


Abstract
Complementation of function after coexpression of pairs of nonfunctional G protein-coupled receptors that contain distinct inactivating mutations supports the hypothesis that such receptors exist as dimers. Chimeras between members of the metabotropic glutamate receptor-like family have been particularly useful because the N-terminal ligand binding and heptahelical transmembrane elements can be considered distinct domains. To examine the utility of a related approach for opioid receptors, fusion proteins were generated in which a pertussis toxin-resistant (Cys351Ile) variant of the G protein Gi1α was linked to the C-terminal tails of the δ opioid peptide (DOP), κ opioid peptide, and μ opioid peptide receptors. Each was functional as measured by agonist stimulation of guanosine 5′-([γ-35S]thio)triphosphate ([35S]GTPγS) binding in Giα immunoprecipitates from membranes of pertussis toxin-treated HEK293 cells. Agonist function was eliminated either by fusion of the receptors to Gi1αGly202Ala,Cys351Ile or mutation of a pair of conserved Val residues in intracellular loop 2 of each receptor. Coexpression, but not simple mixing, of the two inactive fusion proteins reconstituted agonist-loading of [35S]GTPγS for each receptor. At equimolar amounts, reconstitution of DOP receptor function was more extensive than κ or μ opioid receptor. Reconstitution of DOP function required two intact receptors and was not achieved by provision of extra Gi1αCys351Ile membrane anchored by linkage to DOP transmembrane domain 1. Inactive forms of all G protein α subunits can be produced by mutations equivalent to Gi1αGly202Ala. Because the amino acids modified in the opioid receptors are highly conserved in most rhodopsin-like receptors, this approach should be widely applicable to study the existence and molecular basis of receptor dimerization.
Extensive literature now exists on the capacity of a wide range of G protein-coupled receptors (GPCRs) to form dimers and/or higher-order oligomers (Lee et al., 2003; Breitwieser, 2004; Milligan, 2004). Despite this, many of the reports have been predominantly descriptive and provide limited insights into the proportion of different GPCRs that may exist as dimers, the relative propensity of different GPCRs to oligomerize, the molecular basis of dimerization, and whether there are differences in the details of how closely related GPCRs form dimers/oligomers.
The ability of the DOP, KOP, and MOP opioid receptor subtypes to form homodimers and/or higher-order oligomers has previously been investigated using both coimmunoprecipitation and resonance energy transfer techniques (Cvejic and Devi, 1997; George et al., 2000; McVey et al., 2001; Li-Wei et al., 2002; Ramsay et al., 2002). Despite this, little information is available on the issues noted above, although informatic analysis has suggested potential interfaces in transmembrane helices that may contribute to opioid receptor subtype homodimerization (Filizola and Weinstein, 2002).
If coexpression of two nonequivalent and nonfunctional mutants of a GPCR is both able and required to reconstitute receptor ligand binding and/or function, this can provide evidence in favor of direct protein-protein interactions and quaternary structure for the active receptor (Milligan and Bouvier, 2005). For example, coexpression of two forms of the angiotensin AT1 receptor that were unable to bind angiotensin II or related ligands because of point mutations in transmembrane region III or V restored ligand binding (Monnot et al., 1996). Such an approach has also been used to explore mechanisms of dimerization. Theoretical models of GPCR dimerization include both “contact” and “domain swap” dimers. Using the histamine H1 receptor as a model, Bakker et al. (2004) showed that although single point mutations in both transmembrane region III and transmembrane region VI prevented binding of antagonist radioligands, including [3H]mepyramine, coexpression of the two mutants resulted in reconstitution of [3H]mepyramine binding sites with the anticipated pharmacological characteristics. From a conceptual standpoint, this should not be possible for a contact dimer in which transmembrane domains are not exchanged but simply appose each other.
In addition to the restoration of ligand binding, studies that have used pairs of nonfunctional mutants to restore GPCR signaling have produced data consistent with GPCR-GPCR interactions. By generating mutants of the luteinizing hormone receptor that were either unable to bind ligand or unable to signal but able to bind the agonist, Lee et al. (2002) were able to reconstitute agonist-mediated regulation of cAMP levels after coexpression of the two mutants. The luteinizing hormone receptor, as with other GPCRs with related ligands, has an extended N-terminal region involved in ligand binding. As such, Lee et al. (2002) were able to consider the N-terminal “exo-domain” and the seven transmembrane element “endo-domain” as distinct entities in a manner equivalent to the extracellular and transmembrane elements of class C GPCRs, which has allowed elegant chimeric receptor approaches to understand the mechanism of signal transduction through obligate heterodimers (Pin et al., 2005).
As a variant of this, functional complementation was recently observed after the coexpression of pairs of α1b-adrenoceptor-G11α and histamine H1 receptor-G11α GPCR-G protein fusion proteins that were both inactive when expressed individually because they contained specific mutations in either the GPCR or G protein element (Carrillo et al., 2003). All G protein α subunits contain a conserved Gly that, when mutated, prevents effective GDP-GTP exchange and hence activation (Milligan et al., 2005). Furthermore, nearly all class A, rhodopsin-like GPCRs have one or, more usually, two hydrophobic residues in the second intracellular loop homologous to those mutated to generate inactive forms of the α1b-adrenoceptor and histamine H1 receptor (Milligan et al., 2005). We thus wished to test whether equivalent pairs of inactive opioid receptor-Giα fusion proteins could be produced and to assess whether variations in pharmacology and/or reconstitutive capacity could provide insights into the basis of opioid receptor subtype dimerization.
Materials and Methods
Materials/Ligands. [15,16-3H]Diprenorphine (50 Ci/mmol) and guanosine 5′-([γ-35S]thio)triphosphate (1250 mCi/mmol) were from PerkinElmer Life and Analytical Sciences. (Boston, MA). [d-Ala2,d-Leu5]-enkephalin (DADLE), [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO), [d-Pen2,d-Pen5]-enkephalin (DPDPE), naloxone, and pertussis toxin were from Sigma-Aldrich (Poole, Dorset, UK). U69593 was from Tocris Cookson (Bristol, UK). Recombinant, myristoylated rat Gi1α subunit was from Calbiochem (San Diego, CA).
Antibodies/Antisera. The anti-Gαi1-2 antiserum (SG) has been described previously (Green et al., 1990). The mouse monoclonal anti-Flag antibody (M2) was from Sigma-Aldrich. The rabbit polyclonal anti-c-myc antiserum was from Cell Signaling Technology (Nottingham, UK)
Molecular Constructs. hDOP-Gi1αC351I in pcDNA3.1 was generated previously (Moon et al., 2001) and used as a template to introduce mutations in the 2nd intracellular loop of the receptor to produce hDOPV150E,V154D-Gi1αC351I using the QuikChange kit (Stratagene, La Jolla, CA) and the following primers: sense, 5′-GAC CGC TAC ATC GCT GAG TGC CAC CCT GAC AAG GCC CTG GAC TTC-3′; antisense, 5′-GAA GTC CAG GGC CTT GTC AGG GTG GCA CTC AGC GAT GTA GCG GTC-3′. Bold letters indicate altered bases. The PCR product was then digested with DpnI and transformed into bacteria.
hDOP-Gi1αG202A,C351I. In a similar manner, hDOP-Gi1αC351 I was used to introduce the G202A mutation in Gi1α using the following primers: sense, 5′-G TTT GAC GTG GGA GCC CAG AGA TCA GAG C-3′; antisense, 5′-G CTC TGA TCT CTG GGC TCC CAC GTC AAA C-3′. The PCR product was then digested by DpnI and was transformed into bacteria.
