


Abstract
A crucial limitation for structural and biophysical analysis of G protein-coupled receptors (GPCRs) is the inherent challenge of purifying and stabilizing these receptors in an active (agonist-bound) conformation. Peptide ligands, such as the vasoactive, cyclic hormone urotensin-II (U-II), may provide new purification tools, via high affinity, pseudo-irreversible binding suitable for ligand-based affinity purification. We show that the U-II receptor (UT) is resistant to desensitization as a result of low phosphorylation and diminished endocytosis. UT also displays an unusual proclivity to remain active with vasoconstriction sustained despite extensive washout of the ligand. To exploit these properties for ligand-supported purification, we modified the U-II ligand by attaching a biotin moiety and spacer arm to the N terminus, creating a novel affinity ligand (Bio-U-II) to interface with streptavidin media. Bio-U-II bound to UT with pharmacological properties analogous to those of the unmodified U-II ligand (high-affinity, pseudo-irreversible binding). The prebinding of Bio-U-II to UT (before exposure to detergent) facilitated specific capture of UT by stabilizing the receptor structure during solubilization with detergent. Solubilization of UT with the most compatible detergent, n-dodecyl β-d-maltoside, was dependent on the critical micelle concentration, and Gαq/11 protein was copurified with captured Bio-U-II-UT complexes. Furthermore, captured Bio-U-II-UT complexes were resistant to dissociation at elevated temperatures, suggesting that UT is relatively thermostable, making it an ideal candidate for future structural and biophysical studies. This work demonstrates the utility of pseudo-irreversible ligands to support the purification of a GPCR during detergent extraction, resulting in the first successful purification of the UT.
Introduction
Conventional paradigms of GPCR activation predict that the active conformation (R* state) is selected (or induced) by ligand binding and couples to G proteins, which initiate intracellular signaling. These signals are terminated by receptor phosphorylation mediated by GPCR kinases and recruitment of arrestins, preventing further interaction with G proteins and promoting receptor internalization (Oakley et al., 2001). For GPCRs, the balance that exists between receptor activation and deactivation (desensitization) determines the strength and duration of a given stimulus.
Despite recent advances in delineating the mechanism of GPCR activation, our understanding of the conformational landscape that underpins GPCR activation is underdeveloped. This is primarily because GPCRs (particularly the R* forms) are generally recalcitrant to purification and are inherently unstable during membrane extraction. The problems associated with purifying GPCRs are multifactorial: 1) they are expressed at low levels, 2) it is difficult to select and stabilize the R* state(s), and 3) the detergent extraction and separation processes destabilize important hydrophobic interactions. The result of destabilizing these interactions is that large quantities of GPCRs become denatured or irreversibly inactivated, leading to poor purification yields (for review, see Chiu et al., 2008). This is reflected by the lack of structural information on the active conformation of GPCRs. Moreover, most current GPCR structures are stabilized in an inactive (R) conformation (for review, see Topiol and Sabio, 2009), except for an intermediary form of the rhodopsin receptor (Salom et al., 2006). Scheerer et al. (2008) crystallized the complex between an 11-mer synthetic peptide of the Gαt C terminus and opsin, showing slight structural shifts in both the receptor and G protein fragment. Although the study provides clues to the mode of GPCR activation, the structure may not be indicative of an active state, because the interaction between GPCR and G protein is likely to be more complex. Indeed, NMR in combination with molecular modeling has been used to demonstrate structural rearrangement of transmembrane domains 6 and 7 via disruption of a salt bridge in extracellular loop 2, which may play a role in receptor activation (Bokoch et al., 2010).
One approach to overcome barriers associated with GPCR instability is to screen for receptor mutations that increase protein thermal stability (Standfuss et al., 2007; Magnani et al., 2008; Serrano-Vega et al., 2008; Warne et al., 2008; Shibata et al., 2009); however, this mutation approach can also yield an inactive conformer (Standfuss et al., 2007; Warne et al., 2008). As an alternative, or perhaps a supplement, to mutagenesis, we reasoned that some receptor-ligand pairings might be amenable to affinity chromatography, using resins functionalized to capture a related affinity-tag linked to the receptor's ligand. This approach to GPCR purification, termed “ligand-supported purification,” can exploit essentially irreversible GPCR-ligand pairings, in which the ligand is modified and serves to place the GPCR in the R* state. Detergents are then applied to solubilize the plasma membrane and the ligand supports the GPCR structure during extraction and purification by dampening the denaturing effects of the detergent, with the goal of purifying the GPCR to a solid support capture matrix in the active R* state.
The cyclic peptide hormone urotensin-II (U-II) is one of the most potent vasoconstrictors in a variety of species (Douglas et al., 2000). The rat U-II receptor (UT) is a Gαq/11-coupled receptor that activates phospholipase Cβ1 to generate the second messengers inositol triphosphate and diacylglycerol, which release calcium from intracellular stores and activate protein kinase C, respectively (Proulx et al., 2008).
The U-II/UT pairing has features that make it a prime candidate for ligand-supported purification: 1) we confirm here that the UT/U-II interaction is high affinity, being essentially irreversible (Douglas et al., 2000) with long-lasting effects—both at a cellular level and in vivo (Camarda et al., 2002); 2) the N terminus of the U-II peptide is reportedly redundant for high-affinity binding (Coy et al., 2002), allowing modification of the ligand with the affinity group; 3) we show that the U-II bound UT is resistant to the common regulatory mechanisms of phosphorylation, arrestin binding, and internalization that normally lead to desensitization of receptor signals, suggesting that the receptor may exist in a more restricted set of active conformations. Together, these properties indicate that the UT has a strong propensity to remain in an active conformation at the cell surface for prolonged periods of time, increasing the likelihood of purifying a GPCR in the active R* state. We report here the application of ligand-supported purification with a biotinylated U-II ligand (Bio-U-II), enabling the capture of stable ligand-receptor complexes.
Materials and Methods
Materials.
Cell culture and transfection reagents and Protein A agarose beads were purchased from Invitrogen (Mulgrave, VIC, Australia). The rat U-II and Bio-U-II peptides were synthesized by GL Biochem (Shanghai) Ltd. (Shanghai, China) Angiotensin II (Ang II) was synthesized by Auspep Pty Ltd. Peptides were iodinated with a 125I tracer (specific activity, ∼1000 Ci/mmol) by ProSearch International Australia (Malvern, VIC, Australia). Detergents, protease inhibitors, and the streptavidin resin, silanizing agent (Sigmacote) were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia). Goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP were purchased from Bio-Rad Laboratories (Gladesville, NSW, Australia). Rabbit polyclonal anti-ERK 2, rabbit polyclonal Gαq/11 and goat anti-rat IgG-HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Other antibodies used were mouse monoclonal anti-phospho-p44/42 MAPK (Thr202/Tyr204; Cell Signaling Technology, Danvers, MA), mouse monoclonal anti-HA antibody (12CA5), and rat monoclonal anti-HA High Affinity (3F10; Roche Applied Science, Castle Hill, NSW, Australia). Western blots were developed with Supersignal West Pico Chemiluminescent substrate (Pierce Chemical, Rockford, IL). The biotin assay kit was purchased from Biacore Life Sciences/GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK).
