


Abstract
Ligand-mediated activation of the pregnane X receptor (PXR, NR1I2) is postulated to affect both hepatic and intestinal gene expression, because of the presence of this nuclear receptor in these important drug metabolizing organs; as such, activation of this receptor may elicit the coordinated regulation of PXR target genes in both tissues. Induction of hepatic and intestinal drug metabolism can contribute to the increased metabolism of drugs, and can result in adverse or undesirable drug-drug interactions. 2(S)-((3,5-bis(Trifluoromethyl)benzyl)-oxy)-3(S)phenyl-4-((3-oxo-1,2,4-triazol-5-yl)methyl)morpholine (L-742694) is a potent activator of the rat PXR and was characterized for its effects on hepatic and intestinal gene expression in female Sprague-Dawley rats by DNA microarray analysis. Transcriptional profiling in liver and small intestine revealed that L-742694 and dexamethasone (DEX) induced the prototypical battery of PXR target genes in liver, including CYP3A, Oatp2, and UGT1A1. In addition, both DEX and L-742694 induced common gene expression profiles that were specific to liver or small intestine, but there was a distinct lack of coordinated gene expression of genes common to both tissues. This pattern of gene regulation occurred in liver and small intestine independent of PXR, constitutive androstane receptor, or hepatic nuclear factor-4α expression, suggesting that other factors are involved in controlling the extent of coordinated gene expression in response to a PXR agonist. Overall, these results suggest that ligand-mediated activation of PXR and induction of hepatic, rather than small intestinal, drug metabolism genes would contribute to the increased metabolism of orally administered pharmaceuticals.
Orally administered drugs are often subject to first-pass metabolism in the gut and liver. Drugs can elicit induction of drug metabolism enzymes in these organs, potentially resulting in clinically relevant drug-drug interactions. The relative induction of genes in the liver and intestine, however, has not been thoroughly characterized. To understand the potential for new drugs to alter gene expression, genome-wide gene expression analyses that characterize chemical/drug induction of both hepatic and intestinal drug-metabolism enzymes, and drug-transport proteins, has been undertaken in the rat.
Induction of CYP3A4 in human liver or possibly intestine is responsible for an ever-expanding list of drug-drug interactions. Given that the CYP3A4 enzyme metabolizes an extensive array of marketed pharmaceuticals, any drug that induces the expression of the CYP3A4 gene has the potential to alter the pharmacokinetics of a coadministered drug, resulting in a perpetrator-victim scenario (Evans et al., 2003). Furthermore, xenobiotic mediated induction of the CYP3A family of genes occurs through activation of the pregnane X receptor (PXR; NR1I2), irrespective of species (i.e., human, mouse, and rat; Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998; Zhang et al., 1999). Ligand mediated activation of the PXR leads to PXR binding to a distal (-7.8 kilobases) xenobiotic-responsive element of the CYP3A4 transcription start site (Goodwin et al., 1999). PXR binds as a heterodimer with the retinoic acid receptor to a specific nuclear receptor binding motif, where the CYP3A4 binding motif consists of two half-sites (AG(T/G)TCA) separated by six nucleotides in an everted-repeat (ER-6) orientation. Similar distal enhancer elements in various orientations [e.g., ER-6, ER-8, direct repeat (DR)-3, DR-4, DR-5] have been reported in numerous other genes in both humans and rodents, including rat CYP3A (Kliewer et al., 1998, 2002).
The relevance of PXR to understanding xenobiotic induction of hepatic and intestinal gene expression in both humans and rodent preclinical species is derived from the tissue distribution of PXR. Localized expression of PXR to both the liver and intestinal tract (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998; Zhang et al., 1999; Jones et al., 2000), suggests that ligand-mediated activation of PXR and induction of PXR target genes in both tissues would be expected (Maglich et al., 2002). Furthermore, the liver and intestine express a similar compliment of drug metabolism enzymes and drug transport proteins that are regulated by PXR, including human and rodent orthologs of CYP3A, UGT1A, MRP2 (ABCC2), and MDR1 (ABCB1) (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998; Synold et al., 1999; Kast et al., 2002; Maglich et al., 2002; Chen et al., 2003; Xie et al., 2003). Of the genes regulated through ligand-mediated activation of PXR, many are central in the adaptive response of the organism to chemical insult, which include genes that encode for drug metabolism enzymes and xenobiotic transport proteins. Consequently, PXR-mediated induction of these drug-management genes facilitates excretion of potentially harmful chemicals from the organism. However, in cases in which a compound activates PXR, induces PXR target genes (e.g., CYP3A4), and perpetrates a clinically relevant drug-drug interaction, the pharmacokinetics and efficacy of a coadministered drug are compromised as a result of induction of hepatic and/or intestinal genes.
To date, only limited evidence in rodents and humans exists to support coordinated regulation of PXR target genes in liver and small intestine by PXR agonists. Studies in rats have focused on understanding the potential for compounds like dexamethasone (DEX), a rodent PXR agonist, to coordinate induction of hepatic and intestinal CYP3A and P-glycoprotein expression, to explain the effect of high-dose dexamethasone to alter the pharmacokinetics of drugs like cyclosporin A, indinavir, and tamoxifen (Sulphati and Benet, 1998; Lin et al., 1999; Cotreau et al., 2001; Huang et al., 2001; Yokogawa et al., 2002). In each of these previous studies, the authors suggested that induction of hepatic CYP3A was much more significant than that observed in intestine. Recently, Maglich et al., (2002) reported that PXR deficiency could ablate the inductive effect of pregnenolone-16α-carbonitrile on hepatic as well as intestinal PXR target genes other than mouse Cyp3a11, including Aldh1a1, Aldh1a7, Cyp2b10, Gsta1, Gstm1, Gstm2, Ugt1a1, Mdr1a, Mdr1b, and δ-aminolevulinic acid synthase 1. From these studies, activators of PXR in rats and mice are expected to have a broad effect on both hepatic and intestinal gene expression of important drug metabolism genes.
In this article, we report on the finding that two potent rat PXR agonists, L-742694 (Fig. 1) and DEX, elicited pleiotropic effects on hepatic and small intestinal gene expression after oral administration to female Sprague-Dawley rats. Furthermore, these PXR agonists elicited robust effects on hepatic genes encoding for drug metabolism proteins, but this did not occur in small intestine, and this finding was independent of PXR gene expression in the intestine. Extrapolation of these data from rats to humans suggests that PXR-mediated induction of hepatic genes is the major contributing factor to the increased presystemic metabolism, loss of efficacy, and manifestation of drug-drug interactions that occur when many drugs are coadministered with PXR agonists.

