


Abstract
Alzheimer's disease (AD), the major contributor to dementia in the elderly, involves accumulation in the brain of extracellular plaques containing the β-amyloid protein (Aβ) and intracellular neurofibrillary tangles of hyperphosphorylated tau protein. AD is also characterized by a loss of neurons, particularly those expressing nicotinic acetylcholine receptors (nAChRs), thereby leading to a reduction in nAChR numbers. The Aβ1–42 protein, which is toxic to neurons, is critical to the onset and progression of AD. The discovery of new drug therapies for AD is likely to be accelerated by an improved understanding of the mechanisms whereby Aβ causes neuronal death. We examine the evidence for a role in Aβ1–42 toxicity of nAChRs; paradoxically, nAChRs can also protect neurons when activated by nicotinic ligands. Aβ peptides and nicotine differentially activate several intracellular signaling pathways, including the phosphatidylinositol 3-kinase/v-akt murine thymoma viral oncogene homolog pathway, the extracellular signal-regulated kinase/mitogen-activated protein kinase, and JAK-2/STAT-3 pathways. These pathways control cell death or survival and the secretion of Aβ peptides. We propose that understanding the differential activation of these pathways by nicotine and/or Aβ1–42 may offer the prospect of new routes to therapy for AD.
I. Introduction
Alzheimer's disease (AD1) is the most common form of dementia in elderly persons. It is a neurodegenerative disease marked by decline in memory and cognitive performance, including deterioration of language as well as defects in visual and motor coordination, and eventual death (for review, see Cummings, 2004). AD also involves loss of neurons, beginning in the entorhinal cortex and later spreading to the neocortex (Braak et al., 2006); early in the disease, nicotinic acetylcholine receptors (nAChRs) are lost (Kadir et al., 2006). AD is characterized pathologically by the occurrence of intracellular neurofibrillary tangles rich in tau protein and extracellular plaques containing amyloid peptides (Price et al., 1991). It has been estimated that in 1997, the disease affected more than 2 million people in the United States alone (Brookmeyer et al., 1998). One study estimated 4.5 million U.S. cases in the year 2000 and predicted that the number will rise 3-fold to 13.2 million by 2050 (Hebert et al., 2003). This may be an overestimate, because it relies on data extrapolated on the basis of education, rather than race, with which AD more strongly correlates, and because many cases of vascular dementia may be mistakenly counted as AD (Grant, 2004). A global study of AD using a Delphi consensus approach indicated that there were 24 million people with dementia worldwide in 2005 and that this will almost double every 20 years, to 42 million by 2020 and 81 million by 2040 (Ferri et al., 2005). The prevalence of AD differs both racially (Froehlich et al., 2001) and geographically (Hendrie et al., 2001; Ferri et al., 2005). Given an estimated per-patient yearly cost of $20,000 (Zhu et al., 2006), even the lowest estimates show that AD is a large-scale and growing social, medical, and economic burden. In such circumstances, finding new routes to therapy for AD is a matter of urgency.
It is generally agreed that the β-amyloid peptide (Aβ) plays an important role in the development of AD. The brains of patients with AD contain deposits of Aβ, and Aβ is toxic to cultured neurons (Kihara et al., 1997a; Yao et al., 2005). In addition, mice transgenically overexpressing Aβ or with mutations that enhance Aβ aggregation show many of the symptoms of AD (Hsiao et al., 1996; van Groen et al., 2006). There is abundant evidence that Aβ also affects cholinergic signaling in the brain. Recent studies indicate that brain nAChRs are not only affected by Aβ but can also initiate signaling pathways that protect against Aβ toxicity (Kihara et al., 1997b; Takada et al., 2003; Arias et al., 2005; Akaike, 2006; Meunier et al., 2006; Dineley, 2007; Liu et al., 2007). Current licensed pharmacological treatments for AD consist largely of three acetylcholinesterase (AChE) inhibitors: rivastigmine, galantamine, and donepezil (Aguglia et al., 2004; Ritchie et al., 2004), although memantine, a blocker of l-glutamate receptors of the N-methyl-d-aspartate (NMDA) subtype, is also deployed in late stages of the disease. Interpreting studies on the effectiveness of these compounds is complicated by differences in the measured outcomes (life expectancy, cognitive score, as well as other quality of life measures, often used in various combinations), small sample sizes, and differences in trial methods. There is, however, a consensus that cholinesterase inhibitors perform measurably, but modestly, in slowing the progression of AD (Raina et al., 2008), one meta-analysis estimating their efficacy to amount to saving 2 months per year in the progression of the disease (Trinh et al., 2003). This underscores the importance of cholinergic signaling in AD and offers encouragement that an improved understanding of the roles of nAChRs and their associated signaling proteins in Aβ toxicity may provide clues for the discovery of new targets for the treatment of AD. Here we review current understanding of the roles in AD played by nAChRs and the downstream signaling cascades to which they are linked.
II. β-Amyloid: Biophysics and Roles in Alzheimer's Disease
The accumulation of plaques consisting of Aβ is one of the histopathological hallmarks of AD. Aβ is the product of serial cleavage of the amyloid precursor protein (APP) first by β and then by γ secretases to yield Aβ peptides of varying lengths, predominantly the 37-, 40-, and 42-residue forms. An increasing ratio of the full-length, 1–42 peptide to the 1–40 form is associated with disease (Kumar-Singh et al., 2006), and mutations underlying familial forms of AD either increase this ratio or increase the amount of Aβ secreted. Aβ peptides belong to a class of natively unfolded proteins and as a consequence can adopt a wide variety of tertiary and quaternary structures in vivo and in vitro, including monomers, oligomers, and fibrils (Luheshi et al., 2007; Roychaudhuri et al., 2009). Early work on AD considered the fibrillar form to be the toxic species. However, a lack of correlation between plaque burden and cognitive score contrasted with a strong positive correlation between total soluble amyloid and cognitive decline pointing to soluble, oligomeric forms as the primary toxic factor (Walsh and Selkoe, 2007). Considerable interest recently focused upon the discovery that Aβ56*, a form whose molecular weight is consistent with its being a dodecamer, can be isolated from the cerebrospinal fluid of transgenic mice expressing human APP and, when injected into rats, rapidly and reversibly induced impaired maze performance (Lesné et al., 2006). However, other naturally occurring oligomeric forms of Aβ are also toxic (Deshpande et al., 2006; Shankar et al., 2008), and evidence is accumulating that the capacity of Aβ, mutant Aβ, or fragments of Aβ to aggregate into oligomers is directly related to toxicity (Luheshi et al., 2007). The biophysics of Aβ aggregation is very complex: aggregation can take different routes to different end points and is highly sensitive to the ionic environment (Roychaudhuri et al., 2009). It is a striking deficit in current research that in physiological experiments designed to determine the mode of action of Aβ, the conditions under which Aβ is prepared and its state of aggregation when tested are not always comparable. This must be borne in mind when reviewing the conflicting reports on the physiology of Aβ action.
III. Nicotinic Acetylcholine Receptors: Structure, Function and Roles in Neuroprotection
nAChRs are ligand-gated ion channels consisting of five subunits that form a central, cation-permeant channel whose opening is gated in response to the binding of the neurotransmitter acetylcholine (ACh). Mammals have 16 nAChR subunit-encoding genes (Fig. 1), five of which function at the neuromuscular junction while the remaining subunits are neuronal. A subclass of α subunits is defined by a pair of adjacent cysteine residues that play a key role in ACh binding. Neuronal nAChRs are generated from α (α2–10) and β (β2–4) subunits (Dani and Bertrand, 2007); the three most abundant brain nAChR subtypes are composed of α7, α4β2, and α3β4 subunits, although nearly 30 brain nAChR subtypes have been described (Lindstrom, 2003).
Several lines of evidence point to a link between brain nAChRs and the development of AD. Biochemical analysis of brains of patients with AD reveals deficits in nAChRs, an increase in butyrylcholinesterase, reduction in ACh, and attenuated activity of cholinergic synthetic [choline acetyltransferase (ChAT)] and inactivating (AChE) enzymes (Bartus et al., 1982; Francis et al., 1999). Butyrylcholinesterase and AChE help terminate ACh signaling by hydrolyzing the transmitter, thereby inactivating it. These findings have led to the cholinergic hypothesis of AD and the development by pharmaceutical companies of therapies targeting cholinergic molecular components, so far mainly targeting the hydrolytic breakdown of ACh by AChE (Arneric et al., 2007). Genetic association studies investigating single nucleotide polymorphisms point to roles for cholinergic signaling components such as the synthetic enzyme ChAT, the inactivating enzyme AChE, and α4β2 nAChRs in AD (Cook et al., 2004, 2005; Vasto et al., 2006). The most vulnerable neurons in AD seem to be those expressing high levels of nAChRs, particularly those containing the α7 subunit (D'Andrea and Nagele, 2006), and the numbers of nAChRs as well as some of their associated proteins change in AD (Martin-Ruiz et al., 1999; Gotti et al., 2006; Sabbagh et al., 2006). In addition, not only have α7 nAChRs been found colocalized with plaques (Wang et al., 2000b) but α7 and α4 subunits are also positively correlated with neurons that accumulate Aβ (Wevers et al., 1999). The putative roles for nAChRs in AD has led to the development of new candidate AD drugs targeting nAChRs (Arneric et al., 2007), some examples of which are shown (Fig. 2).
Thus, although other mechanisms are also involved in the development of AD, there is abundant evidence that defects in cholinergic synaptic transmission and, in particular, nAChR-mediated signaling plays a major role in the disease and are hence the subject of attempts to generate new routes to therapy.
