


Abstract
The interest in the allosteric modulation of G protein-coupled receptors has grown during the past decade. It has been shown that ligands acting at allosteric sites present in these important drug targets have the ability to modulate receptor conformations and fine-tune pharmacological responses to the orthosteric ligand. In the present study, allosteric modulation of the human gonadotropin-releasing hormone (GnRH) receptor by amiloride analogs [e.g., 5-(N,N-hexamethylene)amiloride (HMA)] and a nonpeptide antagonistic furan derivative (FD-1) was studied. First, the compounds' ability to influence the dissociation of a radiolabeled peptide agonist (125I-triptorelin) from human GnRH receptors stably expressed in Chinese hamster ovary cell membranes was investigated. HMA and FD-1, but not 5-(N-benzyl-N-methylaminomethyl)1-(2,6-difluorobenzyl)-6-[4-(3-methoxyureido)phenyl]-3-phenylthieno[2,3-d]pyrimidine-2,4(1H,3H)-dione (TAK-013), another nonpeptide antagonist, were shown to increase the dissociation rate of 125I-triptorelin, revealing their allosteric inhibitory characteristics. The simultaneous addition of HMA and FD-1 resulted in an additive effect on the dissociation rate. Second, in a functional assay, it was shown that HMA was a noncompetitive antagonist and that FD-1 had both competitive and noncompetitive antagonistic properties. Equilibrium displacement studies showed that the inhibition of 125I-triptorelin binding by FD-1 was not affected by HMA. Furthermore, the potency of HMA to increase radioligand dissociation was not affected by the presence of FD-1. Simulation of the data obtained in the latter experiment also indicated neutral cooperativity between the binding of HMA and FD-1. Taken together, these results demonstrate that HMA and FD-1 are allosteric inhibitors that bind at two distinct, noncooperative, allosteric sites. This presence of a second allosteric site may provide yet another opportunity for the discovery of new ligands for the human GnRH receptor.
The gonadotropin-releasing hormone (GnRH) receptor belongs to the rhodopsin-like subfamily (class A) of G protein-coupled receptors (GPCRs) (Millar et al., 2004). Activation of the GnRH receptor results in the biosynthesis and secretion of the gonadotropins luteinizing hormone and follicle-stimulating hormone. The gonadotropins bind to their respective receptors on the gonadal cells, which stimulates germ cell development and hormone secretion in the ovaries (Rhoades and Pflanzer, 1996). GnRH, also named luteinizing hormone-releasing hormone, is a linear hypothalamic decapeptide (Fig. 1) and was first isolated and characterized by Schally et al. (1971). Several peptidic agonists and antagonists for the GnRH receptor have been approved for the treatment of a variety of sex-hormone-dependent diseases, such as prostate and breast cancer and endometriosis (Conn and Crowley, 1994; Kiesel et al., 2002). Superagonists, a somewhat ambiguous term for continually administered peptidic agonists, are used to desensitize and down-regulate the GnRH receptor, resulting in gonadal suppression. Such use of agonists, however, produces an initial hormonal “flare,” resulting in a temporary activation of the pituitary, which can be prevented by giving peptidic antagonists instead. However, peptidic compounds often need to be administered by parenteral (subcutaneous or intramuscular) injection (Kiesel et al., 2002). Therefore, intensive efforts have been initiated to develop nonpeptidic antagonists, which have the potential to become orally available drugs (Armer and Smelt, 2004).

Sequences of GnRH, triptorelin (agonists), ganirelix (antagonist), and chemical structures of FD-1 (antagonist and allosteric inhibitor), HMA, MIBA, DCB (allosteric inhibitors), and TAK-013 (antagonist).
In the past decade, several classes of nonpeptidic GnRH receptor antagonists have been reported (e.g., Imada et al., 2006; DeVita et al., 1999; Chu et al., 2001; Pontillo et al., 2005). These ligands compete with a peptidic agonist for the same binding site on the receptor, providing evidence that they can be classified as orthosteric ligands. In addition, mutational analysis of the GnRH receptor has shown that these nonpeptidic antagonists have overlapping but nonidentical binding sites (Betz et al., 2006). The orthosteric binding site of a GPCR has been defined as the site that is recognized by the endogenous ligand (May et al., 2007). For several GPCRs, however, another (allosteric) binding site has been identified [e.g., for muscarinic receptors (Class A), the corticotropin-releasing factor1 receptor (class B), and glutamate receptors (Class C) (for reviews, see Christopoulos and Kenakin, 2002; and Soudijn et al., 2004)]. Compared with conventional orthosteric ligands, allosteric modulators can have the therapeutic advantage of greater selectivity and tissue specificity. In addition, the risk of overdose is diminished by their saturability.
In the present study, the allosteric modulation of the human GnRH receptor was examined. Equilibrium and kinetic radioligand binding experiments were performed in the presence and absence of both nonspecific [e.g., 5-(N,N-hexamethylene)amiloride (HMA)] and GnRH receptor-selective allosteric modulators [furan derivative-1 (FD-1)] (Fig. 1). Amiloride derivatives have been well described as allosteric inhibitors for different GPCRs at concentrations in the high micromolar range (Gao and IJzerman, 2000), whereas FD-1 is a derivative of a recently described allosteric inhibitor for the GnRH receptor (Sullivan et al., 2006). The ability of a compound to modulate the dissociation rate of 125I-triptorelin was used as a measure for allosteric modulation. This revealed that there are two rather than one allosteric binding sites on this receptor. This emerging concept of multiple allosteric sites may offer further options to modulate GPCR activity.