Chemical structure of L-742694.
Materials and Methods
Chemicals. Ethinyl estradiol (EE), pregnenolone 16α-carbonitrile (PCN), rifampicin (RIF), DEX, alamethacine, and dansyl chloride [5-(dimethylamino)-1-naphthalenesulfonylchloride[were purchased from Sigma Aldrich (St. Louis, MO). 1,4-Bis[2-(3,5-dichloropyridyloxy)]benzene was purchased from Calbiochem (LaJolla, CA). 17α-[6,7-3H(N)]EE (specific activity, 41 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). 17α-ethinylestradiol-2,4,16,16-d4 was obtained from C/D/N Isotope Inc. (Pointe-Claire, Quebec, Canada). Synthetic derivatives of EE, including the 3-O-glucuronide of EE, 17-O-glucuronide of EE, and the 3-O-sulfate of EE were purchased from Steraloids (Newport, RI). The 2-hydroxy-EE, 4-hydroxy-EE, and 17-sulfate metabolites of EE were synthesized by the Labeled Compound Synthesis Group (Dept. of Drug Metabolism, Merck Research Laboratories, Rahway, NJ). The Merck Medicinal Chemistry department synthesized L-742694 (Fig. 1). All other chemicals were of the highest analytical purity available.
Animals and Drug Treatments. All animal studies were performed in accordance of IACUC guidelines and approved by the Merck Research Laboratory IACUC committee. Female Sprague-Dawley rats (<400 g) were obtained from Charles River Breeding Laboratories (Wilmington, MA). The animals were housed under standard conditions and were maintained under a 12-h light/dark cycle in the Comparative Medicine facilities of Merck Research Laboratories (Rahway, NJ). Rats were allowed access to commercial rodent chow and water ad libitum. For induction studies, female Sprague-Dawley rats (three/group) were given an oral dose of L-742694 (50 mg/kg/day) or DEX (50 mg/kg/day) in 0.25% methylcellulose for 4 consecutive days; control animals received only 0.25% methylcellulose. On day 5, the liver and small intestine (upper two-thirds segment) were dissected from each animal and flash-frozen in liquid nitrogen. All tissue samples were stored at -80°C.
Metabolic Activity in Microsomes from Liver and Intestine. Microsomal and cytosolic subcellular fractions were prepared from liver and small intestinal samples by standard sucrose density centrifugation. Microsomal 6β-hydroxylation of testosterone was assessed as an index of hepatic and intestinal CYP3A activity. Incubation mixtures to monitor CYP3A activity (500 μl) contained 250 μM testosterone (in 5 μl of methanol), 100 mM phosphate buffer, pH 7.4, an NADPH-regenerating system (5 mM glucose 6-phosphate, 1 mM NADP+, and 0.7 IU/ml of glucose 6-phosphate dehydrogenase), 6 mM MgCl2, 10 mM EDTA, and 0.125 mg of microsomal protein. Stock solutions of the substrates in methanol were made such that the final concentration of methanol in the reaction mixture was 1%. The reactions were initiated by adding 50 μl of 10 mM NADP+ and allowed to proceed at 37°C for 10 min. After incubation, the reactions were terminated by the addition of 125 μl of ice-cold methanol containing 2% formic acid. The suspensions were mixed vigorously and spun in a centrifuge at 3000g for 10 min. Formation of 6β-hydroxytestosterone was analyzed from the reaction supernatant (10 μl) by reversed-phase HPLC. Extracts of reaction mixtures were separated on a Zorbax SB-C8 column (4.6 × 150 mm, 4 μm; Agilent Technologies, Inc., Wilmington, DE) and 6β-hydroxytestosterone was monitored at 254 nm. Mobile phase A consisted of 10 mM ammonium acetate in water and mobile phase B consisted of 10 mM ammonium acetate in acetonitrile. The gradient was 25 to 60% B in 7 min and it was maintained at 60% B for 1 min. The flow rate was 1 ml/min at room temperature.
Glucuronidation of EE was evaluated as an index of hepatic and intestinal microsomal UDP-glucuronosyltransferase activity. Reaction mixtures (total volume, 200 μl) containing 10 mM MgCl2, alamethicine (20 μg), 1.5 mM UDP-glucuronic acid, 0.2 mg of liver microsomal protein, 10 mM Bis-Tris propane buffer, pH 7.5, and 4 or 200 nM [3H]EE (in 2 μl of methanol) and were incubated at 37°C for 10, 20, 30, or 40 min. After incubation, the reactions were terminated by the addition of 20 μl of 50% acetic acid and 100 μl of acetonitrile, samples were centrifuged and EE glucuronides were analyzed by HPLC with radiometric detection (see below).
Sulfation of EE was evaluated as an index of cytosolic hepatic and intestinal sulfotransferase activity. Reaction mixtures (500 μl) containing 10 mM MgCl2, 50 mM phosphate buffer, pH 6.5, 0.5 mM 3′-phosphoadenosine-5′-phosphosulfate, 0.05 mg of liver cytosolic proteins, and 200 nM [3H]EE (in 5 μl methanol), was incubated at 37°C for 5, 10, or 20 min. After incubation, the reactions were terminated by the addition of 4 volumes of ice-cold methanol, processed, and analyzed by HPLC.
Glucuronide and sulfate products of EE metabolism formed in microsomal and cytosolic subcellular fractions from liver and intestinal incubates were analyzed on a Zorbax RX-C8 (4.6 × 250 mm; Agilent Technologies) analytical column for in vitro incubations, with UV detection at 210 nm and on-line radioactivity detection. The HPLC system (Shimadzu Scientific Instruments Inc., Columbia, MD) consisted of a controller (SIL-10A vp), two pumps (SIL-10AD vp), an autosampler (SIL-10AD vp), and a UV detector (SPD-10AV vp). The radioactivity was monitored using an on-line β-Ram radio-HPLC detector from IN/US Systems, Inc. (Tampa, FL), equipped with a 1.0-ml liquid cell and ScintFlow software. The scintillant (Ultima-Flo M; PerkinElmer Life and Analytical Sciences) was used at a flow rate of 3 ml/min for detection of radioactivity. The mobile phase consisted of mobile phase A (10 mM ammonium acetate in water) and mobile phase B (7.2 mM ammonium acetate in 7.2% methanol and 92.8% acetonitrile). The eluent flow rate was 1 ml/min using a linear gradient from 20 to 70% B in 40 min.
