


Abstract
2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO) and the corresponding methyl (CDDO-Me) and imidazole (CDDO-Im) esters induce peroxisome proliferator-activated receptor γ (PPARγ)-dependent transactivation in SW-480 colon cancer cells, and these responses were inhibited by small inhibitory RNA for PPARγ. Moreover, in a mammalian two-hybrid assay using the PPARγ2-VP16 fusion plasmid and GAL4-coactivator/corepressor chimeras and a construct (pGAL4) containing five tandem GAL4 response elements, CDDO, CDDO-Me, and CDDO-IM induce transactivation and PPARγ interaction with multiple coactivators. A major difference among the three PPARγ agonists was the higher activity of CDDO-Im to induce PPARγ interactions with the corepressor SMRT. CDDO, CDDO-Me, and CDDO-Im inhibited SW-480, HCT-116, and HT-29 colon cancer cell proliferation at low concentrations and induced cell death at higher concentrations. Growth inhibition at lower concentrations correlated with induction of the tumor suppressor gene caveolin-1 which is known to inhibit colon cancer cell growth. Induction of caveolin-1 by CDDO, CDDO-Me, and CDDO-Im was inhibited by the PPARγ antagonist N-(4′-aminopyridyl-2-chloro-5-nitrobenzamide (T007), whereas higher doses induced apoptosis [poly(ADP-ribose) polymerase cleavage], which was not inhibited by T007. These results illustrate that CDDO-, CDDO-Me, and CDDO-Im induce both PPARγ-dependent and -independent responses in colon cancer cells, and activation of these pathways are separable and concentration-dependent for all three compounds.
2-Cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) is a synthetic triterpenoid-derived compound structurally related to the pentacyclic triterpenoids oleanolic and ursolic acids which exhibit anti-inflammatory and anticarcinogenic activities (Nishino et al., 1988; Huang et al., 1994; Suh et al., 1998). CDDO and related compounds inhibit the growth of multiple cancer cell lines, induced differentiation, and inhibited inducible nitric-oxide synthase and cyclooxygenase 2 activities (Nishino et al., 1988; Honda et al., 1997, 1998, 2000; Suh et al., 1998, 1999; Place et al., 2003). CDDO and the methyl ester derivative (CDDO-Me) bound peroxisome-proliferator–activated receptor γ (PPARγ), and CDDO induced transactivation in CV-1 cells transfected with PPARγ-responsive GAL4-PPARγ or PPARγ response element (PPRE) (promoter) constructs (Wang et al., 2000). CDDO alone or in combination with the retinoid X receptor agonist LG100268 induced 3T3-L1 differentiation, whereas CDDO-Me was inactive in this assay. It was suggested that CDDO was a PPARγ agonist, whereas CDDO-Me exhibited partial antagonist activity (Wang et al., 2000).
CDDO, CDDO-Me, and an imidazole amide (CDDO-Im) decrease cancer cell survival, and their cell context-dependent responses and mechanisms of action have been investigated (Ito et al., 2000; Konopleva et al., 2002; Pedersen et al., 2002; Stadheim et al., 2002; Ikeda et al., 2003; Suh et al., 2003; Konopleva et al., 2004a,b). These triterpenoid compounds are potent inducers of differentiation and apoptosis in leukemia cells; however, their proapoptotic effects were somewhat variable among different cell lines. CDDO-Me induced intrinsic apoptotic pathways in HL-60 cells, and enhanced responses were observed after cotreatment with retinoid X receptor ligands (Konopleva et al., 2002). CDDO-induced apoptosis in HL-60 and U937 cells were inhibited by dominant-negative PPARγ expression and the PPARγ antagonist T007 (Konopleva et al., 2004a). CDDO and related compounds also induced caspase-8–dependent pathways in leukemia cells (Pedersen et al., 2002; Stadheim et al., 2002; Ikeda et al., 2003; Suh et al., 2003; Konopleva et al., 2004a), and this may be caused, in part, by down-regulation of FLIP, an endogenous inhibitor of caspase-8 activation. CDDO also induced apoptosis in leukemia cells through enhanced oxidative stress (Ikeda et al., 2003) and loss of mitochondrial membrane potential (Konopleva et al., 2004b). In breast cancer cells, CDDO inhibits the growth of ER-positive and -negative cells and tumor growth in athymic nude mouse models, and this correlated with the modulation of genes associated with cell-cycle progression, apoptosis, and ER stress (Lapillonne et al., 2003). In COLO 16 human skin cancer cells, CDDO induced apoptosis, and this was in part caused by ER stress and direct mitochondrial effects that disrupted calcium homeostasis (Hail et al., 2004). CDDO-Me induced both the intrinsic and extrinsic apoptosis pathway in lung cancer cell lines (Kim et al., 2002; Zou et al., 2004), and a caspase-8–dependent apoptotic pathway was activated by CDDO in human osteosarcoma cells (Ito et al., 2001). CDDO also inhibited the growth of several ovarian cancer cell lines that express PPARγ, and because cotreatment with the PPARγ antagonist T007 did not block the effects of CDDO, it was concluded that this response was PPARγ-independent (Kodera et al., 2000; Melichar et al., 2004).
