


Abstract
The phosphorylation of μ-opioid receptors (MOPRs) by G protein-coupled receptor kinases (GRKs), followed by arrestin binding, is thought to be a key pathway leading to desensitization and internalization. The present study used the combination of intracellular and whole-cell recordings from rats and mice, as well as live cell imaging of Flag-tagged MOPRs from mouse locus ceruleus neurons, to examine the role of protein kinases in acute desensitization and receptor trafficking. Inhibition of GRKs by using heparin or GRK2-mutant mice did not block desensitization or alter the rate of recovery from desensitization. The nonselective kinase inhibitor staurosporine did not reduce the extent of [Met5]enkephalin (ME)-induced desensitization but increased the rate of recovery from desensitization. In the presence of staurosporine, ME-activated FlagMOPRs were internalized but did not traffic away from the plasma membrane. The increased rate of recovery from desensitization correlated with the enhancement in the recycling of receptors to the plasma membrane. ME-induced MOPR desensitization persisted and the trafficking of receptors was modified after inhibition of protein kinases. The results suggest that desensitization of MOPRs may be an early step after agonist binding that is modulated by but is not dependent on kinase activity.
Introduction
μ-Opioid receptors (MOPRs) belong to the G protein-coupled receptor (GPCR) superfamily and undergo homologous desensitization that is thought to result from phosphorylation of agonist-bound receptors by G protein-coupled receptor kinases (GRKs) and binding to arrestin (Gainetdinov et al., 2004; Schulz et al., 2004; Kenski et al., 2005). It is known, however, that MOPRs are phosphorylated by multiple kinases (Johnson et al., 2005). Several kinase-dependent mechanisms have been proposed to mediate desensitization. The activation of protein kinase C was shown to facilitate the desensitization induced by morphine but not DAMGO, whereas inhibition of GRK blocked the desensitization induced by DAMGO but not morphine (Bailey et al., 2004; reviewed by Kelly et al., 2008). Other studies found that inhibition of GRK2 activity in locus ceruleus (LC) neurons had no effect on the acute desensitization induced by [Met5]enkephalin (ME) (Dang et al., 2011; Quillinan et al., 2011). Dang et al. (2009) found that blockers of both GRK2 and extracellular signal-regulated kinases 1 and 2 were required to reduce desensitization induced by ME. It is also known that phospholipase D2/p38MAPK plays an important role in the internalization of MOPRs, which can reduce the development of receptor desensitization (Koch et al., 2004; Yang et al., 2010). Which specific kinases mediate acute desensitization remains an open question.
This study investigates the role of protein kinases in ME-induced desensitization and trafficking in LC neurons from rats and mice. The desensitization induced by ME in the LC from rats has been well characterized (Harris and Williams, 1991; Bailey et al., 2004; Dang and Williams, 2005; Virk and Williams, 2008). Transgenic mice expressing FlagMOPRs in LC neurons were used to study receptor trafficking in addition to desensitization (Arttamangkul et al., 2008). The nonselective kinase inhibitor staurosporine was selected for study of the role of serine/threonine protein kinases in MOPR desensitization. The results demonstrated that inhibition of protein kinases did not prevent ME-induced desensitization but the rate of recovery from desensitization was enhanced. After treatment with staurosporine, ME induced receptor internalization but the internalized receptors remained close to the plasma membrane and were recycled back to the plasma membrane more rapidly. The results indicated that inhibition of multiple kinases in LC neurons altered the trafficking of receptors but had little effect on acute desensitization.
Materials and Methods
Materials.
All reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO) except 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole (SB203580), which was purchased from Tocris (Ellisville, MO). Alexa Fluor 594 succinimidyl ester was purchased from Invitrogen (Carlsbad, CA). M1-Alexa 594 was conjugated and purified with Bio-Spin 6 Tris columns (Hercules, CA). The nucleotide analog 1-(1,1-dimethyethyl)-3-(1-napthalenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (NaPP1) was a gift from Dr. Kevan Shokat (University of California, San Francisco, CA). All animal experiments were conducted in accordance with the National Institutes of Health guidelines and with approval from the institutional animal care and use committee of the Oregon Health and Science University (Portland, OR).
Electrophysiological Experiments.
Adult, male, Sprague-Dawley rats (150–250 g; Charles River Laboratories, Wilmington, MA), FlagMOPR-transgenic mice (Arttamangkul et al., 2008), and transgenic mice with a GRK isoform (GRK2as5) that is selectively inhibited by the nucleotide analog NaPP1 (Quillinan et al., 2011) were used for electrophysiological experiments. Brain slices were prepared as described previously (Williams et al., 1984). In brief, rats and mice were anesthetized with isoflurane, and the brain was removed and placed in ice-cold artificial cerebrospinal fluid (ACSF) containing 126 mM NaCl, 2.5 mM KCl, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 2.4 mM CaCl2, 21.4 mM NaHCO3, 11 mM glucose, and 0.03 mM dizocilpine maleate [(+)-MK801]. Horizontal slices were prepared (270 μm) by using a Vibratome (Leica, Nussloch, Germany) and were incubated for at least 30 min in warm (34°C) oxygenated ACSF containing (+)-MK-801 (10 μM). Glass electrodes (50–60 MΩ) filled with KCl (2 M) were used for intracellular recording of membrane potentials. Whole-cell recordings were made with pipettes (1.7–2.1 MΩ) with an internal solution containing 115 mM methyl potassium sulfate, 20 mM NaCl, 1.5 mM MgCl2, 10 mM HEPES, 10 mM BAPTA, 2 mM Mg-ATP, and 0.5 mM Na-GTP, pH 7.3. Experiments were performed at 35°C. Data were collected by using Power Lab (Chart version 5.4; ADInstruments, Colorado Springs, CO) and were acquired at 200 Hz.
