


Abstract
Increased energy intake can lead to obesity, which increases the risk for the development of diabetes and cancer. Free fatty acids regulate numerous cellular processes, like insulin secretion, inflammation, proliferation, and cell migration. Dysregulation of these cellular functions by increased lipid intake plays a significant role in the development of diseases like diabetes and cancer. Free fatty acid receptors 1 and 4 (FFAR1 and FFAR4) are two free fatty acid receptors under increasing investigation for their roles in diabetes and more recently also cancer. Both receptors bind medium- to long-chain, saturated and omega-3 unsaturated fatty acids. Increasing evidence shows that enhanced FFAR1 and FFAR4 signaling reduces diabetes symptoms but enhances tumor growth and migration of various cancer types like melanoma and prostate cancer. This review gives an overview of the role of FFAR1 and FFAR4 in diabetes and cancer and discusses their potential to function as targets for treatment.
Introduction
Free fatty acids can function as signaling molecules. Depending on their structure and degree of saturation they can activate various G protein-coupled receptors. The best-studied free fatty-acid receptors are FFAR1, FFAR2, FFAR3, and FFAR4, which are also respectively referred to as GPR40, GPR43, GPR41, and GPR120. FFAR1, FFAR2, and FFAR3 belong to the GPR40 cluster and are highly similar in structure (Milligan et al., 2015; Ulven and Christiansen, et al., 2015; Miyamoto et al., 2016). FFAR2 and FFAR3 are activated by short-chain fatty acids (C2–C8), whereas FFAR1 is activated by medium- to long-chain fatty acids, of which fatty acids with a chain length between 12 and 18 carbon atoms are the most potent. Recently the crystal structure of FFAR1 was resolved (Srivastava et al., 2014). In this publication the authors showed that binding of TAK-875, an FFAR1 agonist, occurs in a lateral accessible binding pocket between transmembrane domains 3 and 4 of FFAR1. Interestingly, binding of GW-9508, a dual FFAR1/FFAR4 agonist, to FFAR1 required different residues than TAK-875 binding, suggesting that the receptor harbors multiple binding sites (Sum et al., 2007). This could potentially explain the large variety of ligands that are able to bind to FFAR1, but further research is needed to investigate how natural ligands bind to FFAR1.
FFAR4 shows little homology with the GPR40 cluster but does share various ligands with FFAR1. FFAR4, like FFAR1, binds medium- to long-chain fatty acids, including omega-3 fatty acids like α-linolenic acid, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA). The shared ligands of FFAR1 and FFAR4 have complicated the early development of receptor-specific agonists and antagonists. Many of the first agonists, like GW9508 and NCG21, activated both receptors, making it difficult to investigate the contribution of a single receptor to a specific biologic process. Recent research has led to the identification of FFAR1- and FFAR4-specific antagonists and agonists (Christiansen et al., 2013; Hudson et al., 2013; Milligan et al., 2015; Hansen et al., 2016). An overview of the ligands described in this review can be found in Table 1.
Overview of FFAR1 and FFAR4 ligands
Summary of all agonists and antagonists discussed in this review and their specificity for FFAR1 and/or FFAR4. *Selective for FFAR4 in vivo but able to bind to both FFAR1 and FFAR4 in vitro. **Selective for FFAR4 in human but dual FFAR1/FFAR4 agonist in mice. ^Selective agonist for FFAR1 with selectivity for PPARs. ^^FFAR4 agonist with agonistic activity at other G protein-coupled receptors and peroxisome proliferator-activated receptor γ.