Rat PXR-LBD Clone and Reporter Constructs. The ligand-binding domain (LBD) of the rat PXR was isolated from a commercially available cDNA library (BD Biosciences Clontech, Palo Alto, CA) using primers designed to amplify the LBD, which corresponded to nucleotides 554 to 1557. Primers for the rat LBD were: forward, 5′-GATCATGGAATTCGCCGCTGTGGAA-3′; reverse, 5′-CTGGGTCTTCTAGACCCATGAGATC-3′ (GenBank accession no. AF151377). The resulting PCR product (rat LBD) was inserted into the EcoRI and XhoI restriction enzyme sites of the pcDNA3.1(+) vector (Invitrogen, Carlsbad, CA) previously constructed with the DNA binding domain of the yeast GAL4 transcription factor. The resulting chimeric construct, rat PXR-GAL4, contained the rat PXR LBD fused to the yeast GAL4 DNA binding domain. The reporter construct was generated through insertion of five tandem copies of the upstream activation sequence for GAL4 into the luciferase reporter vector, pFR-LUC vector (Stratagene, La Jolla, CA), to generate pFR-UASLUC. The rat PXR-GAL4 chimera construct was tested in combination with pFR-UASLUC. Dr. Terri Kelly (Dept. of Metabolic Disorders, Merck Research Laboratories) generously provided the constructs for the rat glucocorticoid receptor trans- activation assay. The LBD of the rat glucocorticoid receptor was inserted into pBIND (Promega, Madison, WI), a GAL4 fusion protein vector, to generate pBindratGR-LBD, where agonist binding is detectable through GAL4 activation of the pG5luc reporter vector (Promega).
Nuclear Receptor Transactivation Assays. Compounds were evaluated in the PXR transactivation assay. HepG2 cells (American Type Culture Collection, Manassas, VA) grown to 90% confluence in 150-cm2 flasks were transfected with LipofectAMINE2000 (100 μl/flask; Invitrogen), 10 μg/flask of the rat PXR-GAL4, and 10 μg/flask of the pFR-UASLUC reporter plasmid all in 15 ml Opti-MEM I (Invitrogen Corp.) After a 3-h incubation, additional Opti-MEM I medium (15 ml/flask) was added to each transfection, and the cells were allowed to incubate overnight. Transfected cells were split onto 24-well plates (150,000 cells/well) on the following day. Immediately after plating, cells were treated with the rodent PXR agonists DEX and PCN, L-742694, or dimethyl sulfoxide as vehicle (0.1%). Forty hours after treatment, the experiment was terminated by aspiration of the media and addition of Glo-Lysis buffer (100 μl; Promega). An equal volume of cell lysate from each sample was combined with luciferase assay reagent (Promega), and luminescence was assessed in a Microbeta 1450 liquid scintillation/luminescence plate reader (PerkinElmer Wallac, Turku, Finland). Similarly, compounds were evaluated in the GR transactivation assay under transfection conditions identical to those described above using the rat pBindratGRLBD and the pG5luc reporter vector. Compounds were assessed in HepG2 cells for transactivation of the mouse CAR receptor with a 5XNR1-secreted alkaline phosphatase reporter gene assay in the presence of 10 μM androstenol. Compounds were also evaluated for Ah receptor activation in a xenobiotic response element-chloramphenicol acetyltransferase reporter gene assay as described previously (Rushmore and Pickett, 1991).
Isolation of RNA. Total RNA from rat liver and small intestinal tissue samples (n = 3 rats/condition) was isolated using the SV Total RNA Isolation System (Promega) according to the manufacturer's instructions. Samples were quantified by spectrophotometry and diluted to a concentration of 15 ng/μl for subsequent analysis by real-time quantitative PCR (RT-qPCR), or 0.2 μg/ml for sample work-up for microarray analysis. Aliquots (500 ng) of RNA were analyzed by agarose/formaldehyde gel electrophoresis to check RNA integrity.
Quantitative RT-PCR. All primers and probes were submitted to the National Center for Biotechnological Information (NCBI) for nucleotide comparison using the basic logarithmic alignment search tool (BLASTn) search for short, nearly exact sequence to ensure specificity. Primers and probes were synthesized by Operon (Alameda, CA), where probes were 5′- and 3′-labeled with the FAM and TAMRA reporter dyes, respectively. The sequences for the rat CYP3A1, Oatp2, UGT1A1, UGT2B1, Mrp2, and Mrp3 primers and probes were as previously reported (Wang et al., 2003). Additional primers and probes were generated as shown in Table 1. The rodent GAPDH primer/probe set was purchased from Applied Biosystems (Foster City, CA) and used per the manufacturer's instructions.
Additional primers and probes
Total RNA (60 ng) was used to generate cDNA by reverse transcription using TaqMan reagents for reverse transcription (Applied Biosystems). A two-step RT-PCR reaction was conducted by reverse-transcribing an aliquot of total RNA (∼50 ng) to cDNA using TaqMan Reverse Transcription Reagents, according to the TaqMan Universal PCR Master Mix protocol (Applied Biosystems), with the random hexamer primer mixture. PCR reactions were then prepared by adding an aliquot of cDNA (5 μl) to a reaction mixture containing the TaqMan Universal PCR Master Mix solution, primers, and probes (300 and 200 nM, respectively) for each target. RT-qPCR was performed using an ABI PRISM 7700 Sequence Detector instrument and Sequence Detector v.1.7 software (PerkinElmer Life and Analytical Sciences). PCR amplification conditions were as follows: 1 cycle at 50°C, 2 min; 1 cycle at 95°C, 10 min; 40 cycles at 95°C, 15 s; and 40 cycles at 60°C, 1 min. PCR amplified cDNAs were detected by real-time fluorescence on an ABI PRISM 7700 Sequence Detection System (Applied Biosystems). Quantitation of the target cDNAs in all samples was normalized to glyceraldehyde 3-phosphate dehydrogenase expression (GAPDH; Cttarget - CtGAPDH = ΔCt), and the effects of each compound on the target cDNA was expressed to the amount in the dimethyl sulfoxide (vehicle) control sample (ΔCtcompound - ΔCtDMSO = ΔΔCt). Fold changes in target gene expression were determined by taking 2 to the power of this value (2-ΔΔCt).
