


Abstract
We constructed a full-length human P2X5 purinoceptor cDNA by incorporating a sequence corresponding to exon 10, which is missing in cDNAs cloned previously from human tissues. We studied the functional properties by patch-clamp recording and fluorescence imaging after expression in human embryonic kidney 293 cells. ATP (1–100 μM; half-maximal current at 4 μM) elicited inward currents at –60 mV; these persisted during brief (2 s) applications but declined during longer applications. The peak current was dependent on the holding potential and showed little rectification; however, both the desensitization during the application and the decline in the current when ATP was washed out were slower at +30 mV than at –60 mV. 2′,3′-O-(4-Benzoyl)-benzoyl-ATP and αβ-methylene-ATP mimicked the action of ATP (half-maximal concentrations 6 and 161 μM, respectively). The currents were inhibited by suramin, pyridoxal-5-phosphate-6-azo-2′,4′-disulfonic acid and Brilliant Blue G, with half-maximal inhibition at 3, 0.2, and 0.5 μM, respectively; 2′,3′-O-(2′,4′,6′-trinitrophenol)-ATP (1 μM) was ineffective. Removing divalent cations did not significantly alter ATP concentration-response curves. Reversal potential measurements showed that the human P2X5 receptor was permeable to calcium (PCa/PNa = 1.5) and N-methyl-d-glucamine (NMDG) (PNMDG/PNa = 0.4); it was also permeable to chloride (PCl/PNa = 0.5) but not gluconate (Pgluc/PNa = 0.01) ions. The permeability to NMDG developed as quickly as the channel opened, in contrast to the P2X7 receptor where the NMDG permeability develops over several seconds. Cells expressing human P2X5 receptors also rapidly accumulated the propidium dye YO-PRO-1 in response to ATP.
A P2X5 subunit cDNA was first isolated from libraries of rat sympathetic ganglia (Collo et al., 1996) and heart (Garcia-Guzman et al., 1996). The RNA is expressed predominantly in heart (Garcia-Guzman et al., 1996) but there is also expression in brain, spinal cord, and adrenal gland (Garcia-Guzman et al., 1996). Immunoreactivity for P2X5 subunits has also been described in developing skeletal muscle of the rat (Meyer et al., 1999; Ryten et al., 2001); recently, the receptor has been implicated in the differentiation of satellite cells into mature multinucleated muscle fibers (Ryten et al., 2002). Mammalian skin (Groschel-Stewart et al., 1999) and endocrine (Glass and Burnstock, 2001) cells also show P2X5 immunoreactivity.
When expressed in HEK293 cells, the rat P2X5 subunit cDNAs gave rise to currents activated by ATP, but these were very small and their properties not extensively studied. The current was elicited by ATP [half-maximal concentration (EC50) about 10 μM] but not by αβmeATP, and it was readily blocked by suramin and PPADS in the 1 to 10 μM concentration range (Collo et al., 1996; Garcia-Guzman et al., 1996). A mouse cDNA has also been cloned and expressed; again, the currents recorded from HEK293 cells were only a few tens of picoamperes and they were not studied in detail (Cox et al., 2001). On the other hand, although these two mammalian receptors do not express well as homomeric channels, their coexpression with P2X1 subunits leads to the appearance of a large membrane current with several distinct properties indicative of a functional P2X1/P2X5 heteromer (Torres et al., 1998; Lê et al., 1999; Surprenant et al., 2000).
Three nonmammalian vertebrate P2X5 receptor subunits have recently been cloned. The zebrafish (Danio rerio) receptor expressed very poorly in HEK293 cells, even though the zebrafish P2X4 receptor gave good currents in parallel experiments (Diaz-Hernandez et al., 2002). The bullfrog (Rana catesbeiana) receptor cloned from larval skin and the chicken (Gallus gallus) receptor cloned from skeletal muscle provided substantive currents in HEK293 cells (Ruppelt et al., 2001) and Xenopus laevis oocytes (Bo et al., 2000; Jensik et al., 2001). These nonmammalian receptors share basic pharmacological properties with their mammalian counterparts (e.g., sensitivity to PPADS and suramin) but also exhibit some interesting features not seen in the limited prior studies of the mammalian receptors. For example, Soto's group (Ruppelt et al., 2001) showed that the homomeric chicken P2X5 receptor was significantly permeable to chloride ions, and Jensik et al. (2001) found that the bullfrog receptor became permeable to NMDG and propidium iodide when divalent cations were removed from the solution.
The human P2X5 receptor cDNA was isolated from brain, and its RNA was particularly enriched in thymus and other immune cells (Lê et al., 1997). However, alignment with other members of the P2X families showed that this cDNA lacked exon 10. This form was originally designated P2X5A; P2X5B also misses exon 3 (GenBank accession number Q93086). Exon 10 encodes a 22-amino acid segment of protein including much of the second of the two membrane-spanning domains. This cDNA did not encode functional channels, but a chimeric receptor in which the C-terminal region of the rat receptor (starting at the position equivalent to the beginning of exon 10) was joined to the human receptor provided robust ATP-activated currents when expressed in X. laevis oocytes (Lê et al., 1997). The human P2X5 gene occupies about 21 kilobases near the end of the short arm of chromosome 17 (p13.3). We found a sequence corresponding to exon 10 of the P2X5 receptor at the appropriate place in the gene (NCBI contig NT 010692, bases 3,048,945–3,049,010). However, the sequence at the 3′ splice site (GGTGCTgggagt) contains gg on the intronic side rather than the gt, which is the consensus for RNA splicing; gt is found at the corresponding position in the mouse (Cox et al., 2001) and chick (Ruppelt et al., 2001) genomic sequences. Remarkably, a single nucleotide polymorphism at precisely this position has been reported (position 3,049,012; NCBI dbSNP ss1321072). In persons with thymidine, exon 10 will be recognized during processing of the P2X5 receptor mRNA and this will be translated to a `full-length' receptor; persons with guanine at this position will make a receptor that does not include exon 10. Most expressed sequence tags in the dbEST database that contain appropriate fragments of the P2X5 receptor show that exon 10 is missing, but there are cases in which it is present (GenBank accession numbers BG116171, duodenal adenocarcinoma; AW402829, B cell germinal center). In the current release of dbEST, 12 ESTs are missing exon 10, and two ESTs contain it; this provides a crude estimate of the frequency of the G > T polymorphism of 14%.
We therefore undertook to construct and express a full-length human P2X5 receptor cDNA. This seemed to be a worthwhile goal in view of 1) the widespread tissue distribution of the subunit, 2) the limited expression studies of other mammalian homomeric P2X5 receptors, 3) the interesting properties of the channels formed by the chick and bullfrog subunits, and 4) the indication from genomic sequence and ESTs that the `exon 10-containing' receptor will in fact be made by a subset of individuals.