The discovery that nicotine, a ligand acting at nAChRs, and its mimetics can protect neurons against Aβ toxicity (Kihara et al., 1998) is of interest, especially in view of the observation that nicotine also enhances cognition (Rusted et al., 2000). Nicotinic receptors play a particularly prominent role in nicotine protection. The protective effect is blocked by the nicotinic antagonists dihydro-β-erythroidine and mecamylamine (Kihara et al., 2001; Takada-Takatori et al., 2006). In addition to nicotine, donepezil and rivastigmine, AChE inhibitors currently used as treatments for mild or moderate AD under the brand names of Aricept and Exelon, also protect cultured neuroblastoma cells from the toxic effects of Aβ. Although there is evidence that, in addition to inhibiting AChE, these ligands are also allosteric modulators of nAChRs (Schrattenholz et al., 1996; Coyle et al., 2007), it has not been established whether these AChE inhibitors protect neurons by their actions on α7 nAChRs rather than by simply inhibiting AChE, thereby elevating ACh in the medium. Curiously, although most studies are in agreement that nAChRs need to be activated to mediate their protective effects, mouse cortical neurons are protected by the α7 antagonist methyllycaconitine (Martin et al., 2004), raising the possibility that neuroprotection by α7 agonists may be through desensitization rather than activation of this rapidly desensitizing receptor. This would be consistent with the α7-dependent activation of intracellular signaling pathways by Aβ (Bell et al., 2004), but the opposite effects on cell survival exerted by Aβ and nicotine means that other mechanisms must be sought, such as ligand-specific coupling to downstream signaling pathways.

Nicotinic acetylcholine receptors (nAChRs) are pentameric proteins composed of homologous nAChR subunits; they are part of the large superfamily of dicysteine loop (Cys-loop) ligand-gated ion channels, which also includes GABA-gated chloride channels, glycine-gated chloride channels, and serotonin (5-HT)-gated cation channels. The tree shows that mammals possess 16 nAChR genes, whereas an additional subunit type (α8) has been found in chicken and pufferfish. nAChRs are expressed in muscle and in the nervous system, with the most abundant brain types consisting of α7, α4β2, and α3β4. The α7 subunits form homomeric nAChRs, whereas α4β2 and α3β4 form heteromeric receptors. α4β2 heteromers can exist as two distinct stoichiometric arrangements.
The action of nicotine may also involve a positive feedback process through the up-regulation of nAChR expression. It is now well established that exposure to nicotine results in increased expression of nAChRs in brain and in cultured cells (for review, see Gentry and Lukas, 2002). Exposure of human neuroblastoma SH-SY5Y cells (which express ganglionic α7 and α3* nAChRs), human TE671/RD cells, or mouse BC3H-1 cells (which express muscle-type nAChRs) to nicotine for up to 120 h induces a dose- and time-dependent increase in surface ACh and α-bungarotoxin (α-BTX) binding not attributable to changes in mRNA levels (Ke et al., 1998). The nicotine-induced nAChR up-regulation in human SH-EP1 cells heterologously expressing α7 nAChRs is mediated by cAMP and protein kinase C (PKC) (Nuutinen et al., 2006). The effects of long-term nicotine treatment on nAChR expression in rat brain differs for receptors of different subtype composition (most pronounced up-regulation being observed for α4β2 receptors) and for different brain regions (Nguyen et al., 2003). Donepezil, which protects cultured rat cortical neurons, when applied for 4 days resulted in an up-regulation of α4 and α7 nAChRs with the result that donepezil was even more potently protective (Kume et al., 2005). The observation that the effectiveness of various continually applied nicotinic ligands to protect PC12 cells from serum-starvation toxicity correlates well with their power to increase 125I-α-BTX binding (Jonnala and Buccafusco, 2001) suggests that such a positive feedback mechanism may play a significant role in nicotine neuroprotection.
A. Which Nicotinic Acetylcholine Receptor Subunits Mediate Nicotine Neuroprotection?
Nicotine neuroprotection via nAChRs can involve several nAChR subtypes and thus can differ between cell types. Nicotine protection of cultured rat cortical neurons against Aβ toxicity is blocked by the α4β2 antagonist, dihydro-β-erythroidine (Kihara et al., 1998). In SH-SY5Y cells, RNA interference (RNAi) knockdown of α7 enhanced Aβ toxicity (Qi et al., 2007), and α7 antagonists, but not α4β2 antagonists, block galantamine protection of cultured rat neurons (Kihara et al., 2004). Donepezil protects cultured rat cortical neurons against Aβ toxicity through both α7 and non-α7 nAChRs (Takada et al., 2003). It is therefore likely that α7 nAChRs are the primary mediators of nicotine neuroprotection, but in some cells, non-α7 subtypes are also likely to contribute.

Several drugs either in current use or in clinical trials for the treatment of Alzheimer's disease are active on nicotinic acetylcholine receptors. The manufacturer is shown in italics.
B. Nicotinic Receptor-Mediated Neuroprotection: a Particular Case of a General Prosurvival Mechanism?
Nicotinic neuroprotection against non-Aβ toxicity is also mediated largely through α7 nAChRs. α7 nAChRs protect PC12 cells against ethanol toxicity (Li et al., 1999a) and from cell death associated with serum depletion (Ren et al., 2005); they protect cultured neurons against glutamate-induced excitotoxicity (Kaneko et al., 1997) and hippocampal slices against oxygen and glucose deprivation (Egea et al., 2007) through the activation of α7 nAChRs (Rosa et al., 2006). Nicotine protects SH-SY5Y cells from cell death induced by thapsigargin, an inhibitor of the sarcoplasmic-reticulum calcium pump (Arias et al., 2004). As in the case of amyloid toxicity, however, non-α7 receptors also play roles in nonamyloid toxicity. This can sometimes be cell-type specific. For example, α4-specific agonists protect porcine small retinal ganglion cells against l-glutamate toxicity (Thompson et al., 2006), whereas α7 nAChRs protect large retinal ganglion cells (Wehrwein et al., 2004) against l-glutamate toxicity. In some cases, only non-α7 nAChRs mediate protection. For example, non-α7 nAChRs protect cultured nigral dopaminergic neurons from toxicity induced by 1-methyl-4-phenylpyridinium, a neurotoxin that selectively damages nigrostriatal dopaminergic neurons (Jeyarasasingam et al., 2002), and this effect is not mediated by α7 receptors. In some cases, non-α7 subunits are necessary for neuroprotection. For example, nicotine effectively protects wild-type mice, but not α4-knockout mice, against methamphetamine-evoked neurodegeneration (Ryan et al., 2001). It is likely, however, that nicotinic neuroprotection can differ according to the toxic chemical (Gahring et al., 2003).
Protection by nAChRs against apoptosis is not restricted to neurons or neuron-like cells. Hepatic vagus nerve activity has recently been shown to protect hepatocytes from Fas-induced apoptosis via activation of α7 nAChRs (Hiramoto et al., 2008). Thus, nicotine seems to exert a general pro-survival action not only on neurons but also on non-neuronal cells, suggesting that the protection offered by nicotine against Aβ toxicity may therefore simply be the result of a general pro-survival response.
Thus, nAChR-mediated protection of neurons and other cell types against Aβ toxicity is a specific instance of nAChR-mediated protection against several toxic compounds and is effected largely through α7 nAChRs. However, non-α7 nAChRs may also play roles with the relative importance of these two subtypes varying between different tissues, indeed even between different cell types within the same tissue, and depending upon the nature of the toxic challenge.
Given the ability of some nAChR antagonists to exert neuroprotection (Martin et al., 2004) and the fact that nicotine neuroprotection has been achieved at concentrations (approximately millimolar) likely to completely desensitize the receptor, the simplest explanation is that such neuroprotective effects are achieved through the receptor in its desensitized state. Whether such a mechanism is effected through a direct (such as an allosteric) link via signaling between the desensitized receptor and downstream signaling molecules (α7 is known to interact physically with cell signaling molecules) or requires conduction of ions through the receptor, or both, remains to be resolved. However, it should be recalled that Aβ also stabilizes the desensitized state (Dineley et al., 2002), leaving open the question of why Aβ has the opposite effect to nicotine. The answer may reside in differences in the downstream signaling evoked by nicotine and the peptide.
C. Nicotinic Acetylcholine Receptor-Linked Neuroprotection Pathways
1. The Phosphatidylinositol 3-kinase/v-akt Murine Thymoma Viral Oncogene Homolog 1 Pathway.
The phosphoinositide 3-kinase/v-akt murine thymoma viral oncogene homolog pathway (PI3K-AKT) is a well established antiapoptotic pathway and has been identified as an important component of nicotine neuroprotection (Kihara et al., 2001). The neuroprotective effects of nicotine are blocked by inhibitors of either PI3K or SRC family kinases, and nicotine evokes an increase in levels of phosphorylated AKT, B-cell chronic lymphocytic leukemia/lymphoma (BCL2), and BCL-2-like protein (Shimohama and Kihara, 2001), which are further downstream in the PI3K/AKT pathway (Fig. 3). Likewise, blocking the PI3K-AKT pathway inhibits the protective effects of AChE inhibitors on neuroblastoma cells or neuronal cells against Aβ (Arias et al., 2005) or l-glutamate neurotoxicity (Takada-Takatori et al., 2006). In all these studies, protection was also inhibited by nAChR blockers, suggesting that these effects are mediated by nAChRs.
How does activating the PI3K pathway by nAChRs protect neurons? One route is through up-regulating the expression of the antiapoptotic protein BCL2. The AD therapeutic AChE inhibitors donepezil, galantamine, and tacrine increase BCL2 expression when applied to cultured neuronal cells (Arias et al., 2004; Takada-Takatori et al., 2006). In these cells, nicotine promotes cell survival and causes the phosphorylation of the proapoptotic protein Bcl2-associated X protein (BAX), through the PI3K/AKT pathway, reducing the movement of BAX from the cytosol to the mitochondria and inhibiting its apoptotic activity (Xin and Deng, 2005). In lung cancer cells, nicotine also exerts an antiapoptotic effect through activating BCL2-antagonist of cell death (BAD), a process that is inhibited by blockers of the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway or the PI3K/AKT pathway (Jin et al., 2004). However, nAChR-mediated neuroprotection may also involve pathways other than those regulating apoptosis. For instance, over-expressing PI3K in Drosophila melanogaster neurons in situ results in an increase in functional synapses as well as synaptic sprouting (Martín-Peña et al., 2006). Thus it is possible that nicotine's activation of the PI3K pathway results in increased synaptic stability, and it would be of interest to explore this further in vertebrates. Thus, the evidence suggests that activation of nAChRs activates the PI3K/AKT pathway to favor antiapoptotic pathways and possibly induce synaptogenesis.