Fluorescent MOPR Internalization.
FlagMOPR-transgenic mice were used for all trafficking experiments (Arttamangkul et al., 2008). All data were collected from male and female hemizygous FlagMOPR-transgenic mice that were crossed with C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) or arrestin 3-knockout (ArrKO) mice (a gift from Dr. Robert Lefkowitz, Duke University Medical Center, Durham, NC). Experiments examining the role of arrestin 3 used knockout (ArrKO) animals that were initially crossed with FlagMOPR-transgenic animals. In a series of subsequent crosses, animals that were hemizygous FlagMOPR-transgenic and homozygous ArrKO(−/−) were obtained (Arttamangkul et al., 2008; Quillinan et al., 2011). Brain slices (200 μm) were prepared as described for electrophysiological experiments. Slices were incubated for 45 to 60 min in a solution containing M1-Alexa 594 (10 μg/ml). The tissue was observed with an upright microscope (Olympus, Center Valley, PA) equipped with a custom-built two-photon apparatus. Data were acquired and collected by using ScanImage software (Pologruto et al., 2003). A z-series was collected at 1-μm intervals for 15 μm. Drugs were applied through perfusion at a rate of 1.5 ml/min. All experiments were performed at 35°C.
Quantification of Receptor Internalization and Reinsertion.
Analysis was performed offline with Image J software (http://rsbweb.nih.gov/ij/). Details of the analysis were published previously (Arttamangkul et al., 2008). In brief, the fluorescence values of stacks of 15 images were summed. Five random regions of interest away from neuronal staining were selected and averaged for background fluorescence. The average background fluorescence was then subtracted from the total fluorescent intensity of the whole frame. The fluorescent intensity obtained from slices before ME application (30 μM) was considered to represent total fluorescent receptors (C). After treatment with ME (30 μM) for 15 min, the slice was treated for 10 min with calcium-free ACSF containing 0.5 mM EGTA, to strip all extracellular antibody binding. The fluorescence that was retained within the cells was considered to represent internalized receptors (I). The proportion of internalization was calculated as (I/C) × 100. In experiments in which the return of receptors to the plasma membrane was measured, slices were perfused with ACSF for 10 or 45 min after treatment with ME (30 μM) for 10 min before the calcium-free solution wash (an additional 10 min). The fluorescence remaining after these recovery periods was calculated as described above.
Drug Application.
Drugs were applied through perfusion at the rate of 1.5 ml/min. Slices were preincubated with staurosporine (10 μM) or SB203580 (10 μM) for 45 min before experiments, followed by the presence of inhibitors at 1 μM throughout. NaPP1 (1 μM) was applied to slices 10 to 15 min before experiments and was kept in test solutions for the whole time.
Data Analysis.
Data analysis was performed with Prism software (GraphPad, San Diego, CA). All results are reported as mean ± S.E.M. Statistical significance was assessed by using a two-tailed, unpaired, Student's t test or two-way ANOVA with a Bonferroni post hoc test. The level of significance was set at P < 0.05.
Results
Staurosporine Had No Effect on Desensitization but Increased the Rate of Recovery from Desensitization.
A number of kinases are known to phosphorylate MOPRs, which suggests that simultaneous inhibition of several independent kinases may be required to affect desensitization (Dang et al., 2009). With this in mind, a high concentration of a nonselective kinase inhibitor was used to examine the kinase dependence of desensitization. Initial experiments examined the effects of staurosporine (10 μM) on the extent of and recovery from desensitization induced by ME. Intracellular recordings were performed in LC neurons in slices from rats (Fig. 1A). ME (30 μM) applied for 10 min resulted in a peak hyperpolarization that declined during the application, which was an indication of desensitization. The decline in the peak hyperpolarization was not different in untreated and staurosporine-treated slices (peak in control slices of 35.1 ± 1.0 mV decreased by 26.7 ± 4.0%, n = 8; peak in staurosporine-treated slices of 29.8 ± 1.2 mV decreased by 30.4 ± 3.0%, n = 8; P = 0.7, Student's t test). Unexpectedly, the recovery after desensitization measured with the hyperpolarization induced by ME (0.3 μM) was significantly increased (P < 0.001, two-way ANOVA) (Fig. 1A). The results indicated that the desensitization measured with the decline in the peak hyperpolarization with a saturating concentration of ME was not affected by a high concentration of staurosporine, whereas the rate of recovery from desensitization was increased.
