


Abstract
The peripheral δ opioid receptor (DOR) is an attractive target for analgesic drug development. There is evidence that DOR can form heteromers with the κ-opioid receptor (KOR). As drug targets, heteromeric receptors offer an additional level of selectivity and, because of allosteric interactions between protomers, functionality. Here we report that selective KOR antagonists differentially altered the potency and/or efficacy of DOR agonists in primary cultures of adult rat peripheral sensory neurons and in a rat behavioral model of thermal allodynia. In vitro, the KOR antagonist nor-binaltorphimine (nor-BNI) enhanced the potency of [d-Pen2,5]-enkephalin (DPDPE), decreased the potency of [d-Ala2,d-Leu5]-enkephalin (DADLE), and decreased the potency and efficacy of 4-[(R)-[(2S,5R)-4-allyl-2,5-dimethylpiperazin-1-yl](3-methoxyphenyl)methyl]-N,N-diethylbenzamide (SNC80) to inhibit prostaglandin E2 (PGE2)-stimulated adenylyl cyclase activity. In vivo, nor-BNI enhanced the effect of DPDPE and decreased the effect of SNC80 to inhibit PGE2-stimulated thermal allodynia. In contrast to nor-BNI, the KOR antagonist 5′-guanidinonaltrindole (5′-GNTI) reduced the response of DPDPE both in cultured neurons and in vivo. Evidence for DOR-KOR heteromers in peripheral sensory neurons included coimmunoprecipitation of DOR with KOR, a DOR-KOR heteromer selective antibody augmented the antinociceptive effect of DPDPE in vivo, and the DOR-KOR heteromer agonist 6′-GNTI inhibited adenylyl cyclase activity in vitro as well as PGE2-stimulated thermal allodynia in vivo. Taken together, these data suggest that DOR-KOR heteromers exist in rat primary sensory neurons and that KOR antagonists can act as modulators of DOR agonist responses most likely through allosteric interactions between the protomers of the DOR-KOR heteromer.
Introduction
Management of pain by opioid analgesics is confounded by central adverse effects that limit clinical dosages and treatment paradigms. Consequently, increased attention has been given to analgesia mediated by peripheral opioid receptors. Opioid receptors are expressed in peripheral primary sensory neurons that transduce pain information (nociceptors). It is noteworthy that peripherally restricted opioids generally do not elicit an analgesic response when administered to normal tissue but can produce antinociception when administered to injured or inflamed tissue (Ferreira and Nakamura, 1979; Stein et al., 1989). This finding suggests that some stimulus from the inflamed tissue interacts with opioid receptor systems in nociceptors to make them functionally competent to inhibit nociceptor signaling.
We have shown previously that functional competence of rat peripheral opioid receptor systems can be induced by brief pretreatment with inflammatory mediators, such as bradykinin (BK). When applied locally to the rat hind paw (intraplantar), the δ-opioid receptor (DOR) agonist [d-Pen2,5]-enkephalin (DPDPE), does not alter PGE2-induced thermal allodynia. However, when administered 15 min after local injection of BK, DPDPE produces a profound antiallodynic response (Rowan et al., 2009). Likewise, in primary sensory neuronal cultures of adult rat trigeminal ganglion (TG), the DOR agonists DPDPE and [d-Ala2,d-Leu5]-enkephalin (DADLE) are ineffective at reducing PGE2-stimulated cAMP accumulation or BK/PGE2-stimulated neuropeptide release. However, after brief (15 min) pretreatment with BK (or other activators of Gq-mediated signaling), DOR agonists become capable of inhibiting adenylyl cyclase activity and neuropeptide release (Patwardhan et al., 2005, 2006). We found similar effects of BK on induction of functional competence to inhibit adenylyl cyclase activity and neuropeptide release by activation of the μ-opioid receptor (MOR) system (Berg et al., 2007a,b) as well as for the κ-opioid receptor (KOR) system (Berg et al., 2011). Several studies have demonstrated that DOR and KOR can form heteromers in heterologous expression systems (Jordan and Devi, 1999; Waldhoer et al., 2005; Xie et al., 2005). Heteromers offer intriguing possibilities for drug development in that heteromer-selective ligands would be expected to have greater tissue specificity; such drugs would be effective only in tissues that coexpress the heteromer receptor pairs. It is noteworthy that 6′-guanidinonaltrindole (6′-GNTI), originally developed as a KOR agonist (Sharma et al., 2001), seems to have selective DOR/KOR heteromer agonist properties (Waldhoer et al., 2005). 6′-GNTI does not activate DOR and has only weak efficacy at KOR when expressed individually in HEK cells. However, it is a potent and efficacious agonist when both receptors are coexpressed, and its effects can be fully blocked by occupancy of DOR with the antagonist naltrindole or KOR with the antagonist nor-binaltorphimine (nor-BNI). 6′-GNTI has been shown to produce analgesia when administered into the spinal cord, but not the brain, supporting the notion that heteromer-selective ligands will provide greater tissue specificity of action (Waldhoer et al., 2005).
An interesting property of heteromers with respect to drug development is the potential for allosteric interactions between the protomers (Fuxe et al., 2010; Smith and Milligan, 2010; Keov et al., 2011). The interaction between two protomers of a heteromeric pair could influence the affinity, efficacy, or both of a ligand for one of the protomers. For example, the potency of orexin A to promote activation of extracellular signal-regulated kinase in Chinese hamster ovary cells was increased 100-fold by the presence of the CB1 receptor (Hilairet et al., 2003). Ligand binding to the orthosteric site of one protomer of a heteromeric pair can alter the function of a second ligand that binds to the orthosteric site of the second protomer of the pair. With respect to DOR-KOR heteromers expressed in HEK-293 cells, the DOR-selective antagonist, naltrindole, increased the binding of the KOR-selective antagonist, nor-BNI, and vice versa (Xie et al., 2005). Furthermore, Han et al. (2009) recently showed that the conformational state of one protomer of a dopamine D2 receptor heteromer could alter agonist efficacy at the second protomer.
An understanding of the allosteric interactions between protomers of heteromeric pairs of receptors may lead to the development of new drugs with improved efficacy and specificity for treatment of disease. However, although heteromers have been well studied using heterologous expression systems, little is known of the role of heteromers in vivo. Here we studied the effect of KOR antagonist ligands on the function of DOR agonist ligands in a behavioral model of inflammatory peripheral pain (PGE2-induced thermal allodynia) and in a primary cell culture model of nociceptor function.
Materials and Methods
Materials.
The following compounds were purchased from Sigma-Aldrich (St. Louis, MO): DPDPE, DAMGO, DADLE, SNC80, naltrindole, 5′-guanidinonaltrindole (5′-GNTI), 6′-GNTI, and nor-binaltorphimine (nor-BNI). PGE2 was purchased from Cayman Chemicals (Ann Arbor, MI). 125I-cAMP was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Collagenase was from Worthington (Lakewood, NJ). Fetal bovine serum and all other tissue culture reagents were purchased from Invitrogen Corp (Carlsbad, CA). All other drugs and chemicals (reagent grade) were purchased from Sigma-Aldrich.
