the range of 10 to 500 ng/μl. All neurons microinjected with different concentrations of CB1R cDNA were checked for CB1R expression by measuring the effect of either WIN, 2-AG, 2-AGE, or AEA on the Ca2+ current (see below).
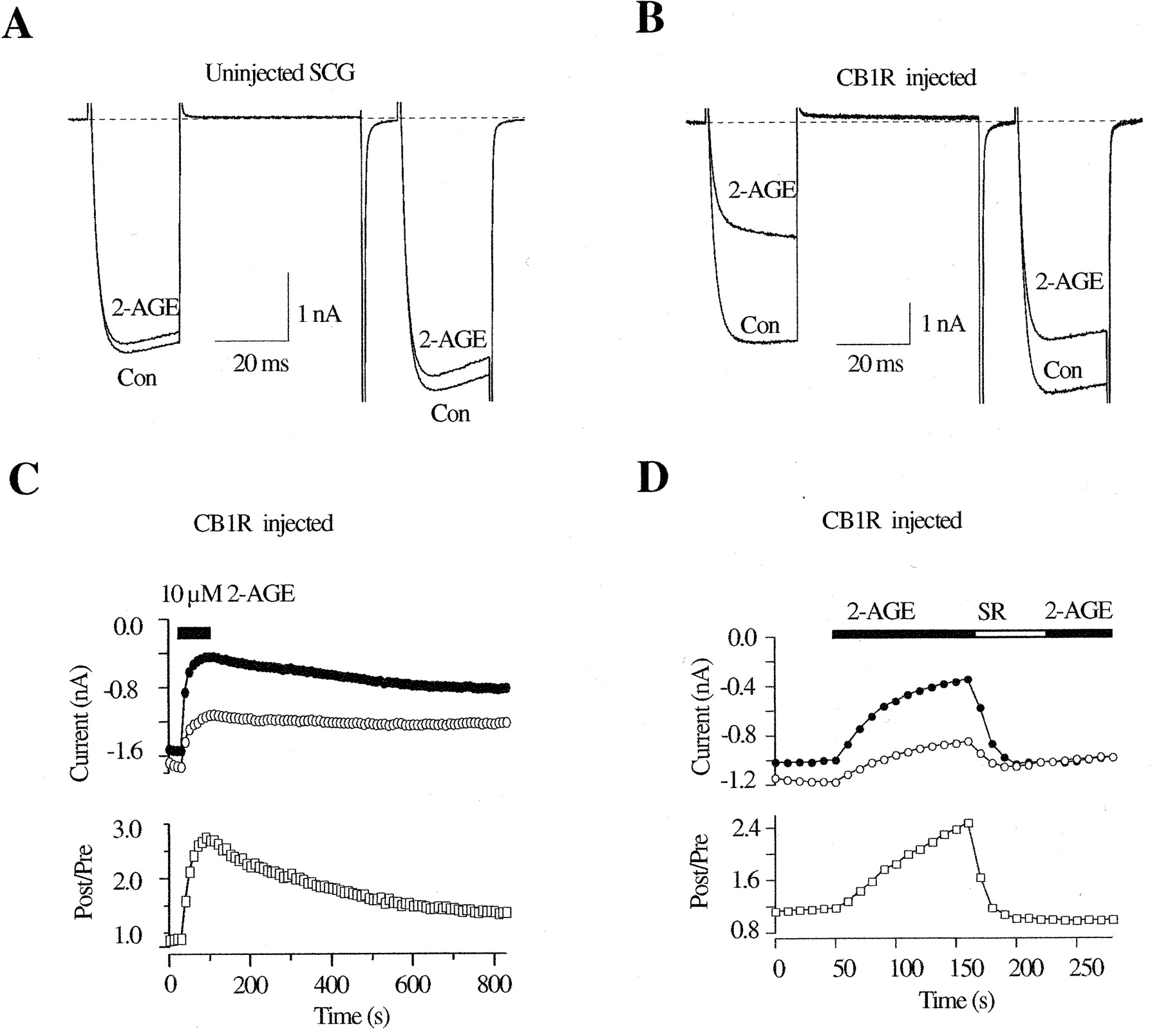
Inhibition of Ca2+ Currents by 2-AGE via CB1 Receptors. 2-AGE is a novel putative endocannabinoid recently isolated from porcine brain (Hanus et al., 2001) that selectively binds to CB1Rs and evokes responses typical of cannabimimetic compounds. Because the effect of 2-AGE on ion channel modulation has not been reported, we sought to examine whether 2-AGE modulates N-type Ca2+ current in CB1R-expressing neurons. In uninjected neurons, 2-AGE (10 μM) did not significantly affect Ca2+-current amplitude (4 ± 2%, n = 4) (Fig. 3A). Like 2-AG, 2-AGE (1–10 μm) inhibited the Ca2+ current in the majority (40 of 62) of neurons injected with CB1R cDNA. Mean Ca2+-current inhibition in the presence of 10 μM 2-AGE was 49 ± 4% (n = 21) (Fig. 3B) for responding neurons (defined as inhibition >15%). 2-AGE–induced Ca2+-current inhibition was voltage-dependent as determined from the slowing of current activation kinetics and increased prepulse facilitation ratio (from 1.38 ± 0.08 to 2.25 ± 0.14, n = 21). Figure 3C illustrates the time course of the current inhibition by 2-AGE. The time needed to reach the steady-state inhibition was variable, usually taking between 60 and 120 s, and the recovery from inhibition was slow compared with the washout of 2-AG–mediated inhibition (Fig. 2C). Pretreatment with SR (1 μM) for 60 s abolished 2-AGE–induced Ca2+-current inhibition (3 ± 1%, n = 10; data not shown), confirming the involvement of CB1Rs in this response. Moreover, application of SR after maximal inhibition was achieved accelerated the recovery of the inhibited current (Fig. 3D), suggesting the rebinding of 2-AGE to CB1Rs. The 2-AGE–induced response was also mediated via a Gi/o-protein because overnight pretreatment of PTX strongly attenuated the voltage-dependent Ca2+-current inhibition (4 ± 2%, n = 7).