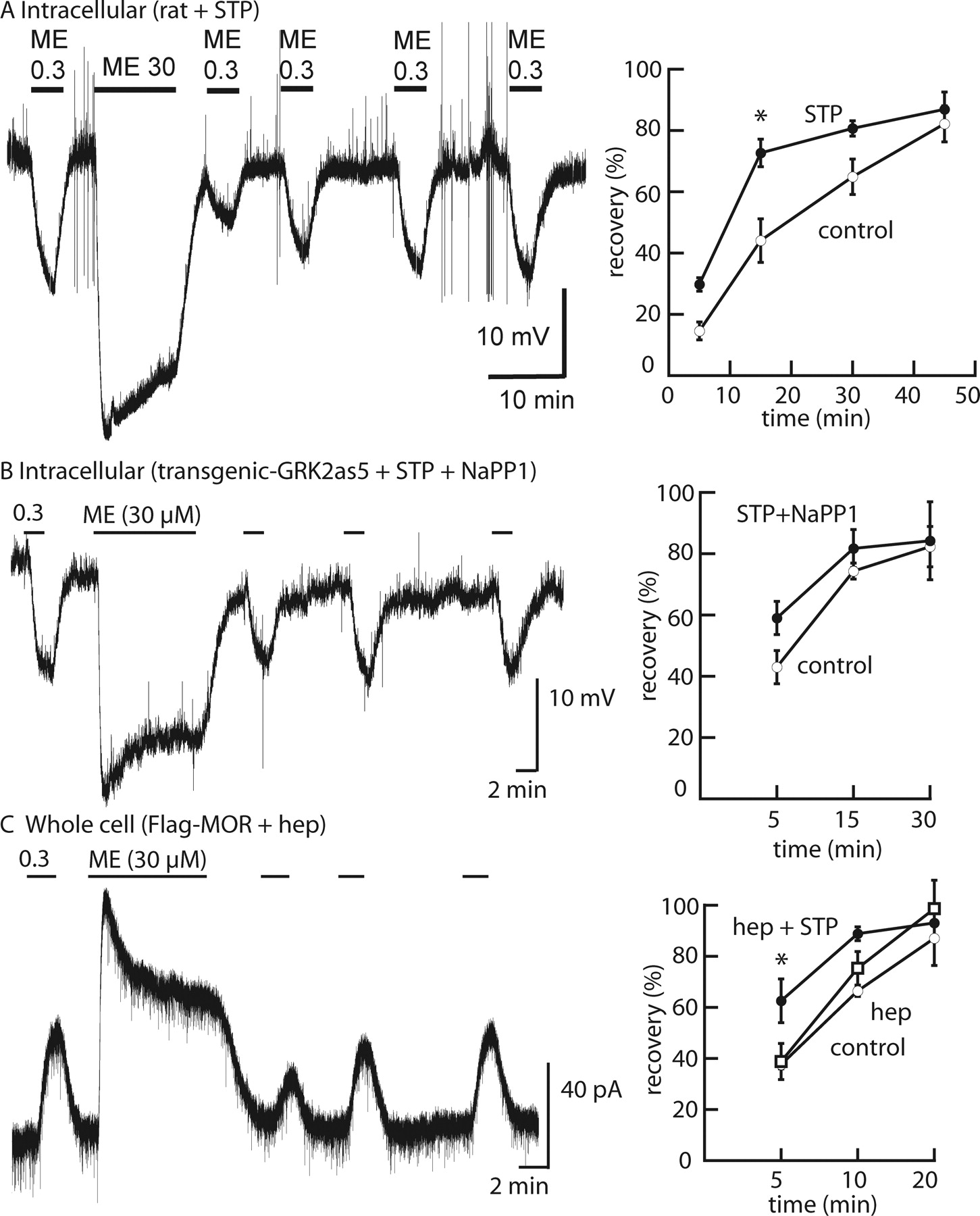
MOPR desensitization in the presence of protein kinase inhibitors. A, left, example trace showing desensitization and recovery from desensitization in a rat slice incubated with staurosporine (STP) (10 μM, 45 min); right, summarized results showing recovery from desensitization. B, left, example trace showing desensitization and recovery from desensitization in a GRK2as5-transgenic mouse slice incubated with staurosporine (10 μM, 45 min) and NaPP1 (1 μM); right, summarized results showing recovery from desensitization. C, left, example trace showing desensitization and recovery from desensitization in a FlagMOPR-transgenic mouse slice, recorded with heparin (hep) (1 mg/ml) in the pipette; right, summarized results showing recovery from desensitization for control, with heparin, and with heparin and staurosporine. *, P < 0.05, two-way ANOVA with Bonferroni post hoc test.
The Desensitization of MOPRs Was Not Blocked by Staurosporine and Inhibitors of GRKs.