Physiologic Roles of FFAR1 and FFAR4
FFAR1 was first identified as a receptor for medium- to long-chain fatty acids by Briscoe et al. (2003). FFAR1 is highly expressed in insulin-producing pancreatic β-cells and it has been shown to enhance glucose-induced insulin secretion leading to glucose uptake in cells (Itoh et al., 2003; Briscoe et al., 2006; Tomita et al., 2006). FFAR1 stimulation in β-cells leads to activation of phospholipase C (PLC), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into diacyl glycerol and inositol 1,4,5-trisphosphate. Inositol 1,4,5-trisphosphate releases intracellular calcium (iCa2+) from the endoplasmic reticulum. This calcium flux releases insulin-containing vesicles from the cells (Feng et al., 2012). In addition to the activation of classic second messenger pathways, FFAR1 and FFAR4 stimulation can also lead to the recruitment of β-arrestins resulting in differential cellular responses (Williams-Bey et al., 2014; Mancini et al., 2015). Furthermore, enteroendocrine cells in the intestine also express FFAR1. Within these cells activation of FFAR1 led to the production of glucagon-like peptide-1 (GLP-1). GLP-1 inhibits glucagon release and stimulates insulin secretion, leading to lower blood glucose levels (Hauge et al., 2014; Reimann and Gribble, 2016). A recent study by Christensen et al. (2015) showed that free fatty acids required uptake in the vascular system before they could activate FFAR1 and elicit GLP-1 secretion in rat intestinal cells. These findings may change the perspective on the mechanism of FFAR1 (and perhaps also FFAR4) activation. In addition, FFAR1 is also expressed in taste buds, where it plays a role in taste preference for lipids (Cartoni et al., 2010).
FFAR4 has been shown to bind medium- to long-chain fatty acids, including omega-3 fatty acids. It is expressed on adipose tissue, macrophages, and taste buds and is involved in insulin signaling, taste perception, and anti-inflammatory functions (Cartoni et al., 2010; Oh et al., 2010, 2014; Ozdener et al., 2014; Milligan et al., 2015). Its expression on adipose tissue and macrophages led to the discovery that this receptor was important in metabolic control. Oh et al. (2010) showed that FFAR4 is a receptor for omega-3 fatty acids and that stimulation of adipocytes with GW-9508 activated phosphatidylinositol-4,5-biphosphate 3-kinase (PI3K), leading to translocation of glucose transporter type 4 to the membrane and subsequent enhanced glucose uptake. Simultaneously, they showed that FFAR4 activation in macrophages could reduce LPS and tumor necrosis factor-α–mediated proinflammatory functions, leading to increased production of cytokines associated with M2 phenotypes like arginase 1, interleukin (IL)-10, and Ym-1, and decreased production of M1 cytokines like IL-6, monocyte chemoattractant protein-1, and IL-1β (Oh et al., 2010). In line with these findings, Li et al. (2013) found that stimulation of FFAR4 with DHA led to reduced cyclooxygenase-2 activation and less prostaglandin E2 (PGE2) production in RAW264.7 peritoneal macrophages. On the contrary, another study showed DHA-mediated upregulation of PGE2 in macrophages (Liu et al., 2014). Nevertheless, both studies report a switch toward an M2 phenotype associated with decreased expression of proinflammatory IL-6. Furthermore, FFAR4 is also expressed in the small intestine, where it, like FFAR1, induces GLP-1 secretion from enteroendocrine cells (Hirasawa et al., 2005).
FFAR1 and FFAR4 in Diabetes
Obesity is a major risk factor for the development of type 2 diabetes. The chronic exposure to increased glucose, triglycerides, and free fatty acid levels generate insulin resistance and eventually exhaust pancreatic functions to the extent that pancreatic β-cells no longer produce insulin. Since FFAR1 and FFAR4 are both important in metabolic control, dysregulation of receptor signaling plays a significant role in diabetes. Various studies have shown that activation of FFAR1 by either natural ligands like α-linolenic acid, EPA, and DHA or chemically synthesized agonists enhances insulin production and glucose uptake both in vitro and in vivo (Salehi et al., 2005; Kebede et al., 2008). Since various agonists activate both FFAR1 and FFAR4, studies using FFAR1−/− or FFAR4−/− mice are essential to distinguishing receptor-specific events. Tan et al. (2008) showed that FFAR1 activation by selective agonists promoted glucose-induced insulin secretion in primary pancreatic islets of wild-type mice but not in FFAR1−/− animals. In addition, glucose tolerance was improved after treatment with FFAR1 agonists in wild-type mice but not in FFAR1−/− mice. Interestingly, FFAR1−/− mice did not respond differently to a high-fat diet compared with wild-type mice. FFAR1−/− mice had body weight, fat tissue distribution, and hyperinsulinemia similar to wild-type mice, suggestive of other compensatory mechanisms (Lan et al., 2008). Li and coworkers (2016a) showed that, in addition to its effects in pancreas, FFAR1 attenuates liver lipid accumulation (hepatic steatosis). Liver steatosis is strongly associated with insulin resistance and type 2 diabetes. FFAR1 activation in liver cells was accompanied by upregulation of 5′-AMP-activated protein kinase and decreased liver X receptor expression, leading to decreased lipid accumulation in hepatocytes in vitro and in vivo.