Organ Bath Studies.
Sprague-Dawley rats (240–400g) were anesthetized with 20% O2/80% CO2 and decapitated; the thoracic aorta was removed, placed in oxygenated Kreb's solution (118 mM NaCl, 4.5 mM KCl, 0.45 mM MgSO4, 1.03 mM KH2PO4, 25 mM NaHCO3, 11.1 mM glucose, and 2.5 mM CaCl2), cleared of connective tissue, and cut into 3-mm rings. The aortic rings were placed in organ baths containing 7 ml of Kreb's solution (37°C) bubbled with 95% O2/5% CO2 to maintain pH 7.4. The rings were stretched to a tension of 20 mN and equilibrated for 60 min. A reference contraction to 80 mM KCl was followed by treatment with either U-II (30 nM) or Ang II (100 nM). Changes in tension were recorded using a Grass FTO3 isometric transducer connected to a MacLab recording system.
Expression Plasmids.
The UT was cloned as described previously (Tzanidis et al., 2003). For efficient immunoprecipitation, three HA epitope tags were inserted after the start codon of the UT using standard polymerase chain reaction-based mutagenesis. All constructs were verified by direct DNA sequencing, and expression was monitored by radioligand binding and immunoprecipitation. Expression plasmids for the HA-tagged Ang II type 1A receptor (AT1A) were as described previously (Thomas et al., 1998). β-Arrestin constructs were kindly provided by Drs. R. J. Lefkowitz and M. G. Caron (Duke University, Durham, NC).
Rational Bio-U-II Design.
The UT binding pocket for the U-II peptide was modeled using bovine rhodopsin (PDB accession number 1U19) as a template. Epi-biotin complex with core streptavidin (PDB accession number 2F01) was used to view the binding pocket of streptavidin for biotin. Swiss-PdbViewer (ver. 3.7; http://spdbv.vital-it.ch/) was used for the rational design of Bio-U-II as outlined in Supplemental Tables 1 and 2 and Supplemental Fig. 1.
Cell Culture and Transfection.
HEK293, CHO-K1, or COS-1 cells were cultured in Dulbecco's modified Eagle's media (DMEM) supplemented with 10% fetal bovine serum and passaged with trypsin into twelve-well plates (150,000 cells/well) or onto 100-mm culture dishes (1.8 × 106 cells/dish). Cells were maintained at 37°C in air enriched with 5% CO2. Cells were transiently transfected with 0.6 μg of DNA per well (in a 12-well plate) using Lipofectamine reagent with expression vectors containing DNA upon reaching 70 to 80% confluence. For transfection in 100-mm culture dishes, volumes for all reagents were increased by a factor of 12. Transfection was stopped by replacing the transfection mixture with DMEM supplemented with 10% fetal bovine serum.
Receptor Phosphorylation.
Receptor immunoprecipitation and phosphorylation was performed as described previously for the AT1A (Thomas et al., 1998). 12CA5 anti-HA mouse monoclonal antibody was used for immunoprecipitation and 3F10 high affinity anti-HA rat monoclonal antibody was used for Western blotting.
Radioligand Binding Assays.
Binding experiments with 125I-U-II and 125I-Bio-U-II were carried out 48 h after transfection. Cells transfected with UT in 12-well plates were washed twice with phosphate-buffered saline (PBS), followed by binding 30 pM radioligand at room temperature for 5 h in U-II binding buffer (20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 10 mM d-glucose, and 0.1% bovine serum albumin). After binding, cells were washed three times with 1 ml of U-II binding buffer to remove unbound ligand and harvested with 1 ml of lysis buffer (0.25% SDS, 0.25 M NaOH). Nonspecific binding was determined in the presence of 1 μM unlabeled U-II.
Competitive binding assays were carried out on cells transfected with UT. Either U-II or Bio-U-II was added to samples at varying final concentrations (0–3 μM). Radioiodinated U-II (30 pM) was added to samples and incubated (5 h, 4°C). Cells were then washed with U-II binding buffer and harvested with lysis buffer.
Quantitative Internalization Assay.
Cells expressing UT or AT1A were washed twice with PBS (4°C) and incubated with receptor binding buffer containing 120 pM 125I-U-II (5 h, 4°C). Unbound 125I-U-II was removed by washing with ice-cold PBS. Warm receptor binding buffer (37°C) was added to cells and incubated at 37°C for specified times. Receptor internalization was terminated by washing with ice-cold PBS, and surface-bound 125I-U-II was stripped using acid wash solution (50 mM acetic acid and 150 mM NaCl) with two 30-min incubations. Internalized 125I-U-II was harvested using lysis buffer. Receptor internalization was calculated as the percentage of radioactivity in the cell lysate relative to the total counts. Data were corrected for nonspecific internalization determined in the absence of a 37°C incubation step.
Pseudo-Irreversible Binding Assay.
For acid wash experiments, cells were incubated with 100,000 cpm of either 125I-U-II, 125I-Bio-U-II, or 125I-Ang II for (5 h, 4°C) as described in the radioligand binding assay method. Acid wash solution was added to cells for 3 min, removed, and then retained for analysis. Several samples were subjected to a second 3-min acid wash to analyze further dissociation of the radioligand. Cells were then washed with U-II binding buffer and lysed with solubilization buffer to determine the level of radioligand associated with receptor.
Analysis of Radioligand Binding Data.
All radioligand binding experiments were repeated in triplicate, with each data point carried out in duplicate or triplicate. Radioactivity was quantified by gamma counting. Analysis of binding data were carried out with Prism 4.0 (GraphPad Software Inc., San Diego, CA).
Biotin Assay.
Accessibility of the biotin moiety within the Bio-U-II was determined using a Biacore 3000 instrument (Biacore Life Sciences/GE Healthcare). The format chosen was an indirect assay with anti-biotin antibodies and a biotin sensor chip. The effective concentration of the Bio-U-II peptide was determined by comparing the response to a calibration curve. The calibration curve was prepared with a 1:2 dilution of biotin in HBS-EP buffer [10 mM HEPES, 150 mM NaCl, 3.4 mM EDTA, and 0.005% (v/v) Tween 20; Biacore] with a constant amount of anti-biotin antibody (50–1.59 ng/ml) as provided in a biotin assay kit. Given that the quantification range of the biotin assay is 2.0 to 70 ng/ml, the concentration of biotin within Bio-U-II chosen for analysis was 25 ng/ml, and samples were prepared in a manner similar to that for the biotin calibration standards (i.e., diluted in HBS-EP buffer). A total of 70 μl of each sample was injected over the biotin chip at a rate of 40 μl/min. Responses were evaluated using GraphPad Prism 4.0 and analysis was corrected for purity of the ligand.