Flag-hDOPV150E,V154D-Gi1αC351I. Flag-hDOPV150E,V154D-Gi1αC351I was constructed using the following primers: sense, 5′-ACT AGT GCT AGC ATG GAC TAC AAG GAC GAC GAT GAT AAG GAA CCG GCC CCC TCC GCC GGC-3′; antisense, 5′-GAA TTT GGA TCC GGC GGC AGC GCC ACC GCC GGG-3′. The sense primer contains a Flag sequence (in bold) and an NheI restriction site (underlined) and corresponds to the N-terminal region of hDOP. The antisense primer contains a BamHI site (underlined) and corresponds to the C-terminal region of hDOP. The PCR product and pcDNA3.1 vector containing hDOPV150E,V154D-G i1αC351I were digested by NheI and BamHI. The digested products were then ligated.
c-myc-hDOP-Gi1αG202A,C351I.c-myc-hDOP-Gi1αG202A,C351I was constructed using the following primers: sense, 5′-CCC TTT GCT AGC ATG GAA CAA AAG CTT ATT TCT GAA GAA GAT CTG GAA CCG GCC CCC TCC GCC-3′; antisense, 5′-GAA TTT GGA TCC GGC GGC AGC GCC ACC GCC GGG-3′. hDOP-Gi1αG202A,C351I was amplified by these primers. The sense primer contains a c-myc sequence (bold) and NheI restriction site (underlined), and the antisense primer contains a BamHI site (underlined). The PCR product and pcDNA3.1 containing hDOP-Gi1αG202A,C351I were digested with NheI and BamHI. The digested products were then ligated.
hMOPV169EV173D-Gi1αC351I. hMOR-Gi1αC351I cDNA in pcDNA3 was generated previously (Massotte et al., 2002) and was used as a template to introduce mutations in the 2nd intracellular loop of the receptor using the following primers: sense, 5′-GAT CGA TAC ATT GCA GAG TGC CAC CCT GAC AAG GCC TTA GAT TTC-3′; antisense, 5′-GAA ATC TAA GGC CTT GTC AGG GTG GCA CTC TGC AAT GTA TCG ATC-3′. The appropriate valines were mutated into glutamate (GAG) and aspartate (GAC), respectively. Altered bases mutated are in bold. The PCR product was digested by DpnI and was transformed into bacteria.
hMOP-Gi1αG202A,C351I. hMOP-Gi1αG202A,C351I was produced as for hDOP-Gi1αG202A,C351I but using hMOP-Gi1αC351I cDNA as the template.
rKOP-GilαC351I. rKOP-Gi1αC351I was constructed using the following primers: sense, 5′-CCC AAA AAG CTT ATG GAG TCC CCC ATC CAG ATT TTC C-3′; antisense, 5′-GGC ATC GGT ACC TAC TGG CTT ATT CAT CCC ACC CAC ATC CCT CAT GGA-3′. Rat KOP was amplified between these primers corresponding to the N and C termini of rKOP and containing HindIII and KpnI restriction sites (underlined). The PCR product and pcDNA3 containing Gi1αC351I were digested by the above enzymes. Because rKOP contains an internal HindIII site, a two-way ligation was performed to ligate the vector and the two elements of the digested PCR product.
rKOP V160E,V164D-Gi1αC351I. rKOP-Gi1αC351I cDNA, as above, was used as a template to introduce mutations in the 2nd intracellular loop of the receptor, using the following primers: sense, 5′-GAC CGC TAC ATT GCC GAG TGC CAC CCT GAC AAA GCT TTG GAT TTC-3′; antisense, 5′-GAA ATC CAA AGC TTT GTC AGG GTG GCA CTC GGC AAT GTA GCG GTC-3′. Bases mutated are in bold.
rKOP-Gi1αG202A,C351I. rKOP-Gi1αC351I cDNA was used as a template to introduce the mutation in Gi1α as for hDOP and hKOP.
Flag-Nt-TM1-Gi1αC351I. Flag-Nt-TM1-Gi1αC351I was constructed using the following primers: sense, 5′-ACT AGT GCT AGC ATG GAC TAC AAG GAC GAC GAT GAT AAG GAA CCG GCC CCC TCC GCC GGC-3′: sense, 5′-CCC ATT GGA TCC GGT GGC CGT CTT CAT CTT AGT GTA CCG-3′. Flag-hDOP-Gi1α C351I was used as template for PCR. The first 252 base pairs were amplified by PCR and were then digested using BamHI and NheI (restriction sites underlined). The same digestion was used on the template, NheI being situated at the end of the receptor sequence. PCR products and vector were ligated.
Cell Transfection and Treatment. HEK293 cells were transfected using Lipofectamine reagent (Invitrogen, Carlsbad, CA) or Gene Juice (Novagen, Madison, WI) and the appropriate cDNA(s) according to the manufacturers' instructions. Cells were treated with pertussis toxin (25 ng/ml) for 16 to 18 h before harvest.
[3H]Diprenorphine Binding. The expression of GPCR-G protein fusions was assessed by measuring the specific binding of [3H]diprenorphine in cell membrane preparations. Nonspecific binding was assessed by the addition of 100 μM naloxone. Samples were incubated for 1 h at 25°C, and bound ligand was separated from free by vacuum filtration through GF/B filters (Whatman, Maidstone, UK) pretreated with 0.3% polyethylenimine in 10 mM Tris/HCL, 0.1 mM EDTA, and 10 mM MgCl2, pH adjusted to 7.5. Bound ligand was estimated by liquid scintillation counting. Competition studies were conducted with 1 nM [3H]diprenorphine and a range of concentrations of other ligands. Data were analyzed using Prism (GraphPad Software, San Diego, CA). Saturation data were fit to nonlinear regression curves.
[35S]GTPγS Binding Studies. Experiments were initiated by adding the assay buffer mix (20 mM HEPES, pH 7.4, 3 mM MgCl2, 100 mM NaCl, 10 μM GDP, and 0.2 mM ascorbic acid) containing 50 nCi of [35S]GTPγS in the presence or absence of agonist to a defined amount of membranes. Nonspecific binding was determined in the presence of 100 μM GTPγS. The reaction was incubated for 15 min at 30°C and terminated by adding 1 ml of ice-cold stop buffer. The samples were centrifuged for 15 min at 16,000g at 4°C, and the resulting pellets were resuspended in solubilization buffer (100 mM Tris HCl, 200 mM NaCl, 1 mM EDTA, 1.25% Nonidet P40, pH adjusted to 7.4) plus 0.2% SDS. Samples were precleared with Pansorbin for 1 h at 4°C and centrifuged for 2 min at 16,000g. Supernatant was added to a mix of protein G and the anti-Gi1α/Gi2α antiserum, SG (Green et al., 1990), and left rotating overnight at 4°C for immunoprecipitation. The immunocomplexes were washed twice with ice-cold solubilization buffer, and bound [35S]GTPγS was measured.
Coimmunoprecipitation. Cells were resuspended in 1 ml of 1× radioimmunoprecipitation assay buffer and rotated for 60 min at 4°C to allow lysis. The samples were centrifuged at 14,000g for 10 min at 4°C, and the supernatant was retained. Fifty microliters of a protein G-Sepharose/phosphate-buffered saline slurry was added to the supernatant and rotated for a further 60 min at 4°C to preclear. Samples were centrifuged at 14,000g for 10 min at 4°C. Supernatant was conserved, and protein concentration was measured using the BCA assay method. Samples were equalized to 1 μg/μl. Target proteins were then immunoprecipitated from 500-μl samples by incubation with 20 μl of protein G-Sepharose and the appropriate antibody/antiserum overnight at 4°C on a rotating wheel. Immune complexes were isolated by centrifugation at 14,000g for 1 min and washed twice with radioimmunoprecipitation assay buffer. Proteins were eluted from the protein G-Sepharose by the addition of 30 to 50 μl of Laemmli buffer and heated for 4 min at 85°C. The eluates were then loaded onto SDS-PAGE gels.
Quantitation of Flag-Nt-TM1-Gi1αC351I Expression Levels. Varying amounts (12.5-50 ng) of recombinantly expressed, myristoylated rat Gi1α were run on SDS-PAGE alongside membranes of HEK293 cells transfected to coexpress Flag-Nt-TM1-Gi1αC351I and hDOP-Gi1αG202A,C351I. After immunoblotting with the anti-Gi1α/Gi2α antiserum SG, densitometry indicated that the signal corresponding to the recombinant Gi1α increased in a linear fashion over this range. Interpolation of the immuno-signal corresponding to Flag-Nt-TM1-Gi1αC351I (molecular mass, 49.26 kDa) in different amount of transfected cell membranes allowed estimation of expression levels.
Results
A fusion protein was constructed between the human DOP (hDOP) receptor and a form of the α subunit of the G protein Gi1 that was rendered resistant to the ADP-ribosyltransferase activity of pertussis toxin by conversion of Cys351 to Ile (Gi1αC351I). The hDOP-Gi1αC351I fusion protein was expressed transiently in HEK293 cells that were also treated with pertussis toxin (25 ng/ml, 16 h) before harvest to cause ADP-ribosylation of the endogenously expressed forms of the Gi/Go group of G proteins. Membranes prepared from these cells were used in saturation [3H]diprenorphine ligand binding assays to measure expression levels of the construct and its affinity for the ligand (Table 1). Expression levels were 1816 ± 209 fmol/mg of membrane protein and the pKd for [3H]diprenorphine was 9.20 ± 0.03 (n = 4, means ± S.E.M.). The functionality of hDOP-Gi1αC351I was assessed by the capacity of the synthetic opioid peptide DADLE to stimulate binding of [35S]GTPγS in membranes containing the construct that were subsequently immunoprecipitated with the anti-Gi1α/Gi2α antiserum, SG (Fig. 1A). Virtually no [35S]GTPγS was recovered in immunoprecipitates from membranes of mock-transfected cells treated with either DADLE or vehicle (Fig. 1A). By contrast, although binding of [35S]GTPγS in immunoprecipitates from hDOP-Gi1αC351I-expressing cell membranes was greatly increased by DADLE, the construct was also able to load [35S]GTPγS in the absence of agonist (Fig. 1A). When membrane amounts corresponding to varying levels of hDOP-Gi1αC351I were used, DADLE stimulation of [35S]GTPγS binding was linear with fusion protein amount over the full range tested and up to at least 60 fmol (Fig. 1B).