2-AGE induced Ca2+-current inhibition in rat SCG neurons expressing CB1R. A and B, superimposed Ca2+-current traces evoked with the double-pulse voltage protocol (illustrated in Fig. 1A) from control (A) and CB1-expressing (B) neurons in the absence or presence of 10 μM 2-AGE. The dashed lines indicate the zero current level. C and D, time courses of the Ca2+ current amplitudes (top) and facilitation ratio (bottom) for CB1R-expressing neurons. The measurement of Ca2+ current and facilitation was illustrated in Fig. 1, C and D. The filled bars indicate the application of 10 μM 2-AGE, and the unfilled bar indicates application of 1 μM SR.
Concentration-Dependent Ca2+ Current Inhibition by 2-AG, 2-AGE, and WIN.Figure 4 depicts concentration-response curves for Ca2+ current inhibition generated from CB1R-expressing SCG neurons. Only one or two concentrations of agonists were tested for each neuron because of the relatively slow reversibility of the compounds. The lower concentration was tested first, the current was allowed to return to a steady-state level, and then the higher concentration was applied. The data points represent the mean inhibition determined from 4 to 54 cells. At all concentrations tested, there was no significant difference between 2-AG– and 2-AGE–induced Ca2+-current inhibition (P > 0.05). The data were fit to a Hill equation (solid lines) using a nonlinear least-squares algorithm. From this analysis, the maximum inhibition, IC50, and Hill coefficient for 2-AG and 2-AGE were 49 and 51%, 480 and 350 nM, and 1.2 and 1.6, respectively. These data suggest that stimulation of CB1Rs by 2-AG and 2-AGE displays a similar efficacy and potency for Ca2+-current inhibition.
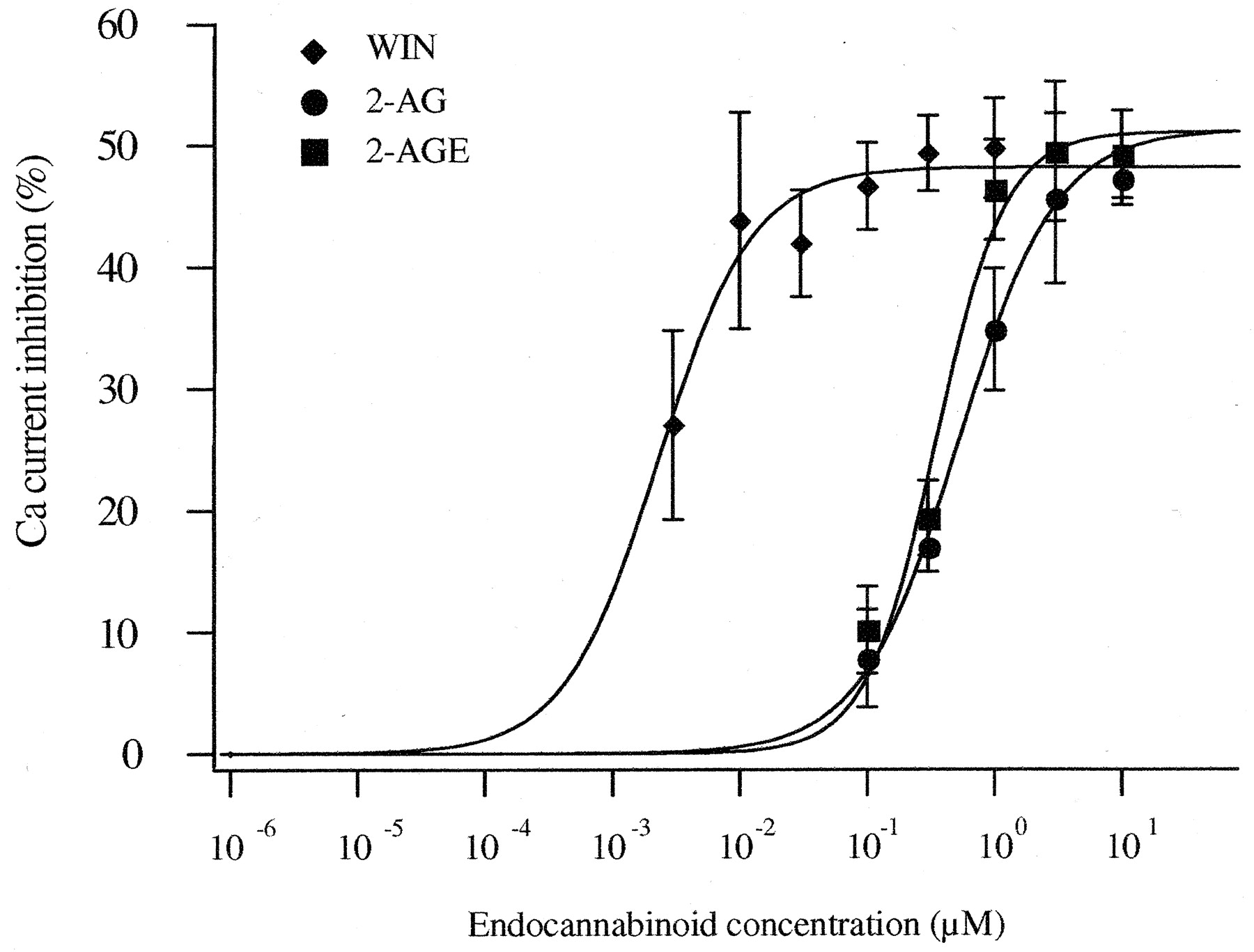
Concentration-response for 2-AG–, 2-AGE–, and WIN-induced Ca2+-current inhibition in CB1R-expressing neurons. The smooth curves were obtained by fitting data to a Hill equation. Data are the mean ± S.E.M. Each point on the dose-response curves represents the mean inhibition from 3 to 54 cells, except for 0.01 μM WIN, where n = 2.