Likewise, FFAR4 has been associated with antidiabetic effects, with the addition that activation of FFAR4 also alleviates diabetes-associated chronic inflammation owing to its function in macrophages. Activation of FFAR4 in adipocytes enhances glucose uptake, and macrophages reduce M1-associated cytokines in response to DHA, GW9508, and EPA. (Oh et al., 2010, 2014; Ichimura et al., 2012). FFAR4 seems to also be expressed in the pancreas, albeit at lower levels than FFAR1. Pancreatic δ cells produce somatostatin upon FFAR4 activation (Stone et al., 2014). In addition, FFAR4 shows redundant functions with FFAR1 in the gut, since FFAR4 can also induce GLP-1 secretion (Anbazhagan et al., 2016; Reimann and Gribble et al., 2016). Iwasaki et al. (2015) showed that FFAR4 also plays an important role in the secretion of gastric inhibitory protein (GIP) from enteroendocrine K-cells in the gut. GIP is an incretin that is released upon meal ingestion. FFAR4−/− mice showed a 75% reduction in GIP secretion upon fat ingestion compared with wild-type mice. Figure 1 summarizes FFAR1 and FFAR4 functions in diabetes.
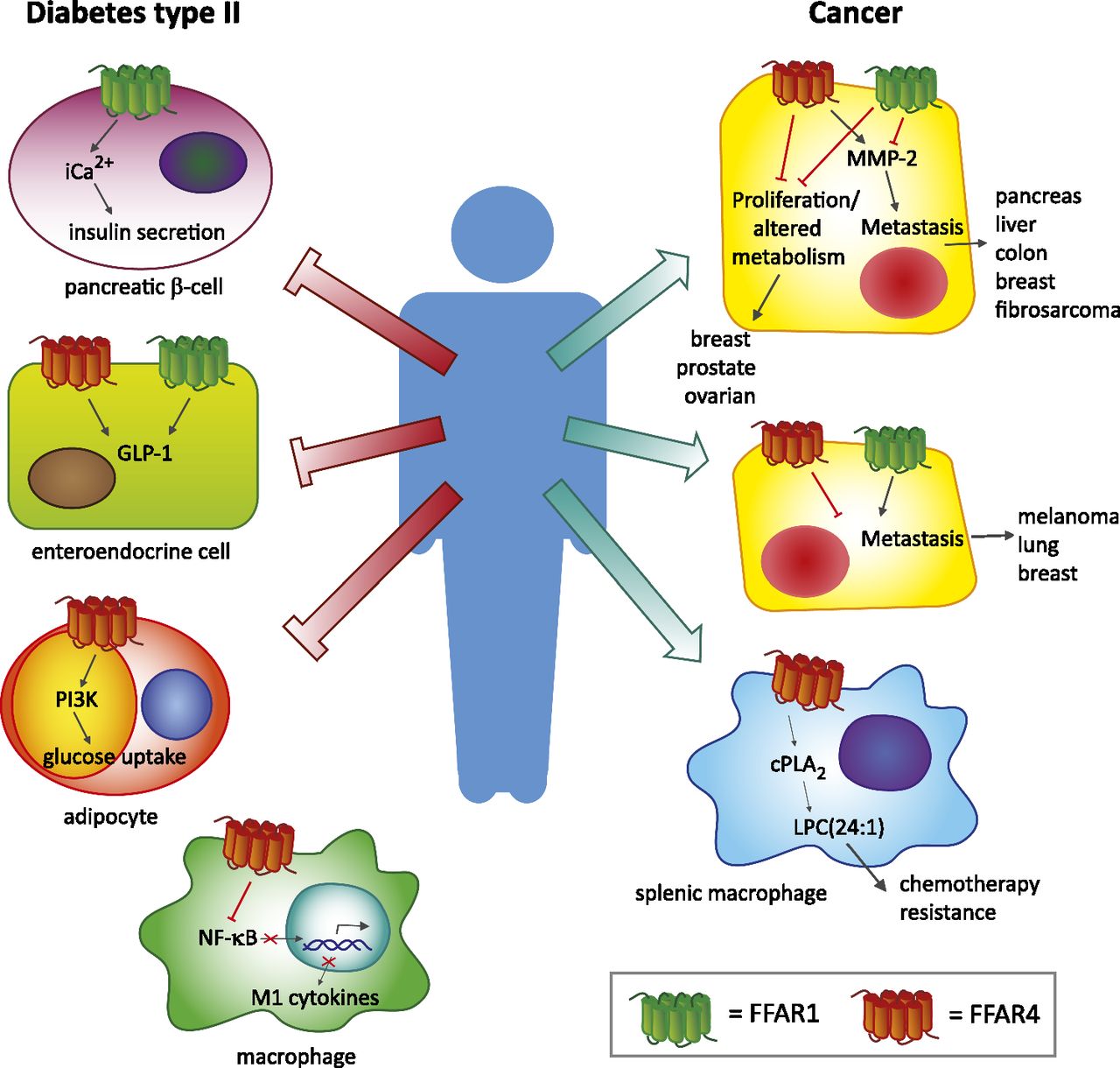
FFAR1 and FFAR4 in diabetes and cancer. Schematic overview of FFAR1 and FFAR4 functions in diabetes and cancer. PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; NF-κB, nuclear factor kappa B; cPLA2, cytosolic phospholipase A2; LPC(24:1), lysophosphatidylcholine 24:1.
Clinical validation of preclinical studies that show relevance of FFAR4 signaling to type 2 diabetes comes from the observation that a loss-of-function mutation in FFAR4 (p.R270H) has been described as a risk factor for obesity, increased blood glucose levels, and type 2 diabetes in humans (Lamri et al., 2016). Taken together FFAR1 and FFAR4 fulfill important metabolic functions and therefore these receptors are under investigation as targets for antidiabetic therapies.
FFAR1 and FFAR4 in Cancer
Increasing evidence shows that FFAR1 and FFAR4 also play a role in tumor genesis, migration, and metastasis. In contrast to its contributions in diabetes, FFAR1 and FFAR4 seem to have opposing functions in cancer biology. Fukushima et al. (2015) showed that loss of FFAR1 in pancreatic cancer cells promoted migration, whereas loss of FFAR4 inhibited migration. The effects on migration were mediated by matrix metalloproteinase 2 (MMP-2). FFAR4 knockdown