Expression levels and [3H]diprenorphine binding affinity of hDOP-Gi1αC351I fusion proteins
Data represent means ± S.E.M. of n = 4 experiments performed on different membrane preparations.
We have demonstrated previously that mutation of Gly208 to Ala in the G protein G11α prevents receptor-mediated guanine nucleotide exchange and hence [35S]GTPγS binding (Carrillo et al., 2002). The α subunit of all heterotrimeric G proteins contains Gly at the equivalent position. To test the general effect of mutating this Gly on the capacity of receptors to enhance guanine nucleotide exchange, we thus generated hDOP-Gi1αG202A,C351I. When this was expressed in HEK293 cells and membranes were prepared from pertussis toxin-treated cells, neither the level of expression of this construct nor the binding affinity for [3H]diprenorphine was different from hDOP-Gi1αC351I (Table 1). However, although 10 μM DADLE caused a 5.2 ± 0.3-fold (n = 4) increase in levels of [35S]GTPγS binding compared with vehicle-treated controls in samples immunoprecipitated from membranes expressing 15 fmol of hDOP-Gi1αC351I (Fig. 2), no significant DADLE stimulation of [35S]GTPγS binding was observed in immunoprecipitated samples derived from membranes containing 15 fmol of hDOP-Gi1αG202A,C351I (Fig. 2). Furthermore, [35S]GTPγS loading in the absence of DADLE was substantially reduced (Fig. 2). Mutation of hydrophobic residues in the second intracellular loop of family A GPCRs can essentially eliminate G protein activation without major effects on antagonist ligand binding (Carrillo et al., 2003, Milligan et al., 2005). To test whether mutation of the equivalent amino acids eliminated G protein activation for hDOP, we also generated hDOPV150E,V154D-Gi1αC351I. hDOPV150E,V154D-Gi1α also was expressed as well as hDOP-Gi1αC351I (Table 1) but bound [3H]diprenorphine with 3-fold lower affinity than hDOP-Gi1αC351I (Table 1). [35S]GTPγS binding studies demonstrated this construct to have much reduced basal guanine nucleotide exchange and not to produce a statistically significant increase in binding of [35S]GTPγS in response to DADLE (Fig. 2). When hDOP-Gi1α G202A,C351I and hDOPV150E,V154D-Gi1αC351I were coexpressed and membranes containing 15 fmol of [3H]diprenorphine binding sites were used in [35S]GTPγS binding studies, DADLE stimulation was partially reconstituted (Fig. 2). With membranes from these cells containing 30 fmol of [3H]diprenorphine binding sites, DADLE-stimulated [35S]GTPγS binding was 60% of that achieved in membranes expressing 15 fmol of the wild-type hDOP-Gi1αC351I fusion construct (Fig. 2). Reconstitution of DADLE-stimulated [35S]GTPγS binding required the coexpression of hDOP-Gi1αG202A,C351I and hDOPV150E,V154D-Gi1αC351I and not simply the presence of both in the assay. When membranes containing 15 fmol of individually expressed hDOP-Gi1αG202A,C351I and hDOPV150E,V154D-Gi1αC351I were simply mixed before the assay to provide 30 fmol of fusion proteins in the assay, no reconstitution of DADLE-stimulated [35S]GTPγS binding was observed (Fig. 2). These data are consistent with a requirement for hDOP interactions to generate function.

A hDOP-Gi1αC351I fusion protein is functional. A, 10 (bars 1 and 3) or 20 (bars 2 and 4) μg of pertussis toxin-treated, HEK 293 cell membranes expressing (bars 3 and 4) or not (bars 1 and 2) hDOP-Gi1αC351I were used to measure the binding of [35S]-GTPγS in the absence (open bars) or presence (filled bars) of 10 μM DADLE. At assay termination, samples were immunoprecipitated with an anti-Gi1α/Gi2α antiserum and counted. B, membranes, as above, expressing different amounts of hDOP-Gi1αC351I, were used to measure DADLE (10 μM) stimulation of [35S]-GTPγS binding. Data are means ± S.E.M. of triplicate assays. Two further experiments produced similar data.
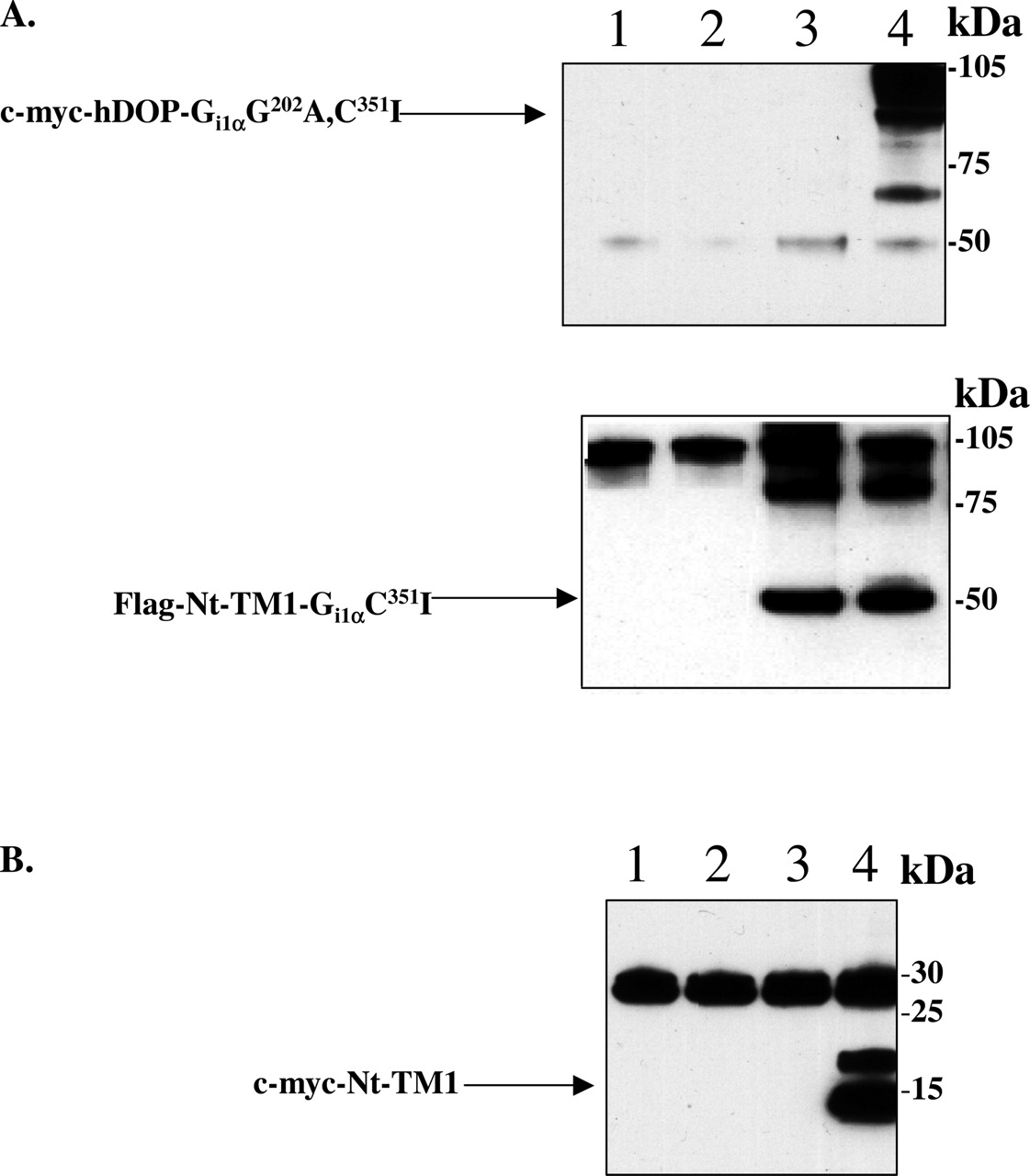
It is interesting that the affinity of [3H]diprenorphine binding in membranes coexpressing hDOP-Gi1αG202A,C351 I and hDOPV150E,V154D-Gi1αC351I was equivalent to the individually expressed hDOPV150E,V154D-Gi1αC351I construct (Table 1). Although this observation might indicate the presence of a substantially greater proportion of hDOPV150E,V154D-Gi1αC351I than hDOP-GilαG202A,C351I in the membranes from coexpressed cells, this is not consistent with the functional reconstitution data (Fig. 2) or with the equivalent levels of expression of these two constructs when expressed individually (Table 1). However, to examine this further and to confirm direct interactions between hDOP-GilαG202A,C351I and hDOPV150E,V154D-Gi1αC351I, we performed coimmunoprecipitation studies using membranes of HEK293 cells transfected to express individually or coexpress N-terminally modified Flag-hDOPV150E,V154D-Gi1αC351I and/or c-myc-hDOP-Gi1αG202A,C351I. Immunoprecipitation with anti-Flag antibody followed by SDS-PAGE and immunoblotting with anti-c-myc antibody resulted in detection of specific c-myc immunoreactivity only when the two fusion constructs were coexpressed (Fig. 3), consistent with a physical interaction between the two variants.