For comparison, the concentration-response relationship for WIN, a potent synthetic CB1R agonist, was determined. In 52 (91%) of 57 cells injected with 10 ng/μl CB1R cDNA, WIN (0.003–10 μM) produced voltage-dependent Ca2+-channel inhibition greater than 15%. As previously reported (Pan et al., 1996, 1998), 1 μM WIN did not significantly affect Ca2+ current recorded from uninjected neurons (4 ± 1%, n = 7). However, 10 μM WIN inhibited Ca2+ currents by 20 ± 4% (n = 8) in uninjected neurons but produced 62 ± 3% (n = 10) inhibition in CB1R-expressing neurons (Fig. 2C). The concentration-response relationship of WIN-induced Ca2+-channel inhibition is shown in Fig. 4. The data were fit to a Hill equation revealing maximal inhibition of 48%, an IC50 of 2 nM, and a Hill coefficient of 1.2. Data obtained at 3 and 10 μM were excluded from analysis because of apparent CB1R-independent effects (see above). Thus, WIN was more potent than 2-AG and 2-AGE, but it had a similar efficacy.
Voltage-Dependent Inhibition by 2-AG and 2-AGE. The voltage-dependence of 2-AG– and 2-AGE–induced Ca2+-current inhibition was characterized in greater detail by generating current-voltage (I-V) relationships in the absence or presence of either 3 μM 2-AG or 2-AGE. Ca2+ currents were elicited at 0.33 Hz from a holding potential of -80 mV with 70-ms test pulses between -120 and +80 mV. I–V curves (Fig. 5A) were normalized to the maximum Ca2+ current in the absence of agonist (normally generated by a step to +10 mV). The mean inhibition of Ca2+ currents produced by 3 μM 2-AG and 2-AGE were plotted against membrane potential over the voltage range shown in Fig. 5B. 2-AG– and 2-AGE–induced maximal inhibition at test potentials that generated the largest Ca2+ current (i.e., approximately +10 mV). The relationship between Ca2+-current inhibition and test-pulse potentials displayed a “bell-shaped” profile clearly indicating the voltage-dependent Ca2+ current inhibition by 2-AG and 2-AGE.
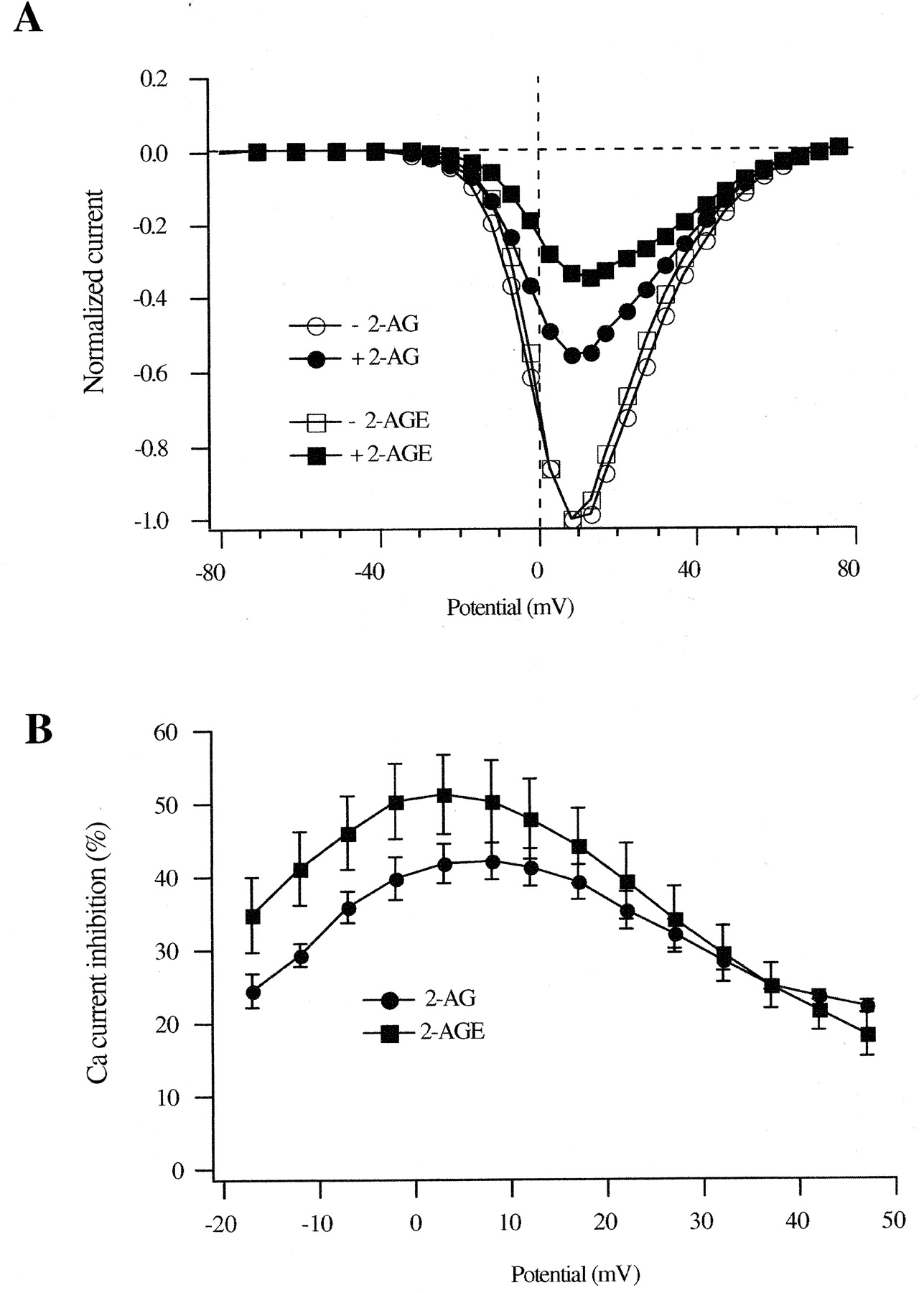
Voltage-dependent Ca2+-channel inhibition by 2-AG and 2-AGE in CB1R-expressing neurons. A, normalized I-V relationship in the absence and presence of 3 μM 2-AG or 3 μM 2-AGE. Ca2+ currents were elicited with 70-ms voltage steps in 5- or 10-mV increments from -120 to +80 mV from a holding potential of -80 mV. The Ca2+ current at each voltage step was normalized to the Ca2+ current elicited by the depolarizing step to +10 mV in the absence of drugs. B, the mean percentage inhibition of Ca2+ currents was plotted against each voltage step to demonstrate the voltage-dependent Ca2+-channel inhibition by 3 μM 2-AG and 3 μM 2-AGE.