2. Nicotinic Acetylcholine Receptors Are Linked to the Phosphatidylinositol 3-kinase/Protein Kinase B Pathway through Tyrosine Kinases.
Tyrosine kinases, such as FYN and SRC [v-SRC sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog], may play a key role in linking activation of nAChRs with the PI3K/AKT pathway. The neuroprotective activation of the PI3K/AKT pathway by nicotine involves the tyrosine kinase FYN, which physically interacts with α7 nAChRs, and the p85 subunit of PI3K in rat fetal cortical neurons in culture (Kihara et al., 2001). The role of FYN in Aβ toxicity and nicotine neuroprotection, however, is complex, and the role of tyrosine kinases in mediating nicotine neuroprotection is complicated by the observation that, in addition to being activated by nAChRs, they also regulate nAChRs (Charpantier et al., 2005). It remains to be resolved whether FYN plays a protective or toxic role in the development of AD. FYN expression is increased in brains from patients with AD, specifically in a subset of neurons with elevated hyperphosphorylated tau protein (Shirazi and Wood, 1993), but it is not known whether this increase in FYN contributes to hyperphosphorylation of tau or is a protective response to it. In extracts of human brains from patients with AD, soluble FYN increases with cognitive score and synaptophysin levels and inversely with the tangle count, suggestive of a pro-cognitive role for FYN (Ho et al., 2005). In a microarray study comparing brains from patients with AD with control brains, FYN was found to be significantly up-regulated in AD (Wang et al., 2003a). In this context, it is of interest that FYN has also been shown to activate the PI3K/AKT cascade, thereby inhibiting apoptosis (Tang et al., 2007). Indeed, FYN is required for phosphorylation of phosphoinositide 3-kinase enhancer (PIKE), which itself regulates AKT (Fig. 3). PIKE binds to AKT and up-regulates its kinase activity, thereby reducing apoptosis. Phosphorylation protects PIKE from caspase cleavage, hence FYN is antiapoptotic (Tang et al., 2007).
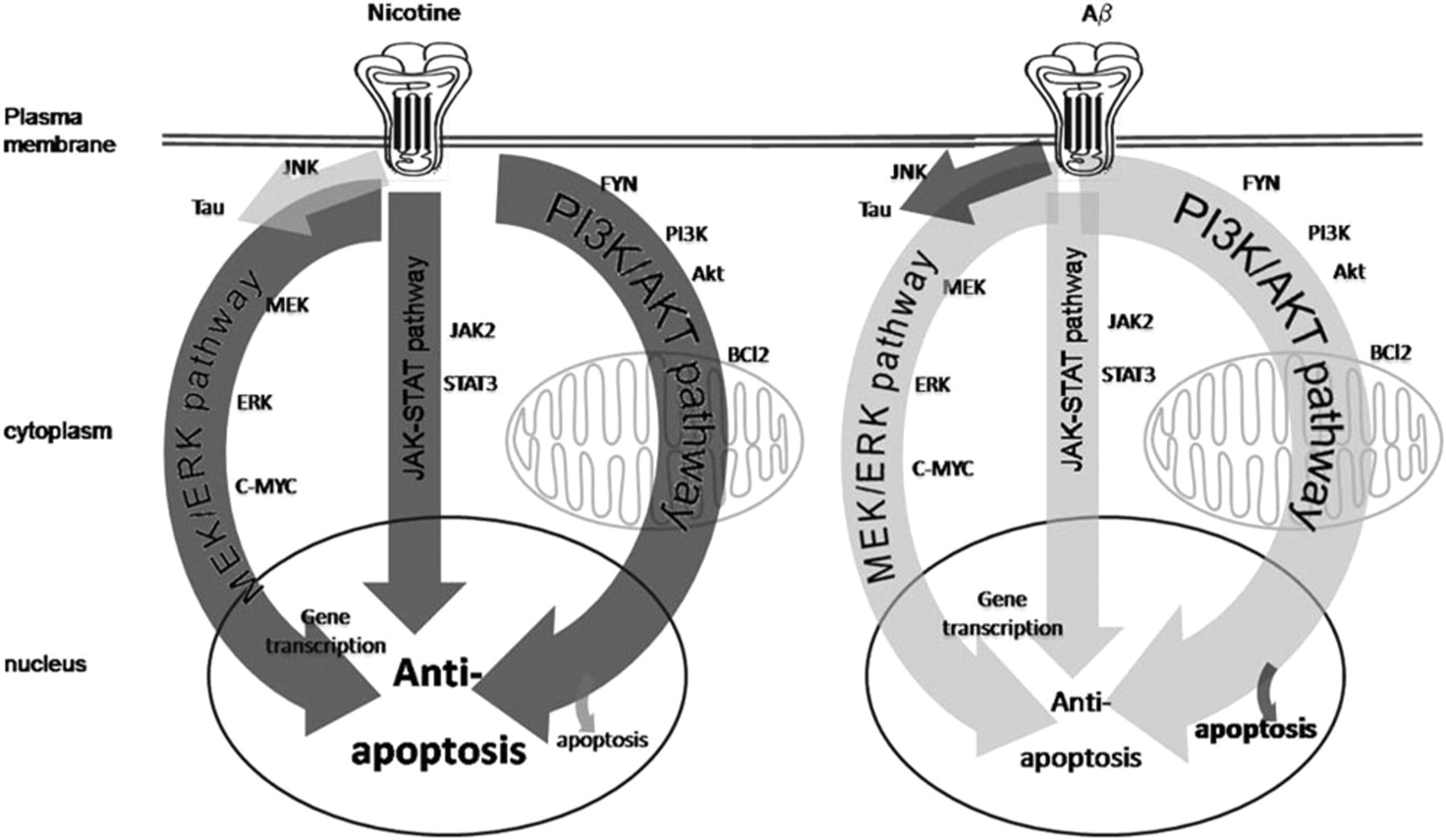
Aβ induces cell death through the activation of nicotinic acetylcholine receptors, as well as through other receptors. Several different signaling cascades, including the ERK/MAPK pathway and the JNK pathway, have been implicated in the actions of Aβ. Aβ also activates AKT and controls the activity of apoptotic proteins, including BAD, BAX, and BCL2. In addition to apoptosis, these pathways also often induce hyperphosphorylation of the tau microtubule protein, providing a link between Aβ-activation of nAChRs and the formation of neurofibrillary tangles. The concentration and time course of Aβ exposure can determine which of these pathways is activated. In addition, several pathways through which nicotine protects cells from Aβ toxicity have been identified, including the JAK-2/STAT-3 pathway, in which nAChR-mediated activation of Janus kinase 2 is transduced into STAT-3 activation; the PI3K/AKT pathway, which results in inhibition of apoptosis; and the ERK/MAPK pathway, which is activated by the Ras/Raf cascade and controls gene expression through c-Myc. There are also direct actions of nicotine on mitochondrial function as well as on the aggregation of Aβ.
From these findings, it would seem that FYN plays a neuroprotective role. However, FYN may also play a paradoxical role in Aβ toxicity. Indeed, Aβ activates both FYN and the PI3K cascade (Williamson et al., 2002), whereas germline knockout of FYN is neuroprotective in mice (Lambert et al., 1998; Chin et al., 2004). FYN knockout protects mature mouse neurons in organotypic central nervous system cultures (Lambert et al., 1998). Furthermore, inhibitors of SRC, a closely related tyrosine kinase, also prevent nicotinic protection of differentiated PC12 cells against serum-deprivation-induced cell death (Li et al., 1999b), and inhibitors of FYN or Janus kinase-2 (JAK-2) block the neuroprotection against Aβ toxicity of therapeutic AChE inhibitors (Takada-Takatori et al., 2006). FYN physically interacts with and phosphorylates tau protein, and the affinity of this physical interaction is enhanced in AD-associated mutations in tau protein (Bhaskar et al., 2005). Aβ rapidly induces tyrosine phosphorylation of many proteins (including tau protein) in human and cultured rat cortical neurons (Williamson et al., 2002). This phosphorylation is concomitant with phosphorylation and inactivation of focal adhesion kinase 1 (FADK1, a major downstream target of FYN), is blocked by inhibitors of SRC kinases and PI3K, and involves FYN associating physically with FADK1 (Williamson et al., 2002). The contradictory nature of these results probably stems from the design of these studies, which pool subcellular compartments that might otherwise have separate and different roles for FYN, which is a highly connected hub molecule in the interactome. More research is therefore needed to resolve the complex roles of FYN and FADK1 in nAChR-mediated neuroprotection and as potential therapeutic targets in AD and neuroprotection.
3. Tyrosine Kinase Regulation of Nicotinic Acetylcholine Receptors May Underlie Phytoestrogen Protection.
Genistein, a phytoestrogen, protects SH-SY5Y cells (Bang et al., 2004) as well as cultured hippocampal neurons (Zeng et al., 2004) from Aβ toxicity. However, in addition to its action on estrogen receptors, genistein is also a general tyrosine kinase inhibitor that protects cultured neurons from l-glutamate toxicity (Kajta et al., 2007). Genistein enhances the amplitude of ACh responses when human α7 nAChRs are expressed in Xenopus laevis oocytes by inhibiting phosphorylation of the receptor in the intracellular loop (Charpantier et al., 2005; Grønlien et al., 2007) and shows similar actions on α7 nAChRs of rat hippocampal and supraoptic nucleus neurons as well as human SH-SY5Y cells (Charpantier et al., 2005). The potentiating effect of genistein on ACh-evoked responses did not involve a large shift in the EC50 for ACh (Grønlien et al., 2007). Thus, although an involvement of estrogen receptors has been demonstrated for the protective action of genistein (Kajta et al., 2007), it is tempting to speculate that the neuroprotective effect of genistein may also involve, at least in part, the enhancement of nAChR function through the inhibition of tyrosine kinase action upon nAChRs. In support of this notion, β-estradiol protects PC12 cells from amyloid toxicity, and this is prevented when α7 nAChRs are blocked with methyllycaconitine (Svensson and Nordberg, 1999).