This study reports the effects of CDDO compound on SW-480 and other colon cancer cell lines and investigates the concentration-dependent induction of PPARγ-dependent and -independent responses. Growth-inhibitory IC50 values for CDDO-Me and CDDO-Im were ≤0.2 μM in SW-480, HCT-116, and HT-29 colon cancer cells, whereas IC50 values for CDDO were ≤0.5 μM after 6 days of growth. CDDO, CDDO-Im, and CDDO-Me also induced PPARγ-dependent transactivation and coactivator-PPARγ interactions in mammalian two-hybrid assays in SW-480 cells. In the same cell lines, CDDO and related compounds induce PPARγ-dependent up-regulation of the tumor suppressor gene caveolin-1 and PPARγ-independent apoptosis, and these responses were activated over distinct and separable concentrations in SW-480 cells.
Materials and Methods
Cell Culture. Human colon cancer cell lines SW-480 and HT-29 were provided by M.D. Anderson Cancer Center (Houston, TX); HCT-116 cells were obtained from the American Type Culture Collection (Manassas, VA). SW-480 and HT-29 cells were maintained in Dulbecco's modified Eagle's medium nutrient mixture with Ham's F-12 (DMEM/Ham's F-12; Sigma-Aldrich, St. Louis, MO) with phenol red supplemented with 0.22% sodium bicarbonate, 0.011% sodium pyruvate, and 5% fetal bovine serum (FBS; Serologicals Corp., Norcross, GA) and 10 ml/l 100× antibiotic antimycotic solution (Sigma-Aldrich). HCT-116 cells were maintained in RPMI 1640 medium (Sigma-Aldrich) supplemented with 0.22% sodium bicarbonate, 0.011% sodium pyruvate, 0.45% glucose, 0.24% HEPES, 10% FBS, and 10 ml/l 100× antibiotic antimycotic solution (Sigma-Aldrich).
Chemicals, Reagents, Plasmids, and Antibodies. The PPARγ antagonist T007 was synthesized in our laboratory and confirmed by gas chromatography-mass spectrometry. CDDO compounds were provided by Dr. Edward Sausville (Developmental Therapeutics Program, National Cancer Institute, Bethesda, MD) through the Rapid Access to Intervention Developmental Program. Horseradish peroxidase substrate for Western blot analysis was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Cell lysis buffer and luciferase reagent were purchased from Promega (Madison, WI), and β-galactosidase (β-gal) reagent was from Tropix (Bedford, MA). Small inhibitory RNA (siRNA) duplexes were prepared by Dharmacon Research (Lafayette, CO). Previous studies in this laboratory have reported oligonucleotide sequences for PPARγ and lamin A/C siRNA (Qin et al., 2004). The Gal4 reporter containing 5× Gal4 DBD (Gal4Luc) was kindly provided by Dr. Marty Mayo (University of North Carolina, Chapel Hill, NC). Gal4DBD-PPARγ construct was a gift from Dr. Jennifer L. Oberfield (GlaxoSmithKline Research and Development, Research Triangle Park, NC), and PPARγ expression plasmid and pM-PPARγ coactivator-1 (PGC1) were gifts from Dr. Bruce M. Spiegelman (Harvard University, Boston, MA). The PPRE-luc construct contains three tandem PPREs with a minimal TATA sequence in pGL2. The PPARγ2-VP16 fusion plasmid (VP-PPARγ) contained the DEF region of PPARγ (amino acids 183–505) fused to the pVP16 expression vector, and the GAL4-coactivator fusion plasmids pMSRC1, pMSRC2, pMSRC3, pMDRIP205, and pMCARM-1 were kindly provided by Dr. Shigeaki Kato (University of Tokyo, Tokyo, Japan). Antibodies for caveolin-1, PARP, Akt, and phospho-Akt were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-β-actin was purchased from Sigma-Aldrich (St. Louis, MO).
Cell Proliferation Assay. Cells (2 × 104) were plated in 12-well plates, and media were replaced the next day with DMEM/Ham's F-12 media containing 2.5% charcoal-stripped FBS and either vehicle (DMSO) or the indicated ligand and dissolved in DMSO. Fresh media and compounds were added every 48 h. Cells were counted at the indicated times using a Coulter Z1 cell counter. The proliferation of SW-480 cells was also carried out as described above using the colorimetric WST1 assay according to the manufacturer's instructions (Roche Diagnostics, Mannheim, Germany). Cells (4000/well) were seeded in 96-well plates and were assayed after 48 h. Each experiment was carried out at least three times, and results are expressed as means ± S.E. for each determination.