ERK1/2 Assay.
Transfected CHO-K1 cells cultured in a 12-well plate were serum starved with DMEM containing antibiotics 12 h before experimentation. Cells were stimulated with U-II or Bio-U-II of varying final concentrations (0–100 nM) and incubated for 10 min (37°C). After incubation, cells were washed twice with PBS (4°C) and lysed with radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2 mM EDTA, 50 mM NaF, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM sodium pyrophosphate, 1 μg/ml aprotinin, 5 μg/ml leupeptin, and 1 μg/ml pepstatin). Samples were centrifuged at 14,000g (15 min, 4°C), supernatants were retained and analyzed with SDS-PAGE followed by transfer. Western blotting was carried out using anti-phospho-p44/42 MAPK antibody and anti-mouse HRP-conjugated antibody. Western blots were then stripped and reprobed with anti-ERK 2, washed, and probed with anti-rabbit HRP-conjugated antibody.
Purification of the UT.
Membrane preparations were generated from cells cultured in six 100-mm dishes, transfected with UT, and serum-starved 16 h before experimentation. Cells were then washed twice with PBS (4°C). Hypotonic solution (50 mM HEPES, pH 7.5, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin, and 1 μg/ml pepstatin) was added to each plate and the cells lifted by scraping. Cells were pooled and homogenized using a Polytron PT3000 device (10,000 rpm, 4 × 5 s, 4°C; Kinematica AG, Littau-Lucerne, Switzerland). The homogenate was centrifuged at 800g (10 min, 4°C) with slow deceleration. The supernatant was decanted and was further centrifuged at 30,000g (30 min, 4°C) to separate the membrane pellet from the cytosolic fraction. The cytosolic fraction was decanted and the membrane pellet was resuspended in solubilization buffer without detergent (50 mM HEPES, pH 7.5, 250 mM NaCl, 2 mM EDTA, 1 mM Na3VO4, 1 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin, and 1 μg/ml pepstatin) and homogenized (10,000 rpm, 4 × 5 s, 4°C).
The crude membrane suspension was incubated in the presence or absence of 1 mM Bio-U-II with agitation (18 h, 4°C). Membranes were evenly aliquoted into presiliconized tubes containing solubilization buffer with detergent, making the final volume up to the desired concentration. Suspensions were solubilized by vortexing (four times for 5 s each, 4°C) followed by gentle agitation on an inverted rocker (20 min, 4°C). Samples were centrifuged at 1000g (5 min, 4°C) to separate solubilized material from the insoluble material.
The supernatant was incubated with streptavidin resin—these samples were placed on an inverted rocker (20 min, 4°C) to allow interaction between Bio-U-II and streptavidin. Samples containing streptavidin and receptor were centrifuged at 500g (2 min, 4°C) to separate captured receptors from the uncaptured flow-through.
Material that was not captured to the streptavidin resin was collected into tubes containing 20 μl of protein A agarose beads and 2 μg of anti-HA (12CA5) mouse hybridoma antibody for immunoprecipitation, and samples were incubated overnight with rocking (4°C). Samples were centrifuged at 8000g (1 min, 4°C), and the supernatant was removed. To the remaining beads, 1× urea-based sample buffer (63 mM Tris-HCl, pH 6.8, 2% SDS, 10% β-mercaptoethanol, 6 M urea, and 20% glycerol) was added to each sample to solubilize protein. Samples were then heated at 65°C for 15min.
Receptor material captured to the streptavidin resin was reduced in 1× urea-based sample buffer and heated (15 min, 65°C). The receptor protein within all samples (captured to the streptavidin resin and uncaptured material within the flow-through) were resolved through SDS-PAGE and silver-stained or Western-blotted using anti-HA high-affinity rat monoclonal antibody (3F10) and anti-rat HRP-conjugated antibody. For the identification of UT-Gαq/11 complexes, streptavidin-captured samples were analyzed by SDS-PAGE and Western blotted with rabbit anti-Gαq/11 antibody and anti-rabbit HRP-conjugated antibody.
Thermostability Testing of the UT.
The UT receptor was solubilized and captured as described under Purification of the UT. Samples of the streptavidin resin containing captured UT were then heated to 30, 37.5, 45, 52.5, or 60°C for 30 min. The heat-treated samples were then centrifuged at 500g (1 min) to separate captured receptors from dissociated ligand-receptor complexes. The streptavidin resin was then washed with solubilization buffer at the temperature corresponding to the specific heat treatment by buffer addition, centrifugation at 500g (1 min), and removal of the supernatant. The streptavidin-captured samples were analyzed by SDS-PAGE and Western blotted with rat anti-HA antibody.
Results
U-II Induces Sustained Vasoconstriction.
U-II reportedly binds to UT pseudo-irreversibly, resulting in unusually sustained cellular responses, leading to potent vasoconstriction (Douglas et al., 2000). To confirm this, we compared the vasoconstriction in thoracic aortic rings treated with U-II and Ang II, which both activate Gαq/11-coupled signaling pathways.
U-II induced vasoconstriction of rat aortic rings is slow to develop (maximal at 30 min, 273 ± 13% of 80 mM KCl) and prolonged, retaining 50% of maximum 2 h after stimulation (Fig. 1). This contrasts with the rapid and transient responses to Ang II, which activates the Gαq/11-coupled AT1A. Contraction to Ang II was maximal at 5 min (171 ± 17% of 80 mM KCl) and returned to near basal at 12 min (Fig. 1).

The contractile action of U-II is sustained. Rat aortic strips were exposed to U-II (30 nM) or Ang II (100 nM) and tension measured every 30 s and normalized to that of 80 mM KCl. The traces are averages (n = 4 for U-II, n = 2 for Ang II).
UT Is Resistant to Classic Desensitization Mechanisms.
We next examined the capacity of the UT to be phosphorylated and internalized after U-II stimulation. The rat UT possesses numerous potential phosphorylation sites—8 of 15 amino acids in the central region of the carboxyl terminus are serine/threonine residues, indicating that the UT might be regulated by canonical phosphorylation, arrestin binding and internalization (Oakley et al., 2001). A UT containing three HA tags at the N terminus was constructed to examine UT phosphorylation (note that introduction of three HA tags did not affect receptor expression, binding affinity, or U-II-mediated accumulation of inositol phosphates; data not shown).