Materials and Methods
Materials. GnRH, triptorelin, guanosine-5′-triphosphate (GTP) and HMA were purchased from Sigma Aldrich Chemie B.V. (Zwijndrecht, The Netherlands). Amiloride, 5-(N-methyl-N-guanidinocarbonylmethyl)amiloride, 5-(N-methyl-N-isobutyl)amiloride (MIBA), phenamil, benzamil, and dichlorobenzamil (DCB) were kindly provided by Dr E. J. Cragoe (Lansdale, USA) and were synthesized as described previously (Cragoe et al., 1967). Suramin was a generous gift from Bayer AG (Wuppertal, Germany). PD81,723 and SCH-202676 were synthesized in our own laboratory as described by van der Klein et al. (1999) and van den Nieuwendijk et al. (2004). Ganirelix was provided by Organon BioSciences (Oss, The Netherlands). TAK-013 and FD-1 were prepared according to literature procedures (Furuya et al., 1997; Sun et al., 2002). Bovine serum albumin (BSA; fraction V) was purchased from Sigma (St. Louis, MO), whereas bicinchoninic acid (BCA) protein assay reagent was from Pierce Chemical Company (Rockford, IL). 125I-triptorelin (specific activity, 2200 Ci/mmol) was purchased from PerkinElmer Life Sciences (Groningen, The Netherlands). Chinese hamster ovary (CHO) cells stably expressing the human GnRH receptor was obtained from Euroscreen (Brussels, Belgium). The CHO-K1 cells expressing the wild-type human GnRH receptor and nuclear factor of activated T cell luciferase reporter gene (NFAT-luc) were provided by Organon BioSciences (Oss, The Netherlands). All other chemicals and cell culture materials were obtained from standard commercial sources.
Cell Culture. CHO cells stably expressing the human GnRH receptor were grown in Ham's F12 medium containing 10% (v/v) normal adult bovine serum, streptomycin (100 μg/ml), penicillin (100 IU/ml), and G418 (0.4 mg/ml) at 37°C in 5% CO2 (Oosterom et al., 2005). The cells were subcultured twice weekly at a ratio of 1:20. For membrane preparation, the cells were subcultured 1:10 and transferred to large 15-cm diameter plates.
Membrane Preparation. Cells were detached from the plates by scraping them into 5 ml of PBS, collected, and centrifuged at 700g (3000 rpm) for 5 min. Pellets derived from 30 plates were pooled and resuspended in 20 ml of ice-cold 50 mM Tris-HCl buffer containing 2 mM MgCl2, pH 7.4. An UltraTurrax (Heidolph Instruments, Schwabach, Germany) was used to homogenize the cell suspension. Membranes and the cytosolic fraction were separated by centrifugation at 100,000g (31,000 rpm) in an Optima LE-80K ultracentrifuge (Beckman Coulter, Fullerton, CA) at 4°C for 20 min. The pellet was resuspended in 10 ml of the Tris buffer, and the homogenization and centrifugation step was repeated. Tris buffer (10 ml) was used to resuspend the pellet and the membranes were stored in 250- and 500-μl aliquots at -80°C. Membrane protein concentrations were measured using the BCA method with BSA as a standard (Smith et al., 1985).
Radioligand Displacement and Saturation Assays. Membrane aliquots containing 5 to 7.5 μg of protein were incubated in a total volume of 100 μl of assay buffer [25 mM Tris HCl, pH 7.4, supplemented with 2 mM MgCl2 and 0.1% (w/v) BSA] at 22°C for 45 min. For saturation experiments, unlabeled triptorelin was spiked with 20% 125I-triptorelin resulting in final concentrations of 0.1 to 3 nM. Nonspecific binding was determined at three concentrations of radioligand in the presence of 100 μM ganirelix. Displacement experiments were performed using 11 concentrations of competing ligand in the presence of 30,000 cpm (∼ 0.1 nM) 125I-triptorelin. Here, nonspecific binding was determined in the presence of 1 μM ganirelix and represented approximately 15% of the total binding. Incubations were terminated by dilution with ice-cold Tris-HCl buffer. Separation of bound from free radioligand was performed by rapid filtration through Whatman GF/B filters presoaked with 0.25% polyethylenimine (PEI) for 1 h using a Brandel harvester. Filters were subsequently washed three times with 2 ml of ice-cold wash buffer (25 mM Tris HCl, pH 7.4, supplemented with 2 mM MgCl2 and 0.05% BSA). Filter-bound radioactivity was determined in a γ-counter (Wizard 1470; PerkinElmer Life and Analytical Sciences).