Animals.
Adult male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 250 to 300 g were used in this study. The animal study protocol was approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio and conformed to International Association for the Study of Pain and federal guidelines. Animals were housed for 1 week with food and water available ad libitum before behavioral testing or harvesting of TG cells.
Rat Trigeminal Ganglia Culture.
Primary cultures of rat TG cells were prepared as described previously (Patwardhan et al., 2005, 2006; Berg et al., 2007a,b). In brief, fresh TG were washed with Hanks' balanced salt solution (Ca2+,Mg2+-free), digested with 3 mg/ml collagenase for 30 min at 37°C, and centrifuged (1000 rpm for 1 min). The pellet was further digested with 0.1% trypsin (15 min) and 167 μg/ml DNase (10 min) at 37°C in the same solution. Cells were pelleted by centrifugation (2 min at 2000 rpm) and resuspended in Dulbecco's modified Eagle's medium (high glucose) containing 100 ng/ml nerve growth factor (Harlan, Indianapolis, IN), 10% fetal bovine serum, 50 U/ml penicillin/50 μg/ml streptomycin, 1× l-glutamine, and the mitotic inhibitors 7.5 μg/ml uridine and 17.5 mg/ml 5-fluoro-2′-deoxyuridine. After trituration to disrupt tissue, the cell suspension was seeded on polylysine-coated 48-well or 10-cm plates. Cells were pooled from three rats to seed 48-well plates, and cells pooled from six rats were used to seed 10-cm plates. Media was changed 24 and 48 h after plating. On day 5 of culture, cells were refed with serum-free Dulbecco's modified Eagle's medium without nerve growth factor. Cells were used on day 6 of culture.
Coimmunoprecipitation.
The ability of DOR to coimmunoprecipitate with KOR was determined using the Cross-link IP kit according to the manufacturer's directions (Thermo Fisher Scientific, Waltham, MA) followed by Western blot analysis. In brief, 4 × 10-cm plates of cells were subjected to cell surface cross-linking with membrane insoluble bis[sulfosuccinimidyl]suberate (1 mM; Pierce) for 30 min at room temperature followed by lysis with 0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% Nonidet P-40, and 5% glycerol, pH 7.4. Lysed material was applied to a spin column (Thermo Fisher Scientific) containing anti-KOR antibody (10 μg; H70; Santa Cruz Biotechnology, Santa Cruz, CA) covalently immobilized to protein A/G agarose beads and incubated overnight at 4°C. After centrifugation, samples (20 μl; 500 μg) were resolved on NuPAGE 4 to 12% SDS-polyacrylamide gradient gels (Invitrogen), and transferred to polyvinyl difluoride (PVDF) membrane using the iBlot transfer system (Invitrogen). Western blots were blocked in blocking buffer (1 h, 23°C; Odyssey; LI-COR Biosciences, Lincoln, NE) and incubated overnight with anti-DOR (1:200; Neuromics) or anti-KOR (1:500; Santa Cruz Biotechnology) antibody, followed by the Goat anti-rabbit IR 800 secondary antibody (1:10,000; IRDye 800CW; LI-COR Biosciences). Samples from rat liver (500 μg), which does not contain KOR, and elution buffer only were applied to spin columns (Thermo Fisher Scientific) containing anti-KOR antibody as control. In addition, samples from TG (500 μg) were applied to spin columns without the anti-KOR antibody. Images were obtained and analyzed with an Odyssey Infrared Western blot imager (LI-COR Biosciences).
Measurement of Cellular cAMP Levels.
Opioid receptor-mediated inhibition of adenylyl cyclase activity was determined by measuring the amount of cAMP accumulated in the presence of the phosphodiesterase inhibitor, rolipram, and the adenylyl cyclase activator PGE2. Cultures in 48-well plates were washed twice with Hanks' balanced salt solution containing 20 mM HEPES, pH 7.4 (wash buffer). Cells were preincubated in 250 μl of wash buffer per well for 15 min at 37°C (room air) with or without BK (10 μM). To assess opioid agonist-mediated responses, cells were incubated with rolipram (0.1 mM) along with opioid receptor ligands in triplicate for 15 min at 37°C, followed by addition of a maximal concentration of PGE2 (1 μM) and incubation for a further 15 min. Incubations were terminated by aspiration of the buffer and addition of 500 μl of ice-cold absolute ethanol. The ethanol extracts from individual wells were dried under a gentle air stream and reconstituted in 100 μl of 50 mM sodium acetate, pH 6.2. The cAMP content of each well was determined by radioimmunoassay.
Behavior Assay.
Opioid agonist-mediated changes in paw withdrawal latency (PWL) to a thermal stimulus were measured with a plantar test apparatus (Hargreaves et al., 1988) as described previously (Rowan et al., 2009). In brief, rats were placed in plastic boxes with a glass floor maintained at 30°C. After a 30-min habituation period, the plantar surface of the hind paw was exposed to a narrow beam of radiant heat through the glass floor. The intensity of the thermal stimulus was adjusted so that baseline PWL values were close to 10 s; cut-off time was 25 s. All drugs were dissolved in phosphate-buffered saline and administered via intraplantar injection (50 μl) into the rat hind paw. To induce functional competence of the opioid receptors (Patwardhan et al., 2005; Berg et al., 2007a; Rowan et al., 2009), BK (25 μg or Veh) was administered via intraplantar injection 15 min before injection of PGE2 with or without a DOR agonist. When indicated, KOR antagonists were administered 15 min before agonist (with BK). The PWL measurements were taken in duplicate at least 30 s apart at 5-min intervals continuing until 20 min after the last injection, and the average was considered for statistical analysis. Observers were blinded to the treatment allocation.
Data Analysis.
For TG cell culture experiments, concentration-response data were fit to a logistic equation (eq. 1) using nonlinear regression analysis to provide estimates of maximal response (Rmax), potency (EC50) and slope factor (n).
 where R is the measured response at a given agonist concentration (A), Ro is the response in the absence of agonist, Ri is the response after maximal inhibition by the agonist, EC50 is the concentration of agonist that produces half-maximal response, and n is the slope factor. Rmax (the maximal inhibition produced by the agonist) was calculated as Ro − Ri. Experiments were repeated at least four times, in triplicate, using cells obtained from different groups of rats. Statistical differences in concentration-response curve parameters between groups were analyzed with Student's paired t test. When only a single concentration was used, statistical significance was assessed using one-way analysis of variance followed by Dunnet's post hoc or Student's t test (paired) using Prism software (Graphpad Software, Inc., San Diego, CA). p < 0.05 was considered statistically significant.
where R is the measured response at a given agonist concentration (A), Ro is the response in the absence of agonist, Ri is the response after maximal inhibition by the agonist, EC50 is the concentration of agonist that produces half-maximal response, and n is the slope factor. Rmax (the maximal inhibition produced by the agonist) was calculated as Ro − Ri. Experiments were repeated at least four times, in triplicate, using cells obtained from different groups of rats. Statistical differences in concentration-response curve parameters between groups were analyzed with Student's paired t test. When only a single concentration was used, statistical significance was assessed using one-way analysis of variance followed by Dunnet's post hoc or Student's t test (paired) using Prism software (Graphpad Software, Inc., San Diego, CA). p < 0.05 was considered statistically significant.