Cells expressing either the UT or the AT1A (as a comparative control) were labeled with 32Pi and stimulated with ligands for the times indicated in Fig. 2. Stimulation of the UT for up to 60 min resulted in negligible phosphorylation of the receptor (Fig. 2A). This refraction to phosphorylation was also observed in HEK293 and COS-1 cells (data not shown), suggesting that the lack of receptor phosphorylation is not cell-specific. In contrast, the AT1A was robustly phosphorylated after Ang II stimulation (Fig. 2A).
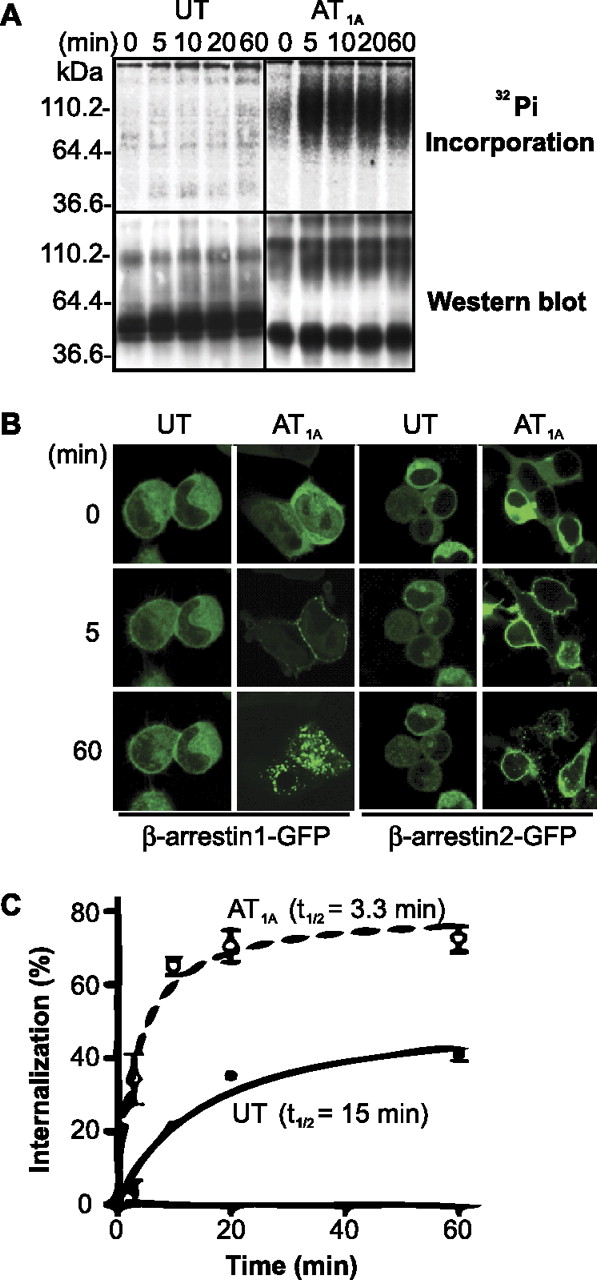
Regulation of the UT. A, CHO-K1 cells expressing UT or AT1A were assayed for phosphorylation, after incubation with U-II (100 nM) or Ang II (100 nM) for indicated times. A representative blot is shown. B, cellular trafficking of β-arrestins upon agonist stimulation. The trafficking β-arrestin-1/2-green fluorescent protein (β-arrestin-1/2-GFP) upon stimulation with agonist in HEK293 cells expressing UT or AT1A (as indicated) was followed using confocal microscopy. C, internalization was measured in CHO-K1 cells expressing UT (●, solid line) or AT1A (○, dashed line), loaded with radiolabeled agonist (5 h at 4°C) and incubated at 37°C for indicated times (n = 6).
Confocal microscopy was used to examine the interaction and trafficking of β-arrestin-1/2-green fluorescent protein in response to activation of UT expressed in HEK293 cells. The trafficking of β-arrestins after activation of the UT was very weak, and most arrestin protein remained cytoplasmic (Fig. 2B). Even after 60 min of U-II stimulation, some weak accumulation of β-arrestin-1/2 in membrane-localized pits was evident, but deep-core vesicles did not form (Fig. 2B). In contrast, β-arrestin-1/2 rapidly trafficked to agonist-activated AT1A with almost all cytoplasmic β-arrestins recruited to the cell membrane by 5 min and trafficked strongly with AT1A into deep-core vesicles by 60 min (Fig. 2B).
The kinetics of UT internalization was measured by determining acid-resistant binding of 125I-U-II to CHO-K1 cells expressing UT (Fig. 2C). Internalization of UT was slow (t1/2 = 15 min) reaching only 41 ± 2% at 60 min stimulation, whereas activated AT1A internalized rapidly (t1/2 = 3.3 min) and robustly (73 ± 4% at 20 min; Fig. 2C). This slower and weaker internalization was also observed in HEK293 cells expressing UT (data not shown).
The prolonged activity of U-II together with the resistance of UT to classic desensitization mechanisms (namely, phosphorylation, arrestin recruitment, and internalization) shows that the UT preferentially adopts an active conformation at the cell surface. These properties were then exploited for ligand-based purification to stabilize the active state of UT.
Bio-U-II Design.
The rat U-II peptide was modified to include an N-terminal biotin tag separated from the U-II sequence by a GSSG spacer arm. The length of the spacer arm was determined by modeling the key binding residues of the UT onto their analogous locations in the rhodopsin (PDB accession number 1U19) crystal structure (Supplemental Table 1 and Fig. 1). Calculations were based upon the distance required to clear both the receptor binding pocket and streptavidin binding pocket (PDB entry 2F01) – this ensures that a) the resin and receptor do not sterically clash and b) the modified ligand can provide a bridge between the receptor and resin. Based on these calculations (Supplemental Table 2), the rat U-II peptide was modified with three repeating GSSG peptide motifs and a biotin affinity group attached to an eight-carbon polylinker group (referred to as Bio-U-II) (Fig. 3). Details of the molecular modeling are more fully described in the supplemental data.

The Bio-U-II peptide. A biotin moiety was linked to the U-II peptide (green) via a spacer sequence.
Bio-U-II Interacts with Both Streptavidin and UT.
A surface plasmon resonance assay was used to investigate whether an anti-biotin antibody binds to Bio-U-II in the same way as free biotin. It was found that anti-biotin antibodies bind to Bio-U-II within ±3.0% of the expected value of free biotin—as corrected based on the purity of Bio-U-II (80.47%) (Fig. 4A), demonstrating that the biotin moiety is accessible to binding partners.