Radioligand Kinetic Association and Dissociation Assays. Association experiments were performed by incubating membrane aliquots containing 5 to 7.5 μg of protein in a total volume of 100 μl of assay buffer (25 mM Tris-HCl, pH 7.4, supplemented with 2 mM MgCl2 and 0.1% BSA) at 22°C with 30,000 cpm of 125I-triptorelin. The amount of radioligand bound to the receptor was measured at different time intervals during incubation for 90 min. Dissociation experiments were performed by preincubating membrane aliquots containing 5 to 7.5 μg of protein in a total volume of 100 μl of assay buffer (25 mM Tris-HCl, pH 7.4, supplemented with 2 mM MgCl2 and 0.1% BSA) at 22°C for 45 min with 30,000 cpm (∼0.1 nM) of 125I-triptorelin. After preincubation, dissociation was initiated by addition of 1 μM ganirelix in the presence or absence (control) of HMA, MIBA, DCB, FD-1, or TAK-013 in a total volume of 5 μl. The amount of radioligand still bound to the receptor was measured at various time intervals for a total of 2 h. Incubations were terminated and samples were obtained and analyzed as described under Radioligand Displacement and Saturation Assays.

Saturation of 125I-triptorelin binding to human gonadotropin-releasing hormone receptors. The specific binding (□) was determined by subtracting the nonspecific binding (♦) from the total binding (▪). The KD value was 0.35 (0.33-0.37) nM and the Bmax value was 217 (207-227) fmol/mg protein. Representative graphs from one experiment performed in duplicate.
Competitive Kinetic Radioligand Dissociation Assays. Dissociation experiments were mainly performed as described above. After preincubation, dissociation was initiated by addition of 1 μM ganirelix in the presence or absence (control) of different concentrations FD-1 (1, 3, or 10 μM) and in the presence or absence (control) of six different concentrations of HMA (5-100 μM) in a total volume of 5 μl. The amount of radioligand still bound to the receptor was measured after 30 min. Incubations were terminated and samples were obtained and analyzed as described under Radioligand Displacement and Saturation Assays.
Luciferase Assays. CHOhGnRH_luc cells were cultured as described under Cell Culture. However, Dulbecco's modified Eagle's medium was added to the culture medium (1:1 with F12). On the day of the assay, cells were washed with PBS and then harvested using trypsol (0.25% (w/v) in PBS containing 4.4 mM EDTA). Cells were resuspended in assay medium consisting of DMEM and Ham's F-12 (1:1) supplemented with 1 μg/ml insulin and 5 μg/ml apo-transferrin. Typically, a well-contained 30 μl of a certain concentration triptorelin, 30 μl of modulator (HMA or FD-1) or assay medium (control), and 30 μl cell suspension containing 7.5 × 105 cells/ml. After 4 h stimulation, 50 μl of luclite (PerkinElmer Life and Analytical Sciences) was added to each well for detection of luciferase protein and plates were left at room temperature for 30 min in the dark. Finally, the luminescence signal was quantified on the Microbeta Trilux 1450 Luminescence Counter (PerkinElmer Life and Analytical Sciences).
Data Analysis. All binding data were analyzed using the nonlinear regression curve-fitting program Prism v. 5.00 (GraphPad Software Inc., San Diego, CA). EC50 values were directly obtained from the dose-response curves and inhibitory binding constants (Ki values) were derived from the IC50 values according to Ki = IC50/(1 + [C]/Kd) where [C] is the concentration of the radioligand and Kd is its dissociation constant (Cheng and Prusoff, 1973). The Kd value of 125I-triptorelin at CHOhGnRH membranes was obtained by computer analysis of saturation curves. Dissociation constants, koff, were obtained by computer analysis of the exponential decay of the percentage of 125I-triptorelin bound to the receptor. Association rates were calculated according to the equation kon = (kobs - koff)/[L], where kobs was obtained by computer analysis of the exponential association of the percentage of 125I-triptorelin bound to the receptor and [L] is the amount of radioligand used for the association experiments. The EC50 from competitive dissociation experiments was obtained from dose-response curves of enhanced dissociation by different concentrations of HMA, where the nonspecific binding was set at 0% and either the true control (buffer) or own control binding (1, 3, or 10 μM FD-1) after 30 min was set at 100%. All values obtained are means of at least three independent experiments performed in duplicate.
Simulation of Cooperativity between FD-1 and HMA. A mathematical model (eq. 1) for two distinct allosteric sites (Lazareno et al., 2000) was implemented in MatLab (version 7.1) to simulate the effects of different cooperativities between HMA and FD-1 on the EC50 of HMA in enhancing 125I-triptorelin dissociation.  in which ECHMA50 is the observed EC50 of HMA in enhancing 125I-triptorelin binding. KtriptorelinFD-1 and KtriptorelinHMA are the affinities on the triptorelin-occupied receptor for FD-1 and HMA, respectively. δ is the parameter defining the cooperativity between HMA and FD-1.
in which ECHMA50 is the observed EC50 of HMA in enhancing 125I-triptorelin binding. KtriptorelinFD-1 and KtriptorelinHMA are the affinities on the triptorelin-occupied receptor for FD-1 and HMA, respectively. δ is the parameter defining the cooperativity between HMA and FD-1.