Materials and Methods
Construction of Full-Length Human P2X5 cDNA. Several hP2X5 receptor-related cDNA sequences were identified by a BLAST search of human dbEST database with the coding region of hP2X5 cDNA (GenBank accession number AF016709). One of the clones from a human lung small cell carcinoma was purchased (GenBank accession number BE791872; Incyte Genomics Inc., Palo Alto, CA) and sequenced. A TBLASTN search of human genomic DNA with the exon 10 sequence of rat P2X5 cDNA identified a corresponding sequence (AF168787, translation is Ala325-Gly-Lys-Phe-Ser-Ile-Ile-Pro-Thr-Ile-Ile-Asn-Val-Gly-Ser-Gly-Val-Ala-Leu-Met-Gly-Ala, which has three conservative differences from the equivalent rat sequence) (Fig. 1A). To produce the full-length hP2X5 receptor, the cDNA of the EST clone was subcloned from the original pOTB7 vector into pRS7T vector with EcoRI and XhoI. The 5′ nontranslated region was removed by digesting with NcoI and the plasmid was religated. To insert the missing exon 10 into the modified EST clone, the following four primers were used for overlap extension-based mutagenesis: 5′-cccaccatcatcaacgtgggctctggggtggcgctcatgggtgctggtgctttcttctgcgacctg, 5′-agagcccacgttgatgatggtgggaatgatgctgaacttccctgccttgccgttcaccatcacgtc, 5′-tattaccgagacgcagccggggt, and 5′-agctcgagtcacgtgctcctgtggggctccag. The fragment obtained with overlap extension, which contained the inserted exon 10, was digested with BamHI and XhoI and was used to replace the sequence between BamHI site inside the P2X5 receptor cDNA and the downstream XhoI site in the vector. This procedure essentially removed all the 3′ nontranslated region of the EST clone. The full-length coding sequence was subcloned into pcDNA3.1Zeo(+) with HindIII and XhoI.
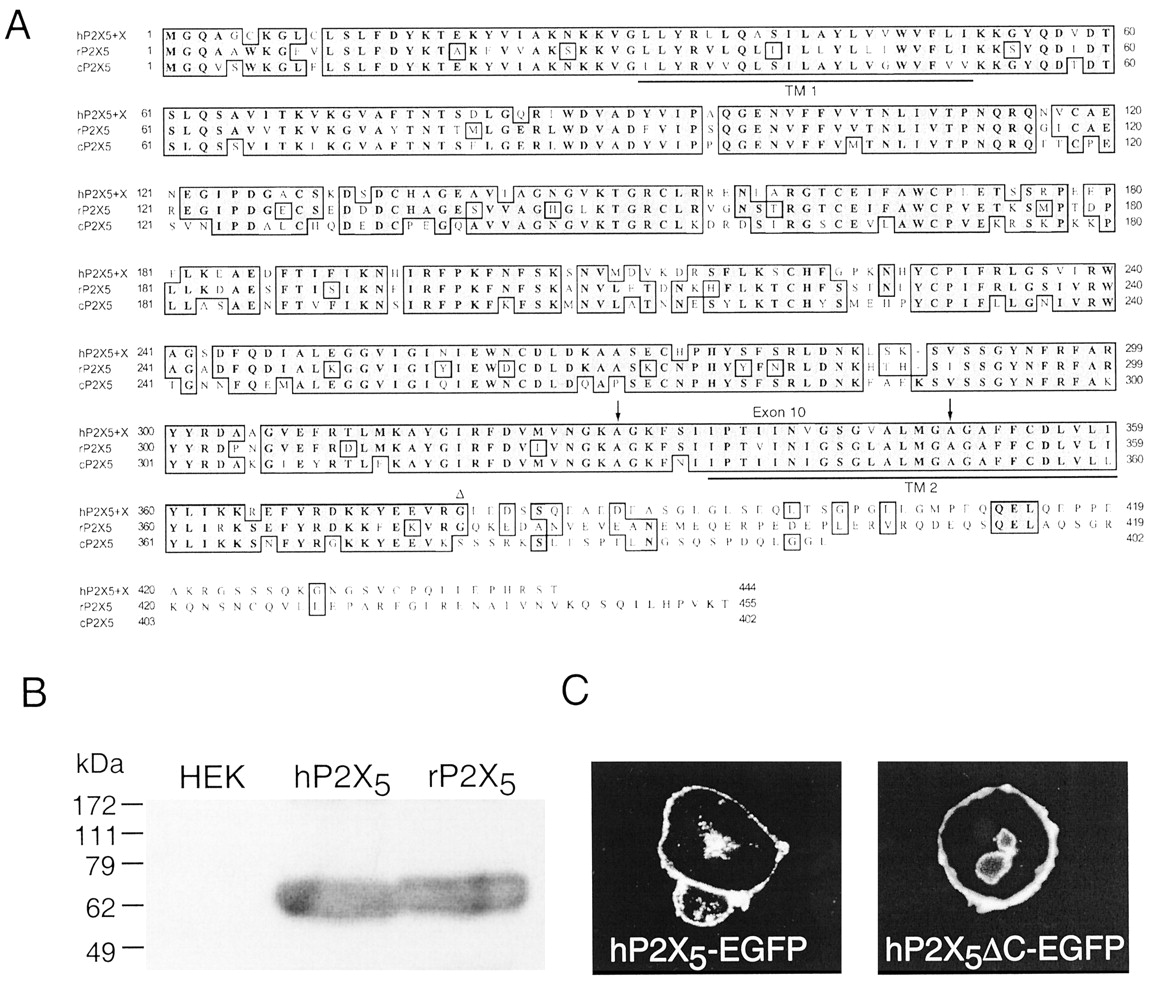
Construction and expression of full-length hP2X5 receptor. A, amino acid sequence alignment of human, rat, and chick P2X5 receptors. Transmembrane domains (TM1, TM2) are underlined, exon 10 is indicated by two arrows, and the C-terminal truncation site on rat and human P2X5 is indicated by Δ. B, Western blot of epitope-tagged human and rat P2X5 receptors; lane 1 is untransfected HEK cells. C, GFP fluorescence in HEK293 cells transfected with full-length hP2X5-EGFP or C-terminal truncated hP2X5-EGFP. Both proteins localize to the plasma membrane.