Amplification, Labeling, and Hybridization to 25K Rat Microarrays. Individual liver samples were profiled on the 25K rat microarray (Waring et al., 2003). Detailed procedures for RNA preparation, amplification, labeling and hybridization are described in our previous publications (Hughes et al., 2000, 2001; Waring et al., 2001). Briefly, total RNA samples were extracted after DNase I treatment. Five micrograms of total RNA from each sample was amplified into cRNA by an in vitro transcription procedure with oligo-dT primer. cRNA was labeled with Cy3 or Cy5 dyes using a two-step process with allylamine-derivatized nucleotides and N-hydroxysuccinimide esters of Cy3 or Cy5 (CyDye; Amersham Biosciences). The labeled cRNAs were fragmented to an average size of ∼50 to 100 nucleotides before hybridization. For each amplified RNA sample, hybridizations were done in duplicate with fluor reversals. After hybridization, slides were washed and scanned using a confocal laser scanner (Agilent Technologies). Fluorescence intensities of the scanned images were quantified, normalized, and balanced. To compensate for the dye bias, the paired control and treated samples were hybridized on a pair of slides with reversed fluorescence, Cy3 versus Cy5 and Cy5 versus Cy3. Each hybridization pair resulted in a profile for the individual sample. The reference cRNA pool was formed by pooling equal amount of cRNAs from vehicle treated control samples
Statistical Analysis. Significantly regulated genes among individual profiles were identified by the combination of the p-value derived from the error model (Hughes et al., 2000; He et al., 2003) and magnitude of regulation. All genes with a p value <0.01 and a log|10(ratio)| > 0.5 of relative change in gene expression in two or more experiments were considered significantly regulated genes. The value of log|10(ratio)| > 0.5 allows consideration of relative induction and suppression of gene expression on an equivalent scale. Signature genes for individual compounds were determined using a nonparametric approach. Specifically, if a gene was identified as significantly regulated among two thirds of profiles for an individual compound, it was chosen as one of the signature genes for that individual compound. These parameters in gene identification were optimally chosen when considering the trade-off between sensitivity and specificity.
Results
Activators of the Rat PXR and Induction of Enzyme Activity. In a rat PXR-GAL4 trans-activation assay used to assess the potential of compounds to activate the rat PXR, L-742694 was found to be a potent PXR activator; it elicited a concentration-dependent activation of PXR that tracked similarly with DEX or PCN (Fig. 2). The human PXR agonist RIF did not activate the rat PXR (Fig. 2). In addition, when tested up to a concentration of 25 μM, L-742694 was not an agonist of the rat GR or mouse CAR when assessed in transient transfection assays, whereas DEX and 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene were potent agonists of these receptors, respectively. L-742694 was a very weak agonist of the Ah receptor (3.0-fold relative to control), compared with the Ah ligands β-napthoflavone or benzo[a]pyrene, which led to a 29- and 91-fold activation of Ah receptor-mediated xenobiotic response element-chloramphenicol acetyltransferase activity, respectively (data not shown). These in vitro results suggested that administration of L-742694 to rats would have the potential to increase hepatic microsomal cytochrome P450 activity via activation of PXR. In female Sprague-Dawley rats treated with either L-742694 (50 mg/kg/day) or DEX (50 mg/kg/day) for 4 days, liver microsomal CYP3A mediated 6β-hydroxylation of testosterone was increased significantly compared with vehicle-treated control animals (Table 2). In addition, L-742694 and DEX significantly induced the microsomal glucuronosyltransferase activity of liver microsomes isolated from induced rats, where both PXR activators increased the 3-O-glucuronidation and 17β-glucuronidation of EE (Table 2). Furthermore, when radiolabeled EE was orally administered to L-742694 treated animals (4 days), a significant increase in the biliary excretion of the hydroxyglucuronide and dihydroxy-diglucuronide metabolites of EE was detected (data not shown). In contrast, CYP3A and UGT activity in intestinal microsomes from control, L-742694, or DEX-induced rats was much less than that observed in liver (≤1/10), and neither L-742694 nor DEX caused a statistically significant increase in intestinal CYP3A or UGT activity (Table 2). Pretreatment of rats with L-742694 or DEX did not elicit a detectable increase in sulfotransferase activity in either hepatic or intestinal cytosolic subcellular fractions supplemented with 3′-phosphoadenosine-5′-phosphosulfate, and sulfate metabolites were undetectable in bile of bile duct cannulated rats challenged with EE (data not shown).

Rat PXR-GAL4 trans-activation assay. HepG2 cells were cotransfected with rat PXR-GAL4 and pFR-UASLUC, treated with varying concentrations of PCN, DEX, RIF, L-742694, or vehicle (0.1% or less) for 40 h. Luciferase activity was evaluated from triplicate determinations and values represent mean luminescence.
Assessment of CYP3A and UGT enzyme activity
Activity was assessed in subcellular fractions prepared from female Sprague-Dawley rats after a daily oral administration of L-742694 (50 mg/kg), DEX (50 mg/kg/day), or 0.25% methylcellulose as vehicle control (CON) for 4 days. All values are presented as mean ± S.E.M.
Genome-Wide Effects of L-742694 and DEX on Hepatic and Intestinal Gene Expression. Genome-wide transcriptional profiling was used to assess hepatic and intestinal gene expression for samples in control, vehicle-treated rats and from rats treated for 4 days with L-742694 and DEX. Hepatic and intestinal RNA samples from control or treated rats were compared against a reference pool of RNA created from all the control samples from a given tissue. As depicted in Fig. 3, the gene expression across the individual control samples did not vary widely between animals. Given that 6β-hydroxytestosterone activity was increased by L-742694 in rat liver (Table 2) and that this compound was an efficacious rat PXR activator (Fig. 1) the effects of L-742694 and DEX on gene expression were assessed. The data demonstrated highly reproducible gene expression profiles obtained between subjects within a given treatment group. One-dimensional hierarchical cluster analysis of significantly regulated genes demonstrated remarkable qualitative differences in intestinal and hepatic gene expression in response to the compounds tested. Relatively few similarities in the response of the intestine and liver to these PXR activators were detected (Fig. 3). DEX and L-742694 elicited gene expression profiles, which overlapped extensively in liver (Fig. 3, clusters c and h) or in intestine (Fig. 3, clusters e and i). However, apparent differences between the dual glucocorticoid receptor/PXR agonist DEX and the specific PXR agonist L-742694 were also evident in liver (Fig. 3, clusters a, b, d, and g) and in small intestine (Fig. 3, clusters a and f).