For behavioral experiments, time course data were analyzed with two-way analysis of variance, followed by Bonferroni's post hoc test. p < 0.05 was considered statistically significant, and data are presented as mean ± S.E.M.
Results
DOR Coimmunoprecipitates with KOR from Primary Sensory Neurons.
Coimmunoprecipitation experiments were done with primary cultures of rat peripheral sensory neurons. After cell surface cross-linking and immunoprecipitation with anti-KOR antibody, a single, 120-kDa immunoreactive band for DOR was visualized via Western blotting (Fig. 1). Likewise, a 120-kDa immunoreactive band for KOR was also visualized along with a lower molecular mass band at 55 kDa. These data suggest that DOR and KOR form heteromeric complexes in primary sensory neurons in culture.
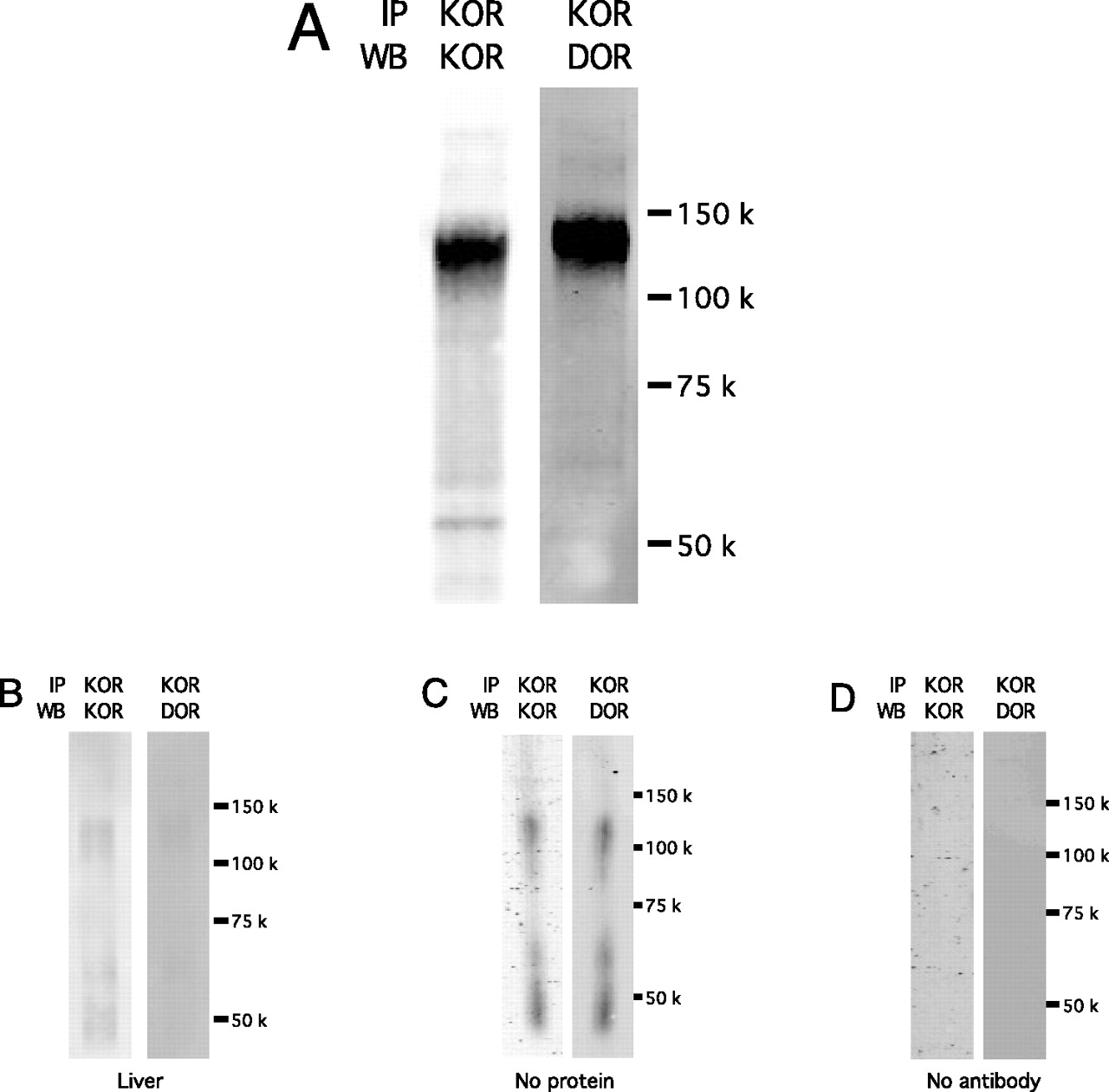
DOR coimmunoprecipitates with KOR in peripheral sensory neurons. A, TG primary cultures in 10 cm plates were treated with membrane insoluble bis[sulfosuccinimidyl] suberate (1 mM) for 30 min at room temperature to cross-link cells surface accessible proteins. Cell lysates were applied to Pierce spin columns containing anti-KOR antibody covalently bound to Protein A/G agarose beads. Samples were eluted, resolved with SDS-PAGE, transferred to PVDF membranes, blotted with anti-DOR or anti-KOR antibody and bands visualized with an Odyssey infrared Western Blot Imager (Licor). After cell surface crosslinking and immunopreciptation with KOR antibody, a single, 120 kd immunoreactive band for DOR was visualized via western blot analysis. The image shown is representative of 3 independent experiments. B to D, negative control immunoblots with anti-KOR antibody. Lysate from rat liver (B), which does not express KOR, or elution buffer only (C) was applied to spin columns containing anti-KOR antibody. D, TG cell lysate was applied to spin columns without anti-KOR antibody. After elution, SDS-polyacrylamide gel electrophoresis, and transfer to PVDF membranes, blots were probed with anti-KOR and anti-DOR antibodies and visualized with the Odyssey Imager.
Responses to the Putative DOR-KOR Heteromer Agonist 6′-GNTI in Peripheral Sensory Neurons Are Blocked by DOR or KOR Antagonists In Vitro and In Vivo.
In accord with previous observations that opioid receptors expressed in primary sensory neuronal cultures derived from adult rat TG do not inhibit adenylyl cyclase activity unless cells are pretreated with an inflammatory mediator, such as BK (Patwardhan et al., 2005, 2006; Berg et al., 2007a,b, 2011), the DOR-KOR ligand 6′-GNTI did not alter PGE2-stimulated cAMP levels unless cells were pretreated for 15 min with BK (Fig. 2A). In cells pretreated with BK (10 μM, 15 min), 6′-GNTI inhibited PGE2-stimulated adenylyl cyclase activity with an EC50 of 2 nM (pEC50 8.72 ± 0.14, n = 4) and a maximal inhibition of 76 ± 8. In the absence of BK, 6′-GNTI, at concentrations up to 1 μM, did not alter PGE2-stimulated cAMP levels. The response to 6′-GNTI in BK-pretreated cells was blocked completely by either the selective KOR antagonist nor-BNI (3 nM, 100 × Ki) or the selective DOR antagonist naltrindole (NTI; 20 nM, 100 × Ki) (Fig. 2B).