Although staurosporine is a nonselective kinase inhibitor that is known to inhibit as many as 300 kinases, particularly at the concentration used in this study (10 μM), it is not a potent inhibitor of GRKs (Karaman et al., 2008). Two approaches were used to determine whether the inhibition of GRKs in addition to staurosporine-sensitive kinases would affect acute desensitization. The first approach was to use GRK2as5-transgenic mice together with the specific nucleotide inhibitor NaPP1 in addition to staurosporine. A recent study demonstrated that acute desensitization induced by ME and recovery after desensitization in slices prepared from morphine-naive transgenic animals were the same in the absence or presence of NaPP1 (Quillinan et al., 2011). In this study, intracellular recordings were made to examine desensitization and recovery from desensitization in the absence and presence of the inhibitors staurosporine (10 μM) plus NaPP1 (1 μM) (Fig. 1B). The peak hyperpolarization and decline from the peak induced by ME (30 μM, 10 min) were not different in the absence and presence of the inhibitors (control, 28.3 ± 1.7 mV, decrease of 31.2 ± 1.6%, n = 6; staurosporine plus NaPP1, 23.7 ± 2.9 mV, decrease of 34.0 ± 2.3%, n = 6; P = 0.4, Student's t test). The recovery from desensitization was not significantly different from that of control slices but trended toward being faster (P > 0.05, two-way ANOVA) (Fig. 1B).
The second approach used whole-cell recordings in slices from the FlagMOPR-transgenic animals with heparin (1 mg/ml) included in the internal solution. Heparin is a polyanionic compound that is known to inhibit all forms of GRKs (Loudon and Benovic, 1994). The internal solution with heparin did not change the amplitude of the outward current induced by ME (0.3 μM; control, 62.7 ± 11 pA, n = 6; heparin, 43.5 ± 5 pA, n = 4; P = 0.2, Student's t test) (Fig. 1C). The decreases in the peak current induced by ME (30 μM) were the same in control and heparin-treated neurons (heparin, 34.3 ± 4.2%, n = 4; control, 42.1 ± 3.2%, n = 5; P = 0.2, Student's t test). The recovery from desensitization in heparin-treated neurons was not different from that in control neurons (P > 0.05, two-way ANOVA). The results indicated that heparin did not alter ME-induced acute desensitization or recovery from desensitization in LC neurons. Addition of staurosporine to the extracellular solution did not change the extent of decline in the peak current induced by ME (30 μM, 10 min; control, 42.1 ± 3.2% of peak, n = 5; heparin plus staurosporine, 43.5 ± 2.1% of peak, n = 4; P = 0.7, Student's t test) but increased the rate of recovery from desensitization (P < 0.05, two-way ANOVA) (Fig. 1C). Experiments aimed at inhibiting the activity of many kinases did not change the extent of desensitization, and staurosporine seemed to have a strong effect on recovery after desensitization.
ME-Induced MOPR Trafficking Changed after Staurosporine Treatment.
The effect of staurosporine to increase recovery after ME-induced desensitization suggested that MOPR trafficking could be modified. Staurosporine (10 μM, 45 min) caused a small reduction in ME (30 μM)-induced FlagMOPR endocytosis (control, 28.9 ± 3.0%, n = 11; staurosporine, 21.4 ± 2.9%, n = 9; P = 0.09, Student's t test). There was a distinct change in the pattern of receptor distribution in the staurosporine-treated cells, however. FlagMOPRs formed clusters that remained closely associated with the plasma membrane (Fig. 2, bottom). In addition, FlagMOPR-bound M1-A594 labeling in these clusters was protected from being stripped by the calcium-free solution. A high-power scanning infrared image of the plasma membrane with labeled FlagMOPRs illustrated the localization of receptors after internalization induced by ME (Fig. 3). In staurosporine-treated slices, clusters of receptors that were not stripped by the calcium-free solutions were located close to the plasma membrane. Insensitivity to the stripping procedure was taken to mean that the receptors were internalized (Puthenveedu and von Zastrow, 2006); however, it is possible that they were not internalized but were protected from the calcium-free solution.