4. Nicotine Protection and the Janus Kinase-2/Signal Transducer and Activator of Transcription-3 Pathway.
JAK-2, another early target in the nicotine neuroprotection pathway that may mediate signaling between the nAChR and the PI3K pathway (Shaw et al., 2002), may link nAChR activation with the JAK/signal transducer and activator of transcription 3 (STAT-3) protective pathway. JAK-2 is also activated by nicotine in non-neuronal cells such as nAChR-bearing keratinocytes (Arredondo et al., 2006). In a microarray study, expression of 8 of 33 JAK/STAT pathway genes was altered when human bronchial epithelial cells were exposed to 5 μM nicotine for 4 to 10 h (Tsai et al., 2006). Thus, the JAK-2/STAT-3 pathway is activated by exposure to nicotine.
Nicotine induced phosphorylation of STAT-3 (signal transducer and activator of transcription 3) in peritoneal macrophages is mediated by an α7-dependent activation of JAK-2, as part of the anti-inflammatory action of vagal nerve stimulation (de Jonge et al., 2005). Short-term nicotine application also induces phosphorylation of p44/42MAPK, p38MAPK, and STAT-3 and was mediated mostly by α7 nAChRs in rat vascular smooth muscle cells (Wada et al., 2007). It is noteworthy that the JAK-2/STAT-3 pathway also mediates the mitogenic effects of insulin, a process recently implicated in AD (Li and Hölscher, 2007). Indeed, in rat vascular smooth muscle cells, insulin and nicotine activate these overlapping pathways (Wada et al., 2007). Nicotine-induced phosphorylation of STAT-3 through activation of α7 has also been demonstrated in human oral keratinocytes (Arredondo et al., 2006).
5. Nicotine Activation of Mitogen-Activated Protein Kinase and JAK/STAT Pathways Leads to Changes in Gene Transcription.
Because STAT-3 is a transcription regulator, its activation by nicotine presumably affects the expression of genes, but so far, little is known of which genes are altered in this way or whether this pathway is required for nicotine neuroprotection. Microarray studies have shown that 24-h incubation in nicotine causes the up-regulation of several genes in SH-SY5Y cells, including ninein (Dunckley and Lukas, 2006), which is known on the basis of a yeast two-hybrid screen to interact with the AD-implicated gene glycogen synthase kinase 3β (Hong et al., 2000). However, alterations in expression of typical antiapoptotic genes, such as the Bcl family, were not detected. Application of nicotine to rat microglia results in the up-regulated expression of cyclooxygenase-2 and prostaglandin E2 (De Simone et al., 2005). Signaling pathways downstream to the MAPK pathway are similarly well placed to effect changes in gene expression. For example, α7-dependent activation of the MAPK pathway is known to activate c-Myc (Liu et al., 2007), a protooncogene whose transcription product sensitizes cells to pro-apoptotic stimuli.
In studies on SH-SY5Y cells and cultured rat hippocampal neurons, nicotine, acting through α7 nAChRs, results in the activation of ERK-1/2 pathways dependent upon calcium and protein kinase A (Dajas-Bailador et al., 2002b). In addition, the α7-specific agonist GTS-21 promotes ERK-1/2 phosphorylation, but not that of c-jun N-terminal kinase (JNK) or p38 (Ren et al., 2005). Furthermore, nicotinic activation of ERK-1/2 promotes survival of cultured murine spinal cord neurons, and the blocking of ERK-1 prevents nicotine's antiapoptotic action (Toborek et al., 2007). Likewise, the α7-specific agonist A-582941 induces phosphorylation of ERK-1/2 in PC12 cells and in mouse brain, and this is completely blocked by the mitogen-activated protein kinase 1 inhibitor SL327 (Bitner et al., 2007). Nicotine also activates ERK in non-neuronal cells such as pancreatic acinar cells (Chowdhury et al., 2007) and vascular smooth muscle cells (Kanda and Watanabe, 2007), although it is not known in those cases which nAChR subtypes are involved. In the cortex and hippocampus of mice, nicotine's inhibition of MAPK (shown by RNAi reduction of α7 expression to be α7-dependent) prevents activation of nuclear factor-κB and c-Myc, also thereby reducing the activity of inducible nitric-oxide synthetase and NO production and decreasing Aβ production (Liu et al., 2007). Paradoxically, Aβ also activates the MAPK pathway through an α7-dependent pathway (Dineley et al., 2001; Bell et al., 2004). In human oral keratinocytes, the Ras/Raf/mitogen-activated protein kinase kinase 1/ERK pathway cooperates with the nicotine activation of the JAK/STAT-3 pathway (Arredondo et al., 2006); the Ras pathway induces STAT-3 up-regulation whereas the JAK/STAT-3 pathway phosphorylates STAT-3.
6. Nicotine Protection and Tumor Necrosis Factor-α Cytokine Signaling.
There is evidence that nicotine's neuroprotective effects can be mediated through tumor necrosis factor-α (TNF-α). Application of either nicotine or TNF-α protects cultured mouse embryonic cortical neurons from N-methyl-d-aspartate (NMDA) toxicity, but coapplication of both does not. However, coapplication of both with α-BTX does lead to neuroprotection, suggesting that TNFα and nicotine protect through antagonistic pathways (Carlson et al., 1998; Gahring et al., 2003). Nicotine may regulate the neuroprotective secretion of TNFα by microglia through enhancement of low-level TNF secretion and suppression of lipopolysaccharide-induced TNFα secretion (Suzuki et al., 2006; Park et al., 2007) via α7-dependent activation of JNK and MAPK pathways.
7. Nicotine Protection and Calcium Signaling Pathways.
Calcium signaling pathways are involved both in the toxic action of Aβ and in the protection against that toxicity offered by nicotinic ligands. Given that α7 homomeric nAChRs are much more permeable to calcium ions than are most other nAChRs (Bertrand et al., 1993), it is to be expected that nicotinic neuroprotection mediated by nAChRs, notably α7, would depend upon the activation of calcium signaling pathways. ABT-418 is a nicotinic agonist that protects primary rat cortical neurons from glutamate toxicity through its activation of α7 nAChRs, and this is blocked when calcium is removed from the extracellular medium (Donnelly-Roberts et al., 1996). Nicotine protects PC12 cells from cell death resulting from serum depletion through a mechanism that depends upon the function of IP3 receptors, L-type calcium channels, ryanodine receptors, and ERK, suggesting that the protective effect of nicotine is mediated by calcium signaling pathways (Ren et al., 2005). Stevens et al. (2003) showed that calcineurin is involved in nicotine neuroprotection. Aβ, through α7 nAChRs, increases Ca2+, which phosphorylates NMDARs via calcineurin and protein tyrosine phosphatase, nonreceptor type 5 (striatum-enriched) (Snyder et al., 2005).
Recent research interest has focused on the role of calcium dyshomeostasis in AD (Green and LaFerla, 2008); for instance, genetic links with the regulation of cytosolic calcium have been identified (Dreses-Werringloer et al., 2008). Thus nAChRs may provide a link between Aβ and disruption of calcium homeostasis. Short-term application of Aβ to the SH-SY5Y human neuroblastoma cell line results in a rapid increase in intracellular calcium ions that is dependent upon both α3β2 and α7 nAChRs (Dajas-Bailador et al., 2002a). This calcium signal in SH-SY5Y cells arises from three sources: influx of extracellular calcium through voltage-gated calcium channels, release of calcium from intracellular stores, and calcium influx through the α7 receptors. It has been shown that the α7 receptors, but not the α3β2 receptors, specifically trigger calcium release from intracellular stores by activating ryanodine receptors. Such a specific functional coupling of α7 receptors and ryanodine-sensitive stores may provide another site of therapeutic intervention. However, the sustained calcium rise seen in these cells upon prolonged nicotine administration, which is more likely to be of relevance to neuroprotection than short-term responses, is more dependent upon the activation of inositol 1,4,5-triphosphate receptors (Dajas-Bailador et al., 2002a), which are also a target for phosphorylation by FYN (Cui et al., 2004). JAK-2, also implicated in the neuroprotective pathway, may play a role in linking nAChR action with calcium signaling, because JAK-2 phosphorylates inositol 1,4,5-triphosphate receptors through its activation of FYN (Wallace et al., 2005). Further research into the relationship between the complex interaction between nAChRs and the calcium signaling machinery is needed to determine the extent to which nicotine exerts its neuroprotective action against Aβ toxicity by counteracting AD-associated disruptions in calcium signaling.
D. Nicotinic Acetylcholine Receptors Mediate β-Amyloid Peptide Actions That Precede Cell Death
The focus of research into the development of AD has shifted in recent years away from toxicity toward earlier events, such as alterations in synaptic function. Consequently, there is mounting evidence that Aβ affects cholinergic signaling independent of its cytotoxic action. For example, Aβ blocks long-term potentiation, a cellular correlate of learning, through activation of JNK and p38MAPK (Wang et al., 2004). APP and APP/presenilin-1 (PS-1) mice do not show neurodegeneration (Irizarry et al., 1997) and yet show several features of AD, including accumulation of plaques and defects in learning (Hsiao et al., 1996), suggesting that many features of AD are not the result of neuronal loss. These animals nonetheless have swollen cholinergic nerve terminals at 12 months, suggesting defective nerve sprouting (Hernandez et al., 2001). It has long been known that cognitive decline in AD correlates well with synaptic loss (Lue et al., 1999), and it has been shown directly that soluble Aβ inhibits synaptic plasticity (Rowan et al., 2004). To date, however, it is not known whether the synaptic actions of Aβ involve nAChRs. Thus, this fertile shift of research focus from toxicity to synaptic mechanisms needs to be matched with similar studies on the nontoxic effects of Aβ on cultured neurons and the effects of nAChR activation on such effects. For example, a recent study has shown that α7-specific ligands rescue the Aβ-induced decrease in neurite outgrowth of cultured mouse neurons (Hu et al., 2007).
E. Apolipoprotein E-ϵ4, the Product of an Alzheimer's Disease Risk Factor Gene, Activates Nicotinic Acetylcholine Receptors
The apolipoprotein E type 4 allele (APOE-ϵ4) encodes the APOE lipoprotein, which through its lipid transport function plays a role in lipid metabolism. APOE-ϵ4 has been found to be a major risk factor for late familial or sporadic AD, with a strong gene-dosage effect such that the number of APOE-ϵ4 alleles correlated positively with the risk of developing AD and the age of onset (Corder et al., 1993). The APOE-ϵ4 gene-dose effect was also found to correlate with the loss of nAChR binding sites in patients with AD, as well as a reduced responsiveness to the therapeutic AChE inhibitor tacrine (Poirier et al., 1995). Within an AD cohort, APOE-ϵ4 dose dependently correlates with higher losses of ChAT but not with losses in α4β2 nAChRs (Lai et al., 2006).