For comparison of expression levels between human P2X5 and rat P2X5 receptors we used a C-terminal EYMPME (EE) epitope (Kim et al., 2001a). We also truncated the receptors at a position corresponding to the end of exon 11 (Cox et al., 2001); the final amino acid of these constructs was Gly378 in both the human and rat receptors; this construct is termed P2X5ΔC (Fig. 1A). The full-length and truncated forms of the human receptor were also subcloned into pEGFP-N2 or pEGFP-N3 vectors (BD Biosciences Clontech, Palo Alto, CA) to make P2X5-EGFP and P2X5ΔC-EGFP. We made point-mutated receptors using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Transient Transfections, GFP Fluorescence, and Immunoblots. The methods were as described in detail previously (Kim et al., 2001a,b). In brief, HEK293 cells were plated in 35-mm Petri dishes, transfected using LipofectAMINE 2000 (Invitrogen) (1 μg of cDNA/2 × 105 cells), and studied 24 to 48 h after transfection. GFP-fused P2X5 receptors were plated on glass coverslips, fixed for 10 min with 4% paraformaldehyde, washed twice with physiological saline, and photographed using a CCD camera and an Olympus BX40 fluorescence microscope. Immunoprecipitation and Western blots were carried out using EE-tagged P2X5 receptors as described previously (Kim et al., 2001a,b). Protein was bound to anti-EE monoclonal antibody (BabCo, Richmond, CA) and precipitated with γ-bind G-Sepharose (Amersham Biosciences, Uppsala, Sweden). Immune complex was then dissociated from γ-bind G-Sepharose, applied to SDS-PAGE, and blotted with anti-EE polyclonal Ab. Molecular weights were calculated using GeneSnap and GeneTools software (Syngene, Cambridge UK).
Electrophysiology and Fluorescent Imaging. Whole-cell patch clamp recordings were obtained using a HEKA EPC9 amplifier and Pulse software (HEKA, Lambrecht Germany) as described previously (Virginio et al., 1998a; Jiang et al., 2001). The internal solution was 172 mM Na+ 148 mM Cl–, 10 mM EGTA, and 10 mM HEPES. An agar bridge (3 M KCl) was used for the indifferent electrode. The standard external solution was 151 mM Na+, 2 mM Ca2+, 1 mM Mg2+, 2 mM K+, 155 mM Cl–, 10 mM HEPES, and 13 mM glucose. We used simplified external solutions for measurements of permeability ratios. Solution 147NaCl contained 151 mM Na+, 0.3 mM Ca2+, and 148 mM Cl–; solution 40NaCl contained 44 mM Na+, 0.3 mM Ca2+, and 41 mM Cl–; solution 110CaCl2 contained 112 mM Ca2+ and 220 mM Cl–; solution 154NMDG contained 154 mM NMDG+ and 146 mM Cl–; and solution 147gluc contained 151 mM Na+ and 147 mM gluconate–. These solutions also contained HEPES and glucose; osmolarity was 295 to 315 mOsm, and the adjustment of the pH to 7.3 with NaOH, Ca(OH)2, or HCl accounts for the difference between the added salt concentrations and the final ion concentrations. Drug applications were made using the RSC 200 system (Biologic Science Instruments, Grenoble, France). In these experiments, solution exchange times were estimated from changes in junction potentials when a solution of 147 mM potassium gluconate was applied; these averaged 80 ms. Antagonists were applied for 1 to 4 min before, and throughout, agonist applications; agonist applications were repeated at 2- to 4-min intervals.
For reversal potential measurements, the whole-cell configuration was established in standard external solution and the external solution was changed to solution 147NaCl; the reversal potential was obtained by a ramp voltage command (–120 to 40 mV; 1-s duration). The solution was then exchanged with one of solutions described above, and the reversal potential was measured again. The reversal potentials reported have been corrected for liquid junction potentials, which were calculated using Henderson's equation (Ives and Janz, 1961), and in any case did not exceed 4 mV. The permeability ratios (PX/PNa) were derived from the reversal potentials (Erev) as follows [where x = Erev F/RT (F is the Faraday constant, R is the gas constant, and T is the absolute temperature)]:
-
For solution 40NaCl: PCl/PNa = {(1 – exp(–x)) ([Na]o – [Na]i exp(x))}/{([Cl]iexp(–x) – [Cl]o) (1 – exp(x))}. The estimate of PCl/PNa (0.52) was used for further estimates of permeability ratios.
-
For solution 110CaCl2:PCa/PNa = (A + B)/4[Ca]o where A = {[Na]i exp(x)(1 + exp(x))} and B = PCl/PNa (1 + exp(x)) ([Cl]o exp(x) – [Cl]i).
-
For solution 147NMDG:PNMDG/PNa = (C + D)/E where C = ([Na]i exp(x))/(1 – exp(x)), D = PCl/PNa ([Cl]iexp(–x) – [Cl]o)/(1 – exp(–x)) and E = [NMDG]o/(1 – exp(x)).
-
For solution 147gluc: Pgluc/PNa = (C+D)/F where F = [gluc]o/(1 – exp(–x)).
Ion concentrations were converted to activities using the following coefficients: γNa = 0.75, γCl = 0.75, γCa = 0.28, γNMDG = 0.81, and γgluc = 0.81.
YO-PRO-1 fluorescence was measured as described previously (Virginio et al., 1999a); we used a Zeiss Axiovert 100 and Fluar 20× objective with Photonics monochrometer imaging (Photonics, Germany).
Data Analysis. Figures show mean ± S.E.M for number of cells and curves fitted from pooled data using Kaleidagraph (Synergy Software, Reading PA). Onset and offset of ATP action were approximated by single exponentials of time constant τon and τoff; the corresponding rate constants (k1 = {(1/τon) – k–1}/[ATP] and k–1 = 1/τoff) were assumed to be related to membrane potential by an expression of the form k(V) = k(0) exp(–z δ V F/R T) where V is the membrane potential, δ is the fraction of the membrane electric field acting on a sensor of valence z, and k(0) is the rate constant at 0 mV. Agonist concentration-response curves were fit by I/Imax = 100 ([A]nH/([A]nH + EC50nH)), where I is the peak current evoked by agonist concentration [A] expressed as percentage of maximal current evoked by ATP, nH is the Hill coefficient, and EC50 is the half-maximal agonist concentration. Antagonist concentration-inhibition curves were fit to I/Io = 100 ([B]nH/([B]nH + EC50nH)), where I is the peak current at a given antagonist concentration [B] as a percentage of current in absence of antagonist (Io) and IC50 is the antagonist concentration that inhibits agonist current by 50%. Numerical estimates of EC50 and IC50 were obtained by curve-fitting to individual cells.