To further explore aspects of pharmacology of the fusion proteins, membranes from pertussis toxin-treated HEK 293 cells transfected to express hDOP-Gi1αC351I, hDOPV150E,V154D-Gi1αC351I, hDOP-Gi1αG202A,C351I, or the combination of hDOPV150E,V154D-Gi1αC351I + hDOP-GilαG202A,C351I were used in [3H]diprenorphine/DADLE competition binding experiments (Table 2). Two-site binding curves reflecting higher and lower affinity binding sites for the agonist DADLE were best fitted in each case. Introduction of the G202A mutation in the G-protein subunit did not alter DADLE binding properties substantially in that similar pKh and pK1 values were observed for hDOP-GilαG202A,C351I and hDOP-Gi1αC351I (Table 2). In contrast, the double mutation in the second intracellular loop of hDOP receptor did alter the binding affinity of DADLE with a ∼30-fold loss of affinity in both high- and low affinity binding sites (hDOPV150E,V154D-Gi1αC351I: pKh, 7.4 ± 0.2; pKl, 5.0 ± 0.4, hDOP-Gi1αC351I: pKh, 9.0 ± 0.2; pKl, 6.8 ± 0.42). In membranes coexpressing hDOP-Gi1αG202A,C351I and hDOPV150E,V154D-Gi1αC351I, there was no significant difference in the percentage of high and low site numbers compared with the wild-type hDOP-Gi1αC351I fusion protein (P > 0.05, one-way ANOVA) (Table 2). A similar reduction in affinity of the high affinity site for the DOP-selective peptide agonist DPDPE was also observed when comparing hDOPV150E,V154D-Gi1αC351I with hDOP-Gi1αC351I or hDOP-Gi1αG202A,C351I (Table 3). Although a similar trend was observed for the low-affinity site (Table 3), this did not achieve statistical significance because of relatively imprecise estimates of pKl. Wild-type DPDPE binding characteristics were again restored after coexpression of hDOPV150E,V154D-Gi1αC351I and hDOP-Gi1αG202A,C351I (Table 3).
Binding affinity of DADLE for individually expressed and co-expressed hDOP-G i1αC351I fusion proteins
Data represent means ± S.E.M. of n = 4 experiments performed in triplicate on different membrane preparations.
Binding affinity of DPDPE for individually expressed and co-expressed hDOP-G i1αC351I fusion proteins
Data represent means ± S.E.M. of n = 4 experiments performed in triplicate on different membrane preparations.
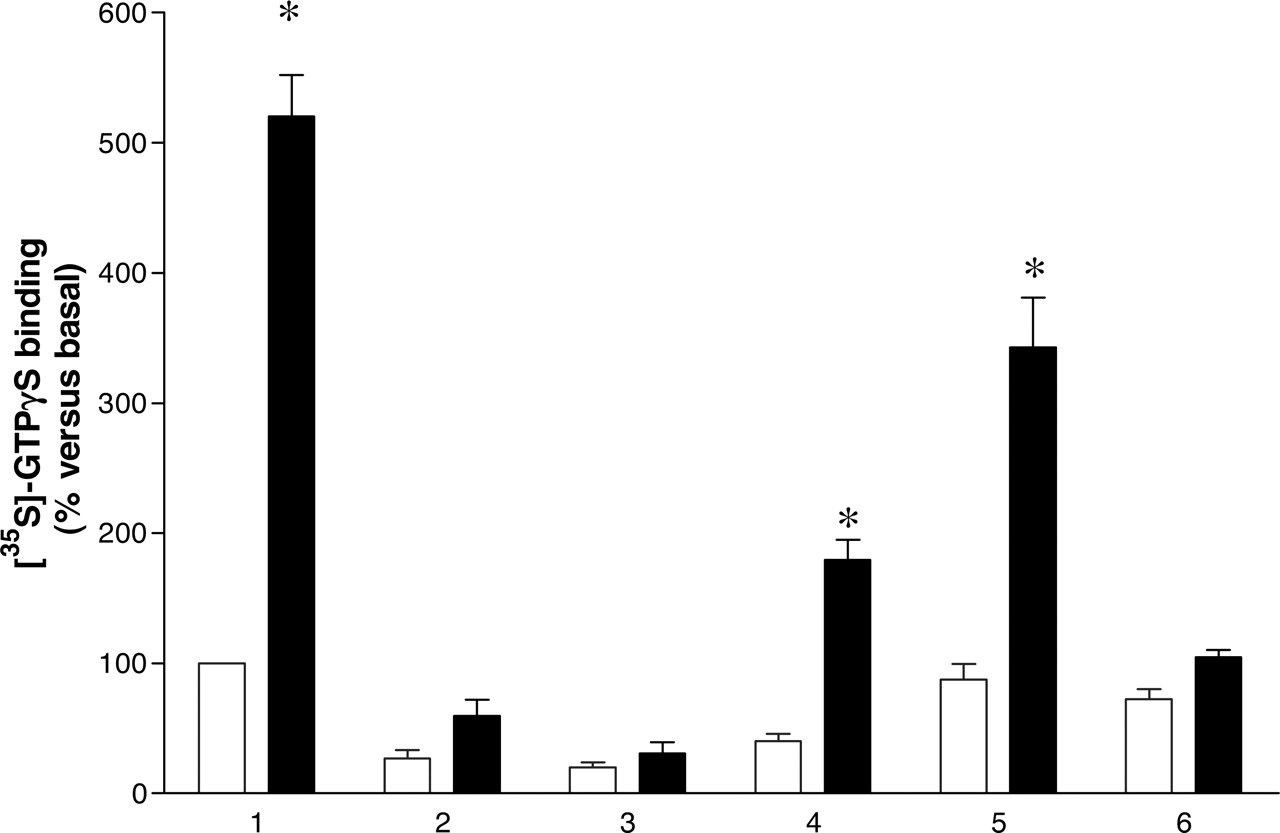
Reconstitution of hDOP-Gi1αC351I function by coexpression of two nonfunctional mutants. Membranes of pertussis toxin-treated HEK 293 cells expressing 15 fmol of hDOP-Gi1αC351I (1), hDOPV150E,V154D-Gi1αC351I (2), hDOP-Gi1αG202A,C351I (3), or hDOPV150E,V154D-Gi1αC351I + hDOP-Gi1αG202A,C351I (4) were used to measure basal (open bars) and 10 μM DADLE (filled bars) binding of [35S]GTPγS as in Fig. 1A. Membranes coexpressing a total of 30 fmol of hDOPV150E,V154D-GilαC351I + hDOP-Gi1αG202A,C351I (5) were also analyzed, as were membranes expressing 15 fmol of hDOPV150E,V154D-Gi1αC351I or 15 fmol of hDOP-Gi1αG202A,C351I that were mixed before assay (6). Data represent n = 5 experiments performed in triplicate. *, significant (p < 0.05) stimulation by DADLE.
Assuming that the predominant form of the hDOP is a dimer rather than a higher-order oligomer, coexpression of hDOPV150E,V154D-Gi1αC351I and hDOP-Gi1αG202A,C351I must be expected to generate hDOPV150E,V154D-Gi1αC351I dimers and hDOP-Gi1αG202A,C351I dimers (which, as shown in Fig. 2, are inactive) as well as the functionally reconstituted hDOPV150E,V154D-Gi1αC351I + hDOP-Gi1αG202A,C351I dimer. Ligand binding studies must reflect the full population of these different hDOP homodimers in the cell membrane. By contrast, in functional assays, only hDOP-Gi1αC351I homodimers and hDOPV150E,V154D-Gi1αC351I + hDOP-Gi1αG202A,C351I homodimers are reported (Fig. 2). The potency of DADLE to stimulate [35S]GTPγS binding via the hDOP-Gi1αC351I dimer and the reconstituted hDOPV150E,V154-D-Gi1αC351I + hDOP-Gi1αG202A,C351I dimer was not different (Fig. 4A). Likewise, the prototypic opioid receptor antagonist naloxone was equipotent in its ability to prevent DADLE-stimulated [35S]GTPγS binding via the hDOP-Gi1αC351I dimer and the reconstituted hDOPV150E,V154D-Gi1αC351I + hDOP-Gi1αG202A,C351I dimer (Fig. 4B).