There is evidence that ApoE directly interacts with nAChRs. An APOE-derived peptide blocks nAChRs on rat hippocampal slices with a submicromolar affinity, and this action is dependent on an arginine-rich segment of the APOE peptide (Klein and Yakel, 2004). Block of heterologously expressed α7 nAChRs is greater than that for α4β2 or α2β2 nAChRs (Gay et al., 2006). This block of α7 receptors is abolished when α7 Trp55 is mutated to alanine, providing strong evidence that it results from a direct interaction between the peptide and the receptors (Gay et al., 2007), and the effects of other substitutions of Trp55 suggests that this interaction is hydrophobic.
ApoE-ϵ4, but not ApoE-ϵ3, disrupts carbachol-stimulated phosphoinositol (PI) hydrolysis and so does Aβ and Aβ/ApoE-ϵ4 complexes in SH-SY5Y cells (Cedazo-Mínguez and Cowburn, 2001). The effect of Aβ and its ApoE complex on PI hydrolysis were blocked by estrogen, and this disruption was itself blocked by wortmannin, suggesting that PI3K mediates estrogen's effect on PI hydrolysis. Because nAChR activation also protects through activation of the PI3K pathway, it would be of interest to determine whether ApoE-ϵ4 also disrupts nAChR-activation of PI3K and nAChR-mediated neuroprotection.
F. Nicotinic Acetylcholine Receptors Regulate β-Amyloid Peptide Secretion and Internalization
In addition to Aβ acting upon nAChRs, nAChRs in turn regulate Aβ secretion. Nicotine or epibatidine applied to the human SHEP1 cell line stably transfected with human α4β2 nAChRs and human APP decreases the secretion and intracellular accumulation of Aβ without significantly affecting the APP mRNA, suggesting that these effects are post-translational (Nie et al., 2007). Nicotine stimulates the secretion of βAPP, which is trophic and neuroprotective against Aβ, from PC12 cells through an α7 and calcium-dependent pathway (Kim et al., 1997) as well as increasing the secretion of soluble APP and lowering the Aβ-containing sAPP-γ in rats (Lahiri et al., 2002), again through nAChR-dependent mechanisms. Galantamine, a nAChR potentiator and AChE inhibitor, also increases the secretion of sAPP from human SH-SY5Y neuroblastoma cells (Lenzken et al., 2007) through the activation of nAChRs. It therefore seems that activation of nAChRs shifts the balance of APP processing away from β-amyloidogenic to soluble APP production.
nAChRs may also regulate Aβ internalization. Intracellular accumulation of Aβ by both APOE transport (Gylys et al., 2003; Sadowski et al., 2004) and APOE-independent endocytotic mechanisms (Saavedra et al., 2007) is believed to contribute to cell death (D'Andrea et al., 2001). The importance of this mechanism to Aβ toxicity is underscored by the recent observation that oligomers, which are the most toxic form of Aβ, are internalized more effectively than the less toxic fibrillar forms (Chafekar et al., 2008). An indication that nAChRs may play a role in Aβ internalization comes from a close inspection of cholinergic neurons in brains from patients with AD, which revealed that neurons with high expression levels of α7 also contained large amounts of intracellular Aβ (Nagele et al., 2002). Addition of Aβ to the culture medium of neuroblastoma cells overexpressing α7 results in more Aβ internalization than in control cells with lower levels of α7 expression (Nagele et al., 2002). It is not clear whether this enhanced uptake is the result of an indirect influence by nAChRs upon internalization or of a direct binding of Aβ to nAChRs. Whatever the mechanism of uptake, it is interesting to note that the signaling pathways evoked by the accumulation of intracellular Aβ resemble those evoked by extracellularly applied Aβ: transgenic rats overexpressing Aβ intraneuronally display elevated levels of phosphorylated ERK2 (Echeverria et al., 2004), as do rat hippocampal slices in response to bath-applied Aβ (Dineley et al., 2001). Again, bath-applied Aβ causes an increase in BAX and a decrease in BCL2 expression in neurons or neuron-like cell lines (Koriyama et al., 2003; Clementi et al., 2006). It is therefore tempting to speculate that the nAChR-dependent activation of signaling pathways by exogenously applied Aβ results from an internalization of Aβ after binding to surface nAChRs. The frequent findings that nAChR antagonists block Aβ-evoked signaling cascades may therefore be due to steric occlusion of the Aβ binding site preventing Aβ internalization. Furthermore, at least some of the reports of an acute partial block of α7 nAChR-mediated responses by Aβ may be the result of Aβ-evoked receptor internalization, resulting in fewer receptors being available at the cell surface. Thus, it seems that nAChRs may play a role in mediating Aβ toxicity through synergistic mechanisms; in addition to possible direct interactions (binding), nAChRs may also result in accelerated cell death through enhancing intracellular Aβ accumulation.
IV. β-Amyloid Peptide Toxicity: Is It Mediated in Part through Nicotinic Acetylcholine Receptors?
Paradoxically, in addition to their neuroprotective action, nAChRs may also partly mediate the toxic action of Aβ. The toxicity of Aβ on SH-SY5Y cells, as measured by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, in which the health of cells is monitored by their ability of cells to reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, was significantly impaired when α7 was silenced by RNAi, suggesting that Aβ may exert its toxicity, at least in part, through a pathway that includes α7 nAChRs (Qi et al., 2007). The toxic action of Aβ may involve α7-dependent internalization of amyloid after Aβ binds with high affinity to the α7 receptor, because the rate of internalization of Aβ in human neuroblastoma SH-SY5Y cells depends upon the level of α7 expression (Nagele et al., 2002). It is noteworthy that this internalization was blocked by α-bungarotoxin, which may indicate that α-bungarotoxin either inhibits binding of Aβ to the α7 receptor (and therefore that Aβ toxicity results from binding of Aβ to α7 nAChRs) or directly inhibits α7 nAChR internalization. However, it has been shown that Aβ1–42 binds with high affinity to α7 nAChRs in several different neuronal tissues (Wang et al., 2000a) and displaces α-bungarotoxin binding (Wang et al., 2000a,b) and, rather than inhibiting receptor internalization, α-bungarotoxin enhances internalization of heterologously expressed nAChRs (Kumari et al., 2008). It is also noteworthy that Aβ-induced tau protein phosphorylation in PC12 cells is inhibited not only by α7 agonists, as would be predicted from the role of α7 nAChRs in neuroprotection, but also by α-bungarotoxin (Hu et al., 2008), as might be predicted if the competition by α-bungarotoxin for the Aβ site blocked a direct action of Aβ on nAChRs. It is therefore possible that the toxicity of Aβ is mediated, at least in part, through a direct physical interaction between Aβ and nAChRs.
A. β-Amyloid Peptide Triggers Signaling Cascades via Nicotinic Acetylcholine Receptors
Aβ initiates intracellular signaling cascades via nAChRs (Fig. 3), including the MAPK kinase signaling pathway, resulting in cell death. In hippocampal slices, Aβ activates ERK-2 isoforms of the ERK MAPK. This is blocked by α7 antagonists, suggesting that Aβ evokes the cascade through α7 nAChRs (Dineley et al., 2001). However, an alternative possibility is that activation of nAChRs is permissive for a process in which Aβ does not directly interact with nAChRs: for instance, calcium entry through nAChRs may be required for Aβ toxicity. A microarray comparison of AD and control autopsy brain tissue revealed a down-regulation of MAPK and ERK activator kinase (Loring et al., 2001), indicating possible compensatory gene regulation aimed at reducing Aβ activation of ERK. Which pathway is activated by Aβ depends upon the time of exposure to the amyloid peptide: chronic applications of oligomeric Aβ to hippocampal slice cultures activate the JNK/MAPK pathway but inhibit the ERK/MAPK pathway, whereas short-term applications of Aβ oligomers do not activate JNK (Bell et al., 2004). This may be one of the routes whereby Aβ impairs memory, because ERK-1 and ERK-2 play key roles in the signaling events central to memory (Satoh et al., 2007). In neuroblastoma cells, as well as cultured hippocampal neurons, Aβ activates JNK and ERK, and blocking these prevents Aβ hyperphosphorylating tau protein, as does α7 antisense oligonucleotides or α7 antagonists, suggesting that Aβ may trigger tau protein phosphorylation through ERK and JNK via α7 receptors (Wang et al., 2003b). Aβ leads to phosphorylation of AKT in cultured mouse neurons through a mechanism that requires α7 nAChRs (Abbott et al., 2008), AKT phosphorylation levels returning to baseline upon prolonged application of Aβ. AKT interacts with BAD to regulate apoptosis and, interestingly, also has many interacting partners in the insulin signaling pathway. Aβ increased activity of BAD, lowered the activity of the antiapoptotic protein BCL2, in rat hippocampal neurons in primary culture (Koriyama et al., 2003) and has been shown to be toxic to human neuroblastoma cells by increasing BAX activity and decreasing BCl-2 activity (Clementi et al., 2006).
