


Abstract
The ATP-driven drug export pump, P-glycoprotein, is a primary gatekeeper of the blood-brain barrier and a major impediment to central nervous system (CNS) pharmacotherapy. Reducing P-glycoprotein activity dramatically increases penetration of many therapeutic drugs into the CNS. Previous studies in rat showed that brain capillary P-glycoprotein was transcriptionally up-regulated by the pregnane X receptor (PXR), a xenobiotic-activated nuclear receptor. Here we used a transgenic mouse expressing human PXR (hPXR) to determine the consequences of increased blood-brain barrier P-glycoprotein activity. P-glycoprotein expression and transport activity in brain capillaries from transgenic mice was significantly increased when capillaries were exposed to the hPXR ligands, rifampin and hyperforin, in vitro and when the mice were dosed with rifampin in vivo. Plasma rifampin levels in induced mice were comparable with literature values for patients. We also administered methadone, a CNS-acting, P-glycoprotein substrate, to control and rifampin-induced transgenic mice and measured the drug's antinociceptive effect. In rifampin-induced mice, the methadone effect was reduced by approximately 70%, even though plasma methadone levels were similar to those found in transgenic controls not exposed to rifampin. Thus, hPXR activation in vivo increased P-glycoprotein activity and tightened the blood-brain barrier to methadone, reducing the drug's CNS efficacy. This is the first demonstration of the ability of blood-brain barrier PXR to alter the efficacy of a CNS-acting drug.
The blood-brain barrier, which resides within brain capillaries, is the primary determinant of drug entry into the central nervous system (CNS) (Begley, 2003, 2004b). Barrier effectiveness reflects both the low permeability of tight junctions between capillary endothelial cells and the high expression of multispecific, ATP-driven drug-efflux pumps on the luminal membrane of those cells (Begley, 2004a). Of these transporters, P-glycoprotein presents the most formidable obstacle to CNS pharmacotherapy. Knocking out P-glycoprotein or reducing its transport function (activity) substantially increases brain levels of chemotherapeutics, HIV protease inhibitors, anticonvulsants, antipsychotics, and glucocorticoids, raising the possibility of devising maneuvers to modulate P-glycoprotein function and thus selectively improve drug access to the CNS (Schinkel et al., 1994, 1996). Indeed, this potential has been realized in recent animal studies with the chemotherapeutic paclitaxel (Taxol), a drug that is normally ineffective against brain tumors. Pretreating mice with a specific P-glycoprotein inhibitor increased brain levels of administered paclitaxel and reduced the mass of an intracerebrally implanted human glioblastoma by 90% (Fellner et al., 2002).
The converse of this argument is that increased P-glycoprotein activity should selectively close the blood-brain barrier, impeding therapy with P-glycoprotein substrates that normally cross the barrier in sufficient quantity to produce beneficial CNS effects (e.g., methadone, morphine, dexamethasone, and some antiepileptics). This has been observed in animals chronically exposed to certain P-glycoprotein substrates (Fromm et al., 1997; Lotsch et al., 2002), but the mechanism underlying the increase in transporter activity has not been identified. We recently detected expression of the ligand-activated, pregnane X receptor (PXR) in isolated rat brain capillaries (Bauer et al., 2004). Previous studies in liver and gut had shown that xenobiotics (e.g., steroids, statins, chemotherapeutics, and endocrine disruptors) acting through PXR transcriptionally up-regulate P-glycoprotein expression in those tissues (Dussault and Forman, 2002). We found similar up-regulation of transporter expression and parallel increases in P-glycoprotein activity in isolated rat brain capillaries exposed to the xenobiotics pregnenolone 16α-carbonitrile (PCN) and dexamethasone, both ligands for rodent PXR (Bauer et al., 2004). We also found substantially increased expression and activity in capillaries isolated from rats dosed with PCN and dexamethasone. These results defined a cause and effect relationship between activation of brain capillary PXR by xenobiotics and increased P-glycoprotein activity. They provide a context within which to examine the pharmacological consequences of activating PXR and increasing P-glycoprotein expression at the blood-brain barrier.
For the experiments reported here, we used a transgenic mouse expressing human PXR (hPXR) rather than mouse PXR (mPXR) (Xie et al., 2000). This choice of animal model is crucial because the ligand specificity of PXR varies substantially with species (Xie and Evans, 2002). The present results for transgenic mice expressing hPXR show increased P-glycoprotein expression and transport activity in isolated brain capillaries exposed to two high-affinity hPXR ligands, rifampin, an antibiotic, and hyperforin, a constituent of the herbal remedy St. John's wort. They also show increased transporter expression and activity in brain capillaries isolated from transgenic mice dosed with rifampin. To determine the consequences of hPXR induction of P-glycoprotein activity, we administered methadone, a CNS-acting P-glycoprotein substrate that normally produces a substantial antinociceptive effect, to control and rifampin-induced transgenic mice. In mice pretreated with rifampin, the methadone antinociceptive effect was substantially reduced, even though plasma methadone levels were unchanged. Thus, hPXR activation increased P-glycoprotein expression and tightened the blood-brain barrier, reducing the efficacy of methadone.
Materials and Methods
Chemicals. Rifampin was purchased from Spectrum Chemical and Laboratory Products (Gardena, CA). PCN, hyperforin, and methadone were purchased from Sigma (St. Louis, MO). NBD-CSA was custom-synthesized by Novartis (Basel, Switzerland) (Schramm et al., 1995). PSC833 was a gift from Novartis. All the other chemicals were of analytical grade and were obtained from commercial sources.
Animals. Male CB6F1 wild-type mice (Charles River Laboratories, Wilmington, MA), CB6F1 hPXR transgenic mice (25-35g), and male CF-1 [mdr1a(+/+) and mdr1a(-/-)] mice (30-40 g; Charles River Laboratories) were used. CB6F1 hPXR (Xie et al., 2000) mice were a gift from Dr. Wen Xie (University of Pittsburgh, Pittsburgh, PA). Animal housing and dosing protocols were approved by the Institutional Animal Care and Use Committee of the University of North Carolina and were in accordance with National Institutes of Health guidelines.