CB1R-Independent and CB1R-Mediated Ca2+ Channel Inhibition by AEA. AEA, the first endocannabinoid to be identified, has complex effects on Ca2+-channel modulation. Unlike 2-AG and 2-AGE, 10 μM AEA produced strong Ca2+-current inhibition in uninjected neurons. In contrast to voltage-dependent inhibition of Ca2+ current, 10 μM AEA inhibited postpulse current (30 ± 3%, n = 11) to a greater extent than prepulse current (18 ± 3%, n = 11), as illustrated in Fig. 6A. As a consequence, the BFR was decreased from 1.25 ± 0.06 to 1.14 ± 0.06 (n = 11, P < 0.05). Figure 6B shows the time course of CB1R-independent inhibition of Ca2+ current by 10 μM AEA. Because the CB1R-independent Ca2+-channel inhibition by AEA was slow to reach a steady-state level, the amount of inhibition was determined ∼200 s after the start of drug application. During drug washout, recovery of current amplitude was slow and incomplete. AEA clearly accelerated the decay of both pre- and postpulse currents, as shown in Fig. 6A. The AEA-induced inhibition was not mediated by covert CB1Rs because 1) pretreatment with 1 μM SR failed to prevent the effect (data not shown); and 2) overnight pretreatment with PTX (500 ng/ml) did not abolish the AEA-induced direct Ca2+-current inhibition (the prepulse currents were inhibited by 15 ± 3%, the postpulse currents were inhibited by 24 ± 6%, n = 4; P > 0.05). Figure 6C shows the normalized Ca2+ current I-V relationships in the absence or presence of 10 μM AEA. There was no “bell-shaped” relationship between the Ca2+-current inhibition and pulse potentials for CB1R-independent Ca2+-channel inhibition (Fig. 6D); in addition, there is a potentiation of Ca2+ current in the range of -30 to -5 mV.

Effect of AEA on Ca2+ current in control neurons. A, superimposed Ca2+-current traces evoked with the double-pulse voltage protocol (illustrated in Fig. 1A) from a control neuron in the absence and presence of 10 μM AEA. The dashed lines indicate the zero current level. B, time course of the effect of AEA on the Ca2+-current amplitude from a control neuron. The filled bar indicates the application of 10 μM AEA. C, normalized I-V relationship in the absence and presence of 10 μM AEA obtained from a control neuron. The measurement of Ca2+ currents is illustrated in Fig. 5. D, the mean percentage inhibition of Ca2+ currents from four cells was plotted against each voltage step to demonstrate the voltage-independent, CB1R-independent Ca2+-channel inhibition by 10 μM AEA.
In 19 (49%) of 39 CB1R cDNA-injected SCG neurons, 10 μM AEA produced Ca2+-current inhibition significantly different from the CB1R-independent inhibition described above. The AEA-induced Ca2+-current inhibition (64 ± 2%, n = 19), although contaminated with the CB1R-independent effects, showed slowing of activation and increased facilitation ratio characteristic of Gβγ-mediated voltage-dependent modulation (Fig. 7A). Figure 7B illustrates the time course of AEA-induced Ca2+-current inhibition in a CB1R cDNA-injected neuron. Like 2-AGE, the onset of current inhibition by 10 μM AEA was variable, reaching a steady-state level within approximately 100 to 250 s. The recovery of current after drug removal was slow and was accelerated by 1 μMSR (Fig. 7B). Pretreatment with SR (1 μM) for 60 s significantly attenuated the voltage-dependent inhibition [prepulse current was inhibited by 14 ± 2% (n = 8), whereas postpulse current was inhibited by 16 ± 3% (n = 8)], indicating that the voltage-dependent Ca2+-channel inhibition is mediated by CB1R.