B. Does β-Amyloid Act Directly on Nicotinic Acetylcholine Receptors?
The finding that cholinergic neurons degenerate in AD and in AD models does not, of course, imply that this is the result of Aβ acting on nAChRs. For instance, in Aβ-overexpressing mice (PDAPP derived from a heterogeneous background comprising the strains C57BL/6J, DBA/2J, and Swiss-Webster), Aβ seems to target the high-affinity choline transporter (Bales et al., 2006). Most studies arguing for a role of nAChRs in Aβ toxicity or neuroprotection against the toxic actions of Aβ do so on the basis of the block of an observed response by selective nAChR antagonists. Such a block, however, does not necessarily imply that a response is initiated by Aβ binding to nAChRs; it could be that nAChR-evoked signaling, such as calcium entry, is permissive for a separate Aβ pathway. To determine whether Aβ acts upon nAChRs, it is necessary to establish that Aβ binds directly to nAChRs. There are two conflicting reports as to whether Aβ binds with high affinity to α7 nAChRs. In both studies, similar approaches are adopted, yet the findings differ. One study showed high-affinity binding of Aβ at picomolar levels to human α7 nAChRs heterologously expressed in cell lines, based both on the ability of Aβ to displace labeled α-bungarotoxin and the ability of α-bungarotoxin to displace fluorescently labeled Aβ (Wang et al., 2000b). In contrast, Small et al. (2007) found no displacement of α-BTX from SH-SY5Y cells (a cell line very closely related to that used by Wang et al.) by either amyloid or methyllycaconitine. Wang et al. (2000b) also showed similar staining of human AD cortical neurons by α7 and Aβ antibodies in double immunofluorescence, suggesting that in human cortical neurons, α7 and Aβ are closely associated, although such an approach does not prove direct binding. However another study (Small et al., 2007) showed no displacement of labeled α-bungarotoxin from cell lines expressing rat α7 nAChRs. In attempting to resolve the conflicting reports, however, two points should be borne in mind. First, it should be noted that the studies by Wang and colleagues critically showed that Aβ coimmunoprecipitated from human hippocampal membranes with α7 nAChRs, the more natural physiological environment for the receptors. Coimmunoprecipitation is a more direct demonstration that two proteins interact than double immunofluorescence because the latter lacks the spatial resolution to establish protein binding. Second, Small et al. (2007) found no inhibition by Aβ of α7 nAChRs expressed in oocytes, in contrast to most similar studies. Taken together, the balance of evidence is that Aβ can bind to nAChRs, although we suspect that the most likely explanation for the discrepancy between these key studies and other studies reporting different actions of Aβ on cells lies in the preparation of Aβ, which, as we argue below, is notoriously sensitive to preparation conditions.
Although there is abundant evidence that Aβ can affect nAChR function, studies disagree as to whether Aβ is an antagonist or an agonist at nAChRs (these findings are summarized in Table 1). For example, Aβ has been reported to inhibit single-channel nicotinic receptor currents in rat hippocampal interneurons (Pettit et al., 2001) as well as currents recorded from human α7 receptors heterologously expressed in Xenopus laevis oocytes (Tozaki et al., 2002; Grassi et al., 2003; Pym et al., 2005). Aβ, however, activates a mutant (L250T) of the α7 receptor—this mutant conducts current in the desensitized state, indicating that Aβ may exert its antagonistic action through receptor desensitization (Grassi et al., 2003). Aβ action on nAChRs depends on subunit composition; it has been reported to block α7, transiently potentiate α4β2 before blocking, and to have no action on α3β4 (Pym et al., 2005). However, in contrast to its reported transient enhancement when expressed in oocytes, an inhibition of α4β2 has been reported when expressed in human SH-EP1 cells (Wu et al., 2004).
Actions of Aβ on nAChRs Direct actions of Aβ on nAChRs. The results of actions of Aβ or Aβ fragments on in situ or heterologously expressed nAChRs are summarized, along with a summary of how the Aβ was prepared (if given in the original report), and which concentrations were tested. Both activation and inhibition are reported by different laboratories, even for the same receptor subtype expressed in oocytes.
Although many studies report the absence of agonist actions of Aβ, there are reports of Aβ1–42 acting as an agonist. Aβ1–42 activates heterologously expressed nAChRs (Dineley et al., 2002), and Aβ25–35 has been shown to activate non-α7 nAChRs in rat basal forebrain neurons (Fu and Jhamandas, 2003) and to evoke a α7-mediated calcium increases in presynaptic terminals isolated from rat hippocampus and neocortex (Dougherty et al., 2003). Perfusion of soluble Aβ into mouse prefrontal cortex increases dopamine secretion through a mechanism that is blocked by α7 antagonists (Wu et al., 2007). Again, despite numerous reports of a block of α7, one study indicated that Aβ failed to block α7, even though it blocked α4β2, α2β2 and α4α5β2 receptors (Lamb et al., 2005). It has also been observed that although Aβ inhibits recombinant human and mouse α7 nAChRs, transgenic mice overexpressing human Aβ express functional α7 nAChRs, and the amplitude of α7-mediated currents is no different from that of wild-type mice (Spencer et al., 2006).
The conflicting evidence for the nature of Aβ actions on nAChRs clearly needs to be resolved. One possible explanation for these discrepancies might be found in the different concentrations of Aβ used. The most comprehensive study of the effects of Aβ at different concentrations showed that at 10 pM, Aβ evoked an inward current mediated by rat α7 nAChRs expressed in X. laevis oocytes, whereas at 100 nM, Aβ blocked nicotine responses through desensitization (Dineley et al., 2002). However, a similar study (Pym et al., 2005) found no evidence of agonist actions of Aβ at 10 pM, although this study used human α7 rather than rat. In addition, complex interactions with receptor-associated proteins and/or intracellular signaling pathways that modulate native nAChRs may also contribute. These may not always be faithfully reproduced in recombinant and cell line studies. Studies differ in the Aβ variant used: at least one study used the more convenient 25–35 fragment (Fu and Jhamandas, 2003), which has been shown to be different in some of its actions from the full-length peptide (Pym et al., 2007). Another intriguing possibility that might explain differences in the findings on the effects of Aβ on nAChRs is that these differences might reflect different splice variants of the nAChR (Severance and Yolken, 2007). A microarray study identified defects in splicing in brains from patients with AD, but nAChRs were not among the gene products highlighted (Heinzen et al., 2007). However, the possibility remains that some differences observed between reports using in vivo preparations may be attributable to different nAChR splice forms in the tissues studied.
The most likely explanation for many of these discrepancies may be differences in the aggregation state of the Aβ used, which is notoriously sensitive to the method of preparation and storage (Teplow, 2006) (see Table 1). Many studies fail to describe how the amyloid was prepared or stored, and others report that they dissolved the amyloid in distilled water. The trifluoroacetic acid salt, the most common form in which commercial preparations are supplied, forms a highly acidic solution, conditions in which aggregation is almost instantaneous (Burdick et al., 1992). This might explain the different findings of Pym et al. (2005), who prepared their Aβ stocks in acetic acid, and Dineley et al. (2002), who prepared theirs in HEPES at pH 8. There is therefore a clear need to resolve the question of different effects of the monomeric, oligomeric, and fibrillar forms of Aβ by systematically examining the effects of carefully prepared solutions on expressed and in situ nAChRs.
V. Loss of Cholinergic Neurons and Nicotinic Acetylcholine Receptors in Alzheimer's Disease
It is clear that AD involves loss of cholinergic neurons in the brain as well as an overall reduction in nAChRs, and it seems that different subunits are differentially up- or down-regulated in AD in different brain regions and different cell types.
Loss of cholinergic neurons has often been demonstrated as lowered ChAT activity in brains of patients with AD. Early post mortem studies indicated a loss of ChAT activity restricted to the neocortex (Slotkin et al., 1990) and this has been confirmed in more recent studies on frontal lobe and temporal cortex (Lai et al., 2006). It is noteworthy that an increase in ChAT activity in the surviving neurons was interpreted as a possible compensatory mechanism (Slotkin et al., 1990).
AD involves loss of cholinergic cells not only in the cortex but also in subcortical nuclei. Up to 50% loss of neurons and of ChAT activity has been reported at autopsy in the locus ceruleus of brains from patients with AD compared with brains from subjects without AD, whereas no change was observed for adrenergic brain-stem nuclei (Strong et al., 1991). A meta-analysis of 67 studies on subcortical nuclei involving more than 1800 patients concluded that the most significant loss in cell numbers occurred in the cholinergic nucleus basalis and in the noradrenergic locus ceruleus, whereas fewer cells were lost in the dopaminergic substantia nigra (Lyness et al., 2003). Selective cell loss in the locus ceruleus has also been confirmed in a double-mutant mouse model ([APPswe, PS1dE9]85Dbo/J; PrP promoter) transgenically expressing the Swedish mutant form of the APP and the mutant human APP/PS-1, suggesting that these mutations can contribute to neurodegeneration (O'Neil et al., 2007).
A. Expression of α4β2 and α7 Nicotinic Acetylcholine Receptors Is Reduced in Alzheimer's Disease
A stereological approach, in which specific, identified regions of cortex were excised as a by-product of therapeutic surgery, revealed an approximately 50% decrease in the number of α7-containing neurons in the temporal cortices of patients with AD, without overall loss in neuron number (Banerjee et al., 2000). In addition to loss of neurons, there are reports of reduced expression of specific nAChR subtypes, particularly of α4β2 and α7 subunits, in many brain areas in AD. However, because these studies rely on the binding of radiolabeled ligands or the levels of protein expression, it is not always known to what extent this represents a loss of nAChR-expressing cells, reduced receptor numbers in the remaining cells, or both. Binding studies using subtype-selective labeled ligands suggest that α4β2 receptors are lost in brains from patients with AD (Warpman and Nordberg, 1995; Martin-Ruiz et al., 1999). Regions showing reduced binding levels include the frontal lobe and the temporal cortex (Lai et al., 2006). The use of subunit-specific (α2–6 and β2–4) antibodies to fractionate [3H]epibatidine (a marker for α4β2 receptors) binding also pointed to a loss of α4 and β2 subunits in the cortex of brains from patients with AD (Gotti et al., 2006). These findings have been confirmed in studies using the recently developed α4β2-specific radioligand, 5-125I-A-85380, which shows reduced expression of α4β2 in the caudate striatum and entorhinal cortex (Pimlott et al., 2004). This same ligand has also been used to demonstrate loss of α4β2 in frontal lobe, striatum, right medial temporal lobe, and pons of patients with AD using single-photon-emission computed tomography (SPECT) (O'Brien et al., 2007). Similar results have been obtained using subtype-specific antibodies. Binding of monoclonal antibodies raised against the α4 or the α7 subunit, for example, was significantly reduced in post mortem cortices of five patients with AD compared with five patients without AD of similar age (Burghaus et al., 2000). In one study, Western blots confirmed that the greatest reduction was in α4 (Guan et al., 2000). Likewise, subunit-specific antibodies reveal a reduced expression of α4 but not α3 or α7 in brains from patients with AD (Martin-Ruiz et al., 1999). One report found no change in α4 in frontal cortex or cerebellum, and attenuated levels of α7 in these tissues in brains from patients with AD (Engidawork et al., 2001). The loss of α4 subunits was suggested to be related to lipid peroxidation, because the loss correlated with the level of peroxidation in the temporal cortex of brains from patients with AD, suggesting that receptor loss may be caused by oxidation of proteins (Yu et al., 2003). Reduction of α7 and α4 subunits in neurons in patients carrying the Swedish mutation has also been reported (Yu et al., 2005). It is noteworthy that a different pattern of changes in nAChRs is seen in non-neuronal cells; expression levels of α7 have been reported to be elevated in astrocytes of brains from patients with AD and in cultured astrocytes (Teaktong et al., 2003; Xiu et al., 2005; Yu et al., 2005). Likewise, studies comparing α7 expression in human AD brain and Swedish-mutant mice found enhanced α7 expression in astrocytes but decreased expression in neurons compared with controls (Xiao et al., 2006).