For in vitro studies, mice were decapitated, and brains were taken immediately for capillary isolation. For in vivo studies, mice were dosed daily for 1 to 3 days with 50 mg/kg rifampin in 0.1% agarose by oral gavage (4 μl/g 0.1% agarose, 12.5 μg/μl 0.1% agarose; agarose at 37°C to keep it liquid); controls received agarose alone. Twenty-four hours after the last dose, mice were decapitated, and brains were taken immediately for capillary isolation. Intestinal mucosa and livers were snap-frozen in liquid nitrogen and stored at -80°C until use.
Capillary Isolation. Mouse brain capillaries were isolated as described previously (Bauer et al., 2004; Hartz et al., 2004) with slight modifications. Mice were decapitated, and brains were immediately put in ice-cold Dulbecco's phosphate-buffered saline (DPBS) buffer (2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, and 8.1 mM Na2HPO4, supplemented with 5 mM d-glucose and 1 mM Na-pyruvate, pH 7.4). Brains were homogenized in buffer, and the homogenate was mixed with Ficoll (final concentration 15%, Sigma) and centrifuged at 5800g for 10 min at 4°C. The resulting pellet was suspended in DPBS containing 1% bovine serum albumin (BSA) and passed over a glass bead column. Capillaries adhering to the glass beads were collected by gentle agitation in DPBS (1% BSA). Capillaries were washed three times in BSA-free DPBS and then used for experiments. For in vitro dosing studies, freshly isolated capillaries were first incubated in BSA-free DPBS buffer with PCN, rifampin, or hyperforin for 6 h at room temperature and then used for transport assays and immunostaining experiments. Capillaries from mice dosed in vivo were used immediately after isolation.
P-Glycoprotein-Mediated Transport. Details of the transport assay are presented in articles by Bauer et al. (2004) and Hartz et al. (2004). In brief, isolated brain capillaries were incubated for 1 h at room temperature in BSA-free DPBS buffer containing 2 μM NBD-CSA, a fluorescent P-glycoprotein substrate. Confocal images of 10 to 15 capillaries were acquired (Zeiss 410 meta laser scanning confocal microscope, 40× water immersion objective, numerical aperture = 1.2, 488 nm line of argon laser; Carl Zeiss Inc., Thornwood, NY), and luminal fluorescence intensity was measured from stored images using Scion Image software (Scion Corp., Frederick, MD) as described previously (Miller et al., 2000).
P-Glycoprotein Immunostaining. Isolated mouse brain capillaries were fixed for 5 to 10 min with 3% paraformaldehyde/0.25% glutaraldehyde at room temperature. After washing with DPBS, capillaries were permeabilized for 15 min with 0.5% (v/v) Triton X-100 and washed with DPBS containing 1% BSA. Then, capillaries were incubated for 1 h at 37°C with a 1:100 dilution (1 μg/ml) of polyclonal rabbit antibody mdr ab-1 (Oncogene Research Products, Cambridge, MA). Capillaries were washed and incubated with Alexa Fluor 488-conjugated anti-rabbit secondary IgG (1:1000; Invitrogen, Carlsbad, CA) for 1 h at 37°C. Nuclei were counterstained with 2.5 μg/ml propidium iodide. Negative controls for each treatment were processed without primary antibody, and these showed only background fluorescence. Immunofluorescence was visualized by confocal microscopy (Zeiss LSM 510 meta laser scanning confocal microscope). For quantitating P-glycoprotein immunofluorescence, confocal images of 10 to 20 capillaries per treatment were acquired. Luminal membrane P-glycoprotein immunofluorescence for each capillary was measured using ImageJ software (ver. 1.29; http://rsb.info.nih.gov/ij/). A 10 × 10 grid was superimposed on each image, and measurements of capillary luminal plasma membrane were taken between intersecting grid lines. The fluorescence intensity for each capillary was the mean of all the measurements.
Western Blotting. Brain capillaries, intestinal mucosa, and livers were homogenized in lysis buffer containing Complete protease inhibitor mixture (Roche, Mannheim, Germany). Homogenized samples were centrifuged at 10,000g for 15 min, and denucleated supernatants were centrifuged at 100,000g for 90 min. Pellets (crude plasma membranes) were suspended in buffer containing protease inhibitor mixture, and protein concentrations were determined. Western blots were performed using the NuPage electrophoresis and blotting system (Invitrogen). After blocking, membranes were incubated overnight with monoclonal mouse C219 primary antibody to P-glycoprotein (1:100; Signet, Dedham, MA). Membranes were washed and incubated with the anti-mouse horseradish peroxidase-conjugated ImmunoPure secondary antibody (1:15,000; Pierce, Rockford, IL) for 1 h. P-glycoprotein was detected using SuperSignal West Pico Chemoluminescent Substrate (Pierce). Bands were visualized with a Gel Doc 2000 gel documentation system (Bio-Rad, Hercules, CA).