Transfection and Luciferase Assay. Colon cancer cells (1 × 105) were seeded onto 24-well plates in DMEM/F-12 media supplemented with 2.5% charcoal-stripped FBS and grown overnight. Transient transfections were performed using LipofectAMINE reagent (Invitrogen, Carlsbad, CA) according to the protocol provided by the manufacturer. For transfections with siRNA for PPARγ, the RNA concentration was 75 nM, and Oligofectamine transfection regent (Invitrogen) was used. Cotransfections were performed using Gal4Luc (0.4 μg), β-gal 0.04 μg), Gal4DBD-PPARγ (0.04 μg), VP-PPARγ (0.04 μg), pMSRC1 (0.04 μg), pMSRC2 (0.04 μg), pMSRC3 (0.04 μg), pMPGC-1 (0.04 μg), pMDRIP205 (0.04 μg), and pMCARM-1 (0.04 μg). After 5 h of transfection, the transfection mix was replaced with complete media containing either vehicle (DMSO) or the indicated ligand for 20 to 22 h. Cells were then lysed with 100 μl of 1× reporter lysis buffer, and 30 μl of cell extract was used for luciferase and β-galactosidase assays. A Lumicount luminator (PerkinElmer Life and Analytical Sciences) was used to quantify luciferase and β-galactosidase activities, and the luciferase activities were normalized to β-galactosidase activity.
Western Blot Analysis. SW-480, HT-29, and HCT-116 (3 × 105) cells were seeded in six-well plates in DMEM/Ham's F-12 media containing 2.5% charcoal-stripped FBS for 24 h and then treated with either the vehicle (DMSO) or the indicated compounds. Whole-cell lysates were obtained using high-salt buffer [50 mM HEPES, 500 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 1% Triton X-100, pH 7.5, and 5 μl/ml Protease Inhibitor Cocktail (Sigma-Aldrich)]. Protein samples were incubated at 100°C for 2 min, separated on 10% SDS-PAGE at 120 V for 3 to 4 h in 1× running buffer (25 mM Tris-base, 192 mM glycine, and 0.1% SDS, pH 8.3), and transferred to polyvinylidene difluoride membrane (PVDF; Bio-Rad, Hercules, CA) at 0.1 V for 16 h at 4°C in 1× transfer buffer (48 mM Tris-HCl, 39 mM glycine, and 0.025% SDS). The PVDF membrane was blocked in 5% TBST-Blotto (10 mM Tris-HCl, 150 mM NaCl, pH 8.0, 0.05% Triton X-100, and 5% nonfat dry milk) with gentle shaking for 30 min and was incubated in fresh 5% TBST-Blotto with 1:1000 (for caveolin-1), 1:250 (for PARP), and 1:5000 (for β-actin) primary antibody overnight with gentle shaking at 4°C. After washing with TBST for 10 min, the PVDF membrane was incubated with secondary antibody (1:5000) in 5% TBST-Blotto for 90 min. The membrane was washed with TBST for 10 min, incubated with 10 ml of chemiluminescence substrate (PerkinElmer) for 1.0 min, and exposed to Kodak X-OMAT AR autoradiography film (Eastman Kodak, Rochester, NY). Band intensities were evaluated by scanning laser densitometry (Sharp Electronics Corporation, Mahwah, NJ) using Zero-D Scanalytics software (Scanalytics, Billerica, MA).
Results
Initial studies investigated the effects of CDDO, CDDO-Me, and CDDO-Im on the growth of SW-480, HT-29, and HCT-116 colon cancer cells. Cells were treated with different concentrations of the test compounds on days 0, 2, and 4 (media and compounds were changed every 2 days), and cell numbers were determined 2, 4, and 6 days after treatment. Significant inhibition of cell proliferation was observed for all compounds after treatment for 2 and 4 days, and IC50 values were ≤0.2 μM for CDDO-Me and CDDO-Im and ≤0.5 μM for CDDO in all three cell lines (data not shown). Cell-survival results after treatment with the CDDO compounds (Fig. 1) were derived from the growth-inhibition curves, and the percentage of cell survival was the ratio of the number of cells in the treated/DMSO (solvent) groups at each time point. Higher concentrations of these compounds decreased cell numbers to levels lower than the initial number of attached cells (i.e., cell death), and after 96 h, the concentrations of CDDO, CDDO-Me, and CDDO-Im that induced cell death in all three cell lines were 1.0 to 2.5, 0.2, and 0.2 μM, respectively. CDDO decreased proliferation of SW-480 cells using the WST-1 colorimetric assay (Fig. 1D), and cotreatment of CDDO plus 5.0 μM T007 (PPARγ antagonist) significantly reversed the growth-inhibitory effects of CDDO. The results demonstrate a role for PPARγ in mediating the effects of CDDO in this assay.
