


Abstract
Clinically, there is a great need for small molecule inhibitors that could control pathogenic effects of transforming growth factor (TGF-β) and/or modulate effects of TGF-β in normal responses. Inhibition of TGF-β signaling would be predicted to enhance re-epithelialization of cutaneous wounds and reduce scarring fibrosis. Selective small molecule inhibitors of the TGF-β signaling pathway developed for therapeutics will also be powerful tools in experimentally dissecting this complex pathway, especially its cross-talk with other signaling pathways. In this study, we characterized 2-(5-benzo[1,3]dioxol-5-yl-2-tert-butyl-3H-imidazol-4-yl)-6-methylpyridine hydrochloride (SB-505124), a member of a new class of small molecule inhibitors related to imidazole inhibitors of p38, which inhibit the TGF-β type I receptor serine/threonine kinase known as activin receptor-like kinase (ALK) 5. We demonstrate that this compound selectively and concentration-dependently inhibits ALK4-, ALK5-, and ALK 7-dependent activation of downstream cytoplasmic signal transducers, Smad2 and Smad3, and of TGF-β–induced mitogen-activated protein kinase pathway components but does not alter ALK1, ALK2, ALK3 or ALK6-induced Smad signaling. SB-505124 also blocks more complex endpoints of TGF-β action, as evidenced by its ability to abrogate cell death caused by TGF-β1 treatment. SB-505124 is three to five times more potent than a related ALK5 inhibitor described previously, SB-431542.
The TGF-β superfamily of ligands includes transforming growth factor (TGF) β, activins, and bone morphogenetic proteins (BMPs) and regulates a wide range of responses including cell proliferation, differentiation, adhesion, migration, and apoptosis (Sporn and Roberts, 1990; Massague, 1998). Signaling by the TGF-β superfamily is mediated by two types of transmembrane receptor serine/threonine kinases, types I and II. Generally, ligand interaction with a homodimer of type II receptors recruits and activates homodimers of type I receptors or activin receptor-like kinases (ALKs). Activated ALKs phosphorylate a subset of downstream signaling molecules, the receptor-activated Smads, which then enables their binding to the common mediator Smad 4. This complex is shuttled into the nucleus and regulates transcription of target genes. There are seven known mammalian type I receptors (ALK1–7) and five type II receptors, and unique combinations of the type I and type II receptors confer specificity of ligand signaling (Piek and Roberts, 2001). Activin- or TGF-β–activated type I receptors, ALK4 and ALK5, respectively, phosphorylate Smad2 and Smad3, whereas BMP-activated ALK1, ALK2, ALK3, and ALK6 phosphorylate Smads 1, 5, and 8 (Heldin et al., 1997). It must be noted, however, that there is evidence to suggest that TGF-β can also activate ALK1 and Smads 1 and 5 in endothelial cells (Oh et al., 2000; Goumans et al., 2002). The more recently identified ALK7 is proposed to mediate signaling by another member of the TGF-β superfamily, Nodal (Reissmann et al., 2001), by activating Smad2 and (Watanabe et al., 1999; Jornvall et al., 2001). Although the Smad pathway has been studied the most intensively, the TGF-β family of ligands also activate other signaling pathways, including MAPK pathways and phosphatidyl inositol 3-kinase, presumably also through their type I receptors (de Caestecker et al., 2000).
Specific small molecule inhibitors developed for therapeutics can be powerful tools in dissecting complex signal transduction pathways involved in both normal and pathological processes. Moreover, better understanding of signal transduction pathways can lead to identification of new points of pharmacological intervention. Dysregulated signaling of TGF-β1, the first discovered and perhaps best-studied member of this TGF-β superfamily, has been implicated in the pathogenesis of a number of diseases, including fibrosis and carcinogenesis (Border and Noble, 1994; Wakefield and Roberts, 2002). TGF-β also plays important roles in wound healing and is possibly dysregulated in the related pathologies of hypertrophic scars and keloids (Bettinger et al., 1996; O'Kane and Ferguson, 1997). Based on this, there is intense interest in development of specific small molecule inhibitors of TGF-β signaling to be used for therapeutics.
SB-505124 was developed as a competitive inhibitor of the ATP binding site of ALK5 (J. Harling, P. E. Bender, J. L. Burgess, J. F. Callahan, J. A. Fornwald, L. M. Gaster, E. T. Grygielko, F. Harrington, N. J. Laping, R. Lehr, et al., submitted). In this study, we investigate the selectivity of this small molecule inhibitor and demonstrate that it inhibits signaling from ALK5 and the closely related receptors, ALK4 and ALK7, but not from other members of the ALK family. Consistent with the specificity of TGF-β and activin for activation of ALK5 and ALK4, respectively, we also show that SB-505124 selectively inhibits signaling from TGF-β and activin but does not affect signaling from BMP, which activates ALKs 3 and 6. SB-505124 also inhibits more complex endpoints that are dependent on TGF-β, such as apoptosis.
Materials and Methods
Plasmids and Reagents. Constitutively active ALK1, ALK2, ALK3, ALK4, ALK5, ALK6, and ALK7 constructs in mammalian vectors have been described previously by Nakao et al. (1997) and Jornvall et al. (2001). Plasmids encoding N-terminally flag-tagged Smad1 and Smad2 have been described previously (de Caestecker et al., 1998). The CAGA12-luciferase plasmid (Dennler et al., 1998) contains 12 repeats of the CAGA Smad binding element driving luciferase expression, and the reporter plasmid ARE-luciferase, which contains three repeats of the activin response element (Liu et al., 1997), was cotransfected with the plasmid encoding the coactivator FAST-1. BRE-luciferase plasmid containing BMP binding sites derived from the Id1 promoter (Korchynskyi and ten Dijke, 2002) was a kind gift from Dr Peter ten Dijke.