Total RNA Isolation and Reverse Transcription-Polymerase Chain Reaction. Total RNA from brain, intestine, and liver of wild-type and hPXR transgenic mice was isolated using TRIzol reagent (Invitrogen) and purified using the RNeasy Mini kit (QIAGEN, Valencia, CA). Reverse transcription (RT) of total RNA was performed using the GeneAmp kit according to the manufacturer's protocol (Applied Biosystems, Foster City, CA). RT products were used for polymerase chain reaction (PCR) of mPXR [GenBank accession no. NM_010936, forward: 5′-CTCTGCCTTGGAAGAGCCCATCAAC-3′, bases 392-416; reverse: 5′-GGTTTGCATCTGAGCGTCCATCAGC-3′, bases 785-809; 418-base pair (bp) amplicon], hPXR (GenBank accession no. AY091855, forward: 5′-GTCTGTTCCTGGAAAGCCCAGTGTC-3′, bases 645-669; reverse: 5′-TCATCATCCGCTGCTCCTCTGTCAG-3′, bases 1009-1033; 389-bp amplicon), and mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (GenBank accession no. BC083149, forward: 5′-GTATGTCGTGGAGTCTACTGGTGTC-3′, bases 309-333; reverse: 5′-GGTGCAGGATGCATTGCTGACAATC-3′, bases 465-489; 181-bp amplicon). PCR for brain, intestine, and liver mPXR, hPXR, and GAPDH was run 35 cycles; PCR for brain capillary GAPDH was run 35 cycles; and PCR for brain capillary mPXR and hPXR was run 45 cycles. All the primers were screened for specificity by using the PubMed BLAST database. Primers were custom-synthesized by Operon Biotechnologies (Huntsville, AL) or MWG Biotech (High Point, NC), respectively. PCR products were separated by agarose gel electrophoresis.
Rifampin Dosing and Plasma Levels. To determine an appropriate rifampin dose for P-glycoprotein induction, unbound rifampin peak plasma concentration (free Cmax) after a single p.o. dose and unbound rifampin average plasma concentration (free Caverage) at steady state after multiple, single daily doses were determined. In initial experiments, hPXR transgenic mice (n = 20) received a single dose of 50 mg/kg rifampin in 0.1% agarose by p.o. gavage. Blood samples (50 μl) were collected by tail nick in heparinized capillary tubes over the next 36 h. Blood was centrifuged (3000g for 10 min), and plasma was obtained. Rifampin plasma concentrations were determined by high-performance liquid chromatography/mass spectrometry (HPLC/MS) (Yang et al., 2003). Total rifampin peak plasma concentration (total Cmax) was obtained directly from the observed concentration time data profile. Free rifampin peak plasma concentration (free Cmax) was calculated from the following equation: free Cmax = (1-0.88) × total Cmax, where 0.88 is bound fraction of rifampin in mouse plasma as determined by us. The area under the concentration time curve from time 0 to infinity (AUC0-∞; i.e., total rifampin exposure) was calculated using the trapezoidal method with WinNonlin Software 4.1 (Pharsight, Mountain View, CA). Average total plasma rifampin concentration (Caverage) at steady state after three single daily doses was calculated using the following equation: Caverage = AUC0-24/τ, where τ is the dosing interval (24 h) and AUC0-24 at steady state after multiple dose is equal to AUC0-∞ after a single dose. The average free rifampin plasma concentration was then calculated from the equation: free Cmax = (1-0.88) × total Cmax.
Methadone Antinociceptive Response. An electrical stimulation vocalization assay (ESV; threshold voltage to elicit vocalization of mice in response to an electrical stimulus) was used to determine the methadone-associated antinociceptive response (Paalzow, 1969a,b). Electrodes were inserted subdermally in the tails of ketamine-xylazine (85 and 0.3 mg/kg, respectively) anesthetized control and rifampin-treated hPXR transgenic mice (50 mg/kg rifampin daily for 3 days by p.o. gavage, five mice per group). Twenty-four hours after the last dose, baseline ESV was determined for each mouse. Thereafter, methadone was administered (3 mg/kg s.c.), and tests were repeated at multiple times for up to 8 h. For each mouse at each time, the antinociceptive effect (%ANE) was calculated as percent increase in voltage threshold using the following equation: %ANE = [(ESVmethadone - ESVbaseline)/ESVbaseline] × 100, where ESVmethadone is the voltage causing vocalization after methadone administration and ESVbaseline is the voltage causing vocalization without methadone administration. The area under the antinociceptive effect versus time curve (0-480 min) was determined using the trapezoidal method.
Methadone Plasma and Brain Disposition. The hPXR transgenic mice in both vehicle- and rifampin-treated groups received a 3 mg/kg s.c. dose of methadone dissolved in saline. Blood samples were collected in heparinized capillary tubes by tail nick before methadone administration and at various times thereafter. Three to four mice were sampled at each time point. Blood was centrifuged at 2000g for 15 min, and plasma was obtained. Samples were immediately frozen and stored at -80°C for later analysis by HPLC/MS.

PXR expression in mouse tissues. A, RT-PCR of brain, intestine, and liver from wild-type mice and hPXR transgenic mice. Top, mPXR (418-bp amplicon, 35 cycles) is expressed in brain (B), intestine (I), and liver (L) of wild-type mice. Middle, hPXR (389-bp amplicon, 35 cycles) is only expressed in hPXR transgenic mice. Bottom, GAPDH loading control (181-bp amplicon, 35 cycles; nc, negative control). B, RT-PCR of brain capillaries from wild-type mice and hPXR transgenic mice. Top, mPXR is expressed in brain capillaries (BC) of wild-type mice (45 cycles). Middle, hPXR is only expressed in brain capillaries of hPXR transgenic mice (45 cycles). Bottom, GAPDH loading control (35 cycles; nc, negative control).
In a separate series of experiments, male CF-1 [mdr1a(+/+) and mdr1a(-/-)] mice (30-40 g; Charles River Laboratories, Inc.) were used to establish a relationship between antinociceptive effect and brain tissue concentration of methadone. In brief, in a dose-response experiment, mice received a single s.c. injection of methadone [0.6-6 mg/kg for mdr1a(+/+) mice and 0.2-1.5 mg/kg for mdr1a(-/-) mice]. Antinociception was evaluated at 30 min postdose; animals were sacrificed for collection of brain tissue and serum (from trunk blood). Additional animals were used to assess antinociception and methadone concentrations at various times after methadone administration. Mdr1a(+/+) and mdr1a(-/-) mice received a single s.c. dose of methadone (4 or 1 mg/kg, respectively), and antinociception, brain tissue methadone, and serum methadone concentrations were determined at selected time intervals through 3 h postdose. To construct the effect versus concentration relationship, antinociception was expressed as percent of maximum, and a sigmoidal Emax model was fit to the effect versus concentration data.