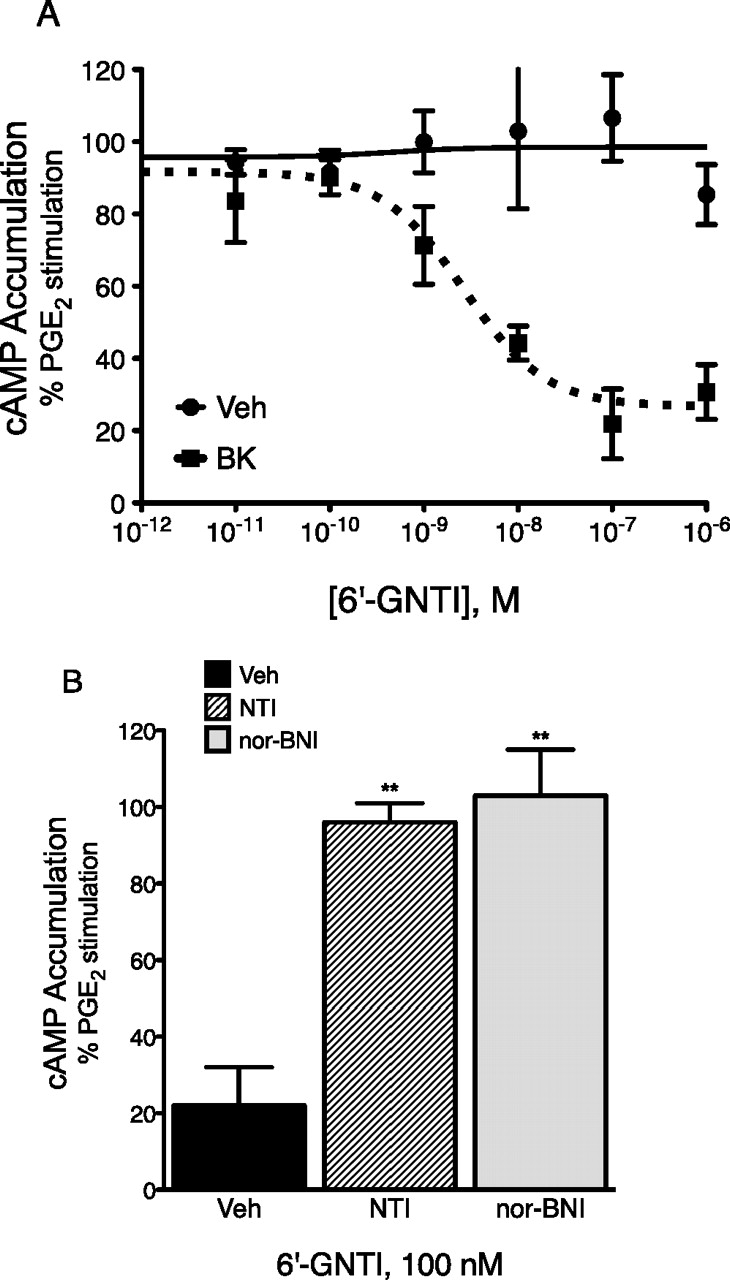
Effect of 6′-GNTI on PGE2-stimulated adenylyl cyclase activity in primary cultures of adult sensory neurons. A, TG primary cultures from adult rats were pretreated with BK (10 μM) or vehicle (Veh) for 15 min. After pretreatment, cells were incubated with various concentrations of 6′-GNTI for 15 min followed by addition of PGE2 (1 μM) and further incubation for 15 min. Cellular levels of cAMP were determined by RIA. Data are expressed as the percentage of PGE2-stimulated cAMP levels and are the mean ± S.E.M., n = 4. Basal (nonstimulated) cAMP levels were 2.76 ± 0.20 pmol/well and PGE2-stimulated cAMP levels were 67% above basal ± 3% (mean ± S.E.M., n = 4). Neither basal nor PGE2-stimulated cAMP levels were altered by BK pretreatment (p = 0.29 and 0.86 for basal and PGE2 cAMP levels, respectively, paired t test). B, the inhibition of PGE2-stimulated cAMP accumulation by 6′-GNTI in BK pretreated sensory neurons was blocked by either the DOR antagonist NTI or the KOR antagonist nor-BNI. TG primary cultures were pretreated with BK (10 μM) in the absence or presence of NTI (20 nM, 100× Ki) or nor-BNI (3 nM, 100× Ki) for 15 min. After pretreatment, cells were incubated with a maximal concentration of 6′-GNTI (100 nM) for 15 min followed by addition of PGE2 (1 μM) and further incubation for 15 min. Cellular levels of cAMP were determined by RIA. Data are expressed as the percentage of PGE2-stimulated cAMP levels and are the mean ± S.E.M., n = 4. **, p < 0.01 compared with Veh, one-way ANOVA with Dunnett's post hoc.
6′-GNTI was also effective in completely blocking PGE2-induced thermal allodynia when administered to BK-pretreated hind paws. As shown in Fig. 3, intraplantar injection of PGE2 (0.3 μg) after vehicle pretreatment produced a prolonged thermal allodynia (□). The injection of 6′-GNTI (1 μg, i.pl.) alone did not alter the PGE2-induced thermal allodynia (Fig. 3, ○). However, when administered 15 min after a intraplantar preinjection of 25 μg BK, 6′-GNTI produced a profound antinociceptive response (▴) that was blocked completely by intraplantar pretreatment with either NTI (400 μg; ▾) or nor-BNI (100 μg; ♦).

Effect of 6′-GNTI on PGE2-induced thermal allodynia in the rat hind paw. Animals received intraplantar preinjection with vehicle, BK (25 μg), BK (25 μg) with nor-BNI (100 μg), or BK (25 μg) with NTI (400 μg) 15 min before intraplantar coinjection with PGE2 (0.3 μg) and either vehicle or 6′-GNTI (1 μg). PWL was measured in duplicate at 5-min intervals until 20 min after the last injection. Data are expressed as the change (seconds) from individual baseline values (9.66 ± 0.21 s) and represent mean ± S.E.M. of 6 to 12 animals per group. ***, p < 0.001; **, p < 0.01 versus other groups by two-way ANOVA with Bonferroni's multiple comparison test post hoc.
KOR Antagonists Regulate DOR Agonist Responses in a Ligand-Dependent Manner In Vitro and In Vivo.