Interactions between coexpressed hDOP-Gi1αG202A,C351I and hDOPV150E,V154D-Gi1αC351I monitored by coimmunoprecipitation. Top, membranes from control HEK 293 cells (1) and cells transiently expressing Flag-hDOPV150E,V154D-Gi1αC351I (2), c-myc-hDOP-Gi1αG202A,C351I (3), Flag-hDOPV150E,V154D-Gi1αC351I + c-myc-hDOP-Gi1αG202A,C351I (4), or a mix of membranes from lanes 2 and 3 (5) were immunoprecipitated with anti-Flag antibody and after resolution by SDS-PAGE were immunoblotted to detect c-myc immunoreactivity. Bottom, samples equivalent to those at the top were directly resolved by SDS-PAGE and immunoblotted to detect Flag immunoreactivity.
To assess whether the reconstitution of function observed upon coexpression of hDOPV150E,V154D-Gi1αC351I + hDOP-Gi1αG202A,C351I could possibly be accounted for simply by the provision of the Gi1αC351I attached to the inactive hDOPV150E,V154D receptor rather than specifically requiring interactions between hDOPV150E,V154D and hDOP, we generated and expressed a construct (Flag-Nt-TM1-Gi1αC351I) in which Gi1αC351I was linked to a sequence comprising the N-terminal domain, transmembrane region 1, and the first intracellular loop of hDOP. This construct did not bind [3H]diprenorphine (data not shown), but its expression as an apparent 48-kDa polypeptide could clearly be detected by immunoblotting transfected HEK293 membranes with the anti-Gi1α/Gi2α antiserum (Fig. 5A). Parallel SDS-PAGE and immunodetection of varying amounts of recombinantly expressed Gi1α, followed by densitometry of the signals, allowed production of a standard curve for Gi1α expression that was linear over the range (0-50 ng) employed. Based on the anti-Gi1α immunological signal in membranes corresponding to Flag-Nt-TM1-Gi1αC351I and its calculated molecular mass (49.3 kDa), we estimated levels of this construct to be 13.8 pmol/mg of membrane protein. Therefore, this construct was present at some six times the level of the hDOP-Gi1α fusion proteins. Cotransfection of Flag-Nt-TM1-Gi1αC351I with hDOP-Gi1αG202A,C351I resulted in very low but statistically significant increases in levels of [35S]GTPγS binding in anti-Gi1α/Gi2α antiserum immunoprecipitates when DADLE was added to such membranes (Fig. 5B). These very small signals did not reflect the possibility that although hDOP-Gi1αG202A,C351I and Flag-Nt-TM1-Gi1αC351I were coexpressed, they were present in distinct membrane compartments. Coexpression of Flag-Nt-TM1-Gi1αC351I with c-myc-hDOP-Gi1αG202A,C351I allowed their coimmunoprecipitation (Fig. 6A), indicating not only proximity but also their capacity for physical interactions. Likewise, coexpression of c-myc-Nt-TM1 with the isolated Flag-hDOP allowed their coimmunoprecipitation, indicating interactions were not a reflection of contacts between the two copies of the G protein (Fig. 6B).
To extend these reconstitution studies to the other opioid receptors we generated equivalent fusion proteins incorporating the human MOP-1 (hMOP) receptor. hMOP-Gi1αC351I, hMOP-Gi1αG202A,C351I, and hMOPV169E,V173D-Gi1αC351I were expressed individually in HEK293 cells and after pertussis toxin-treatment and membrane preparation, expression levels and affinity for [3H]diprenorphine were assessed via saturation binding studies. No significant differences between the three constructs were noted in either parameter (Table 4). Likewise, after coexpression of hMOP-Gi1αG202A,C351I and hMOPV169E,V173D-Gi1αC351I, the characteristics of [3H]diprenorphine binding were equivalent. In functional [35S]GTPγS binding studies (Fig. 7), the selective MOP receptor agonist DAMGO (10 μM) caused a 5.28 ± 0.24-fold (n = 4, mean ± S.E.M.) stimulation in end of assay anti-Gi1α/Gi2α antiserum immunoprecipitates. As with the related hDOP constructs, membranes expressing equal amounts of either hMOP-Gi1αG202A,C351I or hMOPV169E,V173D-Gi1αC351I did not result in DAMGO stimulation of [35S]GTPγS binding (Fig. 7). Cotransfection of hMOP-Gi1αG202A,C351I and hMOPV169E,V173D-Gi1αC351I did result in partial reconstitution of DAMGO-stimulated [35S]GTPγS binding (Fig. 7), an effect not achieved by simple mixing of membranes individually expressing hMOP-Gi1αG202A,C351I or hMOPV169E,V173D-Gi1αC351I (Fig. 7). In comparison with the 60% reconstitution of hDOP function, membranes expressing twice as many hMOP receptor [3H]diprenorphine binding sites after coexpression of the two inactive mutant fusion proteins allowed only 40% of the amount of agonist-stimulated [35S]GTPγS binding as generated by the wild-type hMOP-Gi1αC351I fusion (Fig. 7). A potential explanation for this was uncovered on examining the potency of DAMGO to stimulate [35S]GTPγS binding in membranes expressing hMOP-Gi1αC351I and coexpressing hMOP-Gi1αG202A,C351I and hMOPV169E,V173D-Gi1αC351I. The potency of this ligand was reduced (p < 0.05) by some 2-fold at the functionally reconstituted dimer (pEC50 = 6.1 ± 0.07) compared with the wild-type dimer (pEC50 = 6.5 ± 0.04). It is interesting that although both hMOP-Gi1αC351I and hMOP-Gi1αG202A,C351I displayed both high- and low-affinity binding sites for DAMGO when this ligand was allowed to compete with [3H]diprenorphine (Fig. 8, Table 5), only a low-affinity binding component could be detected for hMOPV169E,V173D-Gi1αC351I (Fig. 8, Table 5), similar to what might be anticipated if GPCR and G protein were uncoupled. When hMOPV169E,V173D-Gi1αC351I and hMOP-Gi1αG202A,C351I were coexpressed, the characteristics of DAMGO binding were akin to a mixture of the two mutant constructs (Fig. 8, Table 5), and analysis of the binding curves was consistent with the presence of the two constructs at a ratio of nearly 1:1.
Expression levels and [3H]diprenorphine binding affinity of hMOP-Gi1αC351I fusion proteins
Data represent means ± S.E.M. from n = 3 experiments performed in triplicate on different membrane preparations. Statistics were performed using one-way ANOVA on Bmax and pKd numbers.
Binding affinity of DAMGO for individually expressed and co-expressed hMOP-G i1αC351I fusion proteins
Data represent means ± S.E.M. from n = 3 experiments performed in triplicate on different membrane preparations. Statistics were performed using one-way ANOVA on pKh and pK1 numbers.

Similar functional pharmacology of hDOP-Gi1αC351I and the reconstituted dimer. A, membranes of pertussis toxin-treated HEK 293 cells expressing 15 fmol of hDOP-Gi1αC351I (open symbols) or hDOPV150E,V154D-Gi1αC351I + hDOP-Gi1αG202A,C351I (closed symbols) were used to measure the ability of increasing concentrations of DADLE to enhance [35S]GTPγS binding as in Fig. 1A. Because the absolute amount of [35S]GTPγS bound was less per [3H]diprenorphine binding site in membranes expressing the functionally reconstituted dimer (see Fig. 2), data are shown as percentage of maximal signal. B, the ability of varying concentrations of naloxone to inhibit [35S]GTPγS binding stimulated by 100 nM DADLE is shown. Data are means ± S.E.M. of n = 3 experiments.