Activin (reconstituted in phosphate-buffered saline/0.1% BSA), epidermal growth factor (EGF; reconstituted in 10 mM acetic acid/0.5% BSA), and TNF-α (reconstituted in phosphate-buffered saline/1 mg/ml BSA) were obtained from R&D Systems (Minneapolis, MN). BMP-2 and TGF-β1 were reconstituted in 4 mM HCL/1 mg/ml BSA. SB-203580 (liquid, 250×) was purchased from Calbiochem (San Diego, CA). Solid anhydrous SB-505124 (J. Harling, P. E. Bender, J. L. Burgess, J. F. Callahan, J. A. Fornwald, L. M. Gaster, E. T. Grygielko, F. Harrington, N. J. Laping, R. Lehr, et al., submitted) was reconstituted in pure molecular grade water to a concentration of 13.45 mM and stored at -20°C. Before being added to cells, SB-505124 was diluted to 1 mM (a 1000× stock concentration).
Cell Culture and Transfection. All cells were maintained at 37°C in DMEM or DMEM/Ham's F12 media (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin in a 5% CO2 atmosphere. COS-1, HepG2, C2C12, Mv1Lu, Hs578t, and FaO cells were grown in supplemented DMEM medium, and NRP-154 cells were grown in supplemented DMEM/Ham's F12 medium. MCF10CA1h cells (a rastransformed derivative of MCF10A breast epithelial cells) were maintained in DMEM/Ham's F12 media containing 5% horse serum. COS-1 cells were transfected with FUGENE (Roche Molecular Biochemicals, Indianapolis, IN), and HepG2 cells were transfected with LipofectAMINE (Invitrogen) according to the manufacturer's instructions.
Western Blotting and p38 Kinase Activity. Cytoplasmic cell lysates were prepared in a Triton X-100 buffer [25 mM HEPES, pH 7.5, 100 mM NaCl, 10% glycerol, 5 mM EDTA, and 1% Triton X-100; protease inhibitors 1 mM phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 2 μg/ml pepstatin, and 2 μg/ml aprotinin; and phosphatase inhibitors sodium fluoride (50 mM) and sodium orthovanadate (1 μM)]. Lysates were cleared by centrifugation and protein concentrations determined using a protein assay (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. Samples (50 μg/lane) were subjected to SDS-polyacrylamide gel electrophoresis on 10% Tris-glycine gels (Novex, San Diego, CA) or 4–12% Bis-Tris gradient gels and electroblotted onto 0.45-μm nitrocellulose membranes (Bio-Rad). The membranes were blocked with 3% BSA in Tris-buffered saline/Tween 20 (10 mM Tris, pH 7.5, 150 mM NaCl, and 0.1% Tween 20), and probed at 4°C overnight with a 1:1000 dilution of primary antibody in 1% BSA-Tris-buffered saline/Tween 20 followed by a 30-min incubation at room temperature with a horseradish peroxidase (horseradish peroxidase)-linked anti-rabbit antibody (Amersham Biosciences, Piscataway, NJ). Horseradish peroxidase activity was visualized by enhanced chemiluminescence (Pierce, Rockford, IL). Primary antibodies used were polyclonal Smad2 (Zymed Laboratories, South San Francisco, CA), phospho-Smad2 (Upstate Biotechnologies, Lake Placid, NY), polyclonal Smad1/5/8 (a gift from Peter ten Dijke), polyclonal JNK, phospho-JNK, p44/42, phospho p44/42, ATF-2, and phospho-ATF-2 (Cell Signaling Technology, Beverly, MA). Typically, blots were probed with phospho-antibodies first, then stripped with Re-Blot Plus (Calbiochem, San Diego, CA) before being reblocked and reprobed for total protein.
p38 kinase activity was determined using a p38 MAPK assay kit (Cell Signaling Technology, Beverly, MA) according to the manufacturer's protocol. Briefly, phospho-p38 was immunoprecipitated from 200 μg of cell lysates with an immobilized phosphor-p38 antibody overnight at 4°C. p38 kinase activity was measured using 2 μg of ATF-2 fusion protein as the substrate with addition of 200 μM ATP. After a 30-min incubation at 30°C, the reaction was terminated with Laemmli sample buffer, and the proteins were boiled and resolved by 10% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, and immunoblotted with phospho-ATF-2 antibody.
Luciferase Assays. These assays were analyzed using the Dual-Luciferase reporter assay system (Promega). Twelve-well dishes were seeded at approximately 50% confluence and transfected with 50 ng of the appropriate luciferase reporter construct and for internal control of transfection efficiency, 5 ng of Renilla reniformis luciferase under a thymidine kinase promoter (pRL-tk). LipofectAMINE was used for cell transfections according to the manufacturer's instructions. After transfection, cells were incubated for 30 min with the indicated concentrations of SB 505124 (from stock concentrations of 1000×) before addition of TGF-β1, activin, or BMP-2. Cell lysates were harvested the following day as indicated by the manufacturer's protocol. Luminescence was measured by VICTOR2 (PerkinElmer Life and Analytical Sciences, Boston, MA).
Microscopy. FaO cells were plated at a confluence of approximately 70% in the wells of a six-well dish and allowed to attach overnight. Cells were treated with or without SB-505124 for 30 min before being treated with the indicated cytokine for 24 h. Images of the live cells were captured at 100× magnification.
In Vitro Protein Kinase Assay. Kinase assays were performed as described previously (Laping et al., 2002) using the kinase domain of ALK5 and full-length N-terminal fused GST-Smad3.
Cell Viability Assays. Cell viability was measured as described previously (Laping et al., 2002) or by using the modified tetrazolium salt WST-1 (Roche, Indianapolis, IN). Approximately 2000 cells were seeded in 96-well dishes in 100 μl of 0.2% FBS phenol red-free media overnight. The cells were treated with 50 μl of SB-505124 (to achieve the final concentrations indicated) for 30 min before being treated with or without TGF-β1 and TNF-α (both from R&D Systems) to a final volume of 200 μl. Cell growth is measured at the indicated time points by incubating each well with 10 μl of WST-1 for 3 h at 37°C. Metabolically active cells cleave WST-1 to water-soluble formazan, which is directly quantitated with an enzyme-linked immunosorbent assay plate reader. Each experiment was done at least twice, and treatment for each cell line was done in triplicate.