The loss of nAChR subunits, as determined by [3H]-epibatidine binding, seems to take place after the transition from mild cognitive impairment (MCI) to AD (Sabbagh et al., 2006), although the loss of epibatidine binding did not correlate with decline in memory, cognitive performance, or with the development of neurofibrillary tangles or plaques (Sabbagh et al., 2001).
Data on changes in levels of α3 subunits in AD are complex. An in situ hybridization study of α3 failed to find any difference in expression in hippocampus, entorhinal cortex, or thalamus of brains from patients with AD compared with control brains (Terzano et al., 1998). However, a reduction of α3 subunits in a Western blot analysis of brains from patients with AD has been observed (Guan et al., 2000), although the loss was not as great as that observed for α4 or α7 subunits. In addition, α3 subunit levels were reduced in the temporal cortex and hippocampus of brains from patients with AD, both smokers and nonsmokers, compared with control subjects (Mousavi et al., 2003). Given the limitations of quantitative results from Western blot studies and that studies on α3 may also be complicated by the overall lower levels of expression of α3 nAChRs in the brain, more work needs to be done to establish whether α3 nAChRs are lost in AD.
Similar effects of Aβ on nAChR expression have been confirmed in studies using cultured cells; Aβ causes a reduced expression of nAChRs in PC12 cells (Guan et al., 2001), and α4, α3, and α7 expression are all increased in cultured rat astrocytes (Xiu et al., 2005). Thus, AD involves opposite expression level changes in astrocytes compared with neurons, and this might reflect different roles for the same nAChR subunit in different cells, in that α7 on glial cells is thought to reduce the intensity of the inflammatory response, whereas it is presumed to have both a fast signaling role and a neuroprotective role in neurons. Thus activation of α7 nAChRs may protect neurons through two routes: 1) pro-survival signaling intrinsic to neurons and 2) inhibition of the inflammatory response.
Findings from in vivo imaging techniques such as real-time measurements based on positron-emission tomography (PET) offer considerable improvements over traditional in vitro ligand-binding studies. Although new ligands for use in PET, such as 5-125I-IA-85380, still have slow kinetics, they offer improved resolution over [11C]nicotine and lower toxicity than epibatidine analogs. PET imaging has been used to show that loss of [11C]nicotine binding in the frontal and parietal cortices of patients with AD correlates with poorer performance in attention tasks but not with defects in episodic memory or visuospatial tasks (Kadir et al., 2006). The deployment of new ligands in PET and SPECT imaging promises to greatly advance our understanding of how the levels of expression of nAChR subtypes change in AD and in the progression from MCI to AD.
Thus, predominantly α4 and α7 subunits, and to a lesser extent α3 subunits, are lost in AD, although there are tissue-specific differences to this pattern, such as the up-regulation of nAChRs on astrocytes. The α7 and α4 nAChR subunits are the most abundant in the central nervous system and are therefore more likely to receive research interest, probably leaving less abundant subunits under-reported. A significant role for these rarer subunits in AD cannot therefore be excluded. The strikingly different responses often reported for the same subunit in neuronal or glial cells also merits further investigation.
B. Loss of α4β2 and α7 Nicotinic Acetylcholine Receptors Is Not Due to Reduced mRNA
Comparison of the binding of subunit-specific antibodies with the abundance of the subunit mRNA suggests that reduced nAChR subunit expression is not accompanied by reduced levels of the corresponding mRNAs (Hellström-Lindahl et al., 1999; Wevers et al., 1999). This may point to a defect in mRNA translation, although other possibilities, such as mRNA sequestration, cannot be excluded. Intracerebral injection of Aβ into rats resulted in a loss of α4 and α7 subunits as measured by Western blotting but an increase in α7 mRNA (Liu et al., 2008), again suggesting that Aβ directly reduces expression of α7 nAChRs through mechanisms other than reduced mRNA production, although caution should be exercised in interpreting quantitative data from Western blot studies. It is noteworthy that a combined patch-clamp and in situ hybridization study of dissociated human brain tissue (obtained as route-of-access tissue removed during surgery) indicated that neurons near Aβ plaques retained α4 and α7 mRNA transcripts, whereas these transcripts were absent in neurons burdened with hyperphosphorylated tau protein (Wevers et al., 1999). Such cell-specific differences might be missed in studies that measure levels of mRNA or protein at the tissue level.
The notion that nAChR loss is not due to reduced gene expression is supported by microarray studies, which confirm changes in expression of signaling genes but not those encoding nAChR subunits (Loring et al., 2001; Colangelo et al., 2002; Wang et al., 2003a; Yao et al., 2003). More recent approaches have confirmed this: RNA profiling of isolated neurons from control brains or brains from patients with AD show no evidence for changes in nAChR RNA (Chow et al., 1998; Ginsberg et al., 2000).
Thus, in brains from patients with AD and in neurons responding to exogenously applied Aβ, there is a reduction in expression of nAChR subunits, especially α4, α7, β4, and possibly α3. Although AD may also involve changes in expression of other ligand-gated ion channels—for example, the expression of NMDA receptors (Bi and Sze, 2002; Jacob et al., 2007), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (Jacob et al., 2007), and β3 GABA receptor subunits are all reduced (Mizukami et al., 1998)—there is abundant evidence of a loss of nAChR subunits in AD possibly caused by the actions of Aβ.
C. Animal Alzheimer's Disease Models Show Loss of Nicotinic Acetylcholine Receptors
Findings of reduced nAChRs observed in human patients with AD have been confirmed in some, but not all, animal models of AD (Table 2). Tg2576 mice expressing human Aβ show reduced [3H]cytisine binding (a label of nAChRs) in the cortex at 17 months after birth (Apelt et al., 2002). In contrast, however, levels of α7 or α4 subunits were unchanged in double-mutant Swedish APP/PS-1 mice as determined by radiolabeled cytosine (α4β2) or α-bungarotoxin (α7) binding (Marutle et al., 2002). In addition, APP or APP/PS-1 double-mutant mice have normal or even enhanced levels of ChAT and an unchanged cholinergic cell count (Hernandez et al., 2001). However, a double-mutant mouse expressing the Swedish APP and overexpressing human AChE showed enhanced mRNA levels of α7 in brain and adrenal medulla, although in brain tissue this enhancement declined with age. In this same mouse, there was no alteration in mRNA levels for α4, and an increase in α3 has also been observed in the brain and the adrenal medulla (Mousavi and Nordberg, 2006), a pattern similar to that seen in Aβ single-mutant mice (Bednar et al., 2002), suggesting that it is not attributable to the human AChE.
Representative list of mouse and invertebrate models used in research on Alzheimer's disease There are several animal models of AD, but no single model mimics all the pathological features of AD, although most display locomotor or learning defects.
VI. Nicotinic Acetylcholine Receptors: Molecular Switches Controlling Survival or Death
Many elements of the pathways involved in either Aβ toxicity or the protective activation of nAChRs have been identified with sufficient consistency to permit a tentative sketch of the interacting pathways involved (Fig. 3) but exercising the appropriate caution necessary when combining findings from different preparations from different phyla and under widely different experimental conditions. There remain a number of important issues to be resolved. 1) The time course of nicotine activation of protective pathways and the effects of long-term exposure to nicotine on nAChRs (especially α7) need to be explored with the aim of developing better models for exploring new therapeutic targets. 2) Many physiological experiments overlook the significance of different aggregation states of Aβ, and very few studies on the effects of exogenous Aβ on the CNS or of nicotine neuroprotection in brains take into account the well documented effects of both of these substances on the vascular system (nicotine, for instance, is angiogenic; some of its protective effects in vivo may be attributable to improved blood supply).
How can nAChRs mediate both the toxic actions of Aβ and the protective actions of nicotine? There must be some way in which these ligands operate different signaling pathways. The simplest explanation might be that Aβ's antagonist actions block the therapeutic effect of nAChR activation. Setting aside the controversy over the actions on nAChRs of Aβ, this explanation is unlikely to offer a full explanation because Aβ alone evokes the ERK/MAPK signaling cascade and causes the phosphorylation of tau proteins through α7 nAChRs (Wang et al., 2003b). Again, both nicotine and Aβ activate the ERK pathway (Dineley et al., 2001; Wang et al., 2003b; Bell et al., 2004) but they do so through different routes (Bell et al., 2004). However, Aβ is toxic to primary cultured neurons from α7-knockout mice, indicating that the α7 receptor is not always required for Aβ toxicity (de Fiebre and de Fiebre, 2003). Thus, either different subpopulations of α7 nAChRs are somehow linked to different signaling pathways or, alternatively, α7 receptors may be differentially coupled to different downstream signaling pathways depending on whether nicotine or Aβ is bound. It must also be borne in mind that α7 receptors in different cellular environments, linked perhaps to different sets of associated proteins, may evoke different signaling cascades.