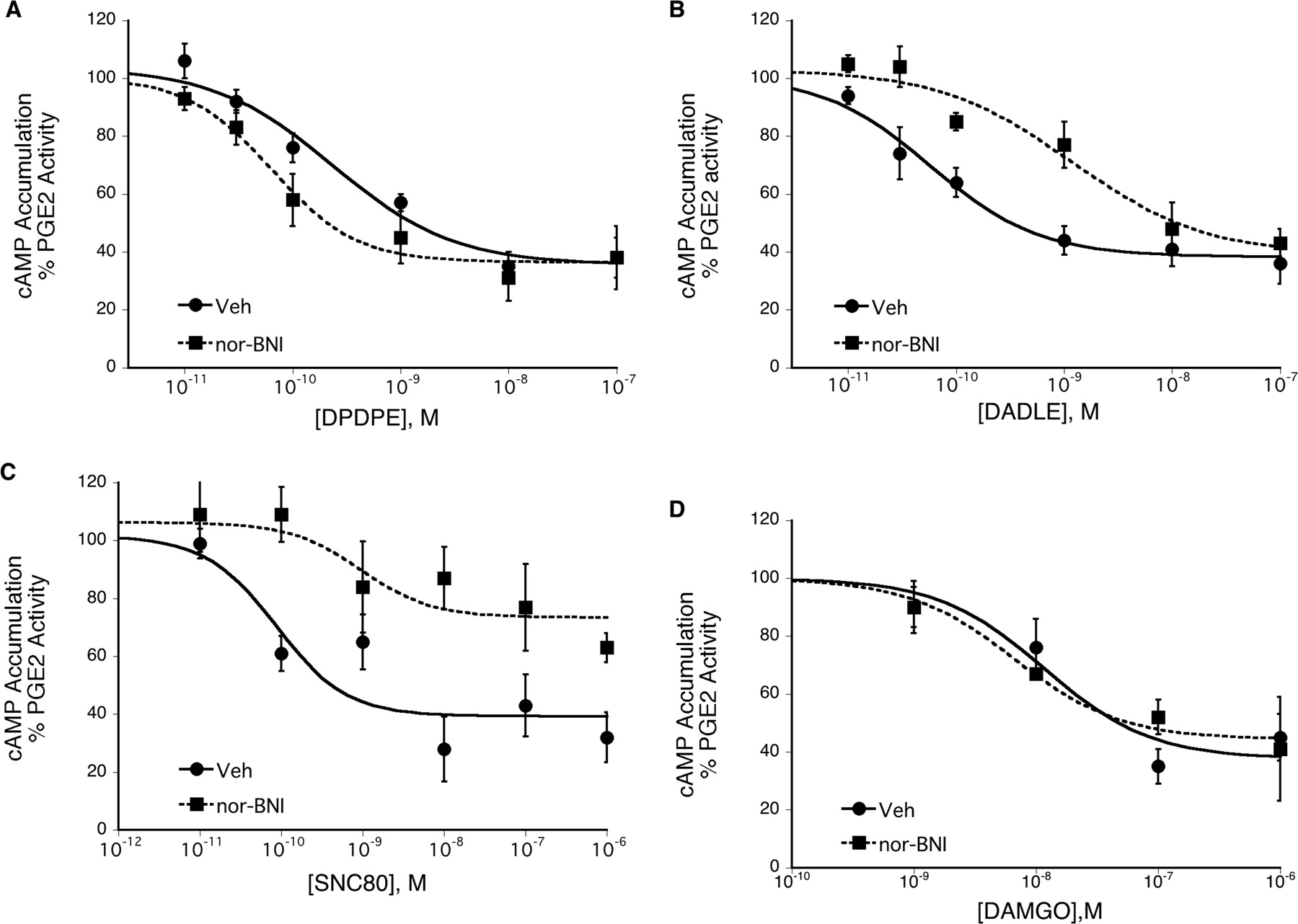
In BK-pretreated TG cultures, the DOR agonist DPDPE inhibited PGE2-stimulated cAMP accumulation with a maximal inhibition of 64 ± 6% and an EC50 of 0.5 nM (pEC50 = 9.31 ± 0.09, n = 6; Fig. 4A). The KOR antagonist nor-BNI (3 nM, 100 × Ki), shifted the concentration response curve to DPDPE to the left by 10-fold, with no change in the maximal response. In the presence of nor-BNI, the maximal inhibition of PGE2-stimulated cAMP accumulation produced by DPDPE was 63 ± 3% and the EC50 was 0.06 nM (pEC50, 10.20 ± 0.06, n = 6; p < 0.05 compared with control, paired t test). Neither basal nor PGE2-stimulated cAMP levels were altered by nor-BNI. Basal levels were 0.75 ± 0.08 pmol/well for vehicle treated cells and 0.82 ± 0.14 pmol/well in the presence of nor-BNI (n = 6, p = 0.67 paired t test). PGE2-stimulated cAMP levels were 166 ± 10% above basal for vehicle and 129 ± 9% above basal with nor-BNI (n = 6; p = 0.357 paired t test).

The effect of the KOR antagonist nor-BNI on the response to DOR and MOR agonists. TG primary cultures from adult rats were pretreated with BK (10 μM) for 15 min followed by incubation with various concentrations of the DOR agonists DPDPE (A), DADLE (B), and SNC80 (C) or the MOR agonist DAMGO (D) in the absence or presence of the KOR antagonist nor-BNI (3 nM) for 15 min. After this incubation, cells were incubated with PGE2 (1 μM) for 15 min. Cellular levels of cAMP were determined by RIA. Data represent the mean ± S.E.M. of four to six experiments each. Basal cAMP levels were 0.85 ± 0.15 pmol/well, and PGE2-stimulated cAMP accumulation was 132 ± 9% above basal, mean ± S.E.M., n = 20.
In contrast to the enhanced sensitivity to DPDPE, nor-BNI decreased the sensitivity to the DOR agonist DADLE to inhibit PGE2-stimulated cAMP accumulation (Fig. 4B). In the presence of nor-BNI, the concentration-response curve to DADLE was shifted to the right by 30-fold, without a change in the maximal inhibition. The pEC50 values for DADLE in the absence and presence of nor-BNI were 10.24 ± 0.05 (0.06 nM) versus 8.74 ± 0.32 (1.8 nM), respectively (n = 6, p < 0.05 paired t test). The maximal inhibition by DADLE in the absence and presence of nor-BNI was 62 ± 3.0 versus 61 ± 11%, respectively (n = 6).
Both the potency and the efficacy of the DOR agonist SNC80 were reduced by nor-BNI (Fig. 4C). The pEC50 values for SNC80 in the absence and presence of nor-BNI were 8.86 ± 0.31 (1.3 nM) versus 7.33 ± 0.32 (47 nM), respectively (n = 4, p < 0.05 paired t test). The maximal inhibition by SNC80 in the absence versus the presence of nor-BNI was 63 ± 7.0 versus 30 ± 7.0%, respectively (n = 4, p < 0.01 paired t test).
nor-BNI did not alter the concentration-response curve to DAMGO, a MOR agonist (Fig. 4D). The maximal inhibition of PGE2-stimulated cAMP accumulation by DAMGO was 72 ± 6.0 versus 66 ± 3.0% in the absence and presence of nor-BNI, respectively (n = 4, p = 0.461 paired t test). The pEC50 for DAMGO in the absence and presence of nor-BNI was 7.85 ± 0.14 (10 nM) versus 8.09 ± 0.06 (7 nM), respectively, (n = 4, p = 0.70 paired t test).