Results
Radioligand Saturation Experiments. Saturation experiments were performed with unlabeled triptorelin spiked with 20% 125I-triptorelin on CHO cells expressing the human GnRH receptor. The results of a representative saturation experiment are shown in Fig. 2. Although the nonspecific binding was high, the receptor binding of 125I-triptorelin was saturable and best characterized by a one-site receptor model. Dissociation constant (KD) and Bmax values of 0.35 (0.33-0.37) nM and 217 (207-227) fmol/mg protein, respectively, were obtained from two independent saturation experiments. The KD value for 125I-triptorelin obtained with these experiments was used to derive Ki rather than IC50 values, as described in the next section.
Radioligand Displacement Assays. Experiments were performed to assess the ability of various ligands to compete with the binding of 125I-triptorelin to CHOhGnRH cell membranes were performed with different ligands. The endogenous agonist (GnRH), a derivative (triptorelin), a peptidic antagonist (ganirelix), and two nonpeptidic antagonists (TAK-013 and FD-1) (Fig. 1), were used to displace radioligand binding. The displacement curves and affinity values are shown in Fig. 3 and Table 1, respectively. All ligands were able to fully displace 125I-triptorelin with affinities ranging from 0.42 nM for triptorelin to 4.9 nM for FD-1. From Fig. 3, it follows that the curve of GnRH had a smaller Hill coefficient than that of the other ligands. Computational analysis indeed showed that it was best described by a two-site competition model with a higher (KH) and a lower affinity (KL) of 0.54 ± 0.004 and 21 ± 10 nM (mean ± S.E.M., n = 3), respectively, with 69 ± 3% of high-affinity receptors.
Receptor affinity of peptidic agonists (GnRH and triptorelin), peptidic antagonist (ganirelix), and nonpeptidic antagonists (TAK-013 and FD-1) Ki represents displacement of specific 125I-triptorelin binding from human gonadotropin-releasing hormone receptors stably expressed in CHO cell membranes. Values are means (± S.E.M.) of at least three separate assays performed in duplicate.
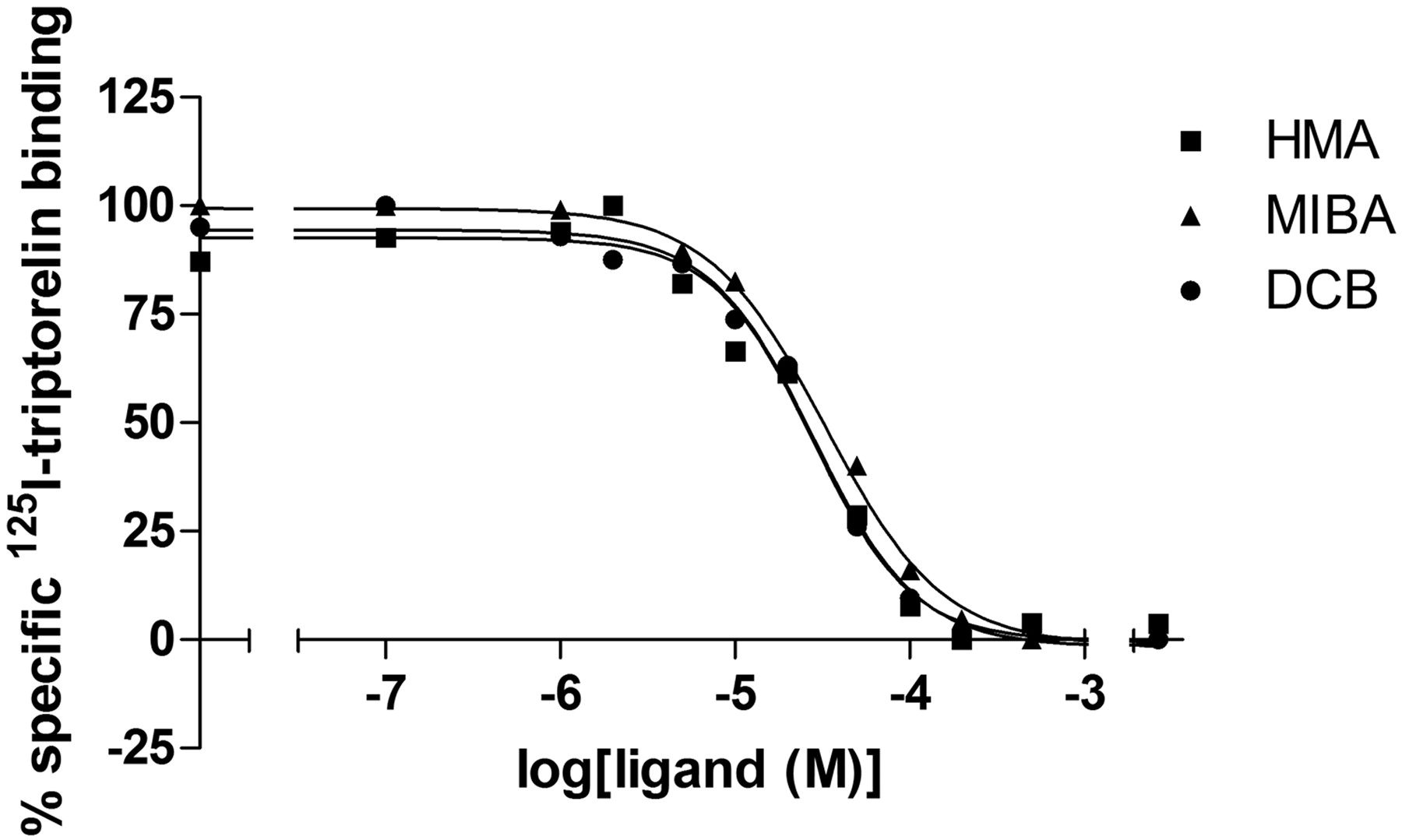
Allosteric Modulation 125I-triptorelin Binding. The effect of some allosteric modulators was tested on equilibrium binding of 125I-triptorelin. As shown in Fig. 4a, PD81,723, a selective adenosine A1 receptor modulator, had no effect on radioligand binding to the GnRH receptor. The addition of GTP, suramin, and sodium ions had a modest effect on the binding of 125I-triptorelin. Both SCH-202676 and HMA (Fig. 1), however, had a detrimental effect on radioligand binding, because almost no radioactivity was detected after incubation with these agents. To investigate the effects of HMA and other amiloride derivatives, a similar experiment was performed with amiloride, 5-(N-methyl-N-guanidinocarbonylmethyl)amiloride, MIBA, phenamil, benzamil, and DCB. From Fig. 4b, it follows that most amilorides had little effect and that only MIBA and DCB were able to inhibit 125I-triptorelin binding. Therefore, displacement of 125I-triptorelin equilibrium binding by HMA, MIBA, and DCB at different concentrations was determined (Fig. 5). The obtained inhibition curves were best described by a one-site receptor model and resulted in similar potencies for HMA (IC50 = 29 ± 3 μM), MIBA (IC50 = 39 ± 7 μM), and DCB (IC50 = 30 ± 3 μM) with pseudo-Hill coefficients of 1.4 ± 0.06, 1.3 ± 0.02, and 1.6 ± 0.2, respectively (Table 2).