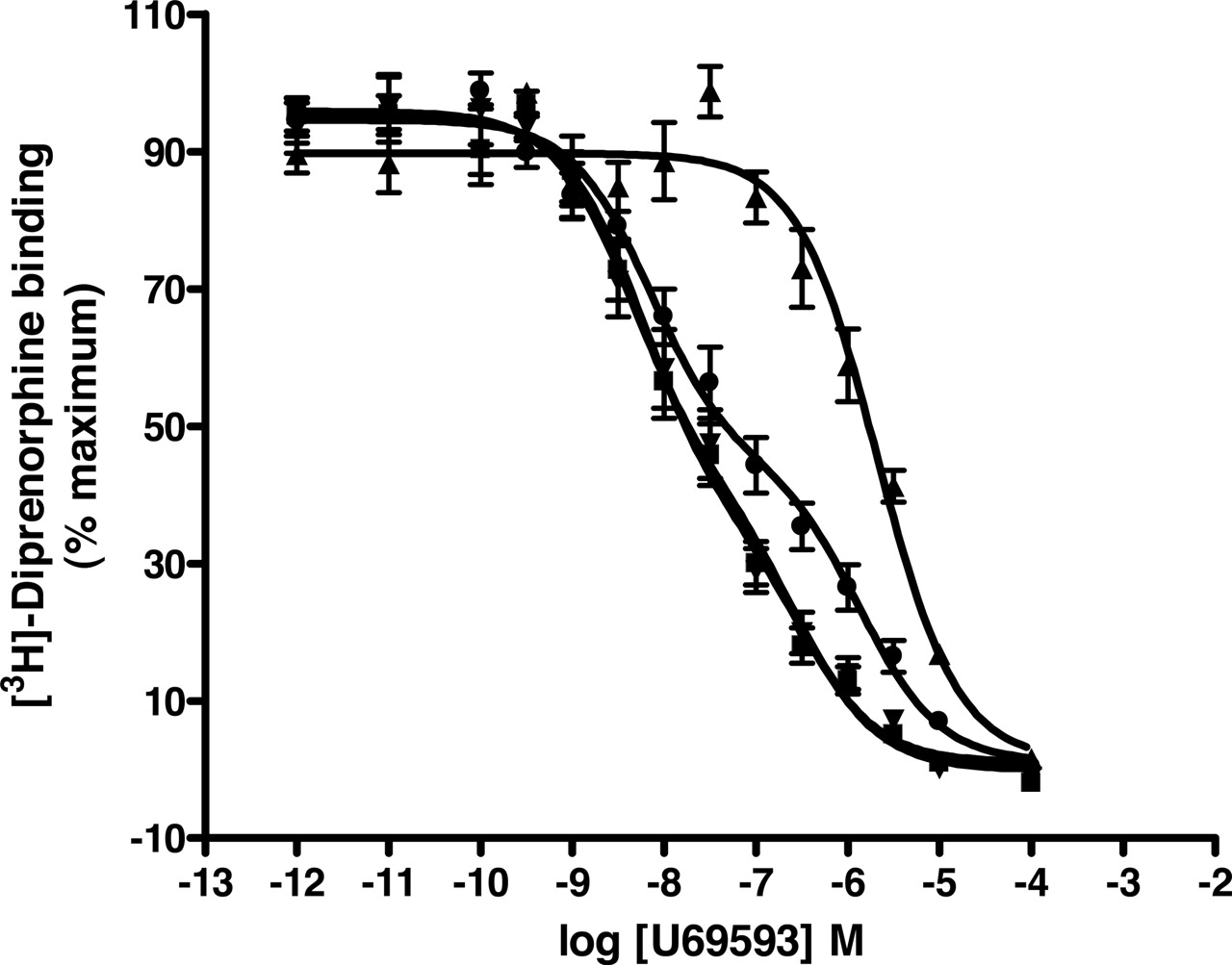
Studies were also performed on the rat (r)KOP receptor. rKOP-Gi1αC351I, and rKOP-Gi1αG202A,C351I, rKOPV160-E,V164D-Gi1αC351I fusions were generated and expressed. These also all bound [3H]diprenorphine with high affinity and expressed to similar levels (Table 6); however, as with the hDOP constructs, a reduction in affinity was recorded for the rKOPV160E,V164D-Gi1αC351I construct that incorporated mutations into the second intracellular loop of the receptor. As with the equivalent hDOP and hMOP constructs, rKOP-Gi1αC351I allowed a large increase in [35S]GTPγS binding in response to agonist treatment (Fig. 9). Individual expression of rKOP-Gi1αG202A,C351I and rKOPV160E,V164D-Gi1αC351I did not result in stimulation of [35S]GTPγS binding in the presence of the KOP receptor-selective agonist U69593, whereas coexpression of rKOP-Gi1αG202A,C351I and rKOPV160E,V164D-Gi1αC351I did (Fig. 9). At a maximally effective concentration of U69593 (10 μM), membranes expressing twice as many rKOP [3H]diprenorphine binding sites, after coexpression of the two inactive mutants, allowed approximately 50% of the amount of agonist-stimulated [35S]GTPγS binding generated by wild-type rKOP-Gi1αC351I fusion (Fig. 9). As with the hMOP constructs, in competition studies between [3H]diprenorphine and U69593, both rKOP-Gi1αC351I and rKOP-Gi1αG202A,C351I displayed both high- and low-affinity binding sites for the agonist. However, rKOPV160E,V164D-Gi1αC351I displayed only a single, low-affinity site for U69593 (Fig. 10, Table 7). In addition, as with the hMOP constructs, coexpression of rKOPV160E,V164D-Gi1αC351I and rKOP-Gi1αG202A,C351I resulted in a pattern of U69593 binding consistent with a mixture of the pharmacology of the two constructs (Fig. 10, Table 7). The potency of U69593 to activate rKOP-Gi1αC351I (pEC50 = 7.3 ± 0.08) was higher (p < 0.05) than that for the reconstituted rKOP dimer (pEC50 = 6.8 ± 0.13).
Expression levels and [3H]diprenorphine binding affinity of r KOP-Gi1αC351I fusion proteins
Data represent means ± S.E.M. from n = 3 experiments performed in triplicate on different membrane preparations. Statistics were performed using one-way ANOVA on Bmax and pKd numbers.
Binding affinity of U69593 for individually expressed and co-expressed rKOP-G i1αC351I fusion proteins
Data represent means ± S.E.M. of n = 4 experiments performed in triplicate on different membrane preparations.
Statistics were performed using one-way ANOVA on pKh and pKl numbers and on high affinity site numbers.

Provision of Flag-Nt-TM1-Gi1αC351I does not reconstitute substantial function to hDOP-Gi1αG202A,C351I. A, membranes from control, pertussis toxin-treated HEK 293 cells (1) and those transfected to express Flag-Nt-TM1-Gi1αC351I (2) or Flag-Nt-TM1-Gi1αC351I + hDOP-Gi1αG202A,C351I (3, 4) were resolved by SDS-PAGE and immunoblotted using the anti-Gi1α/Gi2α antiserum. The polypeptide(s) migrating with apparent Mr near 48 kDa is Flag-Nt-TM1-Gi1αC351I, whereas the polypeptide(s) with apparent Mr near 40 kDa is endogenously expressed Gi1α/Gi2α. Previous studies of HEK293 cells have shown this to be predominantly Gi2α, which is expressed at some 50 pmol/mg of membrane protein (McClue et al., 1992). B, membranes expressing 15 fmol of hDOP-Gi1αC351I (1), hDOP-Gi1αG202A,C351I (2), 10 μg of membranes expressing Flag-Nt-TM1-Gi1αC351I [estimated to contain 138 fmol of this construct (see Results)] (3), membranes coexpressing 15 (4) or 30 (5) fmol of hDOP-Gi1αG202A,C351 I + [estimated as 138 (4) or 276 (5) fmol] Flag-Nt-TM1-Gi1αC351I or a mixture of 10 μg of membranes expressing Flag-Nt-TM1-Gi1αC351I (138 fmol) + 30 fmol of hDOR-Gi1αG202A,C351I (6) were used to measure the binding of [35S]GTPγS in the absence (open bars) or presence of 10 μM (filled bars) or 100 nM (checkered bars) DADLE. Data represent means ± S.E.M. of n = 5 experiments performed in triplicate. *, significant (p < 0.05) stimulation by DADLE.