One-dimensional hierarchical cluster analysis for the effects of L-742694 and DEX on hepatic and small intestinal gene expression in female SD rats. Expression profiles in both liver and small intestine are shown in the heat map, where green, black, and red represent suppression, no change, or induction of gene expression relative to the level of vehicle controls. The color scale for the heat map ranges from 84% suppression (16% of control) to 6.3-fold induction of control level. Control levels of gene expression for each sample are compared with the gene expression profile generated from a pool of all control samples (n = 3) as the reference. Expression profiles are presented by ratios from all the probes representing the same sequence on the chip. Yellow boxes (and corresponding color bar) highlight clusters of differently and commonly regulated genes in liver or small intestine by L-742694 and DEX.
Effects of L-742694 and DEX on Regulation of Drug-Management Genes in the Liver. L-742694 as well as DEX caused a pleiotropic induction of drug metabolism genes in liver. Supervised analyses were performed where all potential drug-management genes (genes with known function in the metabolism and excretion of drugs) represented on the DNA microarrays were queried for significant changes in response to L-742694 or DEX to determine the gene expression signature related to changes in drug-management gene expression (Fig. 4). Each compound elicited many changes in gene expression known to be, or presumed to be, elicited through activation of PXR or, in the case of DEX, the GR as well. Evidence for modulation of gene expression through ligand-activated PXR, was observed in that each compound caused a significant and reproducible induction of known PXR target genes expressed in liver, including the CYP3A family of genes CYP3A1/23 (probe is annotated as CYP3A3 in this version of the DNA microarray; Unigene, Rn.91120) CYP3A2, CYP3A9, and CYP3A18. Other PXR target genes induced by L-742694 and DEX included the CYP2B genes (CYP2B1 is represented as CYP450e phenobarbital-induced; Unigene, Rn.91353), Oatp2 (denoted Slc21a5), UGT1A1 (denoted as UGT1), UGT2B1 (denoted as phenobarbital inducible UGT mRNA), GSTA2, and Carboxylestase 2 (Fig. 4). Furthermore, L-742694 induced ALDH1A1, UGT2A1, the glutathione transferases GSTM2 and GSTYa, and Mrp3 (denoted Abcc3). Induction of Mrp3, but not Mrp2, was detected in response to L-742694, but not DEX (Fig. 4). Two sulfotransferases (SULT1A1 and SULT1A2) were inversely correlated between L-742694 and DEX, where L-742694 suppressed and induced SULT1A1 and SULT1A2, respectively, whereas the opposite occurred in response to DEX (Fig. 4). Both compounds suppressed another sulfotransferase (SULT1C2; Fig. 4). Potential DEX-mediated glucocorticoid effects on hepatic gene expression were observed in which DEX specifically induced hepatic gene expression of CYP2A2, CYP2B19, CYP2C38, SULT1A1, SULT1B1, and UGT2A3 (Fig. 4). The hepatic expression levels of several genes were measured by RT-qPCR and confirmed the DNA microarray findings for those genes common to both assays (Table 3).

Heat map displaying the effects of L-742694 and DEX on hepatic gene expression of genes that function in the metabolism of xenobiotics in female Sprague-Dawley rats. Genes significantly regulated by individual compounds in rat liver were determined by a nonparametric method. All genes significantly regulated greater than 2-fold with p < 0.01 in at least two thirds of the repeats are identified as the signature genes for the individual compound. Each row represents the gene expression profile for liver from an individual rat. Only genes implicated for a role in drug metabolism and regulated by the compounds are displayed in the heat maps. The color scale for the heat map ranges from 50% reduction to 2-fold induction; green, black, and red represent suppression, no change, or induction of gene expression, respectively. Multiple probes exist for individual genes and their profiles are all illustrated in the heat map. All probes are labeled by the gene symbol targeted by the probes (the CYP3A1/23 probes are labeled as Cyp3a3). See Table 4 for all relevant annotation for each probe including probe name, accession number, and a description of each target are listed as they occur from left to right in Fig. 4.
Summary of RT-qPCR data
The effect of daily oral administration of L-742694 (50 mg/kg) or DEX (50 mg/kg) for 4 days on hepatic and small intestinal gene expression in female SD rats. Results shown are fold change in mRNA level relative to vehicle control treated female SD rats, where values > 1.0 and values < 1.0 indicate induction or suppression of the gene, respectively. Values are derived from mean values of n = 3 rats ± S.E.M.; all samples were analyzed in triplicate.
Genes significantly regulated in liver
Genes are listed in the order of first occurrence in Fig. 4 reading from left to right.
Effects of L-742694 and DEX on Regulation of Drug-Management Genes in the Small Intestine. From the heat map (Fig. 5) it was apparent that L-742694 and DEX were in general less potent modulators of drug metabolism genes in small intestine. Modest induction of UGT1A1 (denoted UGT1) and some evidence for induction of CYP1A1 was apparent in small intestine with both treatments. A number of genes were suppressed in small intestine in response to each treatment, most notably CYP3A1/23 (Table 3), but also SULT1C2 and Mrp6 (denoted Abcc6). SULT1A1 was only inducible by DEX, and probably represented a glucocorticoid-specific response (Duanmu et al., 2001). The intestinal expression levels of several genes were measured by RT-qPCR and confirmed the DNA microarray findings for those genes common to both assays (Table 3). L-742694 treatment did not increase intestinal microsomal cytochrome P450 or glucuronosyl transferase activity (Table 2).

Heat map displaying the effects of L-742694 and DEX on small intestinal gene expression in female Sprague-Dawley rats. Genes significantly regulated by individual compounds in rat small intestine were determined by a nonparametric method. All genes significantly regulated greater than 2-fold with p < 0.01 in at least two thirds of the repeats are identified as the signature genes for the individual compound. Each row represents the gene expression profile for small intestine from an individual rat. Genes implicated in drug metabolism and regulated by the compounds are shown in the heat maps. The color scale for the heat map ranges from 50% reduction to 2-fold induction; green, black, and red represent suppression, no change, or induction of gene expression, respectively. Multiple probes exist for individual genes, and their profiles are all illustrated in the heat map. All probes are labeled by the gene symbol targeted by the probes. See Table 5 for all relevant annotation for each probe including probe name, accession numbers, and a description of each target.