For methadone analysis, 25 μl of plasma or brain homogenate was mixed with 100 μl of methanol containing an internal standard (loperamide 50 ng/ml) to precipitate protein. The mixture was vortexed for 2 min and centrifuged at 16,000g for 10 min, and 5 μl of supernatant was taken for analysis using an Agilent 1100 series HPLC/MS (Wilmington, DE) system. The system consisted of a single-quadruple mass spectrometer (G1946D), a capillary pump (G1376A), a micro vacuum degasser (G1379A), and a microALS autosampler (G1389A). The supernatant was injected onto a Luna C8 reverse-phase column 30 × 2 mm (Torrance, CA) at room temperature. Analytes were eluted with an isocratic mobile phase containing 60% methanol and 40% 10 mM ammonium acetate, pH 6.8, at a flow rate of 400 μl/min and a retention time of 0.5 and 0.6 min, respectively. The mass spectrometer was operated in electrospray ionization positive mode. Analysis was carried out using selected ion monitoring for specific m/z 309.2 (methadone) and m/z 477.2 (loperamide); sensitivity of quantitation was 0.01 ng/ml. The calibration curve was linear in the range of 0.1 to 100 ng/ml. The mean of intraday and interday precision was 7.2 to 3.1% (%CV) and 8.4 to 2.2% (%CV), respectively, and the recovery ratio was >98.7%.

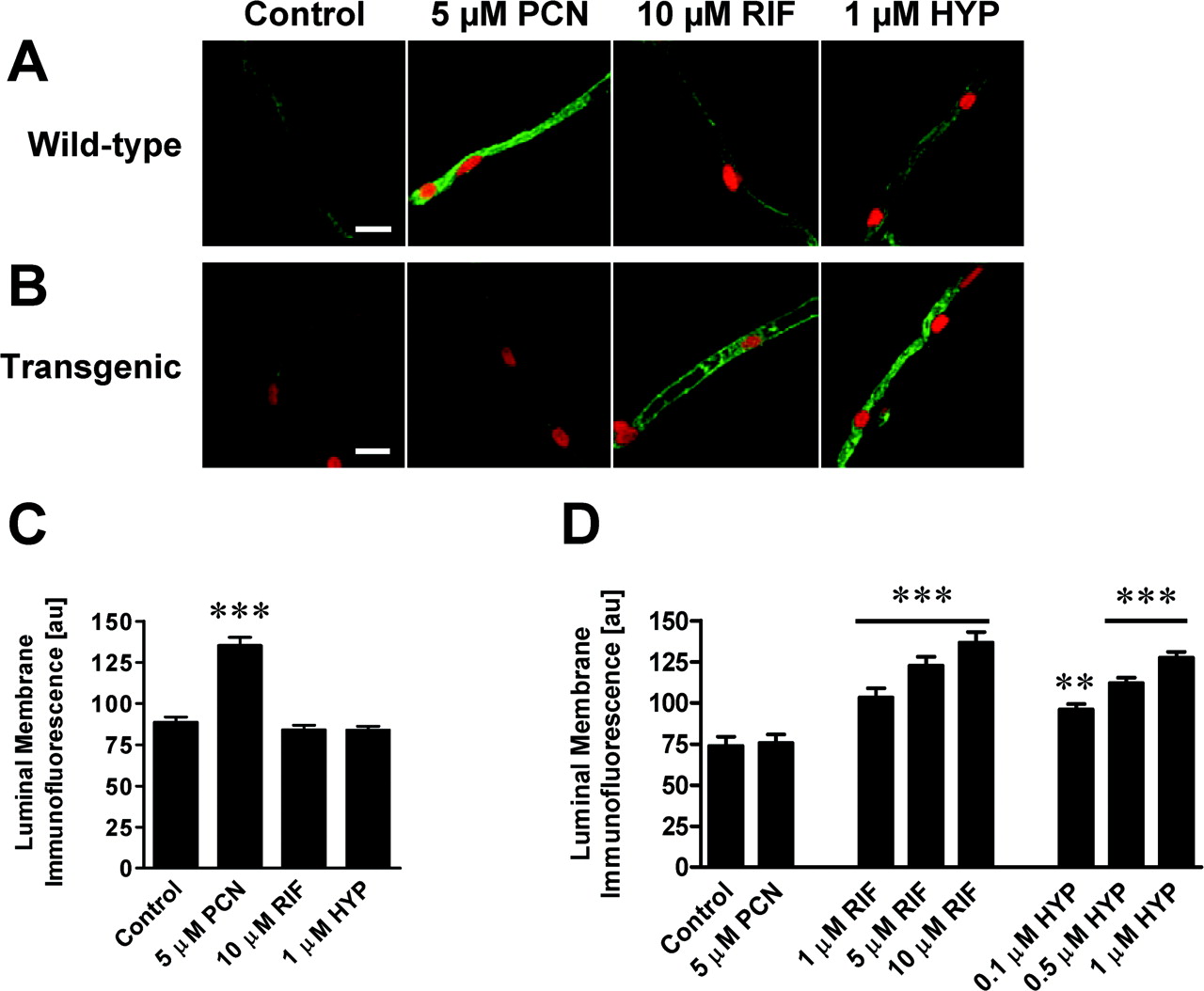
P-glycoprotein expression in brain capillaries after exposure to PXR ligands. A, representative images showing immunostaining of P-glycoprotein in isolated brain capillaries from wild-type mice. Exposing brain capillaries for 6 h to PCN increased P-glycoprotein expression; rifampin (RIF) and hyperforin (HYP) had no effect (scale bar: 10 μm). B, representative images showing immunostaining of P-glycoprotein in isolated brain capillaries from hPXR transgenic mice. Six-hour exposure of rifampin and hyperforin, but not PCN, increased P-glycoprotein expression in capillaries of hPXR transgenic mice (scale bar: 10 μm). C, quantitation of luminal membrane P-glycoprotein immunofluorescence from capillaries of wild-type mice exposed to PCN, rifampin, or hyperforin. D, concentration-dependent increase of luminal membrane P-glycoprotein immunofluorescence from isolated capillaries of hPXR transgenic mice exposed to rifampin or hyperforin; PCN had no effect. Each bar represents the mean value (arbitrary fluorescence units, scale 0-255) of 15 to 20 capillaries from a single preparation (pooled tissue from five wild-type mice or three hPXR transgenic mice); variability is given by S.E. bars. Statistical comparison: **, significantly greater than control, P < 0.01; ***, significantly greater than control, P < 0.001.