Discussion
Fusion proteins between GPCRs and G protein α subunits have been used to examine a wide range of function of these polypeptides (Milligan, 2002; Milligan et al., 2004). The defined 1:1 stoichiometry of the partner proteins is of particular use in measures of agonist-induced GTPase turnover number (Moon et al., 2001) and the regulation [coordinated (Stevens et al., 2001) or otherwise (Barclay et al., 2005)] of posttranslational thioacylation of GPCR and G protein and the effects of mutations in either partner that alter protein steady-state expression levels (Ward and Milligan, 2002). In the current studies, we have generated and explored the function and pharmacology of fusions between each of the DOP, KOP, and MOP opioid receptors with Gi1α. The functionality of each of these mutants was established in [35S]GTPγS binding studies in which immunoprecipitation with an anti-Gi1α/Gi2α antiserum limited nonspecific binding of the nucleotide at assay termination. All commonly used cell lines express members of the Giα G protein family that are substrates for pertussis toxin-catalyzed ADP-ribosylation. To ensure that agonist-driven [35S]GTPγS binding reflected only binding to the fusion proteins under study, they were constructed using Gi1αC351I (Bahia et al., 1998), which is insensitive to the actions of the toxin but able to be effectively activated by receptors, and by treating cells with pertussis toxin before cell harvest to modify the endogenous Giα pool. Mutation of Gly202 to Ala in Gi1α resulted in a form of the G protein that was unable to exchange guanine nucleotide and bind [35S]GTPγS in response to receptor agonists. All G protein α subunits have a Gly residue in the equivalent position, and mutation should therefore be anticipated to produce equivalent lack of function mutants, as shown previously for G11α (Carrillo et al., 2002, 2003). Fusion of wild-type G11α to forms of the α1b-adrenoceptor and the histamine H1 receptor containing hydrophobic-to-acidic residue mutations in intracellular loop 2 also results in lack of agonist-mediated [35S]GTPγS binding without destruction of the ligand binding pocket (Carrillo et al., 2003). Because most rhodopsin-like GPCRs have a pair of homologous hydrophobic residues (Milligan et al., 2005) and in the DOP, KOP, and MOP receptors, both are Val, we converted each of these to either Glu or Asp. This did not alter construct expression levels and had little or no effect on the binding affinity of [3H]diprenorphine. We were thus able to measure and equalize construct expression levels in preparation for functional studies. In each case, coexpression of the pair of nonfunctional opioid receptor-fusion proteins was able to partially reconstitute agonist-mediated [35S]GTPγS binding. Reconstitution did require coexpression; simply mixing membranes expressing the potentially complementary pairs did not generate agonist function. We have previously argued that such results require receptor dimerization (Carrillo et al., 2003) and have provided evidence that the reconstitution reflects an intermolecular rather than intramolecular interaction between GPCR and G protein (Carrillo et al., 2003). Although expression of a single fusion protein, wild-type in both GPCR and G protein sequence, allows agonist mediated signal transduction, like expression of a single GPCR cDNA, this does not allow direct exploration of GPCR quaternary structure. Indeed, the knowledge that a single cDNA was generally sufficient to generate the anticipated function and pharmacology of a GPCR played a significant part in the expectation that GPCRs would be single polypeptide, monomeric structures (Milligan, 2004). Previous studies by Molinari et al. (2003) also noted a capacity of coexpressed of pairs of inactive DOP-G protein fusions to reconstitute a signal. However, although they also concluded that dimerization reflected intermolecular interactions between the coexpressed forms, they did not specifically suggest that dimerization between the pair of DOP receptors was required. This may have been because they also observed an ability of a DOP-G protein fusion to activate a G protein that was membrane-anchored simply by linkage to transmembrane 1 of the vasopressin V2 receptor. In contrast with these observations, we observed only a very limited capacity of the hDOP-Gi1αG202A,C351I construct to activate coexpressed Gi1αC351I when it was tethered to the membrane by linkage to the N-terminal domain and transmembrane domain 1 of hDOP, even though the G protein was provided at levels approximately six times higher in this scenario than when provided by coexpression of the potentially complementary fusion protein. The basis for these differences is unclear but may relate to the high expression levels of the fusion proteins achieved and employed by Molinari et al. (2003), which were in the range in which so called “bystander” interactions and effects have been observed (Mercier et al., 2002), probably simply because of physical proximity rather than direct protein-protein interactions. Although hDOP-Gi1αG202A,C351 I was unable to activate coexpressed Nt-TM1-Gi1αC351I to any substantial extent, these two constructs were able to interact because they could be coimmunoprecipitated after coexpression. This suggests that interaction between two complete receptors might be required for GPCR function and would support other evidence for conformational alterations in the partner GPCR in a dimer induced by ligand binding (Mesnier and Baneres, 2004; El-Asmar et al., 2005). Nt-TM1 could also be coimmunoprecipitated with full-length hDOP, which suggests that TM1 and/or the N-terminal region of hDOP provides a protein-protein interaction interface. Although not explored in detail in these studies, for the α1b-adrenoceptor, symmetrical TM1-TM1 interactions provide key contributions to the quaternary organization of this GPCR (Carrillo et al., 2004), and a series of other reports have supported an important contribution of TM1 to the dimer interface(s) in other GPCRs (Overton and Blumer, 2002; Klco et al., 2003; Stanasila et al., 2003). Because “nonspecific” effects, potentially arising from high level expression in heterologous transfection studies, are an inherent concern, in the current experiments, we maintained fusion construct expression in the range of 1 to 2 pmol/mg of membrane protein, and all “functional reconstitution” experiments were performed under conditions in which agonist-stimulated [35S]GTPγS binding increased linearly with construct amount. This was a key requirement for data analysis, because if opioid receptors exist and function predominantly as dimers, the reconstitution strategy suggests that with 1:1 expression of the two mutant constructs, then, in stochastic terms, 50% of the ligand binding sites should reflect “hetero” interactions that can generate a functional response. One hypothesis, therefore, was that when using membranes coexpressing a pair of potentially suitable mutants, double the number of binding sites would be required to result in the same level of agonist-stimulated [35S]GTPγS binding as with the wild-type fusion. This was not achieved in all cases; the level of reconstitution ranged from 40% for the MOP receptor to 60% for the DOP receptor. This may imply that not all cellular copies of a particular GPCR are present within dimers. This has been an extremely difficult issue to assess quantitatively. The proportion of a GPCR that migrates through SDS-PAGE as an SDS-resistant dimer is almost certainly a lower limit for the native state, and although resonance energy transfer-based estimates of `dimer' proportions have ranged from 25 to 85% (Mercier et al., 2002; Dinger et al., 2003), a considerable number of assumptions are required to allow such calculations (Milligan and Bouvier, 2005). Likewise, there is growing evidence for a requirement of GPCR dimerization for productive signal transduction that is not restricted to the examples of the GABAb and other family C receptors and for greater than dimeric, higher-order quaternary structure (Klco et al., 2003; Carrillo et al., 2004; Fotiadis et al., 2004). Likewise, however, the basic strategy used herein might be restrictive in that a pair of hydrophobic residues from the second intracellular loop were mutated to acidic residues, and this might compromise the effectiveness of GPCR dimerization. It is worth noting, however, that the cytoplasmic face of the opioid receptor subtypes is very highly conserved between DOP, KOP, and MOP, and despite making the equivalent mutations in each, significant differences in reconstitutive effectiveness were observed. This may imply differences in the details of the homodimerization process. Although homodimerization of each of these three receptors has previously been recorded (Cvejic and Devi, 1997; George et al., 2000; McVey et al., 2001; Li-Wei et al., 2002; Ramsay et al., 2002), there is no useful information on the similarities or differences in mechanisms of these interactions that have involved direct experimental study, although this topic has been considered via an informatic approach (Filizola and Weinstein, 2002).
Coexpressed hDOP-Gi1αG202A,C351I and Nt-TM1-Gi1αC351I interact and can be coimmunoprecipitated. A, membranes from pertussis toxin-treated HEK 293 cells (1) and equivalent cells transiently expressing c-myc-hDOP-Gi1αG202A,C351I (2), Flag-Nt-TM1-Gi1αC351I (3), or Flag-Nt-TM1-Gi1αC351I + c-myc-hDOP-Gi1αG202A,C351I (4), were immunoprecipitated with anti-Flag antibody and anti-c-myc immunoreactivity detected after separation of the samples by SDS-PAGE (top). The expression of Flag-Nt-TM1-Gi1αC351I in the appropriate samples was confirmed by immunoblotting membranes with anti-Flag antibody (bottom). B, membranes from pertussis toxin-treated HEK 293 cells (1) or those transiently expressing Flag-hDOP (2), c-myc-Nt-TM1 (3), or Flag-hDOP + c-myc-Nt-TM1 (4) were immunoprecipitated with anti-Flag antibody and detected with anti-c-myc antibody after being resolved by SDS-PAGE.

Reconstitution of hMOP function by coexpression of two nonfunctional hMOP-Gi1α mutants. Membranes of pertussis toxin-treated HEK 293 cells expressing 15 fmol of hMOP-Gi1αC351I (1); hMOPV169E,V173D-Gi1αC351I (2), hMOP-Gi1αG202A,C351I (3), and either 15 (4) or 30 (5) fmol of cotransfected hMOPV169E,V173D-Gi1αC351I + hMOP-Gi1αG202A,C351I were used measure [35S]GTPγS binding were to in the absence (open bars) or presence (filled bars) of 10 μM DAMGO as in Fig. 2. A control was provided by mixing membranes expressing 15 fmol of hMOPV169E,V173D-Gi1αC351I and 15 fmol of hMOPGi1αG202A,C351I before assay (6). Data represent means ± S.E.M. of n = 4 experiments performed in triplicate. *, significant (p < 0.05) stimulation by DAMGO.