Displacement, dissociation, and allosteric modulation of 125I-triptorelin binding by HMA, MIBA, DCB, and FD-1
Inhibitory potency is displacement of specific 125I-triptorelin binding from human GnRH receptors stably expressed in CHO cell membranes. The value of the kinetic dissociation rate constant was obtained by analysis of the exponential dissociation curve of 125I-triptorelin bound to human gonadotropin-releasing hormone receptors in the presence of buffer (control), 0.1 mM HMA, MIBA, or DCB, 3 μM FD-1, or 0.1 mM HMA and 3 μM FD-1. The shift is defined as the ratio of koff values in the presence and absence (control) of modulator, respectively. Modulatory potency is the value for the concentration at half-maximal enhancement of dissociation kinetics. alues are means (± S.E.M.) of at least three separate assays performed in duplicate.

Displacement of 125I-triptorelin from human gonadotropin-releasing hormone receptors stably expressed on CHO cell membranes by GnRH, triptorelin, ganirelix, TAK-013, and FD-1. Representative graphs from one experiment performed in duplicate (see Table 1 for affinity values).

125I-triptorelin equilibrium binding to human gonadotropin-releasing hormone receptors stably expressed on CHO cell membranes in the absence (control, 100%) or presence of GTP, suramin, sodium, SCH-202676, PD 81,723 and HMA (a) and 0.1 mM amiloride derivatives (b). Values are means (± S.E.M.) from at least three independent experiments, performed in duplicate. (*, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control.)
Kinetic Association and Dissociation Experiments. The KD of 125 I-triptorelin in the absence of modulators was also derived from kinetic experiments and the resulting dissociation and association rate constants. Equilibrium binding was reached after approximately 45 min with an association rate constant of 0.28 ± 0.08 nM-1 min-1. Under control conditions, the radioligand dissociated from the receptor with a dissociation rate constant of 0.021 ± 0.002 min-1. Together, this resulted in a “kinetic” KD value of 0.74 nM, which was in good agreement with the KD value (0.35 nM) obtained in the “spiked” saturation analysis. Next, the dissociation kinetics of 125I-triptorelin from CHOhGnRH receptor membranes was determined in the presence of modulator (Fig. 6 and Table 2). All compounds, except TAK-013, increased the dissociation rate compared with the control off-rate, indicative of their allosteric nature and negative modulation of the receptor. The dissociation rate constant of 125I-triptorelin was increased 2.5-fold to 0.053 ± 0.006 min-1 with the addition of 0.1 mM HMA, which was a more potent allosteric inhibitor than MIBA and DCB, although their effect on the equilibrium binding was similar (Table 2). Likewise, the addition of a 3 μM concentration of the nonpeptidic antagonist FD-1 resulted in a 3.2-fold increase of the dissociation rate constant to 0.068 ± 0.009 min-1. The simultaneous addition of HMA and FD-1 in the above concentrations resulted in an additive effect on the dissociation rate constant, which increased 5.2-fold under this condition (koff = 0.11 ± 0.01 min-1).

Inhibition of 125I-triptorelin equilibrium binding to human gonadotropin-releasing hormone receptors stably expressed on CHO cell membranes by HMA and MIBA. Representative graphs from one experiment performed in duplicate (see Table 3 for affinity values).