cells displayed decreased levels of MMP-2, whereas FFAR1 knockdown cells had increased MMP-2 levels. However, the same authors found different results in melanoma. Their results showed that treatment of melanoma cells with the tumor-promoting agent 12-O-tetradecanoylphorbol-13-acetate (TPA) induced expression of FFAR1 and FFAR4. Downregulation of FFAR4 by RNA interference enhanced migration, whereas knockdown of FFAR1 reduced migration (Fukushima et al., 2016). Likewise, treatment of liver epithelial cells with the carcinogen ethionine also enhanced the expression of FFAR1 and FFAR4 comparably to the TPA treatment in melanoma; however, FFAR1 loss led to enhanced migration, whereas FFAR4 loss resulted in reduced migration (Ishii et al., 2015a). Downregulation of FFAR1 enhanced the migratory properties of fibrosarcoma cells. Interestingly, culturing these cells under prolonged cisplatin exposure increased FFAR1 gene expression. Subsequent treatment of these cisplatin-resistant cells with FFAR1 agonists prevented migration and invasion (Ishii et al., 2015b). Furthermore, Kita et al. (2016) demonstrated that knockdown or chemical inhibition of FFAR1 reduced MMP-2 levels, leading to reduced cell motility in three lung cancer cell lines.
The effects of FFAR1 and FFAR4 on migration and metastasis have been described for various tumor types. Liu et al. (2015) showed that FFAR1 and FFAR4 activation in prostate cancer cells inhibits lysophosphatidic acid (LPA)-induced proliferation. Using DU145 and PC-3 cells the authors showed that FFAR1 and FFAR4 activation counteracted LPA-induced proliferation by suppressing the phosphorylation of extracellular signal–regulated kinase, focal adhesion kinase, ribosomal protein S6 kinase beta-1, and expression of matricellular CCN family member 1. In addition, inhibitory effects on migration were found after stimulation of FFAR1 and FFAR4 by either TUG-891, EPA, or GW9508 (Liu et al., 2015). Similar effects were found in the context of breast cancer. Hopkins et al. (2016) showed that FFAR1 and FFAR4 activation in breast cancer cells inhibits LPA- and epidermal growth factor (EGF)-induced proliferation. Both LPA and EGF induced extracellular signal–regulated kinase and Akt activation and upregulation CCN family member 1. These effects were blocked by coadministration of either EPA, GW9508, or TUG-891 (Hopkins et al., 2016).