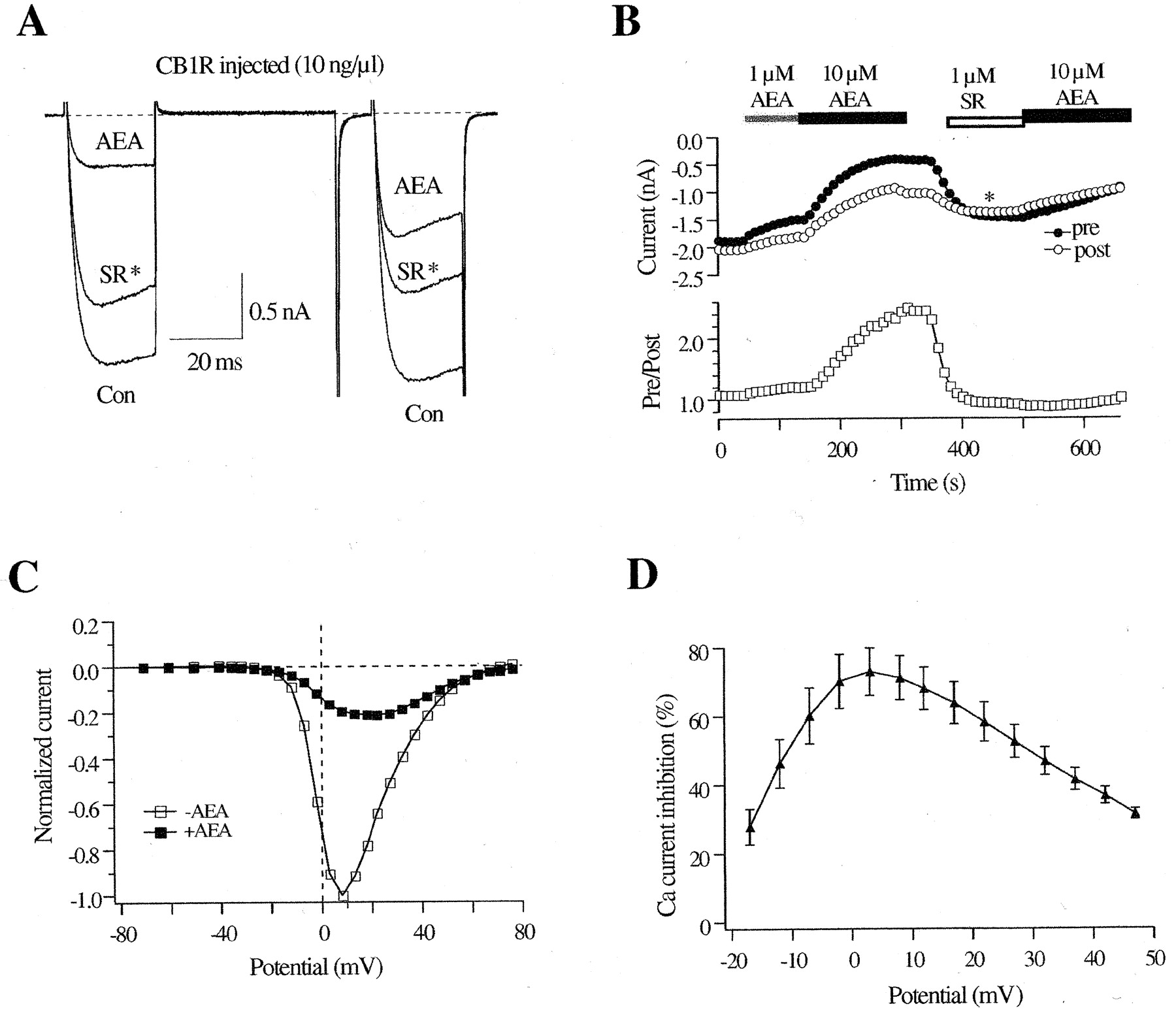
Effect of AEA on Ca2+ current in CB1R-expressing neurons. A, superimposed Ca2+-current traces evoked with the double-pulse voltage protocol (illustrated in Fig. 1A) from a CB1-expressing neuron in the absence and presence of 10 μM AEA. *, the trace was evoked in the presence of 1 μM SR during the washout of 10 μM AEA. The dashed lines indicate the zero current level. B, time course of the effect of AEA on the Ca2+-current amplitude from a CB1-expressing neuron. The filled bars indicate application of 1 μMor10 μM AEA, and the unfilled bar indicates the application of 1 μM SR. C, normalized I-V relationship in the absence and presence of 10 μM AEA obtained from a CB1-expressing neurons. The measurement of Ca2+ currents was illustrated in Fig. 5. D, the mean percentage inhibition of Ca2+ currents from four cells was plotted against each voltage step to demonstrate the voltage-dependent, CB1R-mediated Ca2+-channel inhibition by 10 μM AEA.
Figure 7C shows the normalized Ca2+-current I-V relationships in the absence or presence of 10 μM AEA in CB1R-expressing neurons. In contrast to CB1R-independent response, there was no potentiation of Ca2+ current. The CB1R-mediated Ca2+-current inhibition showed a characteristic “bell-shaped” relationship between the Ca2+-current inhibition and test pulses, with the maximal inhibition occurring at ∼+10 mV (Fig. 7D). Unlike the CB1R-independent response, a Gi/o protein is involved in the CB1R-mediated inhibition because pretreatment of PTX (500 ng/ml) strongly decreased the AEA-induced Ca2+-current inhibition (16 ± 1%; n = 11).
The AEA-induced Ca2+-current inhibition in SCG neurons expressing CB1Rs was contaminated with CB1R-independent Ca2+-current inhibition. Thus, direct comparison of inhibition of Ca2+ current by AEA with that of 2-AG or 2-AGE was challenging. Although it was not clear whether the CB1R-independent and -dependent inhibition were additive in a mechanistic sense, we attempted to estimate the CB1R-mediated portion of Ca2+-current inhibition (at a single potential, +10 mV) by subtracting the mean AEA-induced CB1R-independent inhibition (approximately 18%) from the total inhibition produced by 10 μM AEA. Using this type of analysis, the CB1R-mediated Ca2+-channel inhibition was estimated to be 47 ± 2% (n = 19). There was no significant difference in Ca2+-current inhibition produced by 10 μM 2-AG, 2-AGE, or AEA (p > 0.05). The potency of AEA was less than that of 2-AG and 2-AGE because the estimated IC50 value for AEA was approximately 3 μM[1 μM and 3 μM AEA inhibited Ca2+ current by 16 ± 2% (n = 9) and 32 ± 3% (n = 4), respectively], which is higher than that of 2-AG (480 nM) and 2-AGE (360 nM).