P-glycoprotein transport function in brain capillaries after exposure to PXR ligands. A, left, representative image showing steady state (60 min) NBD-CSA (2 μM) accumulation in two brain capillaries from wild-type mice. The arrows indicate red blood cells (dark) in the capillary lumen (bright). A, right, corresponding image for a capillary exposed to 5 μM PSC833. The arrow shows the capillary lumen with reduced fluorescence (scale bar, 10 μm). B, specific (PSC833-sensitive) NBD-CSA accumulation in the lumen of brain capillaries isolated from wild-type mice. Six-hour exposure to PCN increased luminal NBD-CSA; rifampin (RIF) and hyperforin (HYP) had no effect. C, concentration-dependent increase of specific NBD-CSA accumulation in the lumen of capillaries isolated from hPXR transgenic mice that were exposed to rifampin or hyperforin for 6 h; PCN had no effect. Each bar represents the mean value (arbitrary fluorescence units, scale 0-255) of 10 capillaries from a single preparation (pooled tissue from five wild-type mice or three hPXR transgenic mice); variability is given by SE bars. ***, significantly greater than control, P < 0.001.
Statistical Analysis. Observations are generally reported as mean ± S.E. One- or two-tail unpaired Student's t test was used to evaluate differences between controls and treatment groups; differences were considered to be statistically significant when P < 0.05.
Results
PXR exhibits distinct species differences in ligand specificity (Xie and Evans, 2002). For example, the St. John's wort constituent, hyperforin, and the antibiotic, rifampin, are hPXR ligands but not rodent PXR ligands. On the other hand, PCN is a high-affinity ligand for rodent PXR but does not activate hPXR. To further understanding of the role of hPXR in metabolism-based drug-drug interactions, Xie et al. (2000) generated a transgenic, “humanized” mouse expressing hPXR but lacking mPXR. Using Northern blot analysis, they found that hPXR expression was liver-specific, which was expected because of the albumin-based vector used for transfection. Xie et al. (2000) further showed that these mice responded to hPXR ligands (e.g., rifampin) by up-regulating hepatic Cyp3A11 expression. As a consequence, rifampin-induced mice exhibited reduced sensitivity to two toxicants that are metabolized by hepatic Cyp3A11, tribromoethanol and zoxazolamine. Using a more sensitive assay for gene expression, RT-PCR, we have confirmed some of the initial findings of Xie et al. (2000). As shown in Fig. 1A, livers from transgenic mice expressed hPXR but not mPXR; in livers from wild-type mice, the reverse was true. However, in transgenic mice we detected hPXR expression in intestine, whole brain homogenate (Fig. 1A), and isolated brain capillaries (Fig. 1B), as well as liver; no mPXR expression was detected in any tissue. Thus, RT-PCR showed that hPXR expression in these transgenic mice was not liver-specific as had been originally reported. At present, we have no explanation for this discrepancy other than the high sensitivity of RT-PCR. This technique previously allowed us to detect PXR expression in rat brain homogenates (Bauer et al., 2004) when none had been detected by other laboratories using Northern blot analyses (Kliewer et al., 1998; Zhang et al., 1999; Jones et al., 2000).
We took advantage of this expression pattern to determine the effects of ligands for mPXR (PCN) and hPXR (rifampin and hyperforin) on P-glycoprotein expression and activity in isolated brain capillaries from wild-type and hPXR transgenic mice. When we exposed freshly isolated capillaries from wild-type mice to 5 μM PCN for 6 h, luminal membrane P-glycoprotein immunofluorescence increased significantly (Fig. 2A). Quantitation of luminal membrane P-glycoprotein immunofluorescence revealed a 53 ± 6% increase with PCN (P < 0.001, Fig. 2C). Neither hyperforin nor rifampin increased P-glycoprotein immunofluorescence. This is the same pattern of induction seen in our previous study using rat brain capillaries (Bauer et al., 2004). When we repeated these in vitro dosing experiments with capillaries isolated from hPXR transgenic mice, hyperforin and rifampin increased luminal membrane P-glycoprotein immunofluorescence, but PCN did not (Fig. 2, B and D).

Induction of P-glycoprotein expression and activity in hPXR transgenic mice after in vivo exposure to rifampin (50 mg/kg rifampin daily for 3 days by p.o. gavage). A, P-glycoprotein Western blot from crude membrane fractions of intestine and liver of hPXR transgenic mice (Ctrl, vehicle-dosed control animals; RIF, rifampin-dosed animals; negative control (-), brain homogenate; positive control (+), renal brush-border membranes). B, P-glycoprotein Western blot from plasma membranes of brain capillaries (BCM) isolated from vehicle-dosed controls (Ctrl) and rifampin-dosed (RIF) hPXR transgenic mice (negative control (-), brain homogenate; positive control (+), renal brush-border membranes). C, representative images showing luminal membrane P-glycoprotein immunofluorescence in capillaries from vehicle-dosed and rifampin-dosed hPXR transgenic mice (scale bar: 10 μm). D, quantitation of luminal membrane P-glycoprotein immunofluorescence from capillaries of hPXR transgenic mice. E, specific NBD-CSA accumulation (PSC833-sensitive) in the lumen of brain capillaries isolated from hPXR transgenic mice. Each bar represents the mean value (arbitrary fluorescence units, scale 0-255) of 10 capillaries from a single preparation (pooled tissue from five control hPXR transgenic mice or six rifampin-treated hPXR transgenic mice); variability is given by S.E. bars. Statistical comparison: ***, significantly greater than control, P < 0.001.