Results
Expression of Human P2X5 Receptor. Transfection of HEK293 cells with either human P2X5 or rat P2X5 receptor cDNAs resulted, within 1 to 2 days, in strong expression of the protein (Fig. 1, B and C). Immunoblotting of proteins from these cells showed single bands at 61 and 62 kDa, respectively (Fig. 1B), which is appropriate to glycosylated proteins with calculated molecular masses of the two P2X5 proteins (51 and 52 kDa). This was primarily localized to the plasma membrane by immunocytochemistry (Fig. 1C). Although Western blotting showed equivalent protein expression of the rat and human P2X5 receptor, currents evoked by ATP at the rat P2X5 receptor were extremely small (<50 pA in these experiments; see also Collo et al., 1996; Garcia-Guzman et al., 1996), whereas ATP-evoked currents at the human P2X5 receptor were large (1–5nAat –60 mV) (Fig. 2). We observed no significant differences in the immunohistochemistry or in the ATP-evoked currents between HEK293 cells transfected with an epitope-tagged human P2X5 receptor (hP2X5-EE) or with an EGFP-fused hP2X5 receptor (hP2X5-EGFP) (Fig. 1C); therefore, we have included some experiments with these constructs in the results of the functional experiments.
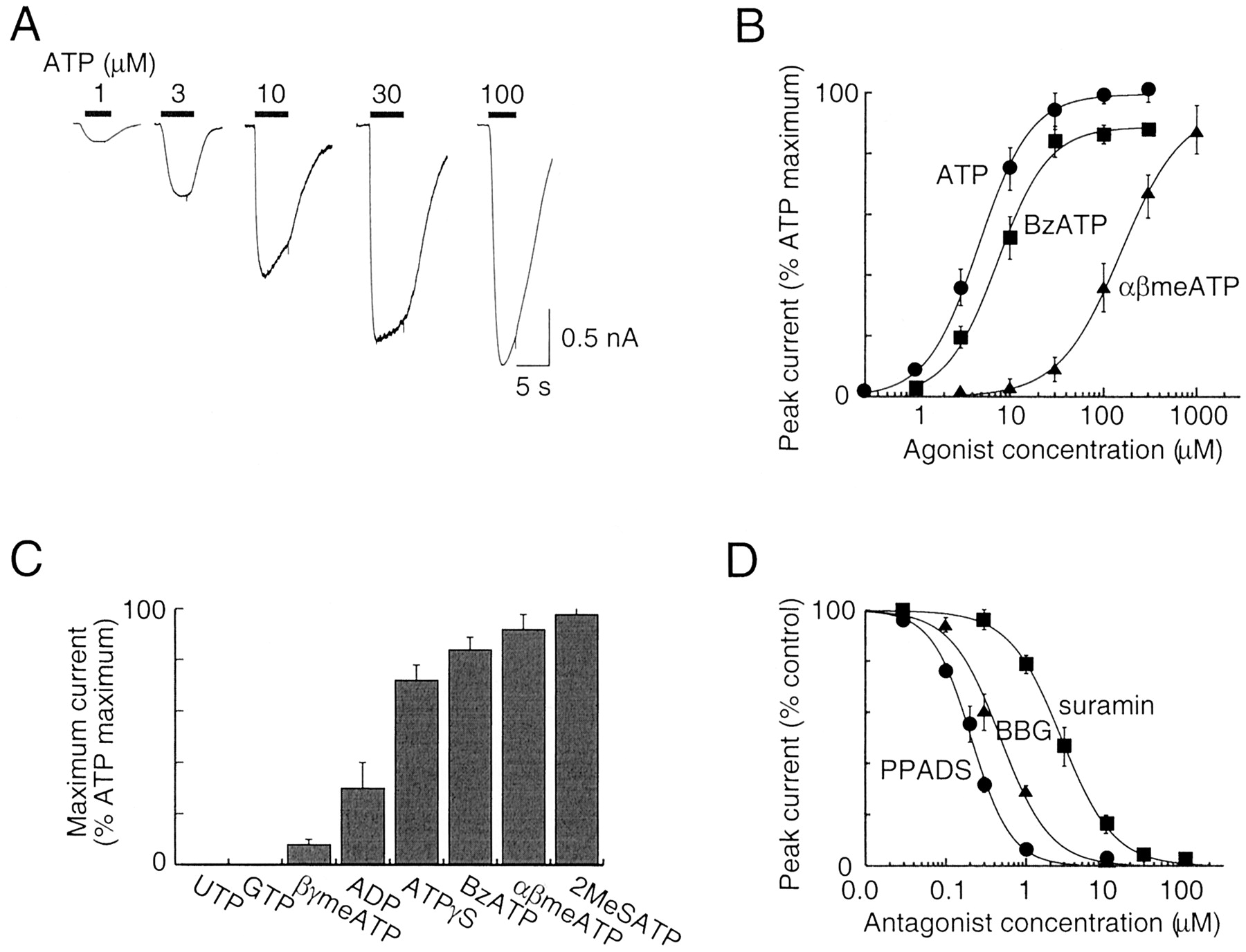
Pharmacological properties of full-length human P2X5 receptor. A, inward currents recorded at –60 mV in response to 5-s applications of ATP. B, agonist concentration-response curves for ATP, BzATP, and αβmeATP. Results are plotted as percentage of maximum ATP current recorded in each cell; each point is mean ± S.E.M. (n = 4–12 points). C, maximum currents caused by various agonists (100 μM) relative to that evoked by ATP; each bar is mean ± S.E.M. (n = 3–12). D, antagonist inhibition curves for PPADS, Brilliant Blue G, and suramin at human P2X5 receptor; ATP (10 μM) was the agonist. Each point is mean ± S.E.M. (n = 3–8).
Agonist and Antagonist Actions at Human P2X5 Receptor. Application of ATP (0.3–3 μM; 1–5 s) evoked inward currents (at –60 mV) that were sustained; the currents elicited by higher concentrations declined during the application (Fig. 2A). Concentration-response curves for ATP, BzATP, and αβmeATP yielded EC50 values of 4.1 ± 0.5 (n = 5), 5.7 ± 0.9 (n = 5), and 161 ± 35 μM (n = 4), respectively (Fig. 2B). BzATP, αβmeATP, and 2-methylthio-ATP elicited >85% of the maximal ATP current; adenosine-5′-O-(3-thio)triphosphate, ADP, and βγ-methylene-ATP acted as less full agonists (Fig. 2C). Thus, the sensitivity to αβmeATP is much greater than that observed for the rat P2X5 receptor (no effect of 300 μM αβmeATP; Collo et al., 1996), somewhat greater than that found for the rat P2X2 receptor (Spelta et al., 2002) but considerably less than that reported for P2X1, P2X3, P2X2/3, and rP2X1/5 receptors (see North and Surprenant, 2000).