Dissociation kinetics of 125I-triptorelin binding to human gonadotropin-releasing hormone receptors stably expressed on CHO cell membranes. Dissociation was initiated by either the addition 1 μM ganirelix mixed with buffer (control) or modulator. Representative graphs from one experiment performed in duplicate (see Table 2 for kinetic parameters).

Concentration-effect curves of triptorelin on NFAT-induced luciferase production through human gonadotropin-releasing hormone receptors in the presence and absence (control) of different concentrations HMA or FD-1. Representative graphs from one experiment performed in duplicate (see Table 3 for EC50 and Emax values).
Allosteric Modulation of Receptor Activation. The effect of HMA and FD-1 on receptor activation by triptorelin was measured using a NFAT-induced luciferase assay (Fig. 7 and Table 3). HMA at three concentrations did not cause a shift in potency of triptorelin (EC50 = 0.24 ± 0.02 nM). However, increasing concentrations of HMA resulted in a dose-dependent lowering of the maximal effect (Emax). For example, the presence of 10 μM HMA resulted in an Emax value of 58 ± 1% compared with control (100%). This indicated noncompetitive antagonism, which agrees with the allosteric inhibition seen in the kinetic dissociation experiments. FD-1 at three concentrations caused parallel rightward shifts in the dose-response curves of triptorelin, proof rather of competitive antagonism. However, addition of FD-1 also resulted in a suppression of the Emax value, indicative for its allosteric nature. For example, addition of 3 μM FD-1 decreased the Emax value to 72 ± 5% of the control value.
Receptor activation by triptorelin in the presence or absence of different concentrations of HMA or FD-1
Ca2+ -mediated luciferase activity in CHO cells that stably express the human gonadotropin-releasing hormone receptor and NFAT-luciferase reporter gene. Values are means (± S.E.M.) of at least three separate assays performed in duplicate.
Effect of HMA on FD-1 Binding. To determine whether the allosteric effects described above occurred through an interaction at different allosteric sites, displacement of 125I-triptorelin by different concentrations of FD-1 was determined in the presence and absence of three concentrations of HMA (Fig. 8 and Table 4). It follows from Fig. 8 that the addition of HMA alone (data points on y-axis) inhibited the binding of 125I-triptorelin dose dependently, as shown by the decrease in Bmax in Table 4 and corresponding to the results shown in Fig. 5. FD-1 potently displaced the binding of the radioligand in a concentration-dependent manner. The addition of HMA, however, did not impede the displacement by FD-1. It is noteworthy that, at 30 μM HMA, the affinity of FD-1 was significantly increased (Table 4), indicating a possible allosteric interaction between these compounds.
Receptor affinity of FD-1 and radioligand binding capacity (in the absence of FD-1) in the presence or absence of different concentrations of HMA
Displacement of specific 125I-triptorelin binding from human gonadotropin-releasing hormone receptors stably expressed in CHO cell membranes. Values are means (±S.E.M.) of at least three separate assays performed in duplicate.
Competitive Dissociation Experiments. Another series of experiments were performed to determine whether FD-1 and HMA bound at a different allosteric site. Because FD-1 also acts as an orthosteric antagonist (Fig. 8), “competitive dissociation” experiments were performed solely to study allosteric interactions. The concentration-dependent effect of HMA on 125I-triptorelin dissociation was studied in the absence and presence of three concentrations of FD-1 (Fig. 9). The data obtained are represented in two formats. Fig. 9a shows that the addition of FD-1 enhanced the dissociation, and under every condition, HMA dose-dependently further enhanced that dissociation. Figure 9b shows that the addition of FD-1 did not affect the modulating potency of HMA (EC50 = 49 ± 7 μM), which indicates a noncompetitive interaction of these two compounds. It is noteworthy that FD-1 has a 10-fold higher modulating potency than HMA: 5.0 ± 1 μM (Table 2).

Displacement of 125I-triptorelin by FD-1 binding at human gonadotropin-releasing hormone receptors stably expressed on CHO cell membranes in the presence or absence (control) of three concentrations of HMA. Representative graphs from one experiment performed in duplicate (see Table 4 for affinity and Bmax values).

Effect of HMA on single point dissociation of 125I-triptorelin from human gonadotropin-releasing hormone receptors stably expressed on CHO cell membranes in the presence or absence (control) of three concentrations of FD-1. The top graph (a) shows data normalized to the control measured in the absence of FD-1 and the bottom graph (b) shows data normalized to the four conditions in the absence of HMA. Graphs are mean ± S.E.M. from at least four independent experiments, performed in duplicate.

Neutral cooperativity between HMA and FD-1 in enhancing 125I-triptorelin dissociation. The experimental data of different concentrations of FD-1 affecting the modulating potency of HMA is displayed with standard deviation. The lines show the fit of the data to eq. 1 (see Materials and Methods), where the situations are simulated that two compounds exhibit positive (δ > 1), neutral (δ = 1) and negative cooperativity (δ < 1).
Simulation of Cooperativity between FD-1 and HMA. Eq. 1 under Materials and Methods, taken from Lazareno et al. (2000), was used to simulate the effects of different cooperativities between HMA and FD-1 on the potency of HMA in enhancing the 125I-triptorelin dissociation. When δ = 1, the binding of two allosteric modulators is noninteracting (neutral cooperativity). When δ < 1 or δ > 1, they exhibit either negative (competitive) or positive (enhancement) cooperativity. These simulations, shown in Fig. 10, demonstrate that the data points we had gathered comply with a δ value of 1, thus indicating a neutral cooperativity between the binding of HMA and FD-1.