Results
SB-505124 Is a Selective Inhibitor of ALK4 and ALK5. SB-505124 was identified as a potent inhibitor of the in vitro kinase activity of ALK5 for its substrate Smad3 with an IC50 of 47 ± 5 nM (Fig. 1A) (J. Harling, P. E. Bender, J. L. Burgess, J. F. Callahan, J. A. Fornwald, L. M. Gaster, E. T. Grygielko, F. Harrington, N. J. Laping, R. Lehr, et al., submitted). This compound was determined to be a reversible ATP competitive inhibitor, because prior incubation of the ALK5 kinase with high concentrations of the inhibitor, followed by dilution to a sub-IC50 concentration had no effect on the kinetics of Smad3 phosphorylation (data not shown). SB-505124 demonstrated no toxicity to renal epithelial A498 cells at concentrations up to 100 μM for 48 h (Fig. 1B). Selectivity of the SB-505124 inhibitor for type I receptors other than ALK5 was first evaluated against ALK4 and ALK2 in vitro (Fig. 2A). SB-505124 inhibited the closely related ALK4 with an IC50 value of 129 ± 11 nM (about 2.5-fold less sensitive than ALK5) but did not inhibit ALK2 at concentrations up to 10 μM. To further assess the selectivity of SB-505124, we used a panel of ALKs mutationally activated to signal independently of their type II receptor or ligand by a single amino change of a threonine to aspartate, to mimic charge effects of phosphorylation near the kinase domain (Nakao et al., 1997; Jornvall et al., 2001). COS-1 cells were cotransfected to overexpress constitutively active forms ALK1, ALK2, ALK3, ALK4, ALK5, ALK6, or ALK7 together with either Smad1 or Smad2. In the canonical signaling pathway, Smad1 is phosphorylated by active ALK1, ALK2, ALK3, and ALK6, whereas Smad2 is activated by ALK4, ALK5, and ALK7 (Watanabe et al., 1999; Piek and Roberts, 2001). Consistent with this, Western blot analysis of Smad2 phosphorylated by the activated ALKs in the presence or absence of 5 μM SB-505124 showed that only constitutively active ALK4, ALK5, and ALK7 resulted in phosphorylation of Smad2, and that this activation was inhibited in the presence of SB-505124 (Fig. 2B). Phosphorylation of endogenous Smad2 in the COS-1 cells was also inhibited by SB-505124 treatment. Constitutively active ALK1, ALK2, ALK3, and ALK6 phosphorylated Smad1, but this activation was not affected by the addition of SB-505124. It is also noteworthy that constitutively active ALK4 and ALK5 also activated Smad1 and, like the activation of Smad2 by these receptors, this activation was also abolished with the addition of SB-505124 inhibitor. This observation is consistent with previously published studies using the related inhibitor SB-431542 (Inman et al., 2002a), suggesting that the preference for receptor-activated Smad is possibly not as stringent as previously thought. However, these results clearly demonstrate selectivity of the SB-505124 inhibitor for ALK4, ALK5, and ALK7 and are consistent with sequence homology data showing that the kinase domains of these three receptors are closely related and differ considerably from the other ALKs.
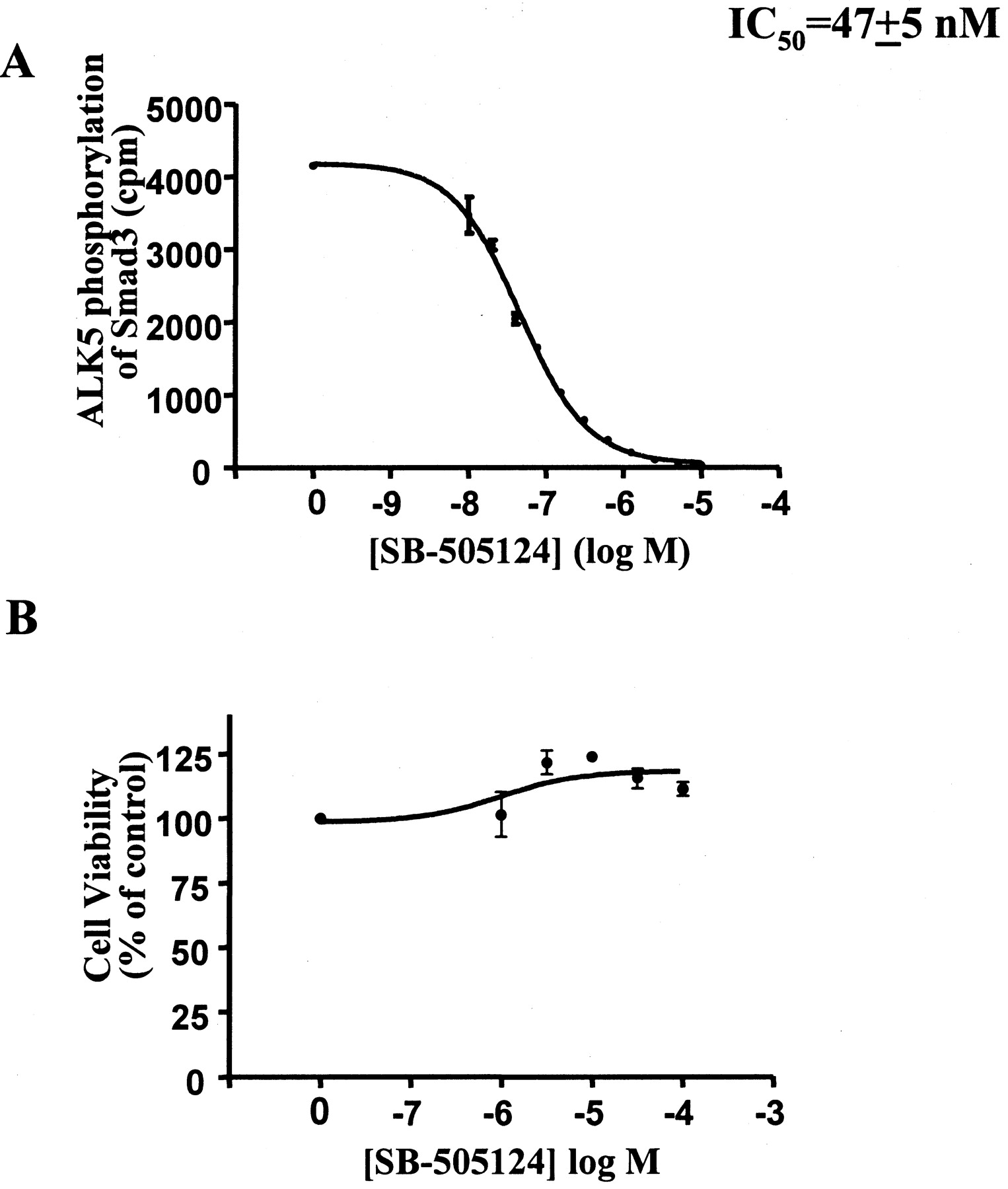
A, in vitro phosphorylation of purified baculovirus expressed GST-Smad3 by the purified kinase domain of ALK5. SB-505124 inhibited ALK5 kinase activity with an IC50 value of 47 ± 5 nM (S.E.M.). B, effect of SB-505124 on cell viability as measured by mitochondrial activity (XTT assay). A498 renal epithelial cells treated for 48 h were not affected by SB-505124 at concentrations up to 100 μM (n = 3 experiments).