Genes significantly regulated in small intestine
Genes are listed in the order of first occurrence in Fig. 5 reading from left to right
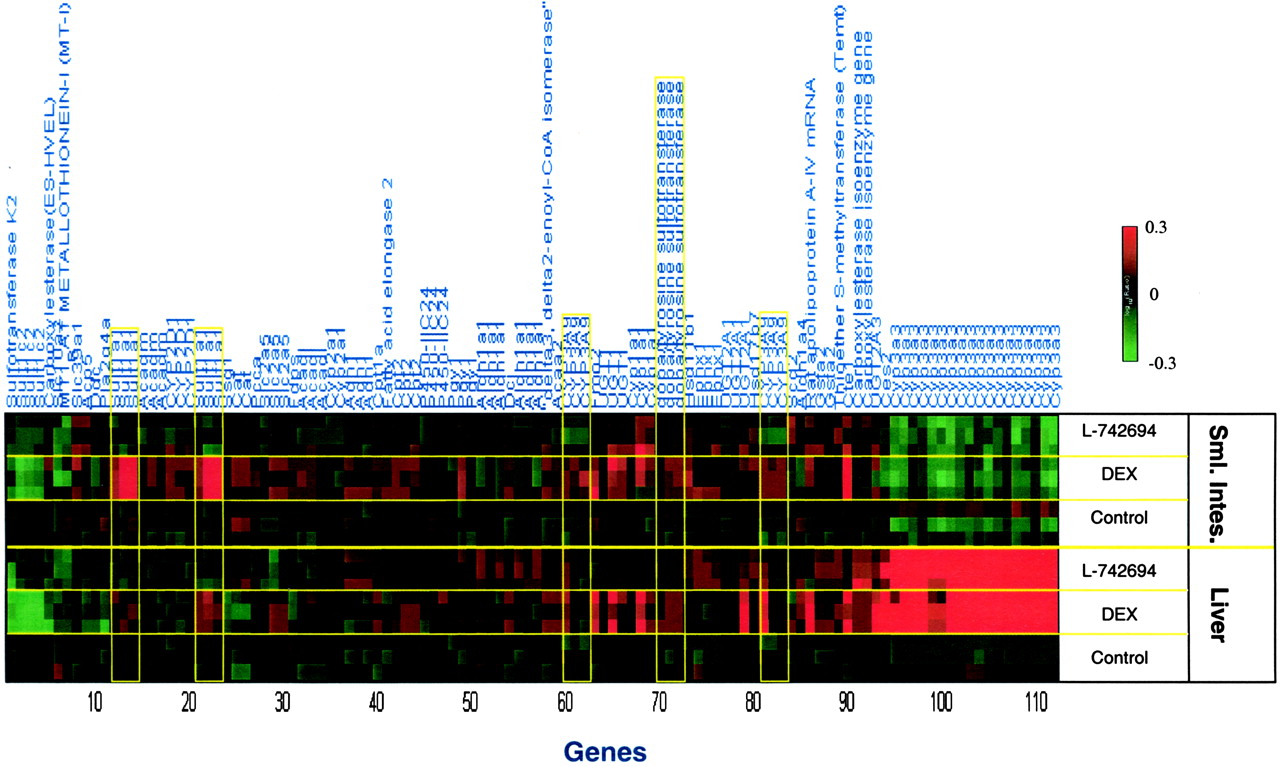
Coregulation of Drug-Management Genes between Tissues. Expression of CYP3A1/23 in liver and intestine were inversely correlated in these studies; for example, CYP3A1/23 was significantly induced in liver but was significantly suppressed in intestine by L-742694 and DEX. This was not caused by differing expression of the PXR, CAR, or HNF-4α in liver and intestine as shown by RT-qPCR (ΔCtPXR-GAPDH liver = 2.1; ΔCtPXR-GAPDH intestine = 2.6; ΔCtCAR-GAPDH liver = 2.3; ΔCtCAR-GAPDH intestine = 2.3; ΔCtHNF-4α-GAPDH liver = 1.4; ΔCtHNF-4α-GAPDH intestine = 0.8). In addition, expression levels of neither PXR nor CAR were affected by any treatment (Table 3). From Fig. 6, it is apparent that very few genes were coregulated between liver and intestine by the compounds used in this study. The expression of SULT1A1 and SULT1B1 (probe identifier dopa/tyrosine sulfotransferase), known glucocorticoid receptor-regulated genes, were induced in both tissues by DEX, but not by L-742694 (Fig. 6, Table 3). Induction of SULT1A1 was more robust and reproducible in intestine (three of three rats and six of six probe reporters) than in liver (Fig. 6, Table 3). The known glucocorticoid responsive tyrosine aminotransferase gene was not significantly altered in liver, and was suppressed in intestine (data not shown). The apparent lack of induction of hepatic tyrosine aminotransferase in these samples (isolated 24 h after the last dose of DEX to the rats) is not surprising given that induction of this gene was reported to be rapid and transient in nature, resolving to baseline at around 24 h after dosing of another synthetic glucocorticoid, methylprednisolone (Almon et al., 2004). Interestingly, CYP3A9 was modestly induced in both liver and intestine by DEX (Fig. 6, Table 3). SULT1C2 was suppressed by both treatments in both liver and small intestine (Fig. 6). Several genes also recognized as part of the aryl hydrocarbon receptor gene battery and antioxidant response element-activated genes were regulated by L-742694 and/or DEX in both liver and intestine in response to these compounds; CYP1A1, epoxide hydrolase (probe identifier, epoxide hydrolase 1), and GSTA2 were induced significantly. These responses were probably caused by activation of PXR rather than the Ah receptor, given that L-742694 and DEX are potent and efficacious activators of PXR with little or no activity on the Ah receptor. Furthermore, these genes were modulated by PXR activators in PXR knock-out mice (Maglich et al., 2002).