PPADS, Brilliant Blue G (BBG), and suramin inhibited the ATP-evoked current; this was concentration-dependent with IC50 values of 0.2 ± 0.02 (n = 3), 0.53 ± 0.04 (n = 3), and 2.9 ± 0.4 (n = 5) μM, respectively (Fig. 2D). The sensitivity to inhibition by PPADS observed for the human P2X5 receptor is about 10-fold higher than previously reported for other heterologously expressed P2X receptors (North and Surprenant, 2000). The inhibition by BBG is intermediate between the high sensitivity of the rat and human P2X7 receptor (IC50, 10 and 200 nM, respectively) and the very low sensitivity of the other P2X receptors (IC50 values >5–10 μM) (Jiang et al., 2000). 2′,3′-O-(2′,4′,6′-Trinitrophenol)-ATP, which is a nanomolar affinity antagonist at the P2X1, P2X3, and P2X2/3 receptors (Virginio et al., 1998b; North and Surprenant, 2000), only minimally inhibited the ATP-evoked currents at the human P2X5 receptor (11 ± 5% inhibition at 1 μM, n = 3).
Kinetics of Agonist Action. There were some features of the action of ATP that differed from those observed previously at other P2X receptors. First, the onset of the current was slower. We measured τon at the human P2X5 and rat P2X2 receptors under the similar conditions using near-maximal ATP concentrations (10 and 30 μM, respectively; holding potential, –60 mV); the values were 410 ± 50 ms (n = 8) and 161 ± 10 ms (n = 4). The rate of onset of the current showed little if any dependence on membrane potential (Fig. 3A), but the rate of the offset of the response upon removal of agonist was almost 10-fold slower at +30 mV than at –60 mV (Fig. 3, B and C). The offset rate constant was 0.2/s at 0 mV, and the fraction of the membrane field sensed by the channel closing (assuming a monovalent sensor) was 0.6. The offset of the current at the rat P2X2 receptor was some 10 times faster and was not voltage-dependent (τoff was about 300 ms at all potentials; Fig. 3C).

Kinetics of human P2X5-mediated currents. A, upper traces show inward currents (holding potential –30 mV) in response to 2- and 10-s applications of ATP (3 μM, approximately EC50). Lower traces show typical outward currents at +30 mV. B, offset of response to ATP (3 μM) at –60, –30, and 30 mV (current at +30 mV inverted; currents scaled for comparison); broken lines are exponential fits (time constants 1, 3.3, and 6.3 s). C, summary data from experiments such as those shown in B. ○, results from parallel experiments on rat P2X2 receptor; ATP was applied at 30 μM, which is close to the EC90 (n = 5 for all points).
The desensitization of the current that occurred with longer agonist applications was also voltage-dependent (Fig. 3A). At a holding potential of –30 mV, the current at the end of a 10-s application of ATP (3 μM) had declined to 48 ± 11% (n = 6) of its peak value; the corresponding value at +30 mV was 86 ± 3% (n = 8). The ATP-evoked current at the chick and bullfrog P2X5 receptor shows rapid desensitization (at –60 mV) that is prevented by removal of external calcium (Bo et al., 2000; Ruppelt et al., 2001). However, in the present study on the human P2X5 receptor, removal of calcium from the superfusion solution did not alter the ATP concentration-response curve (n = 3), the onset or offset kinetics of the ATP current at any membrane potential (n = 6), or the desensitization during the continued presence of agonist (n = 3).
Ion Permeability. We investigated the ion permeability at the human P2X5 receptor by measuring reversal potentials with patch pipettes containing 147 mM NaCl, and changing the composition of the external solutions. The reversal potential in solution 147NaCl was –0.4 ± 1.1 mV (n = 6). We first estimated PCl/PNa with solution 40NaCl (Fig. 4). Table 1 shows that the shift in reversal potential (–12 mV) was much less than expected for a chloride-impermeable channel (the theoretical value would be –33 mV). Thus, the human P2X5 receptor has a very significant permeability to chloride ions (PCl/PNa, 0.5; Table 1), and this was taken into account in estimates of other permeability ratios when chloride ions were present (see Materials and Methods). We substituted extracellular chloride with gluconate (solution 147gluc) and found that gluconate permeability was insignificant (Table 1). By using extracellular NMDG (solution 154NMDG) and calcium chloride (solution 110CaCl2), we found that that the human P2X5 receptor was quite permeable to NMDG (PNMDG/PNa, 0.37) and calcium (PCa/PNa, 1.5) (Fig. 4; Table 1). Parallel experiments were carried out for direct comparison on cells expressing the rat P2X2 and P2X2/3 receptors; we found that these receptors have very low permeability to NMDG (PNMDG/PNa < 0.05) (see also Virginio et al., 1999b) and chloride (PCl/PNa < 0.02) ions (Table 1).

Human P2X5 receptor ion channel is permeable to large cations and chloride. Currents in response to ramp voltage commands in external solutions containing (each control) 147NaCl, and solution 40NaCl, solution 110CaCl2, solution 147NMDG, and solution 147gluc. In each case, arrows indicate change in reversal potential on changing from 147NaCl.
Reversal potential (Erev) and calculated permeability ratios (PX/PNa) for ATP-evoked currents. Values are the means ± S.E.M. for the number of cells indicated in parentheses
The significant difference in chloride permeability between the human P2X5 receptor and the P2X2 receptor led us to search for amino acid residues in or around the transmembrane domains that might differ between the two channels. A possible candidate is Lys52 of the hP2X5 receptor, which corresponds in position to Gln52 of the rat P2X2 receptor; P2X5 receptors are unusual in having lysine in this position, where P2X1, P2X2, P2X3, P2X4, and P2X7 subunits have a negatively charged side chain (Gln, Asp, Asn, or Glu). This position is at the outer end of the first transmembrane domain, and we speculated that the difference in charge might contribute to the difference in anion permeability. We reversed the charge by mutagenesis and measured the PCl/PNa ratios. The value for the human P2X5 receptor was not different from wild type, and the rat P2X2 receptor with the complementary mutation [Q52K] remained impermeable to chloride (Table 1).