A, comparison of in vitro Smad phosphorylation by the purified kinase domain of ALK2, ALK4, or ALK5 shows that SB-505124 inhibited ALK5 and ALK4 with similar IC50 values (47 ± 5 nM and 129 ± 11 nM, respectively) but did not inhibit ALK2 up to concentrations of 10 μM. Error values represent S.E.M. from one experiment done in triplicate. B, SB-505124 selectively inhibits ALK4 and ALK5. COS1 cells were cotransfected with plasmids expressing the indicated constitutively active ALK and plasmids expressing either Smad1 or Smad2. Four hours after transfection, SB-505124 was added directly to the media to a final concentration of 5 μM and cells were incubated overnight. Whole-cell lysates were collected and subjected to Western blot analysis.
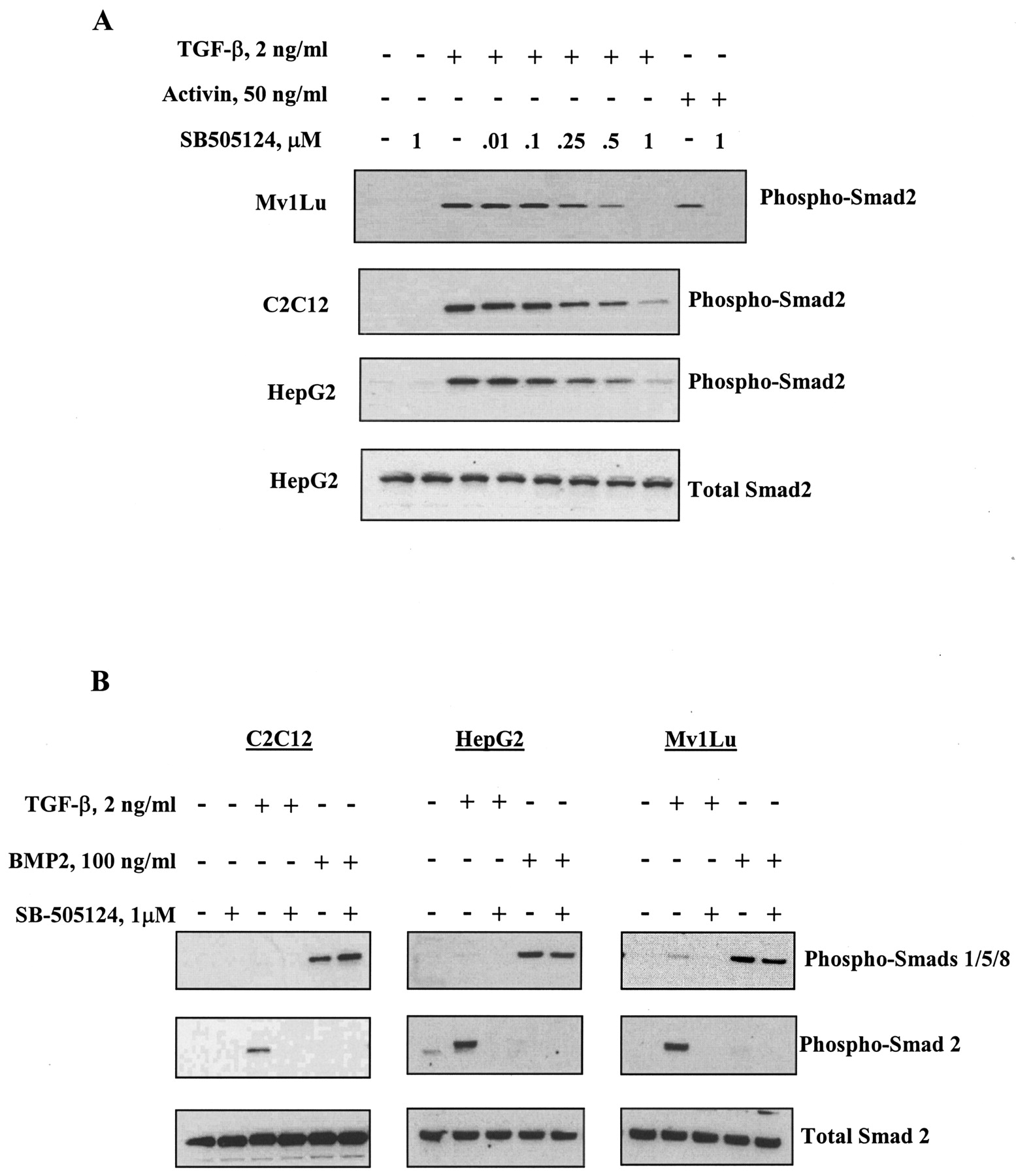
SB-505124 Inhibits TGF-β- and Activin-Induced Smad Activation. Treatment of cells with activin or TGF-β results in phosphorylation of ALK4 and ALK5, respectively, which then enables phosphorylation of their downstream substrates Smad2 and Smad3 (Piek and Roberts, 2001). Treatment of cells with BMP induces activation of ALK3 and ALK6, resulting in phosphorylation of Smads 1, 5, and 8 (Heldin et al., 1997). To further examine the selectivity of SB-505124 for inhibition of ALK4 and ALK5, HepG2 human hepatoma cells, C2C12 mouse myoblasts and Mv1Lu mink lung cells were treated with either activin or TGF-β and endogenous phospho-Smad2 was analyzed by Western blot. SB-505124 inhibited the TGF-β–induced phosphorylation of Smad2 in all three of these cell lines in a concentration-dependent fashion (Fig. 3A). Activin-induced phosphorylation of Smad2 was also abolished by treatment with 1 μM SB-505124. In contrast, BMP-induced activation of Smad1, -5, or -8 was unaffected by pretreatment with the inhibitor (Fig. 3B). This confirms that SB-505124 is a selective inhibitor of TGF-β and activin Smad signaling and does not affect the closely related BMP signaling pathway.