Nonparametric analysis for coordinated hepatic and small intestinal gene expression of drug metabolism related genes in female Sprague-Dawley rats. Significantly regulated and drug metabolism related genes by individual compounds in both rat liver and small intestine are determined by a nonparametric method. To capture additional genes in both organs, all genes significantly regulated greater than 2-fold with p < 0.01 in at least one third of the repeats are identified as the signature genes for the individual compound. Common genes identified in both liver and small intestine were selected from the previous gene sets. The gene expression profile in both liver and small intestine are demonstrated in the heat map with color scale from 50% reduction to 2-fold induction. See Table 6 for all relevant annotation for each probe including probe name, accession numbers, and a description of each target. The information in Table 6 lists the genes as they occur from left to right in Fig. 6.
Genes significantly regulated in liver and small intestine.
Discussion
We have determined that L-742694 is a potent and specific PXR activator in vitro (Fig. 1) and that L-742694 can induce CYP3A-mediated testosterone 6β-hydroxylase activity in vivo (Table 2). In addition, UGT1A1 has been reported as a PXR target gene in rats (Chen et al., 2003) as well as humans (Maglich et al., 2002; Xie et al., 2003); therefore, hepatic glucuronosyltransferase activity was measured to assess the effect of PXR activators on the glucuronidation of EE in liver microsomes. Liver microsomes prepared from rats treated with either L-742694 or DEX increased the glucuronidation of EE to the 3-O-glucuronide and 17β-glucuronide metabolites. To further characterize the effects of L-742694 and DEX on hepatic and intestinal gene expression, we used DNA microarray technology. Overall, the data suggest that L-742694 activates a set of genes recognized as what could be considered part of the PXR responsive gene battery in liver, which was consistent with the activity of L-742694 in the PXR assay. Our data also suggest that L-742694 as well as DEX differentially regulate liver and small intestinal gene expression. Furthermore, the data set as a whole lends itself to the development of a signature gene expression response to PXR activation.
Studies were performed using DNA microarray analyses, where we observed dramatic differential regulation of hepatic and intestinal gene expression. We observed a very broad effect of L-742694 and DEX on regulation of drug metabolism genes, wherein multiple genes reported as so-called PXR target genes were induced. As reported above, robust increases in CYP3A, CYP2B, Oatp2, UGT1A1, and UGT2B1 would probably contribute to an increased capacity of the liver for oxidative and conjugative metabolism in L-742694 treated rats. These data are also supported by recent reports that demonstrate that another rat PXR activator, spironolactone, increased hepatic UGT1A1 and UGT2B1 gene expression and the 3-O-glucuronidation and 17β-O-glucuronidation of EE in microsomes prepared from these rats (Catania et al., 2003). In addition, other PXR target genes that function in the metabolism of drugs were induced in rat liver by L-742694, but not DEX, including ALDH1A1 and GSTA2, and these data are consistent with previous reports in human or rat (Falkner et al., 2001; Maglich et al., 2002).
With respect to the effect of xenobiotic transporters that may function to enhance the clearance of EE-conjugates, a number of transporter genes were increased/decreased to a significant level. The fact that Oatp2, a known PXR target gene (Rausch-Derra et al., 2001; Guo et al., 2002), changes so robustly suggests that the liver is primed to be able to more efficiently extract anionic metabolites formed in the intestine as well as circulating anionic metabolites such as glucuronides. A rifampicin-inducible/PXR-regulated human OATP has yet to be identified, but numerous xenobiotic transporters are constitutively expressed on the sinusoidal membrane of hepatocytes that could function in the plasma clearance and subsequent elimination of extra-hepatic anionic conjugates. It is interesting that hepatic gene expression of Mrp3, and not Mrp2, was induced in these studies but only by L-742694 (Fig. 3 and Table 3). This is an agreement with the report of Staudinger and colleagues (2003) as well as Maglich et al. (2002), which demonstrated in Pxr wild-type mice that the rodent PXR agonist PCN induced Mrp3, but in Pxr-/- mice Mrp3 gene expression did not change. That DEX did not change Mrp3 expression in this study is in agreement with a previous report (Cherrington et al., 2002). Although several reports suggest that Mrp2 expression can be affected at either the transcriptional (Kast et al., 2002) or post-translational level (Johnson and Klaassen, 2002), in the present study, neither hepatic Mrp2 gene expression nor Mrp2 protein levels (data not shown) were increased.
A surprising finding of this study is the lack of concordance between the effect of L-742694 or DEX on liver and small intestinal gene expression, especially with respect to the PXR target gene, CYP3A1/23. In contrast to liver, where DEX and L-742694 elicited a robust induction of CYP3A1/23, this effect was not detectable in the intestinal tract, where expression of these genes was generally suppressed. This effect was not caused by loss of detectable signal for CYP3A1/23; we observed significant constitutive expression of CYP3A1/23 in intestine by RT-qPCR. In addition, testosterone 6β-hydroxylase activity in intestinal microsomes was used as a biomarker for CYP3A1/23, CYP3A9, and CYP3A18 activity (Nagata et al., 1996; Mahnke et al., 1997; Johnson et al., 2000; Wang et al., 2000). In our rat studies, testosterone 6β-hydroxylase activity was not measurable in intestinal microsomes, whereas in liver microsomes CYP3A activity was robustly induced by both L-742694 and DEX (Table 2). Others have reported modest induction of intestinal CYP3A1 in female and male rats treated with DEX (Debri et al., 1995; Salphati and Benet, 1998; Lin et al., 1999; Cotreau et al., 2001; Yokogawa et al., 2002). In contrast, Huang et al. (2001) reported a significant increase in hepatic but not intestinal CYP3A in rats treated with the HIV-protease inhibitors amprenavir and nelfinavir. Although intestinal CYP3A1/23 was suppressed in this report, a modest degree of CYP3A9 induction was observed in the intestine of DEX-treated rats (Fig. 6, Table 3). Recent studies suggest that CYP3A9 as well as CYP3A18 may be the most abundantly expressed cytochromes P450 in rat intestine, whereas CYP3A1 and CYP3A2 may not be expressed at all (Takara et al., 2003). Debri et al. (1995) reported that DEX induced a protein that cross-reacted with a CYP3A2 antibody, which recognized a C-terminal epitope in CYP3A2. This DEX-inducible protein in intestine was probably CYP3A9, given its high degree of similarity with CYP3A2 in the C-terminal region (Wang et al., 1996). Taken together, these findings are also consistent with our finding that intestinal CYP3A9 mRNA levels are increased in response to DEX (Fig. 6, Table 3). Overall, our studies are in agreement with the majority of the findings that suggest CYP3A activity in the small intestine is only a small fraction of the activity found in the liver. However, we did detect appreciable levels of CYP3A1/23 transcript in intestine, as measured by RT-qPCR, that was suppressed in response to L-742694 and DEX. However, we could find no evidence for L-742694 or DEX to induce phenotypically significant intestinal CYP3A expression in this animal model by either DNA microarray, RT-qPCR, or microsomal activity assays.