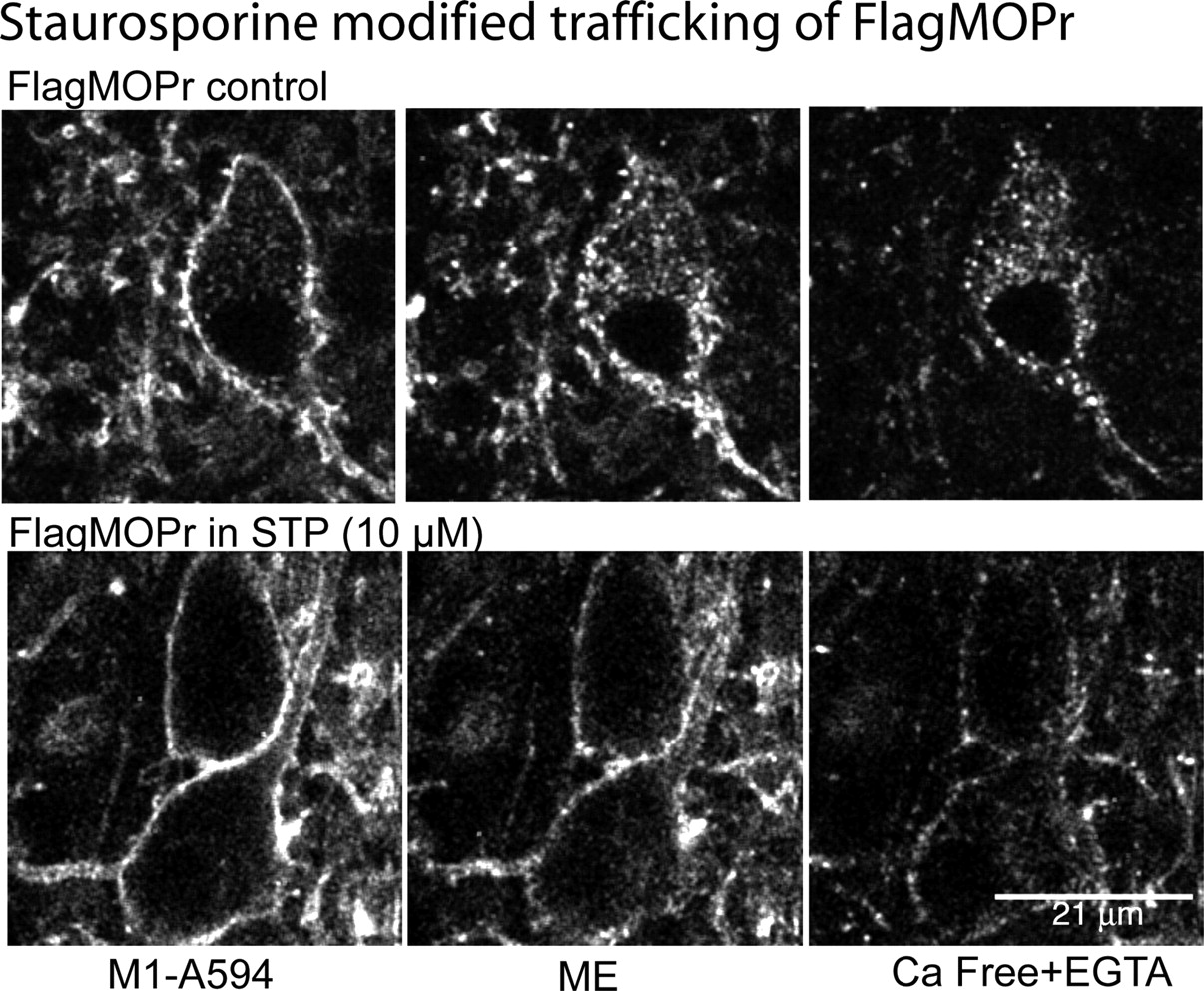
Staurosporine (STP)-induced qualitative changes in the distribution of internalized receptors from FlagMOPR-transgenic mice. Top, sequential images of an untreated slice; left, control staining with M1-A594 (45 min); middle, after treatment of the slice with ME (30 μM, 15 min); right, after treatment of the slice with calcium-free solutions. Bottom, sequential images of a staurosporine-treated slice; left, control staining with M1-A594 after incubation with staurosporine (10 μM, 45 min); middle, after treatment of the slice with ME (30 μM, 15 min); right, after treatment of the slice with calcium-free solutions.
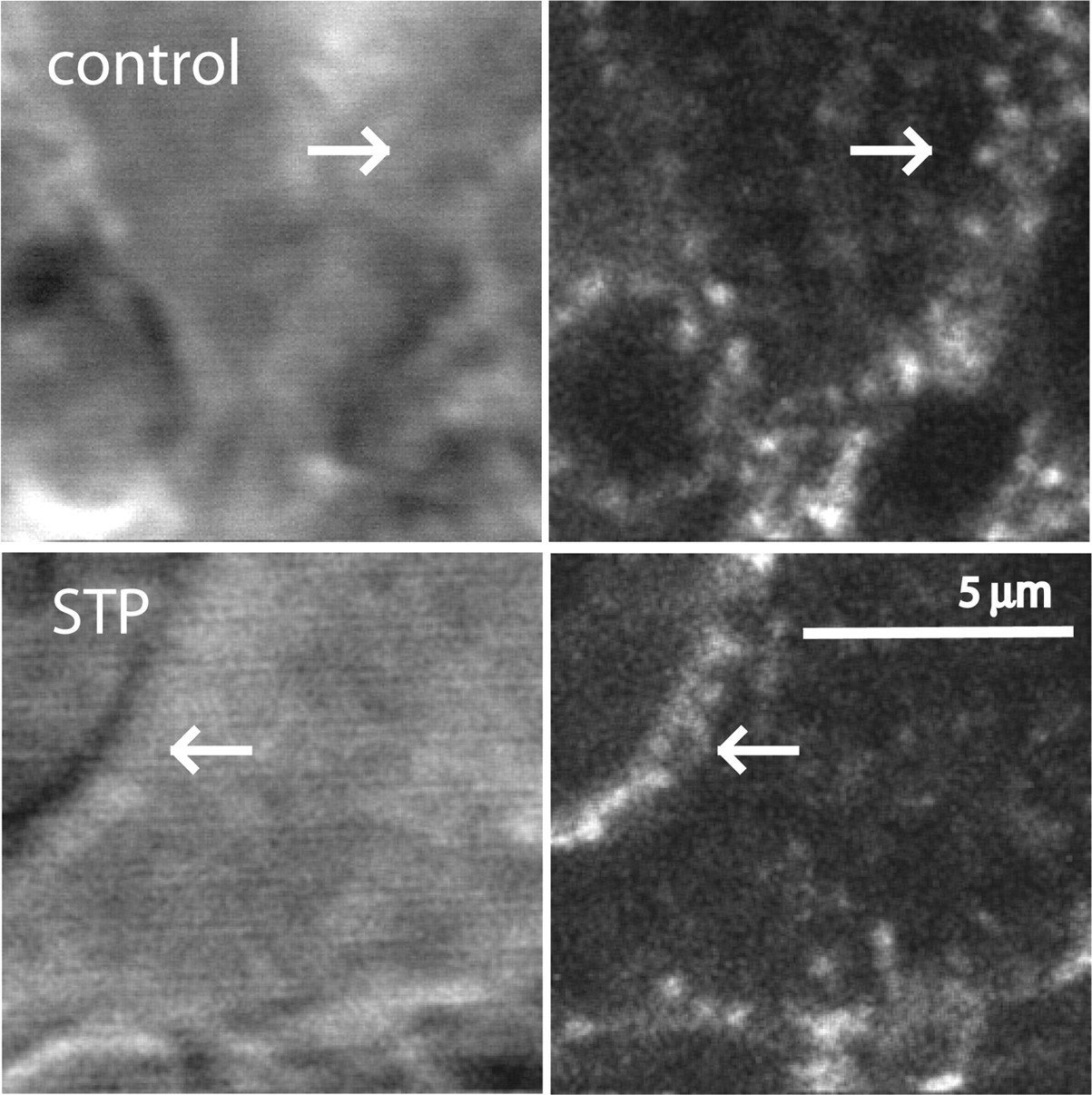
High-power images of staurosporine (STP)-treated cells from a FlagMOPR-transgenic mouse, illustrating receptor clustering in the cytoplasm near the plasma membrane. Top, images of a control slice; left, scanning infrared image showing the location of the plasma membrane (arrow); right, receptor staining with M1-A594 in the cytoplasm. Bottom, images of a staurosporine-treated slice; left, scanning infrared image showing the membrane of the cell; right, receptor staining with M1-A594. Arrows, locations of receptors close to the plasma membrane.
To examine the effect of the GRK/arrestin pathway in combination with staurosporine, the trafficking of MOPRs was examined in FlagMOPR-transgenic mice crossed with the arrestin 3-knockout mice (FlagMOPR-ArrKO mice). Similar to findings in previous reports (Arttamangkul et al., 2008; Quillinan et al., 2011), ME (30 μM)-induced receptor internalization in slices from FlagMOPR-ArrKO mice was 29 ± 2.8% (n = 7), which was not different from that measured in FlagMOPR-transgenic animals (28.8 ± 3.0%, n = 11). After treatment