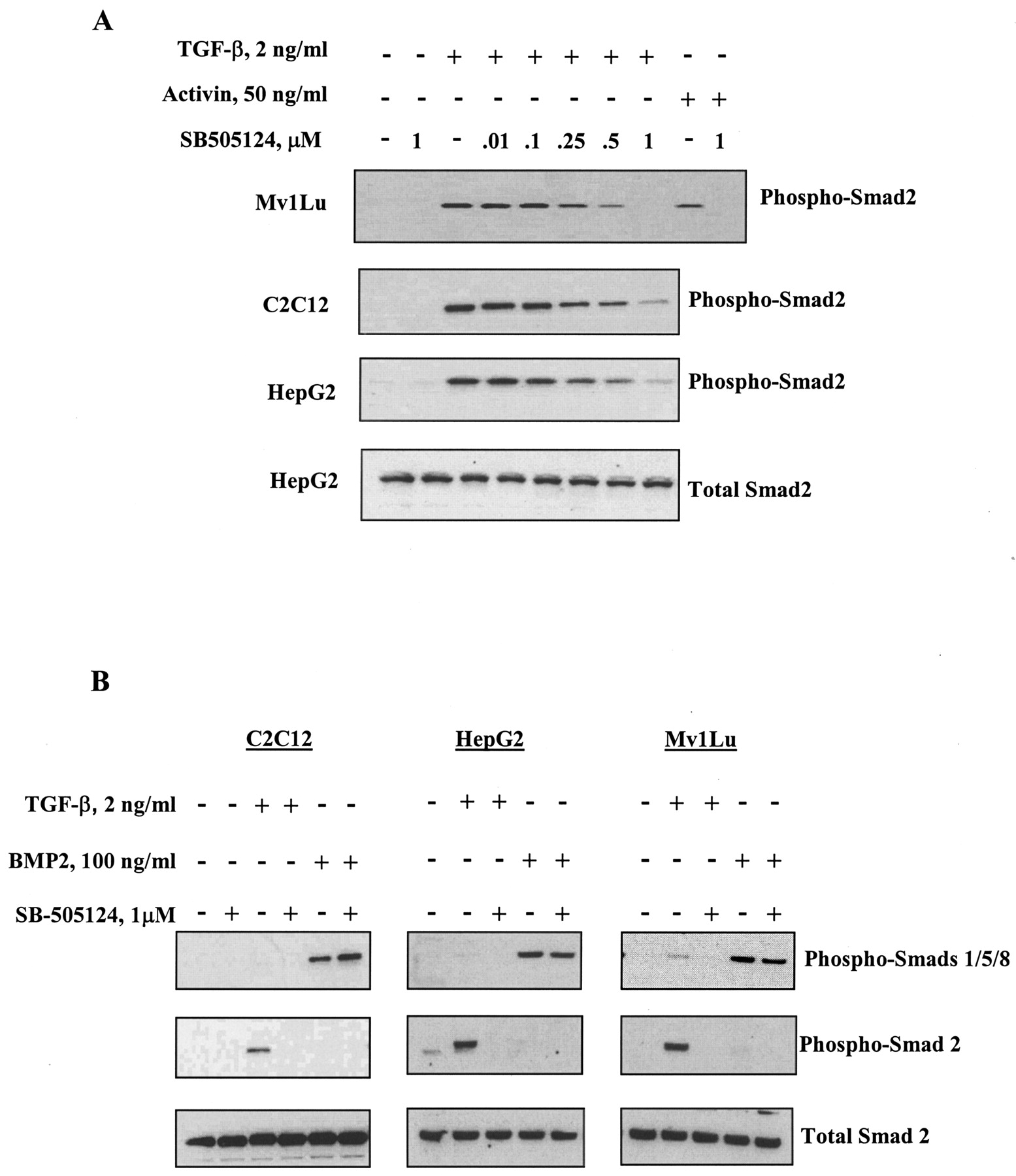
SB-505124 inhibits activin- and TGF-β1–induced phosphorylation of Smad2 in a concentration-dependent manner but does not alter BMP2-induced Smad activation. Cells were serum-starved overnight and treated for 30 min with the indicated concentrations of SB-505124 before being treated with TGF-β1, activin (A), or BMP-2 (B). Whole-cell lysates were collected and subjected to Western blot analysis.