Discussion
In the present study, it was demonstrated that human GnRH receptors are allosterically modulated by amiloride derivatives and a nonpeptidic antagonist (FD-1). Radioligand displacement assays were performed in which four reference compounds were tested (Fig. 3 and Table 1). For GnRH, a shallow displacement curve was obtained that was best fit with a two-site competition model. In the presence of 1 mM GTP, the favored mode of binding for GnRH shifted toward a one-site competition model with a Ki value of 18 ± 0.6 nM (data not shown). Note that the latter affinity equals the affinity found for the low-affinity receptors in the absence of GTP (KL = 21 ± 10 nM). This can be explained by the ternary complex model, in which the presence of GTP causes a shift to a higher Ki value through uncoupling of the receptor from the G protein (Lefkowitz et al., 1981). It is noteworthy that triptorelin binding was best described by a one-site competition model, although the presence of GTP did decrease radioligand binding (Fig. 4a). Beckers et al. (2001) reported the affinity of GnRH obtained in a 125I-triptorelin displacement assay, where they used whole LTK cells transfected with the human GnRH receptor. A 5-fold lower affinity (5.4 ± 1.8 nM) was found that may be caused by a higher amount of endogenous GTP present in whole cells. The affinities reported for triptorelin and ganirelix, however, were in good agreement with the affinities reported here (Table 1). For TAK-013, an IC50 value of 2.5 nM was reported (Sasaki et al., 2003), whereas we found a Ki value of 1.9 ± 0.7 nM. Finally, FD-1 was tested, which belongs to a different class of nonpeptidic antagonists (Table 1). FD-1 had a Ki value of 4.9 ± 1 nM, which was comparable with the affinity reported for an analog of this compound, CMPD-1 (Ki = 6.0 ± 0.8 nM) (Anderes et al., 2003).
The modulation of 125I-triptorelin binding was explored in the absence and presence of different generally known modulators (Fig. 4a). GTP and suramin are compounds that have an effect on G protein coupling. It was shown that they had only a modest effect on 125I-triptorelin binding. The effect of PD81,723 on the adenosine A1 receptor has been extensively studied (Bruns and Fergus, 1990). It has been shown to be a selective allosteric enhancer at the adenosine A1 receptor, and, as might be expected, it did not affect 125I-triptorelin equilibrium binding to the GnRH receptor. The influence of a high concentration of sodium ions was also examined at the human GnRH receptor. On other GPCRs (e.g., adenosine A2A, α2-adrenergic, and dopamine D2 receptors), sodium ions have been shown to regulate ligand binding (Horstman et al., 1990; Neve et al., 1991; Gao and IJzerman, 2000). However, on the GnRH receptor, sodium ions do not have such a profound effect. In contrast, HMA, which has been shown at higher micromolar concentrations to modulate the same receptor subtypes as sodium ions (Hoare and Strange, 1996; Gao and IJzerman, 2000; Leppik and Birdsall, 2000), was able to fully inhibit radioligand binding. In addition, SCH-202676 was shown to have an effect on equilibrium binding similar to that of HMA. However, this compound was recently shown to be a protein modifier rather than an allosteric modulator (Göblyös et al., 2005). To further explore the modulation of 125I-triptorelin binding by HMA, other amiloride derivatives were tested (Fig. 4b). Two other amiloride derivatives, MIBA and DCB, showed inhibition of equilibrium binding. It had been shown previously that MIBA was the most potent of this class of inhibitors next to HMA (Hoare and Strange, 1996; Gao and IJzerman, 2000).