with staurosporine (10 μM), labeled receptors in cells from FlagMOPR-ArrKO mice also formed clusters near the plasma membrane (Fig. 4). As was found in slices from FlagMOPR-transgenic mice (control), the internalization of receptors in slices from FlagMOPR-ArrKO mice was decreased by treatment with staurosporine (staurosporine-treated, 19.0 ± 3.0%, n = 9; untreated, 29.3 ± 2.8%, n = 7; P = 0.03, Student's t test). The time courses of FlagMOPR trafficking after perfusion with ME (30 μM) in slices from FlagMOPR-transgenic and FlagMOPR-ArrKO mice indicated that receptors formed clusters as early as 3 min but they remained in the vicinity of the plasma membrane in the presence of staurosporine, whereas receptors in untreated slices moved into the cytoplasm (Fig. 5). These experiments suggested that no additional effect on receptor trafficking was induced by staurosporine in slices from ArrKO animals.

Staurosporine (STP)-induced MOPR clustering near the plasma membrane in a FlagMOPR-ArrKO mouse. Top, sequential images of an untreated slice; left, control staining with M1-A594 (45 min); middle, after treatment of the slice with ME (30 μM, 15 min); right, after treatment of the slice with calcium-free solutions. Bottom, sequential images of a staurosporine-treated slice; left, control staining with M1-A594 after incubation with staurosporine (10 μM, 45 min); middle, after treatment of the slice with ME (30 μM, 15 min); right, after treatment of the slice with calcium-free solutions.

Time course of FlagMOPR trafficking. Top, control; middle, after treatment with staurosporine (STP) (10 μM, 45 min); bottom, FlagMOPR-ArrKO mouse slice after incubation with staurosporine (10 μM, 45 min). Tg, transgenic.
Staurosporine Facilitated Receptor Reinsertion.