SB-505124 Inhibits Transcription of Reporter Genes Induced by TGF-β and Activin but Not by BMP. Phosphorylated receptor-activated Smads form heterooligomeric complexes with the common mediator, Smad4, and are translocated into the nucleus, where they bind specific DNA sequences and affect gene transcription in a ligand-dependent fashion. To determine whether inhibition of ALK4 and ALK5 by SB-505124 can abrogate Smad-activated transcription, luciferase assays were carried out using artificial receptor constructs known to be specific for Smad2, Smad3, or the BMP Smads. The CAGA12-luciferase reporter is driven by 12 repeats of the CAGACA sequence, or “CAGA box”, which specifically binds the activated Smad3/4 complex (Dennler et al., 1998). The ARE-luciferase reporter consists of three copies of the activin response element sequence derived from the Mix.2 homeobox gene promoter (Chen et al., 1997; Liu et al., 1997). When cotransfected with the forkhead activin signal transducer FAST-1, ARE-luciferase is induced by TGF-β- or activin-activated Smad2/4 complexes (Chen et al., 1996; Liu et al., 1997). In contrast, the BRE-luciferase reporter, consisting of the BMP response elements of the Id1 promoter cloned upstream of a minimal promoter driving luciferase expression, is activated only by BMP and not by TGF-β or activin (Korchynskyi and ten Dijke, 2002). The ability of TGF-β and activin to induce both the CAGA12-luciferase (Fig. 4A) and the ARE-luciferase reporter constructs (Fig. 4B) is potently inhibited in a concentration-dependent manner by SB-505124. However, the ability of BMP2 to induce BRE-luciferase activity was unaffected by treatment up 5 μM SB-505124 (Fig. 4C). Thus, SB-505124 is a highly selective and potent inhibitor of TGF-β- and activin-induced reporter activation with a preference for inhibiting TGF-β over activin signaling.

SB-505124 inhibits TGF-β1- and activin-induced transcriptional activation in a concentration-dependent manner but does not affect BMP-2-induced reporter activity. HepG2 cells were transfected with CAGA12-luciferase (A), ARE-luciferase and FAST-1 (B), or BRE-luciferase (C). Sixteen hours after transfection, the media were replaced with reduced-serum media (0.2% FBS) and SB-505124 was added to cells for 30 min before addition of TGF-β1, activin, or BMP-2. Luciferase activity was measured after an overnight incubation and normalized. Error bars represent S.E.M. of experiments performed in triplicate.
SB-505124 Inhibits TGF-β- but Not EGF-Stimulated Activation of MAP Kinase Pathways. It has recently been demonstrated that activation of ALK 4 and ALK5 by activin and TGF-β can also result in phosphorylation and activation of MAPK pathways (de Caestecker et al., 2000; Mulder, 2000; Derynck et al., 2001), although both the kinetics of activation and the particular type of MAPK pathway activated vary considerably among cell types. To investigate whether SB-505124 also interferes with TGF-β–dependent activation of MAPK signaling, we used FaO cells, a rat hepatoma cell line previously shown to be sensitive to TGF-β–induced activation of MAPKs (Park et al., 2002). Western blot analysis of FaO cells demonstrates that 1 or 5 μM SB-505124 potently inhibits TGF-β–induced activation of JNK/SAP, extracellular signal-regulated kinase 1/2, and p38 despite the different patterns of activation in these cells (Fig. 5). Moreover, as expected, the inhibitor had no effect on the activation of JNK/SAP by treatment of cells with interleukin-1 (Fig. 5B).
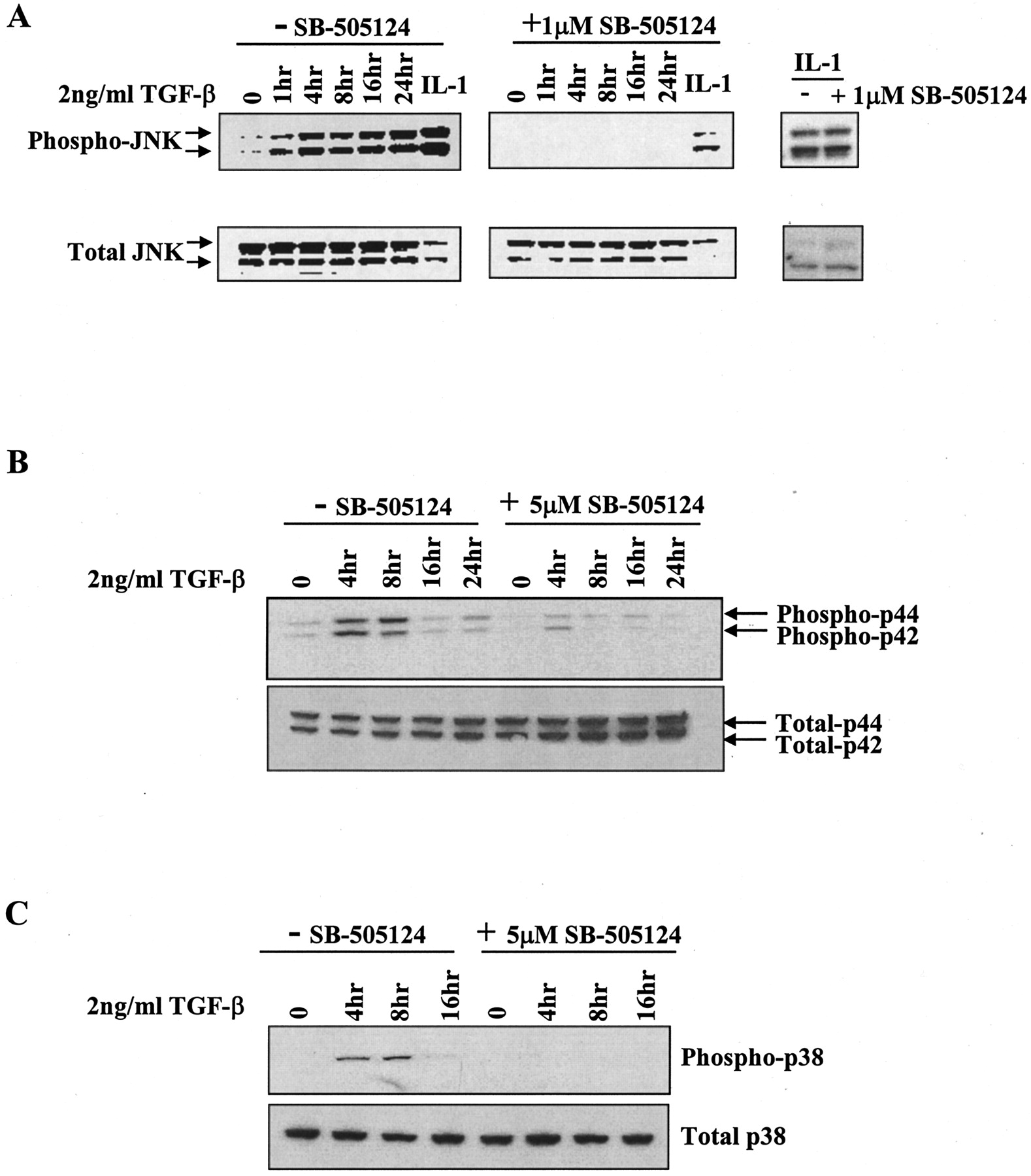
SB-505124 inhibits TGF-β1-induced activation of MAP kinases. FaO cells were serum-starved overnight and treated with SB-505124 for 30 min before being treated with 2 ng/ml of TGF-β1 for various times. Whole-cell lysates were collected at the times indicated and subjected to Western blot analysis for JNK1/2 (A), p44/42 (B), and p38 (C).