The characteristics of binding of DAMGO to individually expressed and coexpressed hMOP-Gi1α fusion proteins. Membranes expressing hMOP-Gi1αC351I (▪); hMOP-Gi1αG202A,C351I (▾), hMOPV169E,V173D-Gi1αC351I (▴) or both hMOP-Gi1αG202A,C351I and hMOPV169E,V173D-Gi1αC351I (•) were used to measure the ability of varying concentrations of DAMGO to compete for binding with 1 nM [3H]diprenorphine. Data represent n = 4 experiments performed in triplicate.

Reconstitution of rKOP function by coexpression of two nonfunctional rKOP-Gi1α mutants. Membranes of HEK 293 cells expressing 15 fmol of rKOP-Gi1αC351I (1); rKOPV160E,V164D-Gi1αC351I (2), rKOP-Gi1αG202A,C351I (3), and 15 (4) or 30 (5) fmol of [3H]diprenorphine binding sites after coexpression of rKOPV160E,V164D-Gi1αC351I + rKOP-Gi1αG202A,C351I were used to measure [35S]GTPγS binding in the absence (open bars) or presence of 10 μM (filled bars) or 100 nM (checked bars) U69593. A control was performed by mixing membranes expressing 15 fmol of rKOPV160E,V164D-Gi1αC351I, and 15 fmol of rKOP-Gi1αG202A,C351I (6). Data represent means ± S.E.M. of n = 4 experiments performed in triplicate. *, significant (p < 0.05) stimulation by U69593.
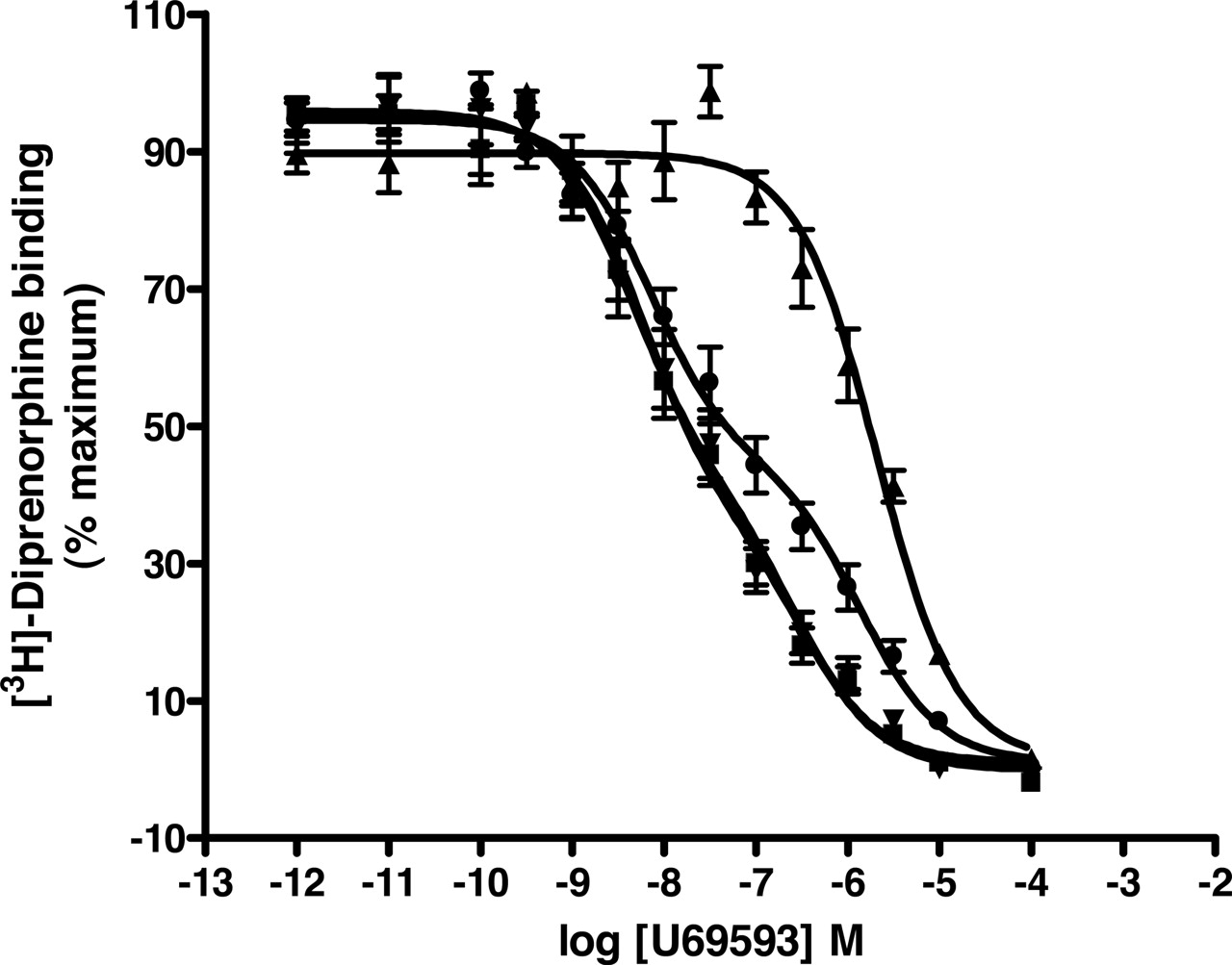
The characteristics of binding of U69593 to individually expressed and coexpressed rKOP-Gi1α fusion proteins. Membranes expressing rKOP-Gi1αC351I (▪); rKOP-Gi1αG202A,C351I (▾), rKOPV160E,V164D-Gi1αC351I and rKOPV160E,V164D-Gi1αC351I (▴) or both rKOP-V160E,V164D-Gi1αC351I and rKOP-Gi1αG202A,C351I (•) were used to measure the ability of varying concentrations of U69593 to compete for binding with 1 nM [3H]diprenorphine. Data represent n = 4 experiments performed in triplicate.
Although the mutation of hydrophobic residues in intracellular loop 2 may have limitations in producing an inactive GPCR, a marked advantage over certain other reconstitutive studies (Monnot et al., 1996; Bakker et al., 2004) is that the orthosteric GPCR ligand binding site was not destroyed. This allowed antagonist binding studies to confirm not only expression of each construct but also that each inactive mutant was expressed at the same level as the wild-type fusion. This was central to the “stochastic” calculations of the potential makeup of the GPCR dimer population generated after coexpression of different proteins. The complete conservation in G protein α subunits of the Gly residue modified herein to generate one of the pair of inactive fusions and the very high conservation of the pair of GPCR intracellular loop hydrophobic residues suggest that this strategy should be widely applicable (Milligan et al., 2005). For example, it is likely to be of considerable use in mutational studies designed to identify key residues involved in the dimerization interface(s) (Hernanz-Falcon et al., 2004). Likewise, there is no reason to limit such studies to GPCR homodimerization and the effectiveness of functional reconstitution may provide quantitative data on the propensity of GPCRs to heterodimerize. Indeed, this has been initiated by studies showing that the histamine H1 receptor and the α1b-adrenoceptor are very poor interaction partners (Carrillo et al., 2003). Finally, because only the reconstituted heterodimer is an active signaling unit, then in true GPCR heterodimerization studies, the functional pharmacology of the heterodimer could be examined without interfering signals generated by the corresponding coexpressed homodimers, which, as shown herein, are essentially inactive.
Footnotes
-
These studies were supported, in part, by a Scottish Enterprise “Proof of concept” award (to G.M.).
-
ABBREVIATIONS: DOP, δ opioid peptide; KOP, κ opioid peptide; MOP, μ opioid peptide; GPCR, G protein-coupled receptor; DADLE, [D-Ala2, D-Leu5]-enkephalin; DAMGO, [D-Ala2,N-Me-Phe4, Gly5-ol]-enkephalin; DPDPE, [D-Pen2, D-Pen5]-enkephalin; U69593, (+)-(5α,7α,8β)-N-methyl-N-[-7-(1-pyrrolodinyl)-1-oxaspirol[4,5]dec-8-yl)benzeneacetamide; h, human; r, rat; PCR, polymerase chain reaction; HEK, human embryonic kidney; GTPγS, guanosine 5′-([γ-35S]thio)triphosphate; SG, anti-Gαi1-2 antiserum; ANOVA, analysis of variance.
- Received April 16, 2005.
- Accepted June 20, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}