PPARγ-Dependent Transactivation Assays. Previous studies showed that CDDO and related compounds activate PPARγ (Wang et al., 2000), and this was further investigated in SW-480 cells transfected with a GAL4-PPARγ chimeric expression plasmid and a reporter construct (pGAL4) containing five tandem GAL4 response elements linked to a luciferase reporter gene. The results (Fig. 2A) show that CDDO, CDDO-Me, and CDDO-Im induced a concentration-dependent increase in luciferase activity, and significant induction was observed at concentrations as low as 0.2, 0.025, and 0.025 μM, respectively. Both CDDO and CDDO-Me induced a maximal 35- to 45-fold increase in luciferase activity; the maximal induced value for CDDO-Im was lower (<30-fold); however, this may be caused by toxicity of this compound at the higher concentrations or differential recruitment of coactivators or corepressors. Ligand-induced transactivation in SW-480 cells transfected with GAL4-PPARγ/pGAL4 was also inhibited by the PPARγ antagonist T007 (Fig. 2B). Maximal induction of luciferase activity by CDDO (2.5 μM), CDDO-Me (0.5 μM), and CDDO-Im (0.5 μM) was inhibited in cells cotreated with 5 μM T007. Similar results were observed at lower concentrations of the CDDO compounds and also with the PPARγ antagonist GW9662 (data not shown). The CDDO compounds also induced transactivation in SW-480 cells transfected with a construct (PPRE3-luc) containing three tandem PPARγ response elements linked to firefly luciferase. The results (Fig. 2C) show that CDDO, CDDO-Me, and CDDO-Im significantly induce transactivation at concentrations of 1.0, 0.2, and 0.2 μM, respectively, and maximal induction (>30-fold) was observed for CDDO-Me. Transactivation induced by CDDO compounds in SW-480 cells transfected with PPRE-luc was inhibited after cotransfection with iPPARγ (Fig. 2D), and this correlates with comparable inhibition of PPARγ-dependent gene expression induced by other PPARγ agonists using iPPARγ (Chintharlapalli et al., 2004). These results confirm that CDDO, CDDO-Me, and CDDO-Im activate PPARγ-dependent transactivation in SW-480 colon cancer cells.
Induction of PPARγ-Coactivator Interactions by CDDO and Related Compounds. Previous reports show that different classes of PPARγ agonists induce structure-dependent interactions of PPARγ with coactivators in mammalian two-hybrid experiments (Kodera et al., 2000; Chintharlapalli et al., 2004); therefore, the effects of CDDO compounds were investigated in a mammalian two-hybrid assay in SW-480 cells transfected with pGAL4, VP-PPARγ and GAL4-coactivators/GAL4-SMRT (corepressor). The results show that CDDO, CDDO-Me, and CDDO-Im induce VP-PPARγ-pM coactivator interactions in SW-480 cells transfected with SRC-1, SRC-2, TRAP 220, CARM-1, and PGC-1. In contrast, CDDO and CDDO-Me but not CDDO-Im enhanced PPARγ-SRC-3 interaction; however, it is possible that higher concentrations of CDDO-Im would induce transactivation. We also investigated interactions with the corepressor SMRT, and the results indicate that all three compounds increased transactivation, but CDDO-Im was significantly more active than CDDO or CDDO-Im. These results confirm that CDDO and related esters induce interactions between VP-PPARγ with pM coactivators in SW-480 cells, and differences between compounds were primarily concentration-dependent.
CDDO Compounds Induce Caveolin-1 in Colon Cancer Cells. Caveolin-1 is a tumor suppressor gene in colon cancer cells, and previous reports show that PPARγ agonists induce this response in some colon cancer cell lines (Burgermeister et al., 2003; Chintharlapalli et al., 2004). The results illustrated in Fig. 4A show that after treatment of SW-480 cells with CDDO (0.25–0.5 μM), CDDO-Me (0.025–0.1 μM), and CDDO-Im (0.5–0.1 μM) for 3 days, caveolin-1 was induced by all three compounds. In contrast, induction of PARP cleavage, a marker of apoptosis was not observed at these concentrations. A slight increase in p27 was observed, and cyclin D1 was unchanged by these treatments (data not shown). In a separate experiment, SW-480 cells were treated with the CDDO compounds or T007 alone and in combination for 3 days, and caveolin-1 protein expression was determined (Fig. 4, B–D). The results show that 5 μM T007 alone did not affect levels of caveolin-1 protein; however, in cells cotreated with CDDO, CDDO-Me, or CDDO-Im plus T007, there was a decrease in caveolin-1 protein expression compared with cells treated with the CDDO compounds alone. These results complement the transactivation studies and demonstrate that induction of caveolin-1 by the CDDO compounds was inhibited by the PPARγ antagonist T007. Induction of caveolin-1 by CDDO, CDDO-Me, and CDDO-Im was also determined in HT-29 (Fig. 5A) and HCT-116 (Fig. 5B) cells, and induction was observed in both colon cancer cell lines. The effects of caveolin-1 induction by CDDO compounds on levels of phospho-Erk in SW-480 cells were minimal (data not shown). However, these compounds induced phosphatidylinositol-3-kinase–dependent phosphorylation of Akt (Fig. 4E), and this was consistent with results of a recent study in human 293 and HeLa cells overexpressing caveolin-1 (Shack et al., 2003). PARP cleavage was minimal or not observed at the concentrations of CDDO compounds used in this experiment.