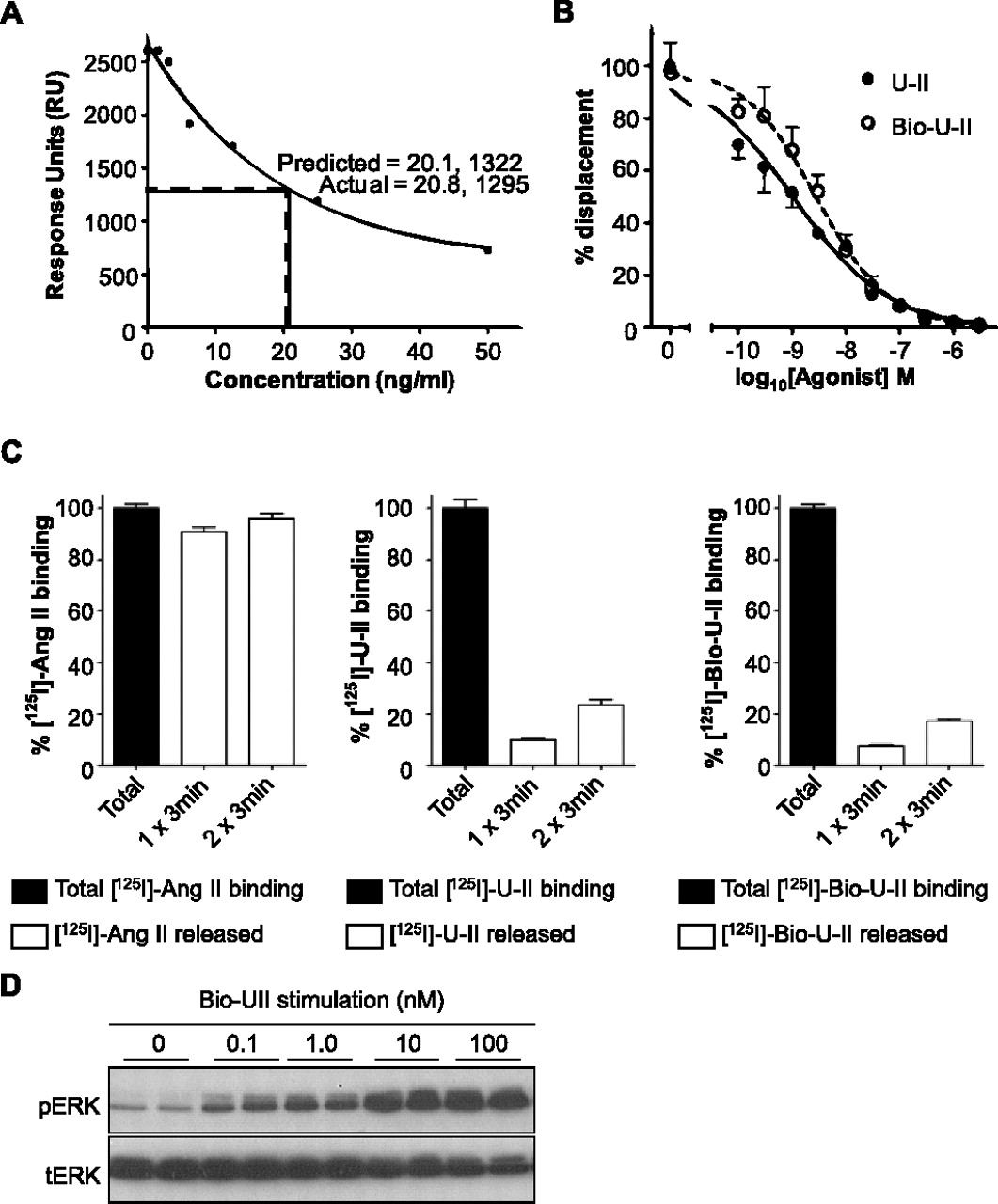
Characterization of Bio-U-II binding and stimulation. A, the biotin-streptavidin interaction was examined using Biacore analysis. A standard curve of known concentrations of biotin is shown. Two samples containing expected biotin concentrations of 25 ng/ml yielded 1295 response units, which corresponds to 20.8 ng/ml when corrected for the purity of Bio-U-II (80.5%). This shows that functionalized biotin (Bio-U-II) deviates by only 3% in recognition by anti-biotin antibody compared with free biotin. B, a competitive displacement assay was carried out to determine the affinity of Bio-U-II for the UT in comparison with U-II. The IC50 values of U-II displacing 125I-U-II and Bio-U-II displacing 125I-U-II were 1.0 and 2.8 nM, respectively; therefore, Bio-U-II has a similar ability to displace 125I-U-II. (n = 3). C, an acid strip protocol (once for 3 min or twice for 3 min each) was carried out on HEK293 cells expressing either the UT or AT1A in which 125I-U-II, 125I-Bio-U-II, or 125I-Ang II was bound. Control cells were not subjected to acid treatment to represent 100% of ligand binding. Binding of 125I-U-II and 125I-Bio-U-II to UT was resistant to washout, whereas acid treatment of AT1A led to almost complete removal of 125I-Ang II from the cell surface (n = 3). D, Bio-U-II dose dependently stimulated UT to promote ERK1/2 phosphorylation (pERK); tERK is total ERK as a loading control.
U-II and Bio-U-II Have Similar Functionality.
Binding assays were carried out to ensure that Bio-U-II retains capacity to bind to UT expressed in HEK293 cells. The affinity of Bio-U-II was characterized through its ability to competitively displace a 125I-U-II radiolabel in comparison with unlabeled U-II. The addition of U-II displaced the specific binding of 125I-U-II with an IC50 value of 1.0 nM, whereas unlabeled Bio-U-II displaced 125I-U-II with an IC50 value of 2.8 nM (Fig. 4B). Thus, the affinities of U-II and Bio-U-II for the UT were comparable.
For successful ligand-supported purification, it is essential that Bio-U-II retains the ability to bind UT pseudo-irreversibly (Douglas et al., 2000). The pseudo-irreversible binding nature of both the U-II and Bio-U-II was tested by the ability of an acid solution to strip the ligand from its binding site (Fig. 4C). After binding radiolabeled 125I-U-II and 125I-Bio-U-II, a 3-min acid wash removed only 10 and 8% of the ligand, respectively (Fig. 4C). Even when cells were subjected to a second 3-min acid wash, only 24 and 17% of the total available ligand was removed. In contrast, binding 125I-Ang II to AT1A was removed to near completion by an acid wash under the same conditions (91% removal after the first acid wash and 96% removal upon two 3-min washes) (Fig. 4C). These results show that, unlike Ang II, Bio-U-II is able to bind strongly and specifically to UT, and Bio-U-II demonstrates similar pseudo-irreversible binding behavior compared with the parent U-II peptide.