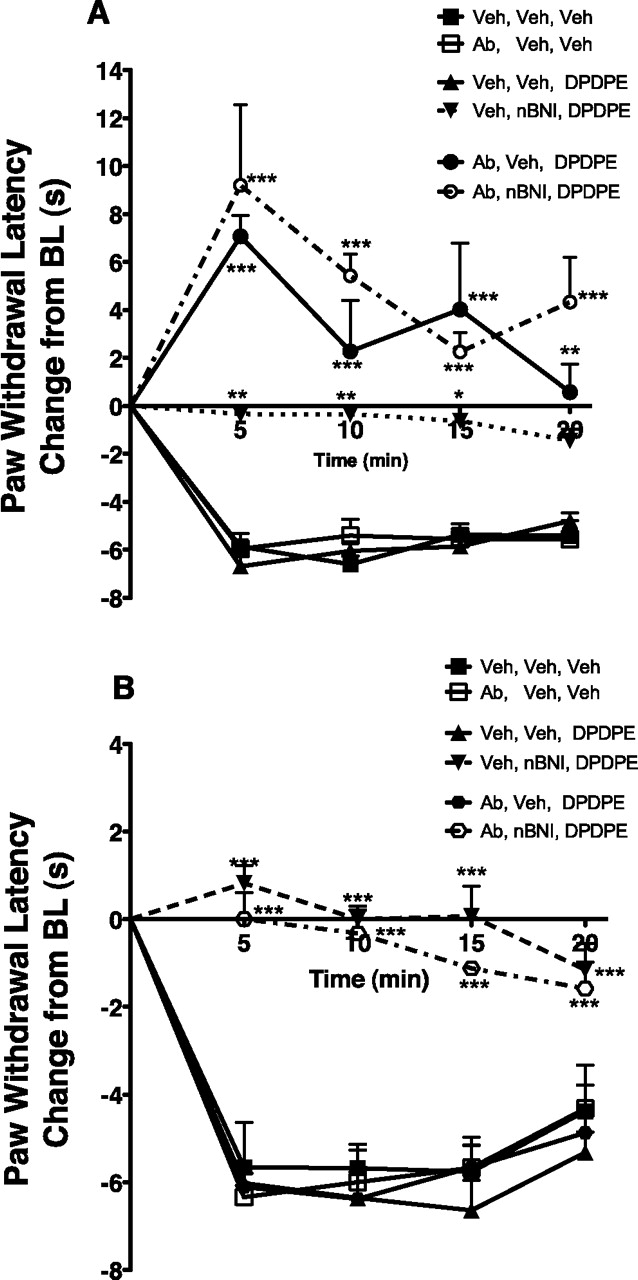
Similar results with nor-BNI were seen in the behavioral experiments. Figure 5A shows that DPDPE (0.2 μg) administered by intraplantar injection into the BK-pretreated rat hind paw did not alter PGE2-induced thermal allodynia. This is consistent with our previous study, in which the ED50 for DPDPE to inhibit PGE2-induced thermal allodynia was approximately 2.0 μg (Rowan et al., 2009). However, nor-BNI (10 μg) enhanced the effect of this subthreshold dose of DPDPE to completely eliminate the allodynia produced by PGE2. In contrast, nor-BNI eliminated the antiallodynic effect of SNC80 (5 μg, intraplantar; Fig. 5B).

The effect of the KOR antagonist, nor-BNI, on DOR agonist mediated inhibition of PGE2-induced thermal allodynia. Animals received intraplantar preinjection with BK (25 μg) with or without nor-BNI (10 μg) 15 min before intraplantar coinjection with DPDPE (0.2 μg, subthreshold) (A) or SNC80 (5 μg) and PGE2 (0.3 μg) (B). PWL was measured in duplicate at 5-min intervals until 20 min after the last injection. Data are expressed as the change (seconds) from individual baseline (A, 9.44 ± 0.23 s, B, 10.80 ± 0.26 s) values and represent mean ± S.E.M. of 6–12 animals per group. ***, p < 0.001; **, p < 0.01: *, p < 0.05 versus other groups by two-way ANOVA with Bonferroni's multiple comparison test post hoc.
The experiments above demonstrate that the nature of effect of nor-BNI was dependent upon the DOR agonist both in primary culture of sensory neurons and in vivo. Figure 6 shows that the effect on DOR agonist-mediated responses also depends upon the KOR ligand used. In contrast to the enhanced sensitivity of DPDPE produced by nor-BNI (Figs. 4A and 5A), the KOR antagonist 5′-GNTI reduced responsiveness to DPDPE in vitro and in vivo. 5′-GNTI did not alter PGE2-stimulated cAMP accumulation (p = 0.91 paired t test) but dramatically reduced the maximal inhibition of PGE2-stimulated cAMP accumulation produced by DPDPE from 53 ± 3 to 5 ± 4% in the absence and presence of 5′-GNTI, respectively (Fig. 6A; n = 6, p < 0.01 paired t test). Likewise, the antiallodynic effect of an ED50 dose of DPDPE (2.0 μg, intraplantar) in BK-pretreated hind paws was completely reduced by 5′-GNTI (Fig. 6B; 2.0 μg, intraplantar).

The effect of the KOR antagonist, 5′-GNTI, on DOR agonist mediated inhibition of PGE2-stimulated cAMP accumulation (A) and PGE2-induced thermal allodynia (B). A, TG primary cultures from adult rats were pretreated with BK (10 μM) for 15 min followed by incubation with various concentrations of DPDPE in the absence or presence of the KOR antagonist, 5′-GNTI (4 nM), for 15 min. After this incubation, cells were incubated with PGE2 (1 μM) for 15 min. Cellular levels of cAMP were determined by RIA. Data represent the mean ± S.E.M. of eight experiments. In the absence of 5′-GNTI, the pEC50 (± S.E.M.) for DPDPE was 9.77 ± 0.067. Basal cAMP levels were 0.82 ± 0.16 pmol/well and PGE2-stimulated activity was 113 ± 21% above basal, mean ± S.E.M., n = 6. Neither basal nor PGE2-stimulated cAMP levels were altered by 5′-GNTI treatment (p = 0.075 and 0.913 for basal and PGE2 cAMP levels, respectively, paired t test). B, animals received intraplantar preinjection with BK (25 μg) with or without 5′-GNTI (2 μg) 15 min before intraplantar coinjection with DPDPE (2 μg) and PGE2 (0.3 μg). PWL was measured in duplicate at 5-min intervals until 20 min after the last injection. Data are expressed as the change (seconds) from individual baseline values (10.84 ± 0.31 s) and represent mean ± S.E.M. of six to nine animals per group. **, p < 0.01; *, p < 0.05 versus other groups by two-way ANOVA with Bonferroni's multiple comparison test post hoc.