In addition to the effects of FFAR1 and FFAR4 on migration and metastasis, other tumorigenic functions have been attributed to FFAR1/FFAR4 signaling. Research by Wu et al. (2013) showed increased expression of FFAR4 in colorectal cancer (CRC) compared with healthy tissue and correlated FFAR4 expression with poor differentiation status and advanced clinical stage. In vitro activation of FFAR4 in CRC cell lines led to enhanced cellular migration. Furthermore, FFAR4 signaling in CRC cells increased angiogenesis of human umbilical vein endothelial cells. This effect was mediated by vascular endothelial growth factor–, IL-8-, and cyclooxygenase-2-derived PGE2 production by the CRC cells (Wu et al., 2013). In ovarian cancer cells, stimulation of FFAR1 by fatty acids derived from adipocytes promoted proliferation. Munkarah et al. (2016) found elevated FFAR1 expression in high-grade serous ovarian carcinomas compared with healthy ovarian tissue. Using a mouse model for ovarian cancer, they showed increased FFAR1 expression upon high-fat diet. Treatment of ovarian cancer cell lines with the FFAR1-specific antagonist GW1100 inhibited tumor growth by decreasing glycolysis and mitochondrial respiration.
Recently, we have shown that activation of FFAR4 by the free fatty acid 16:4(n-3) plays a role in chemotherapy resistance. Splenic macrophages expressing FFAR4 can be activated by the platinum-induced fatty acid 16:4(n-3). Upon activation splenic macrophages produced lysophosphatidylcholine 24:1, which protected tumor cells against DNA-damaging chemotherapeutics in vivo. Inhibition of FFAR4 with the FFAR4-specific antagonist AH-7614 or genetic loss of FFAR4 prevented 16:4(n-3)-mediated chemoresistance (Houthuijzen et al., 2016). An overview of the functions of FFAR1 and FFAR4 in cancer is summarized in Fig. 1 and Table 2. Taken together FFAR1 and FFAR4 have various functions in a wide array of tumor types. Further investigation of their downstream signaling and cellular specificity will show us if these receptors are potential targets for anticancer therapy.
Overview of FFAR1 and FFAR4 functions in cancer
The table summarizes the current literature regarding the biologic processes in which FFAR1 and FFAR4 signaling play a role. An upward arrow (↑) indicates that the receptor promotes/enhances the mentioned process and a downward arrow (↓) indicates inhibition of the process. A dash (–) indicates that the receptor was not involved. N.D. indicates that the receptor was not investigated in the study.
Implications as Drug Targets
The role of FFAR1 and FFAR4 in insulin secretion and inflammation has fueled research to explore their clinical application in the treatment of type 2 diabetes. TAK-875 has been extensively investigated as a therapeutic agent. Preclinical studies in type 2 diabetic rats showed that the FFAR1 agonist TAK-875 was able to enhance insulin secretion and improve hyperglycemia (Tsujihata et al., 2011). Subsequent phase I, II, and III clinical trials showed positive results, but unfortunately further studies were stopped owing to concerns about liver safety (Araki et al., 2012; Leifke et al., 2012; Kaku et al., 2015).
A new FFAR1 agonist, P11187, is currently being assessed in a phase I clinical trial for the treatment of diabetes type 2. In addition, two phase I clinical trials using FFAR1 agonists LY2922470 and SHR0534 have been completed, but no results are published yet (https://clinicaltrials.gov/). As expected, FFAR4 agonists are also under investigation as antidiabetic agents in clinical settings. KDT501, a FFAR4 agonist that also harbors activity at other G protein-coupled receptors and peroxisome proliferator-activated receptor γ, is being investigated for its insulin-sensitizing potential and glucose regulation in a phase II clinical trial. Previous research showed that KDT501 was able to normalize glucose metabolism and insulin levels in a diet-induced obesity mouse model and in diabetic fatty rats (Konda et al., 2014).