Effects of CDDO and related esters on colon cancer cell survival and proliferation. SW-480 (A), HT-29 (B), and HCT-116 (C) cells were treated with different concentrations of CDDO, CDDO-Me, or CDDO-Im for 48 or 96 h, and this percentage of cell survival was determined as described under Materials and Methods. Solvent (DMSO)-treated cell survival was set at 100%, and significantly (p < 0.05) decreased cell numbers after treatment are indicated by an asterisk. The percentage of cell survival is derived from the treated/control (DMSO) cell number ratios. D, effects of T007 on CDDO-induced cell proliferation. SW-480 cells were treated with different concentrations of CDDO and 5 μM T007 for 48 h, and cell proliferation was determined colorimetrically using the cell proliferation reagent WST-1 as described under Materials and Methods. ★, significant (p < 0.05) inhibition of cell proliferation by CDDO; ★★, reversal of these effects by 5 μM T007.
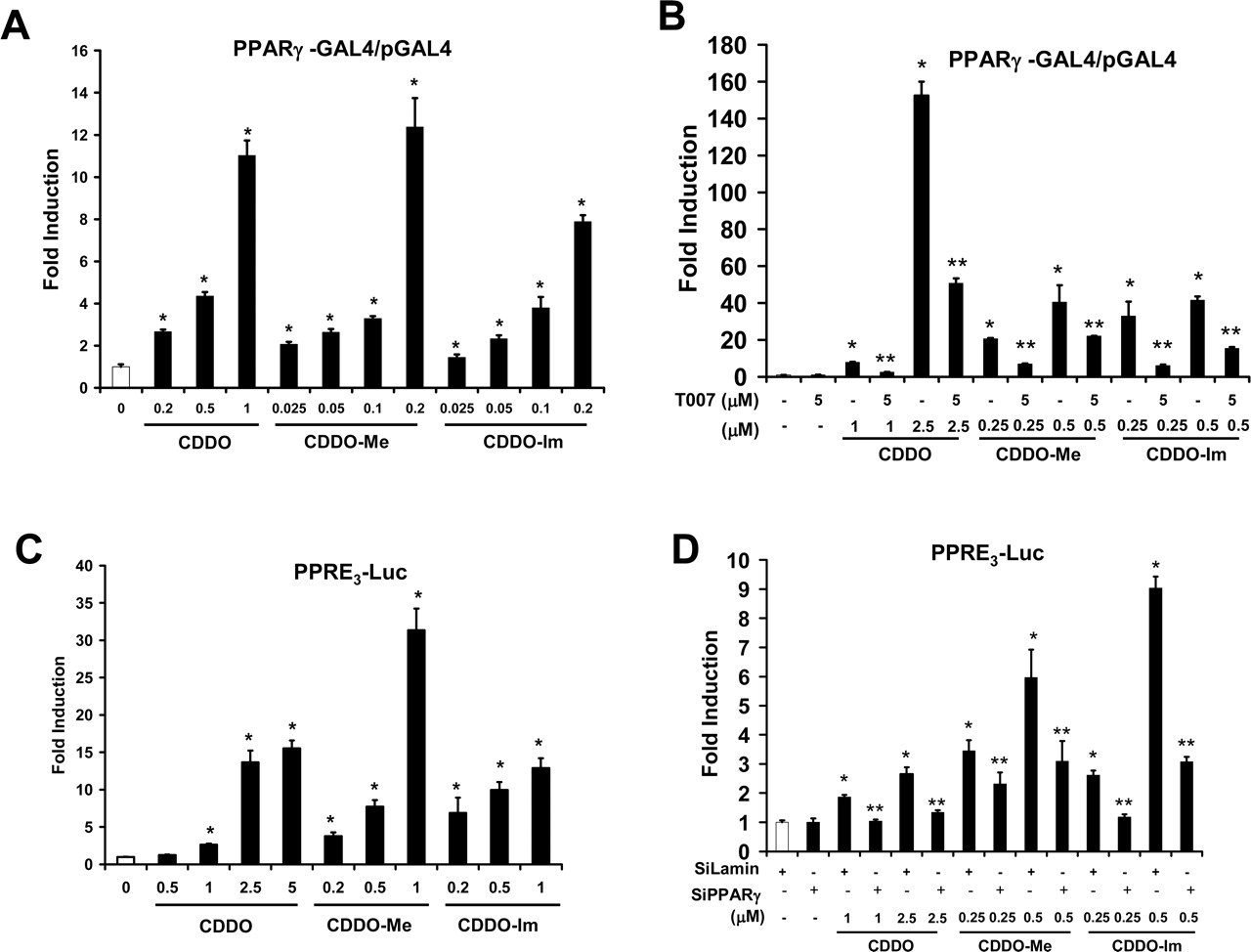
PPARγ-dependent transactivation by CDDO, CDDO-Me, and CDDO-Im in SW-480 cells. Activation of PPARγ-GAL4/pGAL4 (A) and inhibition by T007 (B). SW-480 cells were transfected with PPARγ-GAL4/pGAL4, treated with different concentrations of CDDO compounds, and luciferase activity was determined as described under Materials and Methods. In a separate experiment, cells were treated with CDDO (2.5 μM), CDDO-Me (0.5 and 1.0 μM), or CDDO-Im (0.2 and 0.5 μM) alone or in combination with 5 μM T007. ★, significant (p < 0.05) induction by CDDO, CDDO-Me, and CDDO-Im; ★★, inhibition by T007. C, activation of PPRE3-luc. SW-480 activity cells were transfected with PPRE3-luc, treated with CDDO compounds, and luciferase was determined as described under Materials and Methods. ★, significant (p < 0.05) induction. D, RNA interference assay. Cells were transfected with PPRE-luc, treated with the different CDDO compounds, and small inhibitory RNA for lamin (nonspecific control) or PPARγ and luciferase activity was determined as described under Materials and Methods. ★, significant (p < 0.05) induction by PPARγ agonist; ★★, inhibition after cotransfection with iPPARγ. All experiments (A–D) are expressed as means ± S.E. for at least three separate determinations for each treatment group.