Confirmation of receptor activation by Bio-U-II was tested using phosphorylation of ERK1/2. Stimulation of UT-expressing CHO-K1 cells with the Bio-U-II peptide at 0.1 nM was sufficient to elicit phosphorylation of ERK1/2 (Fig. 4D), indicating that Bio-U-II remains a potent agonist.
Purification of the UT with Detergents.
To test the concept of ligand-supported purification with Bio-U-II, five representative detergents were screened for the purification of the UT. The detergents used for screening were: cetyltrimethylammonium bromide (cationic), sodium cholate (anionic), dodecyl maltoside (DDM) (nonionic;alkyl glucoside), digitonin (nonionic; steroid derivative), and CHAPS (zwitterionic).
Figure 5 shows the extraction of the UT using prebound Bio-U-II at 10 times the critical micelle concentration (CMC) for a panel of detergents. The UT-Bio-U-II complex was captured onto a streptavidin matrix and analyzed by Western blotting for the HA epitope tag on the receptor. Figure 5A illustrates insoluble material after detergent “solubilization” and shows that different detergents had varying solubilization efficiencies for the UT. Figure 5B shows that, of the representative detergents screened, DDM was the only detergent that allowed solubilization and specific capture of the UT to the streptavidin resin. As demonstrated in Fig. 5C, a significant amount of solubilized UT remained uncaptured at this CMC and was detected in the flow-through and analyzed by immunoprecipitation of the HA epitope tag.

Solubilization and purification of UT using Bio-U-II and a variety of detergents. The UT was prebound to Bio-U-II (+) or without Bio-U-II (−). Report points were taken at the solubilization and capture stages; immunoprecipitation was used to detect the uncaptured flowthrough. A, insoluble material; sodium cholate and CHAPS were efficient solubilizers. In contrast, cetyltrimethylammonium bromide (CTAB), DDM, and digitonin seem to be less effective. B, solubilized material captured by streptavidin resin; DDM allows specific capture of UT using Bio-U-II, whereas without ligand (DDM/−) yielded no detectable receptor. Other detergents screened did not allow specific capture of the receptor. C, immunoprecipitated flow-through (not captured by the resin); note that all detergents were used at 10× above the CMC, except CHAPS (2× CMC).
The CMC dependence of purification was further investigated with prebinding of Bio-U-II with DDM at 2, 5 and 10 times the CMC (Fig. 6). Ligand supported purification of the UT was found to be successful at all three detergent concentrations but was most effective at 5 times the CMC (Fig. 6).
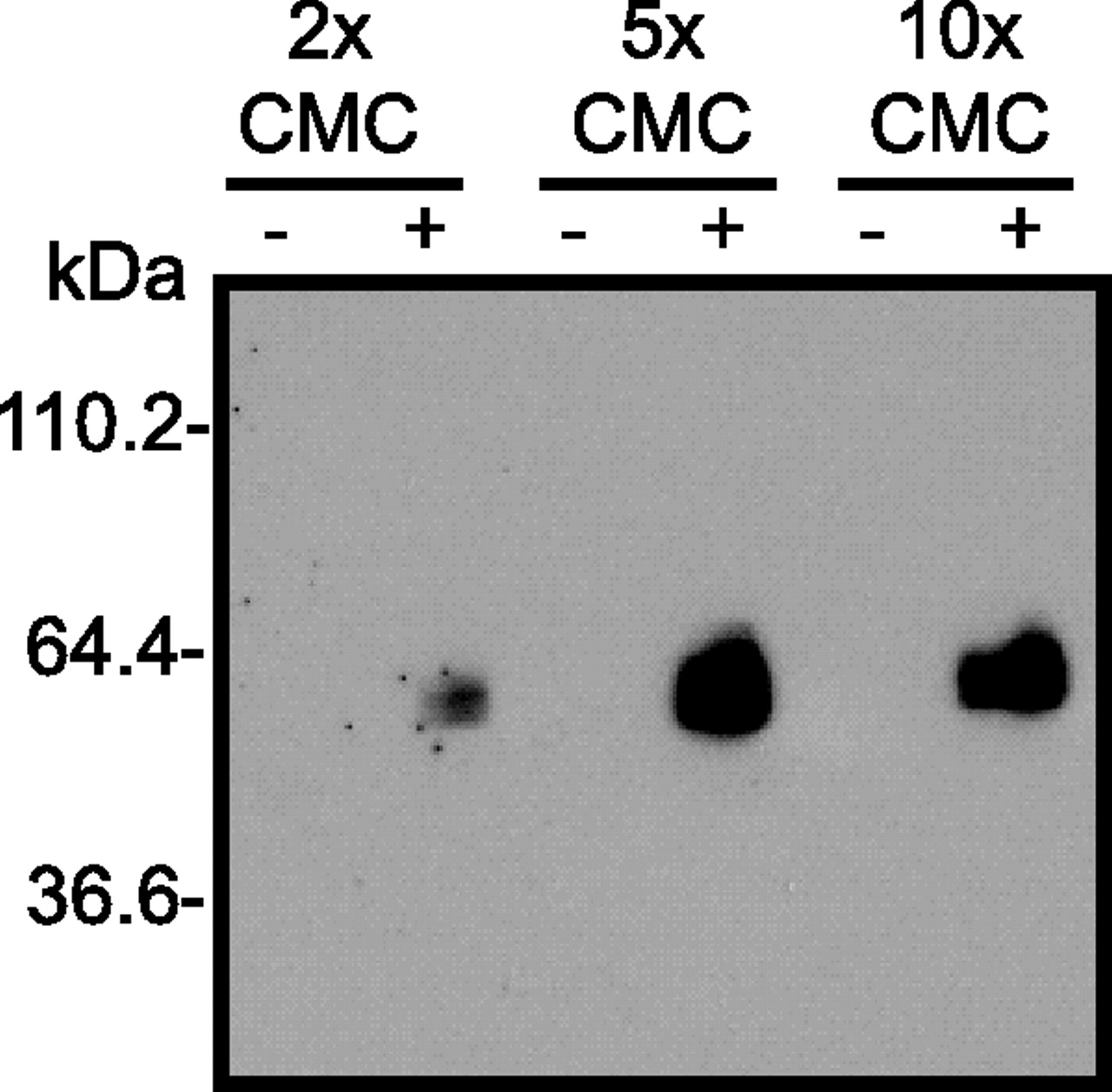
Extraction efficiency of DDM was tested at 2, 5, and 10 times above the CMC. UT was prebound to Bio-U-II (+) or unbound (−) and captured on streptavidin beads at different CMC values.