AEA has been shown to act as a partial agonist in many studies (Mackie et al., 1993; Gonsiorek et al., 2000). However, whether a ligand acts as a full or a partial agonist is strongly dependent on the expression level of receptor, because only a fraction of the receptors present in a cell might be required for a maximum effect of an agonist. Thus, a larger receptor reserve could convert a partial agonist to a full agonist response (MacEwan et al., 1995) as well as induce a leftward shift in the concentration-response curve (Adie and Milligan, 1994). Accordingly, we tested AEA-induced Ca2+-channel inhibition in SCG neurons microinjected with a larger amount of CB1R cDNA (100 and 500 ng/μl). In 22 (85%) of 26 cells injected with 100 ng/μl CB1 cDNA and 28 (88%) of 32 cells injected with 500 ng/μl CB1 cDNA, AEA (1 and 10 μM) produced strong voltage-dependent Ca2+ channel inhibition (Fig. 8). Although the maximal inhibition achieved by 10 μM AEA (61 ± 4, n = 17 and 57 ± 3%, n = 19 for 100 and 500 ng/μl, respectively) was not significantly different from the neurons injected with 10 ng/μl, the potency of AEA was apparently increased in SCG neurons expressing a (presumably) higher density of CB1Rs. 1 μM AEA produced only 16 ± 2% (n = 9) inhibition in neurons injected with 10 ng/μl cDNA, whereas Ca2+ current was inhibited by 36 ± 7% (n = 5) and 34 ± 4% (n = 9) in neurons injected with 100 and 500 ng/μl CB1R cDNA, respectively (Fig. 8). Moreover, the neurons expressing a higher level of CB1 receptors were more responsive to 10 μM AEA—only 49% of neurons injected with 10 ng/μl CB1R cDNA responded to 10 μM AEA, whereas 88% of neurons injected with 500 ng/μl CB1R cDNA responded to the same agonist concentration.
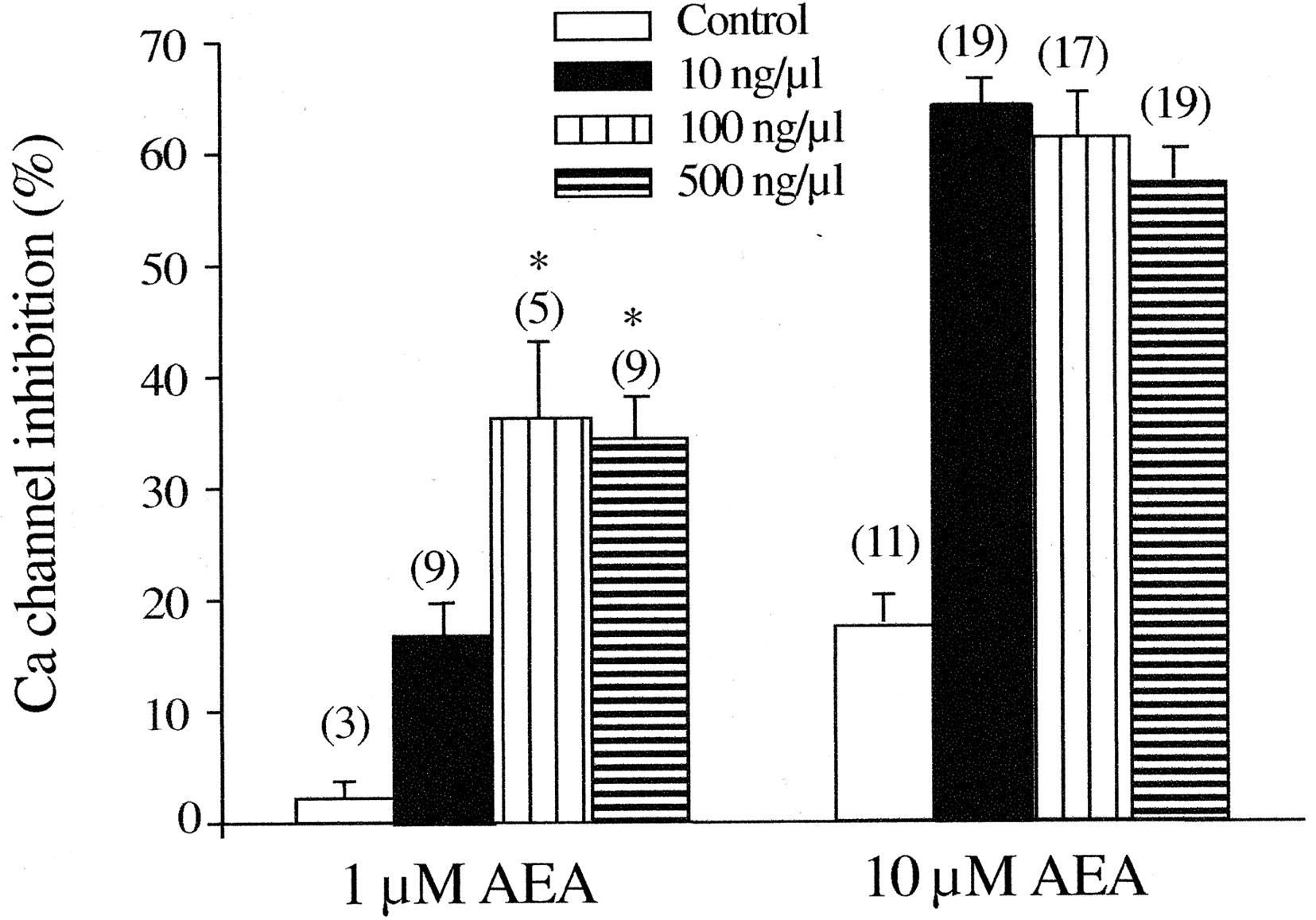
Effect of AEA on Ca2+-channel inhibition in neurons injected with different concentrations of CB1R cDNA. The summary graph shows the AEA (1 and 10 μM)-induced Ca2+ channel inhibition for control neurons and neurons injected with CB1R cDNA at concentrations of 10, 100, and 500 ng/μl. Ca2+-current inhibition was calculated as described in Fig. 2C. Numbers in parentheses indicate the number of neurons tested. *, P < 0.05 compared with neurons injected with 10 ng/μl CB1R cDNA.