Cell-survival studies (Fig. 1) indicated that higher concentrations of CDDO and related esters induced cell death; therefore, we further investigated the induction of PARP cleavage in SW-480 cells treated with 0.5 to 2.5 μM CDDO and 0.2 to 1.0 μM CDDO-Me and CDDO-Im for 1 day (Fig. 5C). Longer treatment (i.e., 3 days) could not be determined because of extensive cell death and minimal cell survival. These results demonstrate that the higher concentrations of CDDO and related esters induced PARP cleavage, and this was consistent with the cell survival studies. In contrast, the PPARγ antagonist T007 did not inhibit induced PARP cleavage, suggesting that this response was PPARγ-independent. Moreover, at these higher concentrations, there was also a significant down-regulation of cyclin D1, and this response was also not affected by T007 (data not shown). These results demonstrate that CDDO and related esters induce PPARγ-dependent and -independent responses associated with growth inhibition/cell death in colon cancer cells, and these mechanistic differences are concentration-dependent.

CDDO-, CDDO-Me–, and CDDO-Im–induced PPARγ-coactivator interactions in SW-480 cells. Ligand-induced interactions with SRC-1 (A), SRC-2 (B), SRC-3 (C), TRAP 220 (D), PGC-1 (E), CARM-1 (F), and SMRT (G). Cells were transfected with VP-PPARγ and GAL4-coactivator chimeric expression plasmids, treated with the CDDO compounds, and luciferase activity was determined as described under Materials and Methods. Results are expressed as means ± S.E. for at least three separate determinations for each treatment group. ★, significant (p < 0.05) induction.

Induction of caveolin-1, Akt phosphorylation, and PARP cleavage by CDDO and related esters in SW-480 cells. A, induction of caveolin-1 and PARP cleavage. SW-480 cells were treated with CDDO, CDDO-Me, and CDDO-Im for 3 days, and caveolin-1 and PARP cleavage proteins were determined by Western blot analysis of whole-cell lysates as described under Materials and Methods. T007 inhibition of caveolin-1 induction by CDDO (B), CDDO-Me (C), and CDDO-Im (D). Cells were treated with the compounds alone or in combination with 5 μM T007 as described in A, and whole-cell lysates were analyzed by Western blot analysis. β-Actin served as a loading control for these experiments. E, CDDO compounds induce Akt phosphorylation. Cells were treated with the CDDO compounds as described in A, and Akt/phospho-Akt was determined by Western blot analysis as described under Materials and Methods.