An experiment comparing prebinding of Bio-U-II against presolubilization was carried out to determine the effectiveness of presolubilization during ligand-supported purification (Fig. 7). It was found that prebinding produced greater yields of UT compared with presolubilization (lanes PB and PS; Fig. 7A) for 5× CMC DDM. Furthermore, a single wash of the streptavidin beads with captured receptors using different DDM concentrations (2×, 5×, and 10× CMC) did not affect purification in prebound or presolubilized samples. Once it was established that the ligand-receptor complex was not susceptible to dissociation after one wash, multiple washes were incorporated at 5× CMC of DDM. After three washes, ligand-receptor complexes dissociated somewhat in samples prepared by presolubilization (Fig. 7A, lanes denoted PS), whereas prebound samples were unaffected (Fig. 7A; see lanes denoted PB). Under the optimized conditions, we estimate that approximately one third of the solubilized receptor can be captured on the resin and that this can be efficiently separated from the nonbound receptor by extensive washing (Fig. 7B).

Testing the stability of Bio-U-II-UT complexes. Dissociation of the Bio-U-II/UT complex was monitored upon multiple washes of the streptavidin beads with 5xCMC DDM (NL, no ligand; PB, prebound ligand; PS, presolubilized receptor). A, UT captured by streptavidin using Bio-UII: PB samples were unaffected by washing whereas PS Bio-U-II-UT complexes dissociated upon extended washing. B, immunoprecipitated flow-through (not captured by resin): UT was present in the flow-through when the resin was not washed. Upon washing, small amounts of UT were released, and smaller amounts were released with each subsequent wash. C, samples were silver-stained (left), the protein bands in PB samples were more intense than those in the NL control. The corresponding Western blot (right) is a molecular mass comparison and shows the capture of the UT to the streptavidin resin. D, Gαq/11 (42 kDa) was copurified with Bio-U-II-UT complexes in PB samples that were washed with 5×CMC DDM as detected by Western blotting.
Figure 7C shows a silver-stained SDS-PAGE gel of prebound UT or control samples (washed three times with 5× CMC DDM). There was a significant enrichment of the protein bands in the prebound samples compared with those in the NL control (Fig. 7C, right). Because the UT was released from the streptavidin resin through boiling instead of elution, both samples show contaminating proteins, which were nonspecifically released throughout. The corresponding Western blot for both samples has been included for molecular weight comparison (Fig. 7C, left).
Gαq/11 Protein Remains Associated to Captured UT-Bio-U-II Complexes.
Prebound UT samples captured to the streptavidin resin were probed with anti-Gαq/11 antibody to determine whether Gαq/11 was copurified with the UT (Fig. 7D). After three washes with 5× CMC DDM, Gαq/11 was found to be complexed to UT-Bio-U-II (Fig. 7D, lane PB). In contrast, Gαq/11 was not detected in control samples that were not captured with Bio-U-II (Fig. 7D).
Thermostability of the UT.
Figure 8 demonstrates the capture of the UT to streptavidin resin followed by incubation at different temperatures (4, 30, 37.5, 45, 52.5, or 60°C) for 30 min. At 4°C (standard purification conditions), sample capture was greatest; even at higher temperatures (30–60°C), the amount of UT captured to streptavidin was maintained and only slightly reduced.

UT purified by ligand supported purification is thermostable. The effect of temperature on the interaction between the prebound (PB) Bio-U-II ligand and UT captured on the streptavidin matrix. A no-ligand control (NL) is also shown for each temperature.
Discussion
The inherent difficulties in purifying active R* conformations of GPCRs prompted the question of whether a ligand-based approach can yield purified R* GPCRs. Here, we introduce a biotin-tagged version of the peptide hormone U-II that allows ligand supported purification of its cognate GPCR, the UT. Our approach to ligand-supported purification involved a ligand that was modified for affinity chromatography without markedly perturbing receptor binding or activation and exploited essentially irreversible receptor/ligand interaction. These features are associated with a number of GPCR/ligand pairings, such as the relaxin RXPF1/2 (Callander et al., 2009) and endothelin ETA receptors (Waggoner et al., 1992), making them ideal candidates for ligand supported purification. If possible, the receptor should have a stable, yet restricted, set of conformers, resistance to the regulatory processes of phosphorylation, and β-arrestin binding and internalization.
Although UT desensitization has been a contentious issue (Giebing et al., 2005; Proulx et al., 2005, 2008), we show here that the UT is refractory to classic GPCR desensitization mechanisms and propose that this relates to its sustained contractile actions. The binding of U-II to the UT seems to induce a receptor conformation capable of activating Gαq/11, which is not readily phosphorylated or bound by β-arrestins. Furthermore, we found UT to have a relatively long internalization half-life (t1/2 = 15 min), which is similar to that previously reported for COS-7 cells (Proulx et al., 2005). For U-II, the initial interaction with the receptor presumably promotes a structure that activates Gαq/11 but does not permit subsequent docking events that promote additional conformation(s) for phosphorylation and desensitization. This may reflect the strong, pseudo-irreversible nature of U-II binding (Douglas et al., 2000). These properties are ideal for ligand supported purification because they show that agonist-stimulated UT preferentially adopts an active R* conformation that is not readily removed from the cell surface.
Two different approaches were investigated for purifying the UT with Bio-U-II. 1) Solubilizing the UT with detergents followed by purification through binding with the affinity ligand, or 2) prebinding the Bio-U-II and then solubilizing the UT with detergents. The rationale for copurifying GPCRs with detergents and a prebound ligand is that the ligand places the receptor in the active state and may remain bound during purification.
GPCRs have been previously purified with presolubilization, either by immobilizing the ligand to a solid support (Couvineau et al., 1990; Hazum, 1990) or by binding the ligand to the solubilized receptor (Hagiwara et al., 1992; Ohtaki et al., 1998; Santos-Alvarez and Sánchez-Margalet, 2000). Few studies have used prebinding during ligand-supported purification with a biotinylated ligand (Desarnaud et al., 1992; Eppler et al., 1992; Brown and Schonbrunn, 1993; Zysk et al., 1996), although, in each case, the authors have demonstrated that prebinding is superior to presolubilization (Desarnaud et al., 1992; Eppler et al., 1992; Brown and Schonbrunn, 1993; Zysk et al., 1996). Indeed, the results presented here demonstrate that prebinding with Bio-U-II produced a greater yield of UT compared with presolubilization with Bio-U-II. Furthermore, prebound complexes seem to be more stable, being more resistant to detergent effects upon washing. It has been suggested that poor recovery of functional receptors upon purification is due either to denaturation upon solubilization (Brown and Schonbrunn, 1993) or the dissociation of the GPCR-G protein complex (Eppler et al., 1992; Brown and Schonbrunn, 1993)—both possibilities lead to reduced yield and produce unstable receptors or low-affinity receptors (Eppler et al., 1992; Brown and Schonbrunn, 1993). Indeed, in the case of somatostatin, prebinding of the affinity ligand was required because of the instability and inactivation of somatostatin upon detergent purification (Eppler et al., 1992; Brown and Schonbrunn, 1993).