No clinical applications of FFAR1 and FFAR4 have been explored for the treatment of cancer. This could be explained by the relatively recent discovery that these receptors are relevant in cancer biology. Furthermore, detailed mechanistic insight into the signaling events leading to the alterations in cell motility and migration is lacking. And finally, their opposing functions in different tissues complicate the generalization of agonistic or antagonistic treatments. Nevertheless, most cancer-related deaths result from therapy resistance and the inability to treat metastatic disease. Multiple research groups have shown that FFAR1 and FFAR4 signaling is important in cell migration and metastasis. The ability to develop treatment strategies to block metastasis in vivo would be of great significance in the clinic.
Discussion
FFAR1 and FFAR4 fulfill multiple functions in various tissues like adipocytes, pancreatic cells, and macrophages and all are related to metabolic control. Dysregulation of their signaling cascades has been shown in type 2 diabetes and reactivation of both receptors by agonists has been shown to alleviate diabetes-associated morbidities in various preclinical and even a few clinical studies. No FFAR1 or FFAR4 agonist has reached the clinic for the treatment of type 2 diabetes yet, but several clinical trials with either an FFAR1 agonists or an FFAR4 agonist are currently ongoing and will soon give more insight into the applicability of these agonist to the treatment of diabetes (Li et al., 2016b).
The role of FFAR1 and FFAR4 in cancer biology has proven more complex. Multiple studies show that FFAR1 and FFAR4 receptor signaling has effects on cell motility and migration but often with opposing outcomes. Furthermore, tissue specificity seems to play an important role. For instance FFAR4 activation may enhance migration of pancreatic tumor cells but inhibits migration of melanoma and prostate cancer cells. Another important factor to keep in mind is that these receptors can be activated by a variety of different ligands and most often different studies use different ligands. Hypothetically these ligands may activate various downstream signaling mechanisms leading to different cellular behavior. Validity for such a hypothesis comes from studies that show that arachidonic acid, EPA, and DHA can activate similar downstream signaling but with different potencies (Mobraten et al., 2013). To add to the complexity, ligands like arachidonic acid, EPA, and DHA can be metabolized by cells, leading to the formation of new ligands that may affect FFAR1 and FFAR4 signaling. In addition, differences are also seen when comparing in vitro versus in vivo studies. We showed that 16:4(n-3) could bind and activate both FFAR1 and FFAR4 in vitro with similar efficiency, as measured by iCa2+ response and β-arrestin2 recruitment assays. However, only signaling via FFAR4 was relevant for 16:4(n-3)-mediated chemoresistance in vivo, despite the fact that both FFAR1 and FFAR4 are expressed by splenic macrophages. These findings indicate that we are just beginning to understand the complexity of fatty acid signaling and that further research is necessary to determine ligand-, receptor-, and tissue-specific effects.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Houthuijzen.
Footnotes
- Received July 8, 2016.
- Accepted August 25, 2016.
Abbreviations
- AH-7614
- 4-methyl-N-(9H-xanthen-9-yl)benzenesulfonamide
- CRC
- colorectal cancer
- DHA
- docosahexaenoic acid
- EPA
- eicosapentaenoic acid
- FFAR
- free fatty acid receptor
- GIP
- gastric inhibitory protein
- GLP-1
- glucagon-like peptide-1
- GW1100
- 4-[5-[(2-ethoxy-5-pyrimidinyl)methyl]-2-[[(4-fluorophenyl)methyl]thio]-4-oxo-1(4H)-pyrimidinyl]-benzoic acid, ethyl ester
- GW-9508
- 3-[4-[(3-phenoxyphenyl)methylamino]phenyl]propanoic acid
- IL
- interleukin
- LPA
- lysophosphatidic acid
- MMP-2
- matrix metalloproteinase 2
- NCG21
- 4-[4-[2-(N-pyridin-2-ylanilino)ethoxy]phenyl]butanoic acid
- PGE2
- prostaglandin E2
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- TAK-875
- fasiglifam (2-[(3S)-6-[[3-[2,6-dimethyl-4-(3-methylsulfonylpropoxy)phenyl]phenyl]methoxy]-2,3-dihydro-1-benzofuran-3-yl]acetic acid)
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- TUG-891
- 3-[4-[[5-fluoro-2-(4-methylphenyl)phenyl]methoxy]phenyl]propanoic acid
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}