We have developed a method to measure P-glycoprotein transport activity in living, intact, isolated brain capillaries. It is based on measuring steady state, concentrative accumulation of the fluorescent, P-glycoprotein substrate, NBD-CSA in capillary lumens using confocal microscopy and quantitative image analysis (Miller et al., 2000; Bauer et al., 2004; Hartz et al., 2004). Figure 3A shows brain capillaries from wild-type mice that were incubated with 2 μM NBD-CSA for 60 min. Relative to bath, NBD-CSA fluorescence intensity was high in capillary lumens (Fig. 3A, left). In agreement with previous studies using capillaries from rat (Fellner et al., 2002; Bauer et al., 2004; Hartz et al., 2004), the P-glycoprotein-specific inhibitor PSC833 reduced luminal NBD-CSA accumulation (Fig. 3A, right). Inhibition was maximal with 5 μM PSC833, resulting in a reduction of approximately 50%; exposing capillaries to 1 mM NaCN produced the same level of transport inhibition (not shown). Thus, the PSC833-sensitive component of NBD-CSA accumulation in the lumens of isolated mouse brain capillaries represents active, P-glycoprotein-specific transport. Remaining luminal fluorescence (PSC833-insensitive component) probably represents simple diffusion and unspecific binding of NBD-CSA to capillary tissue (Bauer et al., 2004; Hartz et al., 2004). In the present experiments, the PSC833-insensitive component averaged 52 to 54 fluorescence units and was not altered when capillaries were exposed to PXR ligands or when animals were dosed with rifampin (not shown).
Figure 3B shows PSC833-sensitive, luminal NBD-CSA fluorescence in capillaries from wild-type mice that were exposed for 6 h to PCN, hyperforin, or rifampin. Consistent with the quantitative immunostaining results and the ligand specificity of mPXR, PCN doubled luminal NBD-CSA accumulation, but hyperforin and rifampin had no significant effects. In contrast, when this experiment was carried out with capillaries from hPXR transgenic mice, both hyperforin and rifampin increased P-glycoprotein-mediated transport in a concentration-dependent manner, but PCN was without effect (Fig. 3C). The observed increases in P-glycoprotein expression and activity in response to rifampin and hyperforin are consistent with the expression of functional hPXR in brain capillaries.
To determine whether in vivo exposure to an hPXR ligand increased P-glycoprotein expression and transport activity, we dosed hPXR transgenic mice daily for 3 days by p.o. gavage with 50 mg/kg rifampin. We chose this dose level based on preliminary experiments in which we measured plasma rifampin levels in dosed mice and compared them with literature values for patients receiving a therapeutic dose of the drug. In mice, after a single p.o. dose of 50 mg/kg rifampin, maximal total plasma levels averaged 7.72 μg/ml. Of this, 88% was bound to plasma proteins, giving a free concentration of 0.93 μg/ml. Corresponding values for patients receiving a single p.o. dose of 6.4 mg/kg rifampin are 5.41 and 1.08 μg/ml (80% plasma protein binding) (Agrawal et al., 2002). Likewise, predicted free Caverage at steady state after multiple doses is also the same for human and mouse, 0.34 and 0.35 μg/ml, respectively (see under Materials and Methods). Thus, dosing mice with 50 mg/kg rifampin daily resulted in peak plasma levels of free drug similar to those seen in patients taking single, daily 6.4 mg/kg p.o. doses (448-mg dose for 70-kg patient).
Figure 4, A and B, shows Western blots of membranes isolated from intestine, liver, and brain capillaries of control and rifampin-dosed hPXR transgenic mice. Rifampin dosing increased P-glycoprotein immunoreactivity in all three tissues. Consistent with this, luminal membrane P-glycoprotein immunofluorescence in capillaries from rifampin-dosed hPXR transgenic mice was significantly higher than in capillaries from control hPXR mice (Fig. 4, C and D). In addition, P-glycoprotein activity (measured as PSC833-sensitive luminal NBD-CSA accumulation) in brain capillaries from rifampin-dosed mice was more than double that found in capillaries from controls (Fig. 4E).

Reduced efficacy of methadone in rifampin-dosed hPXR transgenic mice. Mice were dosed daily for 1 to 3 days with 50 mg/kg rifampin in 0.1% agarose by p.o. gavage (4 μl/g 0.1% agarose, 12.5 μg/μl 0.1% agarose); controls received agarose alone. Twenty-four hours after the last dose, animals were tested to measure the methadone (3 mg/kg s.c.) antinociceptive effect as described under Materials and Methods. B, plasma methadone levels in transgenic mice as a function of time after dosing (3 mg/kg s.c.). Each point represents the mean value for five mice; variability is given by S.E. bars. Statistical comparison: *, significantly less than controls, P < 0.05.