Effects of a DOR-KOR Heteromer-Selective Monoclonal Antibody In Vivo.
A subtractive immunization strategy was used to generate an antibody that selectively recognized endogenous DOR-KOR heteromers, as we have done before (see Supplemental Data and Gupta et al., 2010). Hybridoma clones secreting monoclonal antibodies were screened by enzyme-linked immunosorbent assay against untransfected HEK-293 membranes and HEK-293 membranes expressing similar levels of KOR or DOR or coexpressing DOR and KOR receptors. A monoclonal antibody was chosen based on its ability to recognize an epitope in cells coexpressing DOR and KOR receptors but not in cells expressing either DOR or KOR alone (Supplemental Fig. 1). The selectivity of the antibody for DOR-KOR heteromers was tested by screening against cells expressing DOR, KOR, or CB1 receptors alone or expressing MOR-DOR, DOR-KOR, CB1-type 1 angiotensin II (AT1) receptor, CB1 receptor-CB2 receptor, CB1-MOR, CB1-KOR heteromers. The antibody recognized an epitope in cells expressing DOR-KOR heteromers but not the other receptor complexes (Supplemental Fig. 2).
As shown previously (Fig. 5A), intraplantar injection of nor-BNI, enhanced the antiallodynic effect of a subthreshold dose (0.2 μg) of DPDPE (Fig. 7A). Intraplantar injection of the DOR-KOR antibody (10 μg) also enhanced the antinociceptive effect of DPDPE but to an extent considerably greater than that produced by nor-BNI (Fig. 7A). Whereas DPDPE completely blocked the allodynic effect of PGE2 after nor-BNI administration, the DOR-KOR antibody promoted a profound analgesic response from a subthreshold dose of DPDPE. Injection of an antibody selective for CB1-AT1 receptor heteromers did not alter the effect of DPDPE (Fig. 7B). Neither nor-BNI nor the DOR-KOR or CB1-AT1 antibodies alone altered the allodynic effect of PGE2.
The effect of a DOR-KOR heteromer selective antibody on DPDPE-mediated thermal antiallodynia. Animals received intraplantar preinjection with BK (25 μg) without or with nor-BNI (10 μg) and/or a monoclonal, DOR-KOR heteromer-selective antibody (DOR-KOR Ab, 10 μg) (A) or a monoclonal, AT1-CB1 heteromer-selective antibody (AT1-CB1 Ab, 10 μg) (B). Fifteen minutes later, animals received intraplantar DPDPE (0.2 μg, subthreshold) or vehicle (Veh) and PGE2 (0.3 μg). PWL was measured in duplicate at 5-min intervals until 20 min after the last injection. Data are expressed as the change (seconds) from individual baseline (A, 10.85 ± 0.21 s; B, 10.53 ± 0.12 s) values and represent mean ± S.E.M. of four animals per group. ***, p < 0.001; **, p < 0.01; *, p < 0.05 versus the vehicle-treated group by two-way ANOVA with Bonferroni's multiple comparison test post hoc.
Discussion
In this study, we provide evidence for the presence of DOR-KOR heteromers in peripheral sensory neurons and demonstrate that KOR antagonist ligands regulate DOR agonist function, probably via allosteric interactions between the DOR-KOR protomers. Thus, nor-BNI enhanced the potency with no change in efficacy of DPDPE, decreased the potency with no change in efficacy of DADLE, and decreased both potency and efficacy of SNC80 in primary cultures of adult rat peripheral sensory neurons. In contrast to nor-BNI, the KOR antagonist 5′-GNTI decreased the DPDPE response. The differential effect of KOR antagonists on DOR agonist responses observed in cultured neurons was also observed in a behavioral model of thermal allodynia. nor-BNI enhanced the antiallodynic response produced by DPDPE and decreased the antiallodynic response produced by SNC80. Also consonant with its effect in cultured neurons, 5′-GNTI decreased the antiallodynic response by DPDPE. Collectively, these parallel studies demonstrate profound, ligand-dependent interactions between KOR and DOR in peripheral sensory neurons, which is a hallmark of allosterism.
Heteromerization between DOR and KOR has been shown in a variety of studies. Epitope-tagged DOR and KOR can be coimmunoprecipitated (Jordan and Devi, 1999; Waldhoer et al., 2005) when coexpressed in HEK cells, and DOR-KOR interactions have been observed in bioluminescence resonance energy transfer experiments in live HEK cells (Wang et al., 2005). Although etorphine promotes DOR internalization in HEK cells, it fails to produce substantial internalization in cells that coexpress KOR (Jordan and Devi, 1999). In addition, coexpression of DOR and KOR changes the affinity values for a variety of ligands (Jordan and Devi, 1999; Bhushan et al., 2004; Waldhoer et al., 2005; Xie et al., 2005). Moreover, 6′-GNTI, previously characterized as a KOR agonist (Sharma et al., 2001), has considerably higher efficacy in cells coexpressing DOR and KOR, and its effects can be fully antagonized by either naltrindole (DOR antagonist) or nor-BNI (KOR antagonist) (Waldhoer et al., 2005).
In the present study, the presence of functional DOR-KOR heteromers in peripheral sensory neurons is suggested by several lines of evidence: 1) DOR coimmunoprecipitated with KOR from primary sensory neuronal cultures, and 2) the putative DOR-KOR heteromer-selective agonist 6′-GNTI (Waldhoer et al., 2005) inhibited PGE2-stimulated cAMP accumulation with high potency and efficacy in vitro and elicited a strong antinociceptive response in vivo, both of which were blocked by either a DOR or KOR antagonist. Third, the antinociceptive response to DPDPE was enhanced by injection of a DOR-KOR heteromer-selective monoclonal antibody into the rat hindpaw. Finally, ligand-dependent effects of KOR antagonists on DOR agonist responses in vitro and in vivo were suggestive of allosteric interactions between DOR-KOR protomers.
Although it is possible that the effects of KOR antagonists on DOR agonist responses occurred as a result of cross talk between KOR and DOR signaling systems, this mechanism seems unlikely. First, for a cross-talk mechanism to be responsible for the observed effects, neither nor-BNI nor 5′-GNTI can be true antagonists, but each must regulate some cellular signaling pathway that alters DOR function. This pathway must be independent of the Gi-adenylyl cyclase pathway, because the KOR antagonists alone did not alter adenylyl cyclase activity. Although nor-BNI and 5′-GNTI have weak inverse agonist properties for the adenylyl cyclase pathway (Wang et al., 2007), to our knowledge, there have been no reports of inverse agonist activity for nonadenylyl cyclase signaling. nor-BNI and 5′-GNTI both have agonist activity for c-Jun NH2-terminal kinase phosphorylation (Bruchas and Chavkin, 2010); however, nor-BNI-mediated c-Jun NH2-terminal kinase signaling and its action on DOR would have to be qualitatively different from that of 5′-GNTI, because the actions of these KOR ligands on DPDPE signaling were opposite in direction. Furthermore, for the effect of KOR ligands to be DOR agonist-dependent, the target of the KOR signaling component must be at DOR itself. Changes in any other component of the DOR signaling system beyond the receptor (e.g., adenylyl cyclase) would be expected to regulate all DOR agonists similarly. Moreover, effects of nor-BNI-mediated alterations in KOR signaling on DOR itself must allow for different (opposite) effects on DOR agonist responses (increased versus decreased potency of DPDPE and DADLE by nor-BNI).