Given that recovery from ME-induced desensitization was increased after treatment with staurosporine, the reinsertion of receptors into the plasma membrane during recovery from desensitization was examined. After treatment with ME (30 μM, 10 min), slices were washed with ACSF and then the antibody M1-A594 was stripped with the calcium-free solution (Fig. 6, top). The fluorescence associated with receptors that trafficked back to the plasma membrane would be lost after this treatment, and a decrease in fluorescence would indicate the extent of receptor reinsertion. The internalization and reinsertion of FlagMOPRs were examined in the presence and absence of staurosporine. Two time points for receptor reinsertion were measured (20 and 55 min). After a 55-min wash, the cytoplasmic fluorescence decreased to 19.0 ± 2.5% of control values in control slices (n = 8), whereas the cytoplasmic fluorescence decreased significantly more in slices treated with staurosporine (5.4 ± 2.9%, n = 8; P < 0.01, two-way ANOVA) (Fig. 6). After a 20-min wash, the fluorescence decreased to 19.0 ± 2.5% in control slices (n = 8) and 5.8 ± 2.3% in staurosporine-treated slices (n = 8). These results indicated that, in control slices, some FlagMOPRs were recycled efficiently after the removal of ME, whereas another pool was trapped or recycled more slowly. Treatment with staurosporine altered the extent, distribution, and recycling of receptors, which demonstrated a role of kinases in agonist-dependent trafficking.

Reinsertion of FlagMOPRs after internalization. A, top, left to right, control, after treatment with ME (30 μM, 15 min), after a washout period of 55 min, and after treatment with calcium-free solution; bottom, same experiment performed with a slice incubated with staurosporine (STP) (10 μM, 45 min). B, summarized results showing the amount of fluorescence remaining in slices after washout of ME (30 μM) for different periods. **, P < 0.01, two-way ANOVA with Bonferroni post hoc test.
ME-Induced Desensitization Was Not Affected by Inhibition of p38MAPK.
Studies reported an alternate pathway that regulates MOPR endocytosis (Macé et al., 2005; Yang et al., 2010); p38MAPK was proposed to be the kinase that regulated agonist-bound MOPR trafficking through phosphorylation of the small GTPase Rab5. It was suggested that internalization of MOPRs could reduce desensitization through a rapid recycling process. In this study, acute ME-induced desensitization was examined by using intracellular recordings in the absence and presence of the p38MAPK inhibitor SB203580. In the presence of SB203580 (10 μM), desensitization, as indicated by the decline in the peak hyperpolarization caused by ME (30 μM, 10 min), was not different from control values (control, decrease of 29.5 ± 2.9%, n = 7; SB203580, decrease of 30.5 ± 7.1%, n = 4; P = 0.9, t test) (Fig. 7). The recoveries from desensitization were similar, although recovery was more complete in the presence of SB203580. After 45 min, the hyperpolarization induced by ME (0.3 μM) was 63.2 ± 7.1% (n = 9) of the initial amplitude in control slices and 82.6 ± 3.6% in slices treated with SB203580 (n = 8; P < 0.05, two-way ANOVA).

Evidence that the p38MAPK inhibitor SB203580 (SB) does not block desensitization or change internalization but increases the extent of recovery from desensitization. A, example trace showing desensitization and recovery from desensitization in a slice incubated with SB203580 (1 μM, 45 min). *, P < 0.05, two-way ANOVA with Bonferroni post hoc test. B, summarized results showing the decline and recovery of hyperpolarization induced by ME (0.3 μM) after washout of ME (30 μM, 10 min). C, example images show sequential steps of MOPR internalization and reinsertion; left, control; middle, after treatment with ME (30 μM, 15 min) followed by washout; right, after stripping with calcium-free solutions.
The effect of SB203580 on the internalization of FlagMOPRs was examined next. ME-induced FlagMOPR internalization in the presence of SB203580 was not different from control values (control, 26.1 ± 2.4%, n = 6; SB203580, 32.2 ± 5.9%, n = 7; P = 0.4, t test). The fluorescent receptors were clustered throughout the cytoplasm, as found in untreated slices (Fig. 7). In addition, these receptors were reinserted into the plasma membrane to the same extent as in untreated cells. After a 55-min wash, the remaining cytosolic fluorescence was reduced to 14.9 ± 6.9% of control values (n = 5) in SB203580-treated slices and 14.8 ± 2.8% (n = 9) in untreated slices. In contrast to studies with HEK293 cells (Macé et al., 2005; Yang et al., 2010), the results of the present study suggested that inhibition of p38MAPK alone had no effect on ME-induced FlagMOPR internalization in LC neurons.
Discussion
This study examined the roles of protein kinases in MOPR desensitization, recovery from desensitization, internalization, and reinsertion into the plasma membrane. The results showed that agonist-induced MOPR desensitization was resistant to the inhibition of many serine/threonine kinases. In the presence of nonselective kinase inhibitors (i.e., heparin and staurosporine), desensitization induced by ME was present in LC neurons. The rate of recovery from desensitization was significantly increased in staurosporine-treated slices. This finding correlated with the observation of qualitative changes in receptor trafficking. Although the receptors were internalized, as indicated by insensitivity to the calcium-free wash, they clustered near the plasma membrane and were readily recycled back to the membrane after the washout of ME. The results indicated that kinase activity may not be required for desensitization but is important in receptor trafficking and recovery after desensitization.