Having shown increased P-glycoprotein expression and activity in brain capillaries from transgenic mice after dosing with hPXR ligands, we used these mice to determine the extent to which hPXR activation would alter the efficacy of a drug that is both CNS-active and a P-glycoprotein substrate. Several classes of CNS-acting drugs include members that are P-glycoprotein substrates (e.g., antiepileptics, analgesics, and anti-inflammatory steroids) (Balayssac et al., 2005). To act, these must enter the brain to some extent. One consequence of increased brain capillary expression of P-glycoprotein should be reduced brain penetration of these drugs and thus reduced CNS efficacy. To test this supposition, we assayed the antinociceptive effect of methadone in hPXR transgenic mice dosed p.o. for 3 days with 50 mg/kg rifampin and in vehicle-dosed transgenic mice (controls). Methadone is one P-glycoprotein substrate that enters the CNS in sufficient quantity to have an easily measurable CNS effect (analgesia). This antinociceptive effect is substantially enhanced in P-glycoprotein knockout mice (Zong and Pollack, 2003; Dagenais et al., 2004). The assay we used measured the tolerance of mice to an electrical stimulus before and after administration of 3 mg/kg s.c. methadone (antinociceptive effect; see under Materials and Methods). In agreement with previous studies (Dagenais et al., 2004), methadone substantially increased tolerance in control mice. Thirty-five minutes after methadone dosing, mice tolerated a stimulus almost 3 times greater (271.2 ± 27.7%) than they could in trials without methadone (response set at 100%) (Fig. 5A). In transgenic mice pretreated with rifampin, methadone still had an antinociceptive effect, but this effect was substantially blunted. Thirty-five minutes after dosing, rifampin-treated mice could tolerate a stimulus only 1.5 times greater (151.5 ± 32.5%) than without methadone. Thus, rifampin pretreatment reduced methadone's antinociceptive effect to approximately 30% of that found in mice not given rifampin. In addition, the overall magnitude of antinociceptive effect as indicated by the area under the time-effect curve (0-480 min) was significantly lower in mice pretreated with rifampin (133 ± 17% × min) than in controls (422 ± 65% × min; P < 0.05).
Because measurements of mean plasma methadone levels over the entire time course of the experiment showed no significant differences between control and rifampin-dosed mice (Fig. 5B), the effect of rifampin pretreatment on methadone efficacy seemed to have resulted from reduced methadone access to CNS sites of action rather than from increased methadone excretion or metabolism in peripheral tissues. To define the relationship between antinociceptive effect and brain tissue concentrations of methadone, additional experiments were performed in separate groups of mice. CF-1 mice [mdr1a(+/+) and mdr1a(-/-)] were used for this animalintensive effort. Figure 6A shows dose-response relationships for wild-type and mdr1a(-/-) mice. As one would expect, the curve for mdr1a(-/-) mice was shifted substantially to the left. Plots of brain versus serum methadone concentration were linear. It was clear from these plots that the slope of the line for mdr1a(-/-) mice was substantially higher than for wild-type mice; equivalent plasma methadone concentrations produced roughly 6-fold higher brain concentrations in the mdr1a(-/-) mice (Fig. 6B). These results confirm previous studies on methadone effectiveness and brain levels in mice (Thompson et al., 2000; Dagenais et al., 2004). Figure 6C shows the relationship between antinociceptive effect and brain methadone concentration in these mice. Note that to observe the full response, different methadone dose ranges were used for wild-type (0.6-6 mg/kg) and mdr1a(-/-) (0.2-1.5 mg/kg) mice. Nevertheless, the combined data fit to a single sigmoidal Emax model. The relationship between antinociceptive effect and brain methadone concentration is clearly the same for both wild-type and mdr1a(-/-) mice; i.e., it is independent of P-glycoprotein status. These results indicate that antinociception provides a reasonable surrogate for brain tissue concentrations of methadone in mice; they argue that the decrease in antinociceptive effect seen in rifampin dosing experiments with hPXR transgenic mice was a result of decreased brain methadone accumulation.
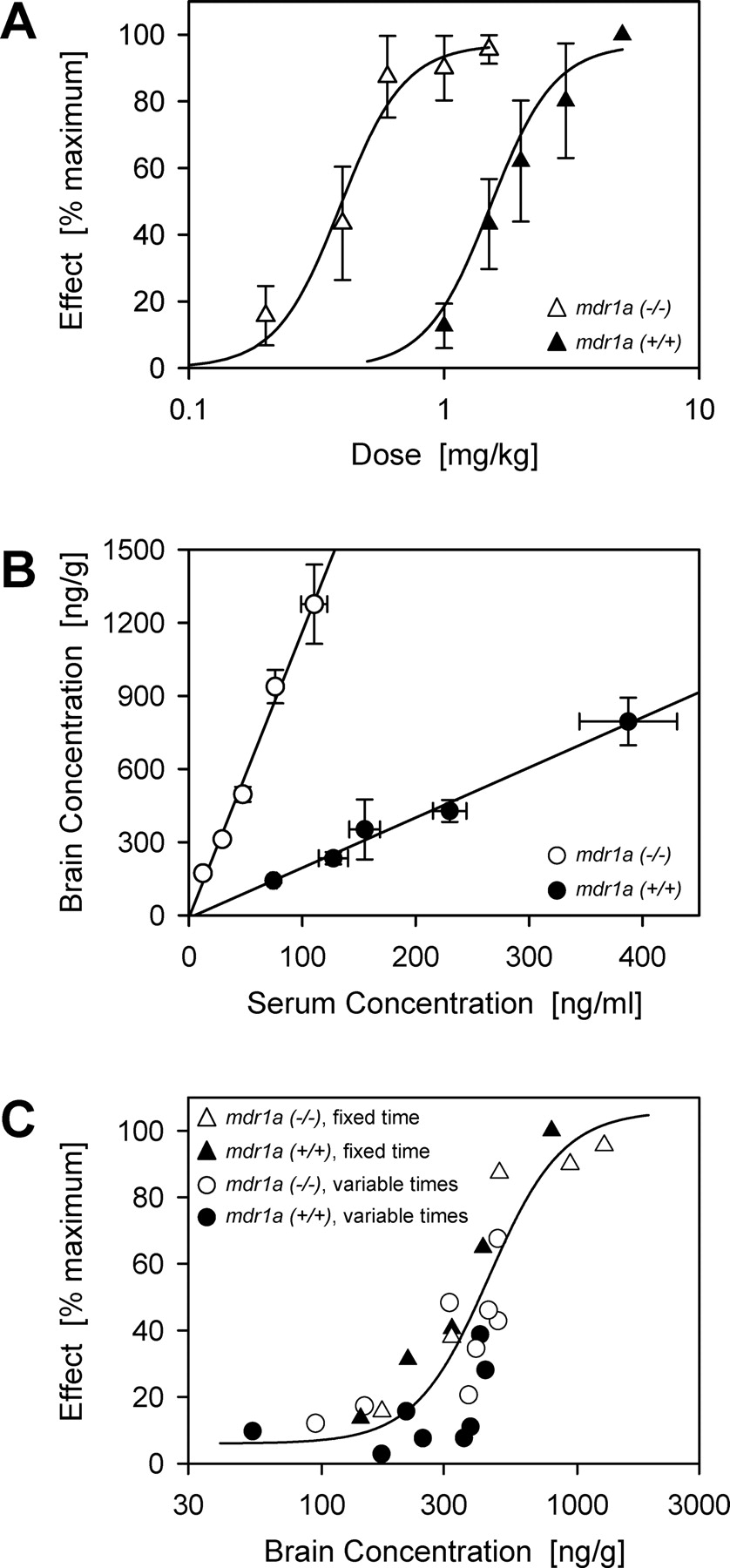
Relationship between antinociceptive effect and brain methadone concentration in mdr1a(+/+) (closed symbols) and mdr1a(-/-) (open symbols) CF-1 mice. A, dose-response curves for methadone antinociception. B, brain and serum methadone levels. Linear regression analysis indicated a good fit for the data from wild-type (r2 = 0.985) and mdr1a(-/-) mice (r2 = 0.991). The regression lines had similar intercepts but very different slopes (2.1 versus 11.7 ml/g for wild-type and mdr1a(-/-) mice, respectively; P < 0.01). C, relationship between antinociceptive effect and brain tissue methadone concentration in mdr1a(+/+) (closed symbols) and mdr1a(-/-) (open symbols) at fixed times (triangles) and at variable times (circles) after methadone. Curve represents fit of a sigmoidal Emax equation to the data (r2 = 0.728); error bars are omitted for clarity. Each point represents the mean value for four or five animals; when shown, error bars indicate SD.