Endocannabinoids Activate GIRK-Type K+ Channels. GIRK-type K+ currents arise from heteromultimers of Kir3 family K+ channels that are activated by Gβγ subunits after receptor stimulation. Because GIRK channels are not natively expressed in rat SCG neurons (Ruiz-Velasco and Ikeda, 1998), the channels were heterologously expressed by intranuclear injection of cDNAs encoding GIRK1 (Kir3.1) and GIRK4 (Kir3.4). GIRK currents were elicited from a holding potential of -60 mV by a voltage ramp that started at -140 and increased to -40 mV over 200 ms. In most GIRK-expressing neurons, there was little tonic activation of GIRK current before agonist application. In the absence of CB1R expression, application of 2-AG, 2-AGE, AEA, or WIN induced negligible inward GIRK current (Fig. 9E), supporting the notion that functional native CB1Rs are absent from the soma of SCG neurons. In neurons expressing CB1R cDNA, application of 10 μM 2-AG (Fig. 9A), 2-AGE (Fig. 9B), AEA (Fig. 9C), or 1 μM WIN (Fig. 9D) resulted in inward currents that exhibited a region of negative slope conductance at very hyperpolarized potentials (approximately -140 to -120 mV) and outward rectification at voltages positive to approximately -80 mV. The current component resulting from agonist application was determined by digitally subtracting traces obtained before and after the application of agonists. Maximal inward current (usually measured near -135 mV) induced by 2-AG, 2-AGE, AEA, and WIN was 0.85 ± 0.3, 0.59 ± 0.16, 1.03 ± 0.50, and 1.13 ± 0.39 nA, respectively (Fig. 9E). Figure 9F summarizes the mean ± S.E.M. maximal inward current density (i.e., current amplitude normalized to the cell capacitance) induced by CB1R agonists for neurons expressing GIRK1/4 and CB1R. The results demonstrate that GIRK K+ channels and CB1Rs heterologously expressed in SCG neurons form functional signaling complexes that are activated by endocannabinoid agonists.

Effect of endocannabinoids and WIN on GIRK1/4 heterologously expressed in SCG neurons. Whole-cell inwardly rectifying K+ currents were evoked by 200-ms voltage ramps from -140 to -40 mV from a holding potential of -60 mV. A to D, superimposed current traces from CB1R and GIRK1/4 coexpressing neurons before (control) and after application of 10 μM 2-AG (A), 2-AGE (B), anandamide (C), and WIN (D). Dashed lines indicate the zero current level. E and F, summary graphs show the mean ± S.E.M. peak current (E) and mean ± S.E.M. peak normalized current (F) activated by 10 μM 2-AG, 2-AGE, AEA and WIN for GIRK1/4 and CB1R-coexpressing neurons. Numbers in parentheses indicate the number of neurons tested.
Discussion
Among G-protein-coupled receptors, CB1Rs are unusual in that they are expressed nearly exclusively in presynaptic terminals and are excluded from the soma of adult neurons. This unique distribution pattern makes it difficult to directly examine the coupling of native CB1Rs to Ca2+ channels. Therefore heterologous expression of CB1Rs by intranuclear injection of CB1 cDNA in an isolated adult mammalian neuron that has well-studied G-protein pathways facilitates in situ investigation of CB1 receptors. In this neuronal expression system, we demonstrated that three putative endocannabinoids, AEA, 2-AG, and 2-AGE, like the cannabimimetic aminoalkylindole WIN, initiate PTX-sensitive, voltage-dependent N-type Ca2+ channel inhibition via CB1Rs.
WIN is a synthetic CB1R agonist widely used in the studies of CB1Rs, including DSI and DSE. Modulation of N-type Ca2+ channel by WIN via CB1Rs was first observed in differentiated NG108-15 neuroblastoma cells and N18 cells (Caulfield and Brown,1992; Mackie and Hille, 1992). Similarly, the Ca2+-current inhibition by WIN was observed in differentiated N18 cells (Mackie et al., 1993) and SCG neurons heterologously expressed with human CB1Rs (Pan et al., 1996). These findings underlie the proposed role for endocannabinoids and Ca2+-channel modulation in the current model of DSI/DSE. However, whether endocannabinoid inhibits Ca2+ channel by activating CB1R has not been fully demonstrated. To our knowledge, AEA is the only endocannabinoid whose effect on Ca2+-channel modulation has been studied. In differentiated N18 cells, AEA acts as a partial agonist to produce a voltage- and concentration-dependent Ca2+-current inhibition with an IC50 of 20 nM (Mackie et al., 1993). However AEA (0.1 μM) had no effect on the Ca2+ current in 25 of 33 SCG neurons expressing rat CB1R (Pan et al., 1996).
We found that endocannabinoids, including AEA, can act as full agonists to inhibit N-type Ca2+ current with an efficacy similar to WIN 55,212-2. What accounts for the differences found among the various studies that have examined AEA effects? First, AEA is less potent than WIN, with an IC50 of ∼3 μM, and possesses a non–CB1R-mediated response that must be accounted for when estimating efficacy and potency. Thus, for example, the lower concentration of AEA (0.1 μM) used in the study by Pan et al (1996) may be a reason that only two neurons injected with CB1R cRNA showed a relatively strong response to AEA. Second, expression levels of CB1R might also account for the discrepancies among different studies. The relationship between receptor occupancy and effector response is related to receptor concentration. At higher levels of expression, less receptor occupancy is required to attain a given response, assuming that signaling intermediates are not limiting. Here we show that increasing the concentration of CB1R cDNA-injected leads results in an increased potency, whereas efficacy remains relatively constant. However, the true intrinsic efficacy of AEA cannot be determined in single-cell systems because the degree of receptor occupancy is unknown. Third, the coupling between CB1R and different G-protein types may explain why AEA is a full agonist in some studies and a partial agonist in others. Glass and Northup (1999) reported that WIN and AEA acted as full agonists in CB1 receptor-mediated Gi activation and partial agonists in Go activation, suggesting that different agonists may induce different CB1R conformations, which in turn selectively interact with different G-proteins. Conversely, Yang and Lanier (1999) also showed that partial agonists of α2-adrenergic receptors clonidine and oxymetazoline behaved as full agonist after expression of Goα1 but not after Giα1. Whether AEA-induced Ca2+-channel inhibition in rat sympathetic neurons is preferably mediated by Go or Gi needs further investigation. Finally, the metabolism of AEA, under some circumstances, may impact the effective final concentration of AEA. Thus, concentrations of AEA used in complex systems (e.g., brain slices) may not be equivalent to those used in single neuron preparations.