Allosteric inhibition of 125I-triptorelin binding was shown by the increase in its dissociation rate from human GnRH receptors in the presence of HMA or MIBA (Fig. 6). CMPD-1 has recently been shown to be an allosteric inhibitor for the GnRH receptor too (Sullivan et al., 2006). Previously, that same compound (named Furan-1 or CMPD-1) had been demonstrated to be a potent nonpeptidic antagonist (Anderes et al., 2003), whereas its allosteric effects occur at higher concentrations. FD-1 and CMPD-1 belong to the same class of nonpeptidic antagonists with only some small structural differences (Fig. 1). It was demonstrated that, as for HMA, MIBA, and DCB, FD-1 was also able to increase the dissociation rate (Fig. 6). As mentioned above, HMA was shown to be an allosteric inhibitor on different GPCRs [e.g., at the adenosine A2A receptor (Gao and IJzerman, 2000)]. The selectivity of FD-1 was therefore tested on this receptor; FD-1 did not modulate the dissociation rate of the A2A receptor radioligand (data not shown). FD-1 is therefore a selective allosteric inhibitor, unlike HMA. The simultaneous addition of HMA and FD-1 resulted in an additive effect on the dissociation rate. However, addition of a high concentration (10 μM) of FD-1 further enhanced the dissociation (Fig. 9a). Therefore, this did not indicate per se that the observed additive effect was due to the presence of two allosteric binding sites, although the two compounds are structurally different. The effect on in vitro functional efficacy was also determined (Fig. 7 and Table 3). The functional data showed that HMA is a pure noncompetitive antagonist (allosteric inhibitor) of the effects of triptorelin. On the other hand, FD-1 showed a mixed type of antagonism, indicating both orthosteric and allosteric characteristics. In this assay, HMA and FD-1 showed the same effects when the endogenous ligand GnRH was used (data not shown), even though the binding sites of triptorelin and GnRH are not identical (Fromme et al., 2001). Furthermore, FD-1 seemed to be a more potent allosteric inhibitor than Furan-1 (Sullivan et al., 2006). To prove that the allosteric characteristics of FD-1 were specific for this nonpeptidic antagonist only, TAK-013 was also examined. It was shown that TAK-013 had no effect on the dissociation rate of 125I-triptorelin (Fig. 6). This suggests that the allosteric nature of FD-1 is not a general feature of all nonpeptidic antagonists but was due to structural aspects of FD-1 itself. It is noteworthy that trypan blue exclusion tests showed that cell viability always exceeded 95%, which ruled out that the decrease in maximal response was caused by any cytotoxic effects of the relatively high concentrations of HMA or FD-1. In addition, reversible binding was shown in a luciferase assay, where the cells were preincubated with the highest concentrations used of HMA and FD-1. After washing of the cells according to a method described by Lu and coworkers (2007), full agonist responses were obtained, while unwashed preincubated cells still showed a decreased maximal response.
Finally, we examined whether HMA and FD-1 exert their effect through two distinct allosteric sites on the GnRH receptor. Lazareno et al. (2000) and Lanzafame et al. (2006) have reported two allosteric sites for the M1 and M4 muscarinic receptor, respectively. In addition, three distinct allosteric sites were reported by Schetz and Sibley (2001) for the dopamine D4 receptor. To explore whether HMA and FD-1 compete for the same allosteric binding site, further experiments were conducted. First, the effect of HMA on the displacement of 125I-triptorelin by FD-1 was determined (Fig. 8 and Table 4). It was shown that HMA has no competitive interaction with FD-1. However, the allosteric nature of FD-1 only occurs at high concentrations (micromolar range), which makes it difficult to observe an effect of HMA. Second, a competitive dissociation assay was performed in which the effect of FD-1 on HMA-induced dissociation was examined. Especially from Fig. 9b, it follows that the presence of FD-1 had no effect on the modulatory potency of HMA. This suggests that FD-1 acts at a site distinct from the binding site of HMA and that there is no interaction between the binding of FD-1 and HMA. To strengthen this, a simulation was performed using a model according to Lazareno et al. (2000). As demonstrated in Fig. 10, HMA and FD-1 indeed have neutral cooperativity (δ = 1). It is quite feasible that other GPCRs modulated by amilorides can also be modulated by a receptor-specific modulator from a second allosteric site. For example, the dopamine D2 receptor, which was earlier shown to be modulated by amiloride analogs (Leppik and Birdsall, 2000), is also influenced allosterically by the tripeptide l-prolyl-l-leucyl-glycinamide (Verma et al., 2005).
In conclusion, we have demonstrated that the GnRH receptor can be allosterically modulated by amiloride analogs. In addition, FD-1 was shown to have both orthosteric and allosteric binding properties. Furthermore, we demonstrate that these two chemically unrelated compounds have two distinct allosteric binding sites on the human GnRH receptor and that these sites show neutral cooperativity. The allosteric sites revealed in this study may provide novel targets at the GnRH receptor for orally available low molecular weight compounds.
Acknowledgments
We thank Dr. Guido J. R. Zaman (Organon BioSciences, Oss, The Netherlands) for helpful comments and critical reading of the manuscript. We are grateful to Top Institute Pharma (TI Pharma, The Netherlands) for continuation of this project.
Footnotes
-
ABBREVIATIONS: GnRH, gonadotropin-releasing hormone; GPCR, G protein-coupled receptor; HMA, 5-(N,N-hexamethylene)amiloride; FD-1, furan derivative-1 [5-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-naphthalen-2-yloxy)-furan-2-carboxylic acid (2,4,6-trimethoxy-pyrimidin-5-yl)-amide]; MIBA, 5-(N-methyl-N-isobutyl)amiloride; DCB, dichlorobenzamil; PD81,723, (2-Amino-4,5-dimethyl-3-thienyl)-[3-(trifluoromethyl)phenyl] methanone; SCH-202676, (N-(2,3-diphenyl-1,2,4-thiadiazol-5(2H)-ylidene)methanamine; TAK-013, 5-(N-benzyl-N-methylaminomethyl)1-(2,6-difluorobenzyl)-6-[4-(3-methoxyureido)phenyl]-3-phenylthieno[2,3-d]pyrimidine-2,4(1H,3H)-dione; BSA, bovine serum albumin; CHO, Chinese hamster ovary; NFAT, nuclear factor of activated T cell; PBS, phosphate-buffered saline; CMPD-1, 5-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthalenyl)methyl]-N-(2,4,6-trimethoxyphenyl)-2-furamide.
- Received November 15, 2007.
- Accepted March 14, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}