The classic model of GPCR desensitization follows the sequential events of agonist-bound receptors being phosphorylated by GRKs to increase affinity for arrestin and to terminate signaling. Multiple phosphorylation sites on GPCRs are reported to be important for arrestin/receptor interactions (reviewed by Gurevich and Gurevich, 2006). One phospho-specific site, Ser375, on the C-terminal tail of MOPRs has been identified and proposed to be an initial site leading to receptor desensitization (Schulz et al., 2004; Doll et al., 2011). A more-detailed quantitative analysis using mass spectrometry demonstrated multiple phosphorylation clusters induced by different opioid agonists (Lau et al., 2011). Although these phosphorylated receptors are important for the process of arrestin binding and internalization, their role in desensitization remains unclear. Studies in LC neurons and HEK293 cells demonstrated that MOPR internalization was not required for desensitization or recovery from desensitization (Arttamangkul et al., 2006; Dang et al., 2011; Doll et al., 2011; reviewed by Dang and Christie, 2011). Inhibition of GRK2 in LC neurons by using GRK2as5-mutant mice or a specific peptide inhibitor did not block ME-induced MOPR desensitization (Dang et al., 2011; Quillinan et al., 2011). Although other isoforms of GRK (i.e., GRK3, GRK5, and GRK6) are able to phosphorylate MOPRs (Kovoor et al., 1998), desensitization in LC neurons was not changed when heparin was applied intracellularly through the recording pipette. All isoforms of GRK are known to be sensitive to heparin (Benovic et al., 1989; Loudon and Benovic, 1994), and the protocol used to introduce heparin is known to inhibit desensitization of δ-opioid receptors in NG108-15 cells and inositol-1,4,5-triphosphate signaling in dopaminergic neurons (Morikawa et al., 1998; Cui et al., 2007). The results suggest a limited role of GRKs in the desensitization of MOPRs and indicate that other kinases may be involved in the phosphorylation of MOPRs.
The present study used a high concentration (10 μM) of staurosporine, which has been shown to inhibit more than 300 different protein kinases (Karaman et al., 2008). We were surprised that desensitization, as indicated by the decline in the peak hyperpolarization or outward current induced by ME (30 μM), was not affected by staurosporine or staurosporine in combination with heparin. When desensitization was measured as the decrease in the response to ME (0.3 μM, EC50) after application of a saturating concentration, the extent of desensitization was attenuated, but this might have resulted from more-rapid recovery from desensitization in staurosporine-treated tissues. Imaging experiments supported this interpretation. Staurosporine caused FlagMOPRs to form clusters that were not distributed into the cytoplasm, and the receptors were reinserted into the plasma membrane more rapidly. Because staurosporine is not a potent inhibitor of GRK, the concentration used in this study may allow receptors to form clusters via the GRK/arrestin pathway. Another possible explanation is that arrestin might transiently bind to unphosphorylated receptors and initiate endocytosis (DeFea, 2011); however, the pronounced effect of staurosporine on MOPR redistribution was not dependent on the level or the isoform of arrestin present in the cells. The increase in the rate of recycling in the staurosporine-treated cells may explain a small reduction in the extent of internalized receptors.
The observation that desensitization was not blocked with the inhibition of many kinases suggests that the desensitized state of the receptor might occur before phosphorylation and trafficking. This might be a receptor conformation that is stabilized by an agonist but is unable to activate G proteins efficiently. Structural studies indicated that agonist-bound receptors have multiple affinity states, one of which might represent a desensitized conformation (Rosenbaum et al., 2009). The intrinsic mechanism that underlies the desensitization state of ionotropic receptors, in particular the AMPA receptor, has been elegantly described (Armstrong et al., 2006; Sobolevsky et al., 2009). It is also possible that interactions between MOPRs and G proteins are dependent on the lipid environment and agonist-dependent movement between lipid microdomains affects signaling (Zheng et al., 2008; Levitt et al., 2009). Mechanisms beyond the classic concept of GPCR phosphorylation and arrestin binding are needed to explain acute MOPR desensitization.
Authorship Contributions
Participated in research design: Arttamangkul and Williams.
Conducted experiments: Arttamangkul, Lu, and Williams.
Contributed new reagents or analytic tools: Lau.
Performed data analysis: Arttamangkul, Lu, and Williams.
Wrote or contributed to the writing of the manuscript: Arttamangkul, Lau, Lu, and Williams.
Acknowledgments
We thank Drs. Erica Levitt and William Birdsong for comments on the manuscript.
Footnotes
This work was supported by National Institutes of Health National Institute of Drug Abuse [Grants DA08163, DA026617].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
ABBREVIATIONS:
- MOPR
- μ-opioid receptor
- NaPP1
- 1-(1,1-dimethyethyl)-3-(1-napthalenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- ME
- [Met5]enkephalin
- LC
- locus ceruleus
- ArrKO
- arrestin 3-knockout
- ACSF
- artificial cerebrospinal fluid
- FlagMOPR
- Flag-tagged μ-opioid receptor
- ANOVA
- analysis of variance
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- MAPK
- mitogen-activated protein kinase
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]enkephalin
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- (+)-MK801
- dizocilpine maleate
- SB203580
- 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole.
- Received October 4, 2011.
- Accepted November 23, 2011.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}