In the case of the P2X7 receptor (Surprenant et al., 1996; Virginio et al., 1999b), the permeability to NMDG is very low at the beginning of the ATP application and progressively increases over several seconds. Indeed, when the application of agonist is kept brief (≈2 s), there is no significant permeability to NMDG (Surprenant et al., 1996). NMDG permeability has also been found for the P2X2 and P2X4 receptor, but in these cases, it also develops during several seconds of agonist application (Khakh et al., 1999; Virginio et al., 1999b). This progressive increase in permeability is detected as a time-dependent change in reversal potential during a sustained application of agonist. We therefore directly compared the kinetics of NMDG permeability at the rat P2X7 and the human P2X5 receptors under the same conditions (Fig. 5). As reported previously (Virginio et al., 1999a,b), the reversal potential in NMDG chloride shifted over an exponential time course from about –90 mV (PNMDG/PNa < 0.02) in the first several seconds of agonist application to a steady-state value of approximately –21 mV (PNMDG/PNa 0.25) after 30 s (Fig. 5, B and C). However, for the human P2X5 receptor, the reversal potential in NMDG chloride was –15 ± 2 mV (n = 7) as soon as it was feasible to measure it (1–4 s after ATP application) and did not change during the subsequent 30 s of receptor stimulation (Fig. 5, A and C). Indeed, the onset kinetics of the inward current were no different in solutions containing sodium or NMDG as the only extracellular cation (Figs. 3 and 5), indicating that, within our resolution, the channel is NMDG-permeable from the time at which it opens. The permeability to NMDG was observed in external solutions containing no divalent cations (used for more accurate measurement of reversal potentials), but it was also observed in solutions containing NMDG, calcium (2 mM), and magnesium (1 mM) (n = 4).

NMDG permeability is not time-dependent at the human P2X5 receptor. A, left, with NMDG as the extracellular cation, application of ATP evokes an inward current that declines during 30 s. Holding potential, –60 mV. Right, the reversal potential of the current remains at about –15 mV throughout. B, left, a similar experiment on a cell expressing rat P2X7 receptor. The current is initially outward, because the reversal potential is negative to the holding potential (–60 mV). Right, the reversal potential shifts in a positive direction during 20 s of BzATP application. C, summary of results from experiments shown in A and B [n = 4 (human P2X5), n = 3 (rat P2X7)].
YO-PRO-1 Permeability of Human P2X5 Receptor. YO-PRO-1 is a propidium dye that becomes fluorescent when it intercalates nucleic acid. We followed its entry into cells by fluorescence imaging. We compared the rate of YO-PRO-1 uptake induced by ATP and BzATP at the human P2X5 and rat P2X7 receptors (Fig. 6). Neither BzATP (100 μM) nor ATP (1 mM) evoked any YO-PRO-1 fluorescence in mock-transfected HEK293 cells (n > 40 cells from three separate experiments; Fig. 6A). The absolute fluorescence measured 60 s after the addition of a maximal concentration of either ATP or BzATP was ∼2-fold greater at the human P2X5 receptor (Fig. 6A), whereas the rate of YO-PRO-1 uptake was almost 4-fold greater at the human P2X5 receptor (Fig. 6B). For the rat P2X7 receptor, the rate of YO-PRO-1 uptake was greater when the external solutions contained no divalent cations (see Surprenant et al., 1996), but this difference was not present for the P2X5 receptor (Fig. 6B).
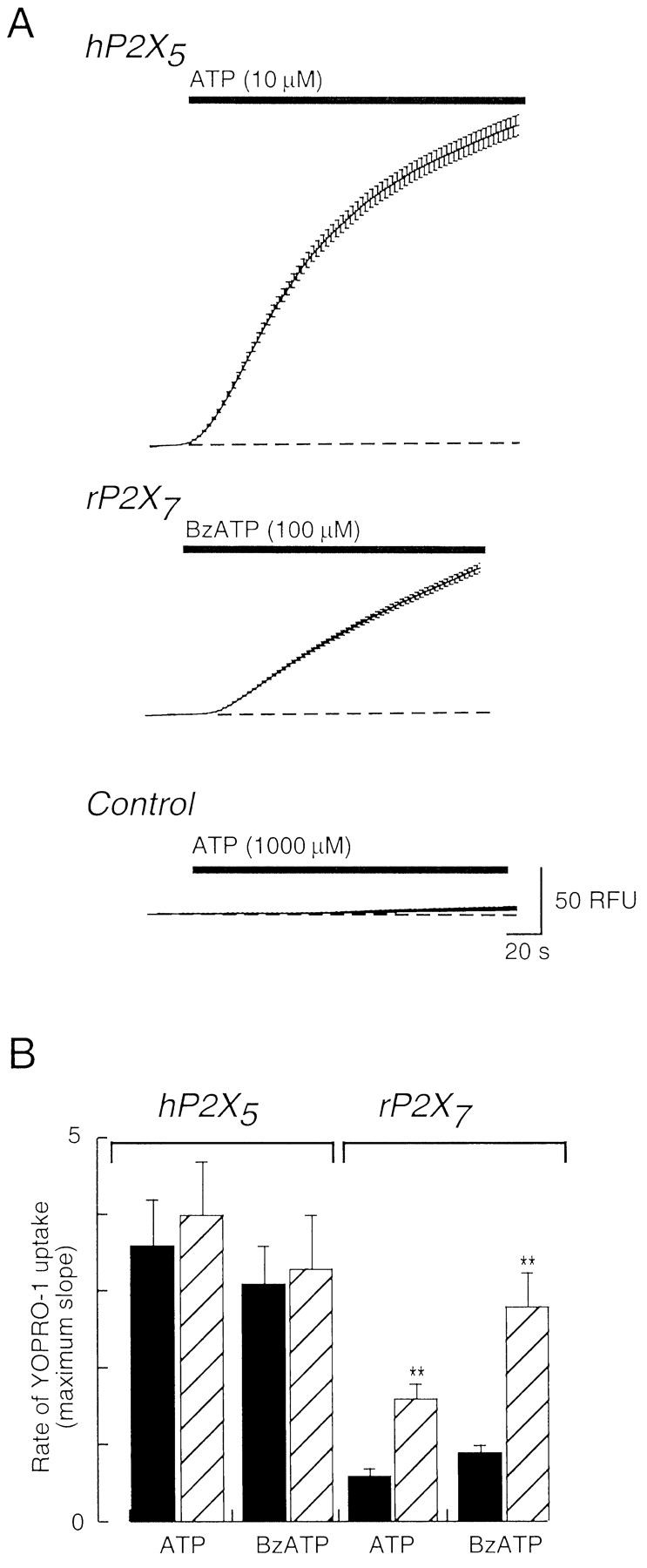
Uptake of YO-PRO-1 by cells expressing human P2X5 receptors. A, fluorescence of individual HEK cells was measured, and average values are plotted with S.E.M. Top, increase in fluorescence occurring during application of ATP as shown. Middle, a similar experiment on HEK cells expressing rat P2X7 receptors. Bottom, a similar experiment on mock-transfected HEK cells. B, summary of results from experiments such as those shown in A. ▪, experiments in standard extracellular solution; ▨, experiments in solution containing 0.3 mM calcium and 0 mM magnesium. Agonist concentrations used were hP2X5, 10 μM ATP, 100 μM BzATP; rP2X7, 1 mM ATP, 100 μM BzATP (n = 3–5). **, p < 0.05.