Discussion
PPARγ is expressed in tumors from multiple tissues and cancer cell lines (Ikezoe et al., 2001), and chemicals that activate PPARγ have been extensively investigated for their efficacy in cancer chemotherapy (Desvergne and Wahli, 1999; Escher and Wahli, 2000; Murphy and Holder, 2000; Fajas et al., 2001; Willson et al., 2001; Grommes et al., 2004). PPARγ agonists typically decrease cancer-cell survival caused by the induction of differentiation genes and apoptosis and modulation of genes/proteins linked to G1 → S phase progression. It is somewhat paradoxical that the induction of these responses by PPARγ agonists, such as the thiazolidinediones (TZDs), 15-deoxy-Δ12,14-prostaglandin J2 (PGJ2), and CDDO compounds, seems to be both PPARγ-dependent and –independent, and the relative contributions of these pathways are often not well-defined. For example, troglitazone inhibits growth and induces apoptosis in colon cancer cell lines and induces early growth response-1 gene expression, which in turn activates downstream growth-inhibitory effects (Baek et al., 2003). This response was unique to troglitazone among PPARγ agonists and was induced via PPARγ-independent pathways.
CDDO, CDDO-Me, and CDDO-Im activate PPARγ in transactivation assays and CDDO-induced apoptosis was inhibited by dominant-negative PPARγ in myeloid HL-60 cells and by T007 in myeloid U937 cells (Konopleva et al., 2004a). In another study, CDDO-Im induced differentiation in leukemia cells that was not inhibited by the PPARγ antagonist GW9662 (Ito et al., 2001), and T007 did not affect inhibition of SKOV3 ovarian cancer cell growth by CDDO (Melichar et al., 2004). Thus, it is clear that CDDO and related compounds activate PPARγ-dependent and -independent pathways that inhibit cancer-cell growth, and this dual action may be beneficial for cancer chemotherapy. Results of this study demonstrate that CDDO, CDDO-Me, and CDDO-Im also inhibit growth and survival of colon cancer cells (Fig. 1), and we have primarily used SW-480 cells as a model for determining the activation of PPARγ-dependent and -independent pathways by CDDO and related esters.
CDDO, CDDO-Me, and CDDO-Im activate PPARγ-dependent transactivation (Fig. 2) and coactivator interactions (Fig. 3), and differences between compounds were primarily associated with the fold-induction response in which induction was lower for CDDO-Im because of the higher toxicity of this compound. Previous studies indicate that induced transactivation in the mammalian two-hybrid assay using VP-PPARγ and GAL4 coactivators was both structure- and coactivator-dependent (Burgermeister et al., 2003; Chintharlapalli et al., 2004). For example, a series of 1,1-bis(3′-indolyl)-1-(p-substitutedphenyl)methanes [methylene-substituted 1,1-bis(3′-indolyl)methanes (C-DIMs)], which activate PPARγ, induced interactions of PPARγ primarily with PGC-1 in colon cancer cell lines (Chintharlapalli et al., 2004), whereas CDDO, CDDO-Me, and CDDO-Im induced interactions with several coactivators, including SRC-1, SRC-2, CARM-1, TRAP 220, and PGC-1. The main distinguishing feature among the CDDO compounds was the failure of CDDO-Im to induce PPARγ-SRC-3 interactions with VP-PPARγ and the relatively high activity of CDDO-Im to induce PPARγ interactions with the corepressor SMRT. These differences among CDDO compounds in PPARγ-dependent interactions with coactivators might result in some tissue/cell selectivity in their PPARγ-dependent responses.

Induction of caveolin-1 and PARP cleavage by CDDO compounds in colon cancer cell lines. Induction by CDDO, CDDO-Me, and CDDO-Im in HT-29 (A) and HCT-116 (B) cells. Cells were treated with different concentrations of CDDO and related esters for 3 days, and whole-cell lysates were analyzed by Western blot analysis as described under Materials and Methods. C, higher concentrations of CDDO, CDDO-Me, and CDDO-Im induce PPARγ-independent PARP cleavage in SW-480 cells. SW-480 cells were treated for 1 day with the indicated doses of CDDO compounds alone or in combination with 5.0 μM T007, and PARP cleavage was determined by Western blot analysis of whole-cell lysates as described under Materials and Methods. β-Actin served as a loading control.