We postulated that this differential regulation of CYP3A1/23 in liver and intestine was caused by the differential expression of PXR, CAR, or HNF-4α in these organs given the reported role of these transcription factors in ligand-mediated induction (Tirona et al., 2003). However, suppression of intestinal CYP3A1/23 gene expression was independent of expression of PXR, CAR, or HNF-4α expression in small intestine. In fact, abundant levels of these transcripts were detected in small intestine and liver and did not change after the 4-day treatment with L-742694 or DEX (Table 3). Both PXR and HNF-4α were previously reported to be highly expressed in both liver and small intestine (Sladek et al., 1990; Zhang et al., 1999). We cannot rule out that these nuclear receptors and transcription factors may play a role in the regulation of intestinal drug metabolism, as suggested previously (Maglich et al., 2002; Tirona et al., 2003). However, our studies suggest that in rats, intestine is less responsive than liver to the effects of compounds like L-742694. In addition, our data collectively suggest that there is not a robust coregulation of genes common to both liver and intestine in response to PXR activators (Fig. 5). Clearly, these differences in hepatic and intestinal response to PXR activators require further study. We can only speculate that these differences in hepatic and intestinal gene expression in response to these PXR activators may be caused by differences in the half-life of hepatocytes compared with enterocytes or that the coactivator(s)/corepressor(s) differs between the two tissues.
The current data set supports the notion that a battery of PXR signature genes exists, at least in liver. Maglich et al. (2002) observed that PCN induced Aldh1a1, Cyp2b10, Cyp3a11, Gsta1, Gstm1, Gstm2, Ugt1a1, Mdr1a, Mdr1b, Mrp3, and Oatp2, in the livers of wild-type but not PXR receptor-null mice. In addition, these authors used human hepatocytes to demonstrate that rifampicin induced human ALDH1A1, CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP3A4, GSTA2, and ABCB1. These data were further corroborated in the report by Watkins et al. (2003), wherein hyperforin and rifampicin significantly induced CYP3A4, CYP2B6, CYP1A1, CYP1A2, CYP1B1, CYP2C8, CYP2C9, P450 oxidoreductase, ABCB1, epoxide hydrolase, ALDH1A1, and GSTA2. Herein, we report comparable findings for the effect of PXR ligands on rat liver gene expression for a number of orthologous genes, including rat CYP3A, CYP2B, Oatp2, ALDH1A1, GSTA2, GSTM2, UGT1A1, Mrp3, and CYP1A1. Thus, although the ligand binding domains for rodent and human PXR share only 75% amino acid sequence homology and have slightly different substrate specificity (Jones et al., 2000), extrapolation from rodent models may reasonably predict the effect of PXR activators on the regulation of orthologous drug metabolism genes in humans. This is a particularly salient point given that the rat is the preferred preclinical species for evaluation of new drug entities in drug development. As such, drug-drug interaction studies in the rat that predict an interaction through activation of PXR, such as that presented here for L-742694, may in fact predict a drug-drug interaction in the human. Obviously, one must consider the species differences beyond PXR (e.g., species differences in pharmacokinetic parameters, uptake transporters, and plasma protein binding), but if a drug activates both the rat and human PXR receptor, a similar program of orthologous genes may be modulated in liver regardless of species, resulting in a gene expression signature typical for activation of the PXR receptor.
In conclusion, L-742694 is a potent activator of rat PXR and elicits pleiotropic, but non-overlapping, effects on hepatic and small intestinal gene expression in female Sprague-Dawley rats. It is apparent from these observations that the robust effects of PXR agonists (L-742694 and DEX) on the expression of drug metabolism genes in liver do not occur in small intestine in the rat and that this finding occurs independently of nuclear hormone receptor gene expression in these tissues. These data suggest that activation of PXR in rats yields a gene expression profile in liver that is similar to previous findings from human model systems and that may be useful in defining the signature gene expression profile for pharmacological activation of PXR irrespective of species. Further studies in human model systems need to be performed to gain more insight into the regulation of hepatic and intestinal drug-management genes that occurs in response to activation of PXR, in that this knowledge has significant ramifications for interpreting drug-drug interaction studies in humans.
Acknowledgments
We thank Dr. M. Braun for supplying synthetic samples of metabolites 2-hydroxy-EE, 4-hydroxy-EE, and 17-O-sulfate of EE; Dr. Terri Kelly for supplying the plasmid constructs for the rat GR assay; Dr. Mark Abramovitz for assessing activity of these compounds in the mouse CAR assay; and the Gene Expression Laboratory at Rosetta for carrying out all the hybridization reactions.
Footnotes
-
ABBREVIATIONS: PXR (NR1I2), pregnane X receptor; ER, everted repeat; DR, direct repeat; L-742694, 2(S)-((3,5-bis(trifluoromethyl)benzyl)-oxy)-3(S)-phenyl-4-((3-oxo-1,2,4-triazol-5-yl)methyl)morpholine; DEX, dexamethasone; EE, ethinyl estradiol [(17α-ethinyl-1,3,5(10)-estratriene-3,17α-diol); CAR, constitutive androstane receptor; HNF-4α, hepatic nuclear factor-4α; PCN, pregnenolone-16α-carbonitrile; RIF, rifampicin; HPLC, high-performance liquid chromatography; LBD, ligand-binding domain; PCR, polymerase chain reaction; GR, glucocorticoid receptor; RT-qPCR, real-time quantitative polymerase chain reaction; FAM, 5-carboxyfluorescein; TAMRA, 5-carboxytetramethylrhodamine; Ct, cycle threshold; Ah, aryl hydrocarbon receptor; ALDH/Aldh, aldehyde dehydrogenase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GST/Gst, glutathione S-transferase; UGT/Ugt, glucuronosyltransferase; SULT, sulfotransferase; MDR/Mdr, multidrug resistance protein; Oatp, organic anion transporting polypeptide; ABC, ATP-binding cassette; Mrp, multidrug resistance protein.
- Received December 10, 2003.
- Accepted February 18, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}