Comparison of Human and Rat P2X5 Receptors. We were struck by the robust expression of ionic currents by HEK cells transfected with the human P2X5 receptor, compared with the rat P2X5 receptor (Collo et al., 1996; Garcia-Guzman et al., 1996). Moreover, there were clear functional differences between the properties of the two homomers (e.g., increased sensitivity to ATP and αβmeATP, voltage-dependent kinetics). We sought to determine whether these differences might be related to the different C-terminal regions between the two receptors (Fig. 1A).
In general, the C-terminal regions of the P2X receptors show little conservation of sequence among the seven subtypes, but within each subtype, there is significant homology among species orthologs. However, for the P2X5 receptor, the C-terminal regions are related in sequence only between rat and mouse but not in the case of bullfrog, zebrafish, chicken, and human; in these species, all relatedness ends at the splice site between exons 11 and 12 (Cox et al., 2001). We therefore compared the properties of the human and rat receptors truncated at this point (Fig. 1A). Both truncated receptors (with C-terminal EGFP fusions) localized to the plasma membrane in a manner similar to that of the wild-type receptors, although they both showed a lower level of immunofluorescence than did the wild type. The maximum current amplitudes were slightly lower in each of the truncated receptors compared with their respective wild-type receptors. The maximum ATP-evoked currents at wild-type and truncated rat P2X5 receptors were 49 ± 14 pA and 28 ± 9 pA (n = 10 each); at wild-type and truncated human P2X5 receptor, maximum currents were 2.9 ± 0.5 and 0.95 ± 0.1 nA (n = 9 each). However, no other significant differences in the functional properties were observed. The EC50 for ATP was 3.3 ± 0.2 μM (n = 3) at the truncated human P2X5 receptor; αβmeATP (100 μM) did not evoke any current at the truncated rat P2X5 receptor but was almost a full agonist at the truncated human P2X5 receptor (n = 4), and the truncated human P2X5 receptor also showed a voltage-dependence time course of current offset (n = 5).
There are three amino acid differences between human and rat receptors in the part of the protein encoded by exon 10. We made a chimeric receptor in which the sequence coded by exon 10 from the rat receptor was used to replace the equivalent sequence of the human receptor. The resulting human receptor [P2X5, V337I/V340I/V344L] had properties indistinguishable from those of the human P2X5 receptor. There are only minor differences between the human and rat P2X5 receptors in the regions before and including the first transmembrane domain (1–50) and between the end of the second transmembrane domain and the truncation point (334–379); it is likely that different residues in the ectodomains may account for the different properties.
Discussion
Our work shows that a P2X5 receptor containing all 13 exons encoded by the human gene expresses well in HEK293 cells and produces robust currents in response to ATP. It has previously been well documented that the form of the receptor that does not contain exon 10 can not form homomeric channels despite apparently good membrane expression (Lê et al., 1997). Human tissues seem to contain more P2X receptor forms that are spliced so as to produce nonfunctional proteins than are found in rodent orthologs (e.g., P2X2, Lynch et al., 1999; P2X6, Urano et al., 1997; see North, 2002); this might reflect a loss of function during evolution. The presence of a polymorphism at a critical position in the human P2X5 gene strongly indicates that a subset of humans will process and translate their RNA to make the protein that we have expressed. Presumably, mice and chickens also make a protein corresponding to the `full-length' 13-exon form (see Introduction).
When expressed in mammalian cells or oocytes from cDNAs, the human (this study), chick (Bo et al., 2000; Ruppelt et al., 2001), and bullfrog (Jensik et al., 2001) P2X5 receptors express well; it is less clear why currents at the rat (this study; Collo et al., 1996; Garcia-Guzman et al., 1996), mouse (Cox et al., 2001), and zebrafish receptors (Diaz-Hernandez et al., 2002) do not. The rat sequence contains in its C terminus a sequence Arg404-Val-Arg; this is similar in the mouse (Arg404-Val-His). The RXR motif is a well known endoplasmic reticulum retention motif and has been implicated in the trafficking of several ion channels to the plasma membrane (Ma and Jan, 2002). We mutated this sequence to Ala-Ala-Ala in the rat P2X5 receptor but found that the currents recorded from transfected HEK293 cells were still very small (L.-H. Jiang, unpublished observations). In any event, using Western blotting and immunohistochemistry, we detected no obvious difference between the expression of rat and human P2X5 receptors.
Compared with other homomeric P2X receptors, the pharmacological profile of the human P2X5 receptor is most similar to the P2X2 receptor (North and Surprenant, 2000). It is rather more sensitive to the agonists ATP (EC50 ≈ 5 μM) and αβmeATP (EC50 ≈ 150 μM) and the antagonist PPADS (IC50 < 1 μM). The current shows much less rectification than is observed for the P2X2 receptor; in this respect, it is more similar to homomeric P2X4 and P2X7 receptors (see North, 2002). Compared with P2X5 receptors from other species [chick (Bo et al., 2000; Ruppelt et al., 2001) and bullfrog (Jensik et al., 2001)], the most noticeable difference is the relatively slower desensitization of the current at the human receptor. As seen for the chick receptor (Ruppelt et al., 2001), we observed that holding the membrane potential at positive could largely prevent desensitization. In the chick, calcium entry might contribute to the desensitization because removing the extracellular calcium ions also prevented it; we did not find this to be the case for the human P2X5 receptor.
The kinetics of channel opening, as best we could estimate with the present approach, corresponded to a macroscopic k+1 of 1 μM/s, which is approximately similar to that observed for P2X2 receptors (Ding and Sachs, 1999); this rate showed only little voltage dependence. On the other hand, the rate of decline of the current ranged from 1/s at –60 mV to 0.15/s at +30 mV (Fig. 2). This is in contrast to the rat P2X2 receptor, where the offset of current shows little or no voltage-dependence (Fig. 2). Taken together, the results indicate that depolarized membrane potentials tend to stabilize the human P2X5 receptor in an open state, from which it less easily enters either a desensitized or a closed state.