PPARγ agonists such as TZDs, PGJ2, and C-DIMs typically inhibit genes/proteins associated with cell-cycle progression and differentiation in cancer cell lines, and these responses may be receptor-dependent or -independent. Previous studies in colon cancer cell lines with C-DIMs showed that these compounds had minimal effects on critical cell-cycle proteins cyclin D1, p21, and p27 (Chintharlapalli et al., 2004); and similar results were observed in this study at the lower concentrations of CDDO compounds (data not shown). The induction of caveolin-1 by PGJ2, C-DIMs, and TZDs has been observed in colon cancer cell lines (Burgermeister et al., 2003; Chintharlapalli et al., 2004), and overexpression of caveolin-1 inhibits colon cancer cell growth in vitro and tumor growth in athymic nude mice (Bender et al., 2000; Burgermeister et al., 2003; Chintharlapalli et al., 2004). Results in Figs. 4 and 5 show that CDDO, CDDO-Me, and CDDO-Im significantly induce caveolin-1 in colon cancer cells at concentrations that also inhibit cell growth (Figs. 1). Moreover, in SW-480 cells, the induced caveolin-1 expression is inhibited by the PPARγ antagonist T007. The relatively low concentrations of CDDO, CDDO-Me, and CDDO-Im that inhibit SW-480 cell proliferation induce minimal to nondetectable apoptosis as indicated by PARP cleavage (Figs. 4 and 5, A and B). We also observed that CDDO compounds induced phosphatidylinositol-3-kinase–dependent Akt phosphorylation, which is normally associated with increased cell survival. However, a recent report (Shack et al., 2003) showed that overexpression of caveolin-1 in human 293 and HeLa cells also resulted in up-regulation of the PI3-K/Akt pathway, which sensitized the cells to stress-induced cytotoxicity.
In contrast, higher concentrations of CDDO compounds induce apoptosis and PARP cleavage; however, this response was PPARγ-independent and was not inhibited by T007 (Fig. 5C). Moreover, at the higher concentrations of CDDO, CDDO-Me, and CDDO-Im, there was also a marked decrease in cyclin D1 protein expression that was also not inhibited by T007 (data not shown). Similar results have also been observed in our laboratory after treatment of breast cancer cells by PGJ2, TZDs, and C-DIMs (Qin et al., 2003, 2004). These data demonstrate that CDDO and related esters inhibit colon cancer cell growth through both PPARγ-dependent and -independent pathways, which are observed at different concentrations and are linked to growth inhibition and cell death, respectively.
Previous reports of mechanistic differences regarding the role of PPARγ agonists in mediating growth-inhibitory responses in different cancer cell lines may be explained, in part, by the results of this study. CDDO and related esters are unique examples of a specific structural class of PPARγ agonists that induce both PPARγ-dependent (caveolin induction) and -independent (apoptosis) responses over a relatively narrow dose range in colon cancer cells. It is possible that the antimitogenic and anticarcinogenic activities of PPARγ agonists may be enhanced when the dose ranges for their PPARγ-dependent and -independent responses are similar, and this may contribute to the high potencies observed for CDDO, CDDO-Me, and CDDO-Im in colon cancer cell lines. We are now investigating the dose-dependent effects of thiazolidinediones, CDDO, and related esters and C-DIMs in different cancer cell lines to determine their relative potencies in activating PPARγ-dependent and -independent pathways. This approach will provide important mechanistic insights into the activities of these compounds and their potential clinical applications for cancer chemotherapy.
Footnotes
-
This work was financially supported by the National Institutes of Health (ES09106 and CA112337), the M.D. Anderson Cancer Center Pancreatic Cancer Spore (P20 CA10193), and the Texas Agricultural Experiment Station.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.011437.
-
ABBREVIATIONS: CDDO, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid; T007, N-(4′-aminopyridyl-2-chloro-5-nitrobenzamide; PPARγ, peroxisome proliferator-activated receptor γ; -Me, methyl; -Im, imidazole; PARP, poly(ADP-ribose) polymerase; PPRE, peroxisome proliferator-activated receptor γ response element; iPPARγ, small inhibitory RNA for peroxisome proliferator-activated receptor γ; ER, endoplasmic reticulum; FBS, fetal bovine serum; β-gal, β-galactosidase; siRNA, small inhibitory RNA; DMSO, dimethyl sulfoxide; DMEM, Dulbecco's modified Eagle's medium; PVDF, polyvinylidene difluoride; TBST, Tris-buffered saline/Tween 20; TZD, thiazolidinedione; C-DIM, methylene-substituted 1,1-bis(3′-indolyl)-methane; VP-PPARγ, peroxisome proliferator-activated receptor γ2-VP16 fusion plasmid; LG100268, 3-pyridinecarboxylic acid, 6-(1-(5,6,7,8-tetrahydro-3,5,5,8,8-pentamethyl-2-naphthalenyl)cyclopropyl)-; PGJ2, 15-deoxy-Δ12,14-prostaglandin J2; GW9662, 2-chloro-5-nitro-N-phenylbenzamide.
- Received January 24, 2005.
- Accepted March 18, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}