To further evaluate the selectivity of SB-505124, the effect of the inhibitor on the in vitro activity of various protein kinases was assayed (Table 1). The only kinase inhibited more than 50% by 10 μM SB-505124 was p38 MAPKα. Further analysis showed that SB-505124 inhibits p38 MAPKα with an IC50 value of 10.6 μM (Fig. 6A), which is more than 200 times the IC50 value for ALK5. This suggests that SB-505124 would not interfere with downstream responses to p38 MAPKα at the concentrations of inhibitor used in this study. To test this directly and ensure that p38 activity induced by pathways independent of ALK5 was not altered by SB-505124 treatment, we assessed the effects of EGF on activation of ATF-2, a transcription factor that is a target of MAPK signaling pathways including the p38 signaling pathway. Cells were treated with 100 ng/ml EGF, with or without SB-505124, and the levels of p38 kinase activity was determined by in vitro kinase assay using ATF-2 as substrate or by direct Western blotting of whole-cell lysates. We show that phosphorylation of ATF-2 induced by EGF was reduced by treatment with the p38 inhibitor SB-203580 but was unaffected by SB-505124 at concentrations up to 10 μM (Fig. 6, B and C). These results, together with the results shown in Fig. 5, demonstrate selectivity of SB-505124 for TGF-β–induced activation of p38 MAPK at the concentrations used in this study.
Effect of SB-505124 on the activities of protein kinases in vitro Protein kinases were assayed with 10 μ M SB-505124 in the presence of 0.1 mM ATP. Activities are given as the mean of duplicate determinations relative to control incubations with DMSO vehicle.

A, SB-505124 inhibits in vitro p38 phosphorylation of ATF-2 with an IC50 value of 10.6 ± 1.8 μM. B, p38 kinase assay performed using ATF-2 as substrate for immunoprecipitated phospho-p38 from EGF-stimulated MCF10Ca1h cells. C, Western blot analysis of lysates from cells treated with EGF show that SB-505124 does not inhibit EGF-activated p38 phosphorylation of ATF-2.
SB-505124 Abrogates Cell Death Caused by TGF-β Treatment. TGF-β induces apoptosis in a number of cell lines, including FaO cells and NRP 154 cells (Hsing et al., 1996; Larisch-Bloch et al., 2000; Kim et al., 2002; Park et al., 2002). Light microscopy images show that FaO cells round up and detach from the tissue culture surface (morphological evidence of cell death) in response to treatment with TGF-β or TNF-α for 24 h (Fig. 7A). Pretreatment of the cells with SB-505124 blocked TGF-β–induced cell death but had no effect on TNF-α–induced toxicity. These results are further supported by cell viability assays demonstrating that FaO cells or NRP 154 cells are protected from TGF-β–induced apoptosis by SB-505124 in a concentration-dependent manner, whereas there was no effect of the drug on loss of cell viability caused by treatment with TNF-α (Fig. 7B,C). These results again demonstrate selectivity of SB-505124 for TGF-β–induced and therefore ALK5-dependent responses.

SB-505124 selectively inhibits TGF-β1–induced apoptosis. A, FaO cells were serum-starved overnight and treated with 1 μM SB-505124 for 1 h before addition of 5 ng/ml TGF-β1 or 10 ng/ml TNF-α for 24 h. FaO cells (B) or NRP 154 cells (C) were plated in 96-well dishes in triplicate and treated with the indicated concentrations of SB-505124 before being treated with TGF-β1 or TNF-α. At day 3 (B) or 6 (C), WST-1 cell viability assay was performed. The error bars represent the S.E.M. of experiments performed in triplicate.
SB-505124 Is More Potent Than ALK5 Inhibitor SB-431542. The SB-505124 inhibitor was compared with the chemically related ALK5 inhibitor SB-431542 (Callahan et al., 2002; Inman et al., 2002a) by luciferase assay using the CAGA12-luciferase reporter construct. It was determined that SB-505124 is more potent than SB-431452 in this assay (Fig. 8), consistent with the reported IC50 values of 47 ± 5nM (Fig. 1A) and 94 nM (Callahan et al., 2002), respectively, for in vitro phosphorylation of immobilized Smad3.

SB-505124 is more potent than SB-431542. HepG2 cells were transfected with CAGA12-luciferase. Sixteen hours after transfection, the media were replaced with reduced-serum media (0.2% FBS) and SB-505124 or SB-431542 (both resuspended in DMSO) were added for 30 min. TGF-β1 was added directly to the media, and cells were incubated overnight; then, luciferase activity was measured. Error bars represent S.E.M. of triplicate values.