We suggest the most parsimonious explanation for the mechanism by which KOR antagonists differentially regulate DOR agonist responsiveness is via allosteric interactions between the protomers of DOR-KOR heteromers. One of the hallmarks of allosterism is that effects are dependent upon the nature of both the allosteric modulator and the orthosteric ligand (Kenakin, 2009; Smith and Milligan, 2010; Keov et al., 2011). The same allosteric modulator can either increase, decrease, or not alter the activity of an orthosteric agonist, depending upon the agonist (Jakubík et al., 1997), Thus, a conformational change in KOR elicited by occupancy with nor-BNI or 5′-GNTI would lead to a ligand-dependent conformational change in DOR, resulting in a change in affinity and/or efficacy of DOR agonists. Our estimation of allosteric constants α (affinity) and ξ (efficacy) by fitting the concentration-response curve data for DOR agonist inhibition of cAMP accumulation in the absence and presence of nor-BNI or 5′-GNTI to an allosteric model of agonism (Kenakin, 2005; Supplemental Data) suggests that the differential actions of KOR ligands can be accommodated by models of allosteric mechanisms.
Allosteric interactions between protomers of heteromeric pairs have been previously reported (see Fuxe et al., 2010; Smith and Milligan, 2010; Keov et al., 2011). In heterologous expression systems, the simple presence of one receptor can alter the affinity and/or efficacy of a ligand for a second receptor (Jordan and Devi, 1999; Waldhoer et al., 2005). It is noteworthy that the conformation of one receptor of a heteromeric pair can influence the affinity and/or efficacy of a ligand at the other protomer. Xie et al. (2005) reported that the DOR antagonist naltrindole increased the affinity of nor-BNI and vice versa. Furthermore, the conformational state of one protomer of a dopamine D2 receptor heteromer differentially altered the efficacy of quinpirole at the second protomer (Han et al., 2009). Here we found that nor-BNI differentially altered the potency and/or efficacy of DOR agonism, depending upon the specific DOR agonist used. In addition, the effect on one DOR agonist, DPDPE, was dependent upon the nature of the KOR antagonist used. Consequently, the most likely mechanism for the ligand-dependent effects of KOR antagonists in peripheral sensory neurons is allosteric modulation between the protomers of a DOR-KOR heteromer.
It has been reported that nor-BNI antagonized the analgesic effect of DPDPE in the mouse tail-flick assay when applied intracerebroventricularly or intrathecally (Portoghese and Lunzer, 2003; Lunzer and Portoghese, 2007). Here we found that intraplantar injection of nor-BNI to rats and direct application of nor-BNI to cultures of rat sensory neurons enhanced DPDPE effects. Whether the differences here relate to differences in route of administration (Lunzer and Portoghese, 2007) or differences in phenotype between peripheral sensory neurons versus CNS neurons or species differences remains to be determined.
It is noteworthy that a monoclonal antibody that selectively binds to DOR-KOR heteromers potentiated the antinociceptive effects of DPDPE in the rat hind paw, similar to but greater than the effect of nor-BNI. In the presence of this antibody, a subthreshold, ineffective dose of DPDPE became capable not only of inhibiting the thermal allodynia produced by PGE2 but also producing close to the maximal possible antinociceptive response in this system. Characterization of this antibody as selective for the DOR-KOR heteromer is presented in the Supplemental Data. Injection of this antibody alone did not alter the transient thermal allodynia produced by BK (not shown) or that produced by PGE2. These data suggest that the binding of the antibody alters the conformation of the DOR-KOR heteromer such that the affinity and/or efficacy of DPDPE is enhanced.
Although there is evidence for MOR-KOR heteromers (Wang et al., 2005; Chakrabarti et al., 2010; Yekkirala et al., 2011), we did not observe an effect of nor-BNI on the potency or efficacy of the MOR agonist DAMGO. It was recently shown that expression of MOR-KOR heteromers in spinal cord was very low in male rats but high in females and regulated by female sex hormones (Chakrabarti et al., 2010). Thus, the lack of interaction between nor-BNI and DAMGO could be due to low prevalence of MOR-KOR heteromers in the male rats in this study. It is also possible that MOR and KOR are expressed in different cells of the trigeminal ganglion and therefore would not interact allosterically. Alternatively, given the ligand-dependent nature of allosterism, it is possible that visualization of interactions between MOR-KOR heteromers requires the use of different ligands.
In summary, our data suggest that DOR-KOR heteromers exist in rat primary sensory neurons and that KOR antagonists can modulate DOR agonist responses, most likely through allosteric interactions between the protomers of the DOR-KOR heteromer. Allosteric regulation of opioid agonist responses may provide opportunities for development of analgesic drugs with greater selectivity, because effects will occur only where both protomers are coexpressed in the same cells. Given the ligand dependence of allosteric interactions, it is important to identify optimal pairs of ligands such that low doses of the analgesic agonist can be used, thereby reducing the incidence and severity of dose-related adverse effects.
Authorship Contributions
Participated in research design: Berg, Devi, and Clarke.
Conducted experiments: Berg, Rowan, Gupta, Sanchez, Silva, Gomes, and McGuire.
Performed data analysis: Berg, Rowan, Gupta, Gomes, Devi, and Clarke.
Wrote or contributed to the writing of the manuscript: Berg, Portoghese, Hargreaves, Devi, and Clarke.
Acknowledgments
We thank Dr. Jonathan Javitch and Dr. Marta Filizola for helpful discussions.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant DA024865] (to W.P.C.), [Grant DA026619] (to K.A.B.), [Grant DA008863] (to L.A.D.); and [Grant DA01533] (to P.S.P.); and the National Institutes of Health National Institute of Dental and Craniofacial Research [Grant DE14318] (to M.P.R., B.A.M.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
ABBREVIATIONS:
- BK
- bradykinin
- DPDPE
- [d-Pen2,5]-enkephalin
- PGE2
- prostaglandin E2
- TG
- trigeminal ganglion
- DADLE
- [d-Ala2,d-Leu5]-enkephalin
- MOR
- μ opioid receptor
- KOR
- κ opioid receptor
- DOR
- δ-opioid receptor
- 6′-GNTI
- 6′-guanidinonaltrindole
- nor-BNI
- nor-binaltorphimine
- CB
- cannabinoid
- HEK
- human embryonic kidney
- PVDF
- polyvinyl difluoride
- BK
- bradykinin
- PWL
- paw withdrawal latency
- NTI
- naltrindole
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- AT1
- angiotensin II type 1.

- Received April 4, 2011.
- Accepted November 9, 2011.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}