Another question relates to the whether CB1Rs are active in the absence of ligand (i.e., tonically active). Pan et al. (1998) reported that, in SCG neurons expressing the CB1Rs, the BFR, and indication of tonic G-protein activation, was significantly increased in neurons microinjected with CB1 cRNA at a concentration of 1.5 to 2.0 μg/μl. They suggested that the increased BFR was caused by tonic G-protein activation mediated by constitutively active CB1Rs because SR increased the Ca2+ currents. However, in our experiments, the average BFR was not significantly enhanced in SCG neurons injected with CB1 cDNA up to 500 ng/μl. Our studies also indicate that activation of CB1R can inhibit Ca2+ channels without greatly sequestering G-protein or preventing other proteins from coupling to G-proteins, as has been reported earlier (Vasquez and Lewis, 1999). These differences almost undoubtedly arise from differences in the concentration of expressed CB1R, because the expression system and the response measured were the same. It is difficult to know which situation represents the physiological one, because the effective CB1R concentration in a restricted compartment, such as a presynaptic nerve terminal, is difficult to ascertain. Moreover, in more complex systems, it is difficult to ascertain whether tonic activity of the receptor, as indicated by changes caused by antagonist application, arise from unliganded receptor or the presence of endocannibinoids.
The observation that AEA produces a CB1R-independent Ca2+-channel inhibition is another example of direct effects of AEA on ionic channels. AEA directly inhibits Kv1.2 K+ channels (Poling et al., 1996), activates vallinoid VR1 receptors (Zygmunt et al., 1999), blocks a background potassium current TASK-1 (Maingret et al., 2001), and attenuates both cloned and native T-channels (Chemin et al., 2001). The AEA-induced direct Ca2+-current inhibition differs from the CB1R-mediated effect in that it is PTX-insensitive and voltage-independent. In addition, AEA causes a small leftward shift of current-voltage relationship of Ca2+ current, resulting in current potentiation at approximately -10mV. Interestingly, arachidonic acid, a product of AEA degradation, is also reported to shift the I-V leftward and enhance Ca2+ current at negative potentials (Liu and Rittenhouse, 2003). Whether the CB1R-independent effect reported here has physiological significance remains uncertain. However, the effect is experimentally relevant because the AEA concentration used in the present study (10 μM) is in the range used in other studies. Thus, the CB1R-independent modification of Ca2+ current should be considered when interpreting data obtained with AEA in a similar concentration range. Although the mechanism of the CB1R-independent Ca2+ channel (and other channels) modulation remains uncertain, AEA might regulate neuronal excitability by both CB1R-dependent and -independent–mediated mechanisms.
GIRK channels are another type of channel modulated by Gβγ subunits that play important roles in regulating neuronal excitability. Our data illustrate that the three endocannabinoids activate the recombinant GIRK1/4 expressed in SCG neurons via CB1Rs consistent with results obtained from other expression systems. The physiological implication of endocannabinoid-induced GIRK channel modulation remains uncertain because there is limited evidence for GIRK expression at presynaptic sites (Luscher et al., 1997). However, the modulation of GIRK by endocannabinoids provides a useful strategy to further investigate the role of endocannabinoids in DSI/DSE. For example, although endocannabinoids are proposed to be retrograde messengers mediating DSI/DSE, the mechanism underlying endocannabinoid release from the postsynaptic neurons remains unclear. A patch of membrane-expressing CB1Rs and GIRK channels might therefore serve as a biosensor to directly detect the release of endocannabinoid(s). In the same vein, the coupling of heterologously expressed CBR1Rs (to force somal expression) to neuronal ion channels might allow reconstitution of the events underlying DSI/DSE in a single neuron.
Taken together, our studies indicate that three putative endocannabinoids, 2-AG, 2-AGE and AEA, are capable of inhibiting N-type Ca2+ channel, suggesting a role in DSI/DSE, and activating inwardly rectifying K+ channels. Both the actions of endocannabinoids and the intrinsic tonic activity of CB1R are dependent on receptor expression levels. Because CB1R expression levels vary in different brain regions, the pharmacology of endocannabinoids and the CB1R may exhibit regional differences as well.
Footnotes
-
ABBREVIATIONS: CB1R, CB1 cannabinoid receptor; DSI, depolarization-induced suppression of inhibition; DSE, depolarization-induced suppression of excitation; CB2R, CB2 cannabinoid receptor; 2-AG, 2-arachidonoylglycerol; PTX, pertussis toxin; GIRK, G-protein-coupled inwardly rectifying potassium channel; SCG, superior cervical ganglion; BFR, basal facilitation ratio; AEA, anandamide; 2-AGE, 2-arachidonyl glycerol ether; WIN, WIN 55,212-2, 2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl-1-naphtalenylmethanone mesylate; SR, SR141716R, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride; CP 55,940, (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol; TEA-OH, tetraethylammonium hydroxide; NE, norepinephrine; I-V, current-voltage.
- Received September 10, 2003.
- Accepted November 18, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}