Evidence that G proteins may be copurified with GPCRs using ligand-supported purification has been suggested to indicate that the purified receptor adopts an active R* conformation (Eppler et al., 1992; Brown and Schonbrunn, 1993; Zysk et al., 1996; Santos-Alvarez and Sánchez-Margalet, 2000). Brown and Schonbrunn (1993) showed that somatostatin remains sensitive to guanine nucleotides and exploited the cocapture of G proteins using GDP to dissociate receptor-ligand complexes during elution (causing a notable decrease in agonist affinity), which purified the receptor in an inactive conformation (Brown and Schonbrunn, 1993). We show that Gαq/11 protein is cocaptured with UT-Bio-U-II complexes suggesting that the R* state has been isolated. However, we do not rule out the possibility that the Gαq/11 protein is bound to an intermediary form of UT that is separate from its active R* conformation (i.e., capable of signaling). Indeed, studies using fluorescence or bioluminescence resonance energy transfer show compelling evidence that precoupled GPCR-G protein complexes exist in the absence of agonist stimulation (Nobles et al., 2005; Galés et al., 2006). Hence, further testing is required to more fully determine whether the UT is in an active R* conformation or if Gαq/11 is precoupled to UT.
The UT-U-II system is interesting because of the duration and magnitude of U-II-mediated responses as well as the atypical regulation of UT—indicating that it may be conformationally restricted compared with other GPCRs of known structure [rhodopsin (structures reviewed in Palczewski et al., 2000; Teller et al., 2001; Topiol and Sabio, 2009)], β1/2-adrenergic (Cherezov et al., 2007; Warne et al., 2008), and A2A adenosine receptors (Jaakola et al., 2008). Indeed, there is only 19% sequence homology between rat UT and bovine rhodopsin, making a structure of UT of broad interest to GPCR structural biologists and modelers. It is noteworthy that these crystallized GPCRs are in an inactive (R) conformation.
Structural and biophysical studies of GPCRs are a challenge because of their instability upon detergent extraction. Recent GPCR crystallography successes has been attributed to significant protein engineering such as C-terminal truncation (Cherezov et al., 2007; Jaakola et al., 2008; Murakami and Kouyama, 2008; Park et al., 2008; Warne et al., 2008), fusion protein generation (Cherezov et al., 2007; Rasmussen et al., 2007; Rosenbaum et al., 2007; Jaakola et al., 2008), and introduction of thermostabilizing mutations (Standfuss et al., 2007; Warne et al., 2008). However, these changes are not representative of a wild-type receptor in an active conformation. C-terminal truncations and formation of fusion proteins act to remove what are believed to be highly flexible regions of GPCRs that may impede the crystallization process, whereas thermo-stabilizing mutations improve receptor stability in detergents by potentially locking the GPCR in a single conformation.
In the case of the β1-adrenergic receptor (Magnani et al., 2008; Serrano-Vega et al., 2008; Warne et al., 2008) and adenosine A2a receptor (Magnani et al., 2008; Serrano-Vega et al., 2008), alanine-scanning mutagenesis was conducted on the full-length protein to identify amino acid residues that altered the receptor's thermostability as determined by heating the GPCR followed by radioligand binding (Magnani et al., 2008; Serrano-Vega et al., 2008; Warne et al., 2008). It is noteworthy that Serrano-Vega et al. (2008) showed that preincubation of an agonist with subsequent detergent-solubilization of the thermostable β1-adrenergic receptor increased the melting temperature by 23°C compared with non–ligand-bound wild-type receptor (Magnani et al., 2008; Serrano-Vega et al., 2008; Warne et al., 2008).
In a similar approach, we captured UT to streptavidin using a prebound ligand and examined the thermostability as a measure of UT retention on the resin. Although there is a reduction (∼50%) in specific capture of Bio-U-II-UT between 4°C and the tested temperatures, UT was strongly retained on the streptavidin resin between 30 and 60°C. The resistance to ligand-receptor dissociation at these temperatures suggests that UT has good thermostable properties when prebound to Bio-U-II. This may be attributed to four factors: 1) the inherent high stability of the receptor-ligand interaction, which forms a pseudo-irreversible complex and remains active for extended periods; 2) the selection of a stable conformation of UT; 3) a naturally compact intracellular third loop; and 4) the retention of G protein on the receptor, which may contribute to global as well as localized stability. For these reasons, the application of ligand-supported purification to the UT provides an exciting possibility to study a GPCR system that does not need to be modified to obtain structural information on the active conformation.
In conclusion, we report the first successful purification of the UT; it was physically associated with Gαq/11 protein. Although ligand-supported purification can be applied to other GPCRs for protein stabilization during detergent extraction and potentially tailored for a wider range of GPCRs [such as the relaxin RXPF1 and RXPF2 (Callander et al., 2009) and endothelin ETA (Waggoner et al., 1992) receptors, which also display pseudo-irreversible binding], we attribute appreciable yields of UT to the long-lasting, pseudo-irreversible binding of U-II, the resistance of UT to classic desensitization mechanisms, and selection of a stable receptor conformation. Using this study as a basis, we have embarked upon scaled expression and purification of the UT to allow large-scale production of purified UT in the active R* state for biophysical and structural studies.
Acknowledgments
β-Arrestins were kindly provided by Drs. R. J. Lefkowitz and M. G. Caron. The use of animal tissue was approved by the University of Melbourne Animal Experiment Ethics Committee.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by National Health and Medical Research Council of Australia [Grants 268925, 251646, 334049] and by a Ph.D. scholarship from the Baker Heart Research Institute (to D.O.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065151.
-
ABBREVIATIONS:
- GPCR
- G protein-coupled receptor
- U-II
- urotensin-II
- UT
- U-II receptor
- Bio-U-II
- biotinylated U-II
- HRP
- horseradish peroxidase
- Ang II
- angiotensin II
- HA
- hemagglutinin
- AT1A
- type 1A angiotensin receptor
- PDB
- Protein Data Bank
- HEK
- human embryonic kidney
- CHO
- Chinese hamster ovary
- DMEM
- Dulbecco's modified Eagle's medium
- PBS
- phosphate-buffered saline
- ERK
- extracellular signal-regulated kinase
- PAGE
- polyacrylamide gel electrophoresis
- DDM
- n-dodecyl β-d-maltoside
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate
- CMC
- critical micelle concentration.

- Received March 30, 2010.
- Accepted July 20, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}