Discussion
Polypharmacy is a fact of life in the clinic. With it comes the potential for drug-drug interactions that alter dose-response relationships and thus can affect drug efficacy and safety. Such interactions can be direct (e.g., when two drugs compete for common sites of action, metabolism, or transport) or indirect (e.g., when one drug alters the expression of genes that determine the action, metabolism, or distribution of a second drug). The present results for a humanized, transgenic mouse provide an example of the latter. Using isolated brain capillaries, we showed increased expression and transport activity of the drug efflux pump, P-glycoprotein, after exposure to the hPXR ligands, rifampin and hyperforin. Similar increases in P-glycoprotein expression and activity were found in capillaries from mice dosed p.o. with rifampin. Moreover, rifampin dosing substantially reduced the effectiveness of the CNS-acting drug and P-glycoprotein substrate, methadone, without changing plasma methadone levels. In these experiments, plasma rifampin levels were similar to those measured in patients undergoing antibiotic therapy, suggesting that similar effects would be seen in patients exposed to therapeutic levels of rifampin.
In previous in vitro and in vivo experiments with the rodent-specific PXR ligand, PCN, we found maximal induction of P-glycoprotein expression and activity was approximately 2-fold in rat brain capillaries (Bauer et al., 2004). The present data for mouse indicate roughly the same extent of maximal induction in vitro in capillaries from wild-type (with PCN) and transgenic (with rifampin) animals. Thus, the extent to which activation of PXR increases P-glycoprotein expression in brain capillaries from rodents is consistent. In the present in vivo dosing experiment with transgenic mice, the dose of rifampin used was selected to give plasma levels that matched those found in patients. Although the increase in P-glycoprotein activity and expression was comparable with that seen in vitro, it is not clear whether this dose produced a maximal effect in vivo. These experiments with an animal model do not tell us with quantitative certainty the extent to which rifampin will induce P-glycoprotein expression in patients or how that change in transporter expression will translate into a pharmacodynamic effect. Taken together, these in vitro and in vivo experiments indicate that hPXR ligands can up-regulate expression of a key element of the blood-brain barrier and produce a selective tightening of that barrier.
PXR-driven up-regulation of P-glycoprotein at the blood-brain barrier (and peripheral tissues) has important clinical implications. The wide specificity limits of both P-glycoprotein (roughly half of commonly prescribed drugs) and hPXR (a growing list of metabolites, drugs, and dietary constituents) argue that hPXR-based changes in selective barrier function should be widespread in the population. They are likely contributors to the difficulties encountered in chemotherapy of brain tumors, to the multidrug resistance seen in epilepsy patients, and to patient-to-patient variability in response to CNS-acting drugs (Loscher and Potschka, 2005a,b). On the other hand, recognition of the important role that hPXR plays in determining the level of P-glycoprotein expression at the blood-brain barrier, and thus of barrier selective permeability, may be a first step to devising simple treatments that can be used to prevent or reverse selective barrier tightening (e.g., restrictions in diet or careful choice of prescribed drugs). The extent to which individual hPXR ligands will alter blood-brain barrier transport function in patients clearly will depend on exposure levels, ligand affinity for the receptor, and ligand pharmacokinetics.
Footnotes
-
This research was supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences, and NIH grant GM61191 (to G.M.P.).
-
ABBREVIATIONS: CNS, central nervous system; PXR, pregnane X receptor; PCN, pregnenolone 16α-carbonitrile; hPXR, human pregnane X receptor; mPXR, mouse pregnane X receptor; NBD-CSA, [N-ϵ(4-nitrobenzofurazan-7-yl)-d-Lys8]-cyclosporine A; DPBS, Dulbecco's phosphate-buffered saline; BSA, bovine serum albumin; RT-PCR, reverse transcription-polymerase chain reaction; bp, base pair(s); GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HPLC/MS, high-performance liquid chromatography/mass spectrometry; AUC, area under the concentration-time curve; ESV, electrical stimulation vocalization.
- Received February 24, 2006.
- Accepted July 11, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}