Discussion
SB-505124 is a small molecule inhibitor (Mr 335) identified on the basis of its ability to inhibit the in vitro kinase activity of ALK5 (J. Harling, P. E. Bender, J. L. Burgess, J. F. Callahan, J. A. Fornwald, L. M. Gaster, E. T. Grygielko, F. Harrington, N. J. Laping, R. Lehr, et al., submitted). Our results, using cell-based assays, demonstrate that SB-505124 selectively inhibits ALK5 and the closely related ALK4 and ALK7 without affecting the other type I receptors, ALK1, ALK2, ALK3, or ALK6. Given the specificity of TGF-β and activin signaling through ALK5 and ALK4, respectively, it therefore seemed likely that SB-505124 would selectively abrogate cellular responses resulting from treatment with these particular ligands. Supporting this hypothesis, we show that 1 μM SB-505124 treatment inhibits both Smad signaling and gene transcription induced by activin or TGF-β treatment but does not alter BMP-induced transcription or signaling. SB-505124 treatment also blocked activation of MAPK pathways stimulated by TGF-β but did not affect these same kinases when induced by EGF. Furthermore, 10 μM SB-505124 had little or no effect on a panel of protein kinases tested in vitro except for p38MAPKα. However, we demonstrate by Western blot analysis that up to 5 μM SB-505124, the maximal concentration used in this study, showed no significant decrease in EGF-induced p38 activity. Together, these data demonstrate the selectivity of SB-505124 to abrogate activin- and TGF-β–induced responses by blocking the kinase activity of ALK4 and ALK5. Furthermore, because we also demonstrate that SB-505124 inhibits ALK7 activity, it is likely that this inhibitor will block not only TGF-β- and activin- but also Nodal-dependent signals.
The availability of soluble small molecule inhibitors such as SB-505124 provides an important new tool with which to investigate the mechanism of TGF-β signaling. The previously described chemically related ALK5 inhibitor SB-431542 has already been used to investigate aspects of Smad2 and Smad3 nuclear shuttling and stability of their activated forms (Inman et al., 2002b). However, whereas SB-431542 shows cellular toxicity at 100 μM in vivo, the SB-505124 inhibitor showed no cellular toxicity at concentrations up to 100 μM and was well tolerated at concentrations that should prove effective in inhibiting ALK4, ALK5 and ALK7 in in vivo models (B. A. Olson, M. Spengler, and K. Salyers, unpublished observations).
Aberrant TGF-β signaling has been implicated in numerous animal models of fibrosis as well as in several human fibrotic diseases, including radiation fibrosis (Border and Noble, 1994; Martin et al., 2000). Antagonizing TGF-β signaling using neutralizing antibodies, soluble type II receptors, or antisense oligonucleotides has been shown to perturb various types of TGF-β–mediated fibrosis (Border and Noble, 1994; Hakenjos et al., 2000; Martin et al., 2000). These data provide a strong rationale for clinical testing of type I receptor kinase inhibitors, such as SB-505124, that interfere with TGF-β/activin-dependent signaling.
TGF-β signaling has a central role in tissue repair and cutaneous wound healing, regulating the influx of inflammatory cells and the deposition of extracellular matrix in the granulation tissue as well as the migration of keratinocytes to restore the cutaneous barrier (Singer and Clark, 1999). Our laboratory has demonstrated that loss of Smad3 results in reduced elaboration of TGF-β in the wound bed and both enhances re-epithelialization of the wound and reduces inflammation (Ashcroft et al., 1999; Flanders et al., 2002). Thus, inhibition of TGF-β signaling can be predicted to increase re-epithelialization of wounds and to reduce inflammation, with potential clinical applications for treatment of both surgical and chronic wounds. Our laboratory is currently conducting experiments with SB-505124 to explore its effects on in vivo models of wound healing.
The results presented in this study demonstrate that SB-505124 is a selective, nontoxic, water-soluble inhibitor of ALK4 and ALK5 and suggest that it may have potential as a therapeutic agent to modulate a variety of pathogenic effects of TGF-β. Regardless, SB-505124 will prove to be a valuable tool in elucidating the complex pathway and cross talk of the TGF-β signaling pathway both in vitro and in animal models in vivo.
Acknowledgments
We sincerely thank Drs. Stephen P. Davies, Helen Reddy, and Professor Sir Philip Cohen (Division of Signal Transduction Therapy and MCR Protein Phosphorylation Unit, University of Dundee, UK) for providing data on selectivity of SB-505124 for various protein kinases in vitro (Table 1). We also thank Drs. Jeff Wrana (Samuel Lunenfeld Research Institute, Toronto, Canada) and Liliana Attisano (University of Toronto, Toronto, Canada) for TGF-β-receptor constructs, Dr. Caroline Hill (Cancer Research UK London Research Institute, London, UK) for the ALK7 DNA construct, and Drs. Seong-Jin Kim and Byung-Chul Kim (National Cancer Institute, Bethesda, MD) for FaO cells.
Footnotes
-
ABBREVIATIONS: TGF, transforming growth factor; BMP, bone morphogenetic protein; ALK, activin-like kinase; MAPK, mitogen activated protein kinase; SB-505124, 2-(5-benzo[1,3]dioxol-5-yl-2-tert-butyl-3H-imidazol-4-yl)-6-methylpyridine hydrochloride; BSA, bovine serum albumin; EGF, epidermal growth factor; TNF, tumor necrosis factor; SB-203580, 4-(4-fluorophenyl)-2-(4-methylsulfinyl phenyl)-5-(4-pyridyl) 1H-imidazole; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; JNK, c-Jun NH2-terminal kinase; ATF, activating transcription factor; SB-431542, 4-(5-benzo[1,3]dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)-benzamide.
- Received June 13, 2003.
- Accepted December 12, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}