The permeability of the human P2X5 receptors also shows several unique features. Our measurements of reversal potential when the concentration of extracellular sodium chloride ions was reduced to 40 mM indicate clearly that the channel has substantial chloride permeability (PCl/PNa ≈ 0.5). Although it has been widely assumed that P2X receptors are cation-selective, this has often not been tested directly in heterologous expression systems. In the present study, we measured the reversal potential for currents at the homomeric P2X2 and heteromeric P2X2/3 receptors; these corresponded to the theoretical value for a channel that was impermeable to chloride (Table 1). However, the chick P2X5 receptor has substantial chloride permeability (Ruppelt et al., 2001); their value for PCs/PCl of about 2 corresponds well with our present measurement for the human P2X5 receptor (if we assume that PNa ≈ PCs). The molecular basis for the relative high permeability to chloride ions is not understood. We noticed a lysine residue near the outer end of the first transmembrane domain of the P2X5 receptor sequences. Of the other P2X receptor subunits, lysine is found only in P2X6 and no permeability information is available; the residue at this position is negatively charged in all the other subunits. However, we were unable to change the chloride permeability by making the appropriate amino acid exchanges at this position. Further knowledge of the anion permeabilities of other (homomeric) P2X receptors would be very helpful to inform the continuation of such a mutagenesis approach to the structural basis of chloride permeability in the human P2X5 channel.
The chloride permeability of the receptor is of particular interest because ATP-activated currents in skeletal muscle have been repeatedly shown to involve a chloride permeability. The original evidence that ATP directly gates the opening of ion channels was based on whole-cell and single-channel recordings from embryonic chick skeletal muscle myotubes (Kolb and Wakelam, 1983). Subsequent studies in chick and frog showed striking developmental regulation of these channels; they disappear during late embryogenesis but reappear in the adult after denervation (Hume and Honig, 1986; Igusa, 1988; Thomas and Hume, 1990a; Thomas et al., 1991; Wells et al., 1995). They are also characterized by both cation permeability and a “substantial increase in chloride permeability” (Hume and Thomas, 1988; Thomas and Hume, 1990b). More recently, chick embryonic skeletal muscle has been shown to express high levels of both P2X5 and P2X6 receptor immunoreactivity, which disappears with development (Meyer et al., 1999).
All these observations might be consistent with the notion that ATP-gated channels in embryonic chick skeletal muscle are composed of P2X5 receptors. There is, however, evidence against the view that the chick receptor is homomeric P2X5. For example, αβmeATP was ineffective at the receptor on muscle cells cultured from 10- or 11-day-old chick embryos (Hume and Honig, 1986; Thomas et al., 1991), whereas it does activate currents in oocytes expressing chick P2X5 receptors (EC50 ≈ 30 μM; Ruppelt et al., 2001). A recent study of ATP-gated currents and intracellular calcium responses in mouse and human skeletal muscle has shown a similar developmental pattern of expression of ATP-activated responses as that observed in chick. The ATP responses activated by ATP and BzATP, but they were activated only very weakly by αβmeATP, and they showed no desensitization (Cseri et al., 2002). As for the chick, these observations suggest that the native mammalian receptor is not likely to be homomeric P2X5. The predominant expression of P2X6 subunits in skeletal muscle (Urano et al., 1997) suggests that possibility that the native receptor might include both P2X5 and P2X6 subunits. A functional role for ATP in the differentiation of rat skeletal muscle has recently been suggested (Ryten et al., 2002). The regeneration of skeletal muscle cells after damage involves resident satellite cells being stimulated to differentiate into myotubes. ATP stimulated this differentiation, and evidence from immunohistochemistry, reverse transcription-polymerase chain reaction, and electrophysiology strongly implicated involvement of the P2X5 subunit.
The cation permeation properties of the human P2X5 receptor also exhibit some novel features. Permeability to the large cation NMDG has been demonstrated in the case of homomeric P2X2, P2X4, and P2X7 receptors and heteromeric P2X2/3 receptors (Virginio et al., 1999a,b; Khakh et al., 1999). The human P2X5 receptor is also permeable to NMDG and the relative permeability (Table 1; PNMDG/PNa = 0.37) is similar that described previously for the other receptors. The striking difference for the human P2X5 receptor is that there is no detectable time delay in the development of the NMDG-permeable state. NMDG permeability was also reported for oocytes expressing the bullfrog P2X5 receptor, but this was only observed when the extracellular solution contained no divalent cations (Jensik et al., 2001); it seemed to develop over several seconds, but it is difficult to make direct kinetic comparisons when whole oocytes and small mammalian cells are used as expression systems. YO-PRO-1 is a larger cation than NMDG (19 × 10 × 5.5 Å compared with 10 × 7.6 × 5.5 Å) and divalent rather than monovalent. It has the advantage that its permeation can be followed in physiological solutions, which is of course not possible when reversal potentials are measured. Human P2X5 receptors exhibit uptake of YO-PRO-1, which is at least as robust as that observed for P2X7 receptors (Fig. 5; also Surprenant et al., 1996; Rassendren et al., 1997). It is not possible to say from these experiments how rapidly the permeability to YO-PRO-1 develops in human P2X5 receptors, but it would seem to be considerably slower than that observed for NMDG.
In summary, we have constructed and expressed a human P2X5 receptor cDNA and characterized some of its pharmacological and biophysical properties. Several of these properties are quite distinct from those of other mammalian P2X receptors. These include a significant permeability to chloride ions, which is of interest because skeletal muscle expresses abundant P2X5 receptor subunits and because ATP-activated currents in skeletal muscle are also chloride-permeable. There is permeability to the large cation NMDG that seems to develop as quickly as that to sodium ions; in other P2X receptors, the increase in NMDG permeability occurs only several seconds after the initial increase in sodium permeability. Finally, we have drawn attention to a single-nucleotide polymorphism that will effectively determine whether an individual organism makes a functioning P2X5 receptor or a nonfunctional form that lacks exon 10.
Footnotes
-
↵ 1 Present address: Section of Functional Genomics, Division of Genomic Medicine, University of Sheffield, Royal Hallamshire Hospital, M-floor, Sheffield S10 2JF, United Kingdom.
-
This work was supported by financial assistance from The Wellcome Trust (to A.S.) and AstraZeneca (to R.A.N.).
-
X.B. and L.-H.J. contributed equally to this work.
-
ABBREVIATIONS: HEK, human embryonic kidney; αβmeATP, αβ-methylene-ATP; PPADS, pyridoxal-5-phosphate-6-azo 2′,4′-disulfonic acid; NMDG, N-methyl-d-glucamine; EST, expressed sequence tag; EE, EYMPME epitope; GFP, green fluorescent protein; EGFP, enhanced green fluorescent protein; YO-PRO-1, quinolinium,4-[(3-methyl-2-(3H)-benzoxazolylidene)methyl]-1-[3-(triethylammonio)propyl]di-iodide; BzATP, 2′,3′-O-(4 benzoyl)benzoyl-ATP.
- Received August 26, 2002.
- Accepted March 6, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}