The question of how two or more signaling pathways can pass through a common node and yet preserve their specificity is relevant to several signaling systems, and several solutions have been proposed. In the case of nicotine and Aβ signaling at nAChRs, spatial separation of pathways is an unlikely explanation because in the cell-line and cell culture experiments, at least, both nicotine and Aβ have similar access to the receptors. A more engaging explanation is that cross-talk can be prevented using “kinetic insulation” (Behar et al., 2007). In this model, a common signaling node is envisioned with two downstream effectors in which adaptation and response kinetics are different. A rapidly adapting effector acts as a high-pass filter in the time domain and is therefore unaffected by slowly changing inputs, whereas the second effector responds slowly to a stimulus and does not adapt, and therefore acts as a low-pass filter that is unaffected by transient signals. A mathematical model of such a system shows that rapid, transient signals or slow, sustained signals can pass through a common node and specifically activate different signaling pathways (Behar et al., 2007).
The Aβ and nicotine-evoked signaling pathways contain elements that might contribute to such a process. Aβ phosphorylation of AKT in cultured mouse neurons via α7 nAChRs returns to baseline levels upon prolonged application of Aβ (Abbott et al., 2008). Furthermore, the results of activating the same receptors can depend upon the time course of exposure to the agonist and its concentration. The α7 partial agonist 3-(2,4)-dimethoxybenzylidine anabaseine, has a dual effect on PC12 cells depending on concentration and exposure time (Li et al., 1999b). Lower concentrations elevate PKC and protect cells from serum depletion, whereas higher concentrations do not elevate PKC but are toxic. Furthermore, blocking PKC resulted in attenuated neuroprotection, whereas blocking tyrosine kinases attenuated toxicity. Methyllycaconitine blocked neuroprotection only when added 10 min after 3-(2,4)-dimethoxybenzylidine anabaseine addition, but not when added before, a time scale noted by the authors as corresponding to the time scales of α7 activation and desensitization (Li et al., 1999b). Very few studies on nicotine neuroprotection have explored the effects of timing or measured the time course of downstream signals evoked by either nicotine or Aβ, so there is clearly scope for future developments in this area to resolve whether kinetic insulation plays a role in determining the outcome of nAChR activation.
VII. Future Directions
A. Adjusting the Balance between Survival/Death Pathways
It is the consensus view that Aβ targets neurons at least partly via actions on nAChRs and that activation of nAChRs by nicotine protects neurons from Aβ toxicity. It is also clear that nicotinic agonists protect neurons from Aβ toxicity through several intracellular signaling pathways including the ERK/MAPK, PI3K/AKT, and the JAK/STAT pathways. Thus, these pathways could perhaps be exploited as possible sources for novel therapies. To date, treating AD using anticholinesterases, some of which can both prolong ACh at the synapse and act directly on nAChRs, has had limited success. However, nicotine and Aβ activate similar, but not identical, intracellular signaling pathways, raising the possibility of developing therapies based on diverting the pathways elicited by Aβ toward the survival outcome. Although it is clear that Aβ kills neurons and nicotine protects them against Aβ toxicity, much remains to be resolved to unravel the mechanisms by which death or survival is the outcome. Intracellular signaling initiated by Aβ and nicotine is complex and involves much cross-talk between diverse signaling pathways, arguing for a systems-based approach to understanding precisely how nicotine or Aβ can influence neuronal survival. Such a task could be facilitated by the deployment of mutant suppressor and RNAi screens, as well as the ease of transgene production, afforded by invertebrate model genetic organisms such as D. melanogaster and C. elegans (Culetto and Sattelle, 2000; Buckingham et al., 2004; Crowther et al., 2004; Jones et al., 2005; Link, 2006). Identifying new components of these pathways offers the prospects of new targets for the treatment of AD. In addition, because α7 receptors are present in human lymphocytes, AD-related alterations in expression of the receptor and/or key associated proteins in these cells may form the basis of a biomarker for early detection of AD or evaluating progression of the disease and responsiveness to drugs (Jones et al., 2006). Furthermore, while the role of nAChR-dependent intracellular signaling has been explored in the context of protection against Aβ toxicity, the roles of the same pathways in promoting synapse function has not been fully explored. The effect of PI3K on promoting synapses in D. melanogaster (Martín-Peña et al., 2006), for instance, suggests that this might be a potential target for drugs preventing the early adverse actions of Aβ.
B. Toward Improved Animal and Cell Models
There is scope for the development of improved animal models for AD: in particular, experiments at the cellular level could benefit from improved vertebrate (mouse, rat) and invertebrate (Caenorhabditis elegans or D. melanogaster) models. Transgenic mouse models include the Tg2576 (Hsiao et al., 1996) and APP23 (Sturchler-Pierrat et al., 1997) models that express human Aβ with the Swedish familial AD mutation, as well as the presenilin models expressing mutated presenilin genes (Table 1). None of the models currently available fully recapitulate all the features of AD, prompting the development of double mutants such as the APP/PSEN models (McGowan et al., 2006). Even these models lack the presence of neurofibrillary tangles. The recent generation of a triple mutant mouse (APP/PSEN/tau) addresses many of the earlier shortcomings (Oddo et al., 2003), but even though the endpoint of the disease is mimicked, such models are not necessarily instructive on the development of the disease.
The contributions from future studies using mouse AD models will undoubtedly be complemented by the development of new invertebrate models. Although there are limitations with these models accruing from their phylogenetic distance from humans, these limitations are offset by the ease of access, low cost, and speed that they offer. Along with vertebrate cultured cells, they can be deployed in large-scale RNAi studies aimed at identifying genes involved in nicotine neuroprotection and Aβ toxicity. A number of D. melanogaster models of AD are available, including several transgenically expressing human Aβ (Crowther et al., 2004, 2005; Iijima et al., 2004) and those mimicking presenilin mutations (Struhl and Greenwald, 1999; Ye et al., 1999). Although these models exhibit neurodegeneration, reduced lifespan, and defective learning, the effects on nAChR expression or on the cholinergic system have not been described. A transgenic C. elegans model of AD is available in which microarray studies have shown that expression of three nAChR genes is altered (Link et al., 2003). In this model, the Aβ is expressed in muscle, thus being perhaps a better or at least equally effective model for inclusion body myositis (Rebolledo et al., 2008). A C. elegans model in which Aβ is expressed in neurons would be of value in large-scale screens for compounds that rescue the worms or in suppressor/enhancer screens for identifying new genes in the Aβ toxicity pathway.
VIII. Conclusion
The cholinergic hypothesis of AD has spawned 2 decades of research into the roles of nAChRs in this disease, resulting in a widely held view that Aβ can act on, among other targets, nAChRs to result in alterations in synaptic function and eventually cell death. Considerable advances have also been made in our understanding of the roles of the nicotinic cholinergic system in the development of AD. There remain, however, some outstanding gaps in our understanding to be filled and some difficult issues to be resolved. Despite the widely held view, based on block by nicotinic antagonists, that nAChRs mediate neuroprotection, a permissive role for nAChR activation has yet to be ruled out. More seriously, the critical test of the common belief that Aβ exerts at least part of its toxic action through a direct action on nAChRs—the demonstration of high affinity binding to an nAChR—is based on a single report, and further studies are needed. Another serious shortfall in current research into the mechanisms of Aβ toxicity and nicotine neuroprotection stems from our limited understanding of the mechanism of aggregation of the Aβ peptide and its regulation. Aβ aggregation is a complex process, highly sensitive to preparation conditions, so much so that even the same source of material prepared in the same lab can aggregate at different rates even under apparently identical conditions (Teplow, 2006). Many articles on the actions of Aβ on nAChRs prepare stocks of the protein at low pH and dilute to physiological pH, thus passing through the isoelectric point at which aggregation is maximized (Teplow, 2006; Finder and Glockshuber, 2007). Others promote aggregation by incubating the peptide for several hours at 37°C. To date, despite evidence that oligomers are particularly toxic to neurons and interfere with synaptic transmission and learning, there has been no investigation into how monomers, oligomers and protofibrils interact with nAChRs. The time is therefore ripe for a systematic analysis of the physiological actions of different aggregate forms of Aβ on nAChRs of known composition and on native nAChRs, as well as of the binding of these aggregate forms to nAChRs.
Acknowledgments
This work was supported by core Medical Research Council funding to the MRC Functional Genomics Unit.
Footnotes
-
↵1 Abbreviations: AD, Alzheimer's disease; α-BTX, α-bungarotoxin; Aβ, β-amyloid peptide; A-582941, 2-methyl-5-(6-phenyl-pyridazin-3-yl)octahydro-pyrrolo(3,4-c)pyrrole; A-85380, 3-(2-azetidinylmethoxy)-pyridine; ABT-418, 3-methyl-5-(1-methyl-2-pyrrolidinyl)isoxazole; ACh, acetylcholine; AChE, acetylcholinesterase; AKT, v-akt murine thymoma viral oncogene homolog; APOE, apolipoprotein E type 4; APP, amyloid precursor protein; BAD, BCL2-antagonist of cell death; BAX, Bcl2-associated X protein; BCL2, B-cell chronic lymphocytic leukemia/lymphoma 2; ChAT, choline acetyltransferase; ERK, extracellular signal-regulated kinase; FADK1, focal adhesion kinase; JAK, Janus kinase; JNK, c-jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MCI, mild cognitive impairment; nAChR, nicotinic acetylcholine receptor; NMDA, N-methyl-d-aspartate; PET, positron-emission tomography; PI, phosphoinositol; PI3K, phosphoinositol-3-kinase; PIKE, phosphoinositide 3-kinase enhancer; PKC, protein kinase C; PS-1, presenilin-1; RNAi, RNA interference; SPECT, single-photon-emission computed tomography; SRC, v-SRC sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog; STAT, signal transducer and activator of transcription; TNF-α, tumor necrosis factor-α.
-
This article is available online at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.108.000562.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. β-Amyloid: Biophysics and Roles in Alzheimer's Disease
- III. Nicotinic Acetylcholine Receptors: Structure, Function and Roles in Neuroprotection
- IV. β-Amyloid Peptide Toxicity: Is It Mediated in Part through Nicotinic Acetylcholine Receptors?
- V. Loss of Cholinergic Neurons and Nicotinic Acetylcholine Receptors in Alzheimer's Disease
- VI. Nicotinic Acetylcholine Receptors: Molecular Switches Controlling Survival or Death
- VII. Future Directions
- VIII. Conclusion
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters