


Abstract
Modulation of neurotransmission by the monoamines dopamine (DA), norepinephrine (NE), and serotonin (5-HT) is critical for normal nervous system function. Precise temporal and spatial control of this signaling in mediated in large part by the actions of monoamine transporters (DAT, NET, and SERT, respectively). These transporters act to recapture their respective neurotransmitters after release, and disruption of clearance and reuptake has significant effects on physiology and behavior and has been linked to a number of neuropsychiatric disorders. To ensure adequate and dynamic control of these transporters, multiple modes of control have evolved to regulate their activity and trafficking. Central to many of these modes of control are the actions of protein kinases, whose actions can be direct or indirectly mediated by kinase-modulated protein interactions. Here, we summarize the current state of our understanding of how protein kinases regulate monoamine transporters through changes in activity, trafficking, phosphorylation state, and interacting partners. We highlight genetic, biochemical, and pharmacological evidence for kinase-linked control of DAT, NET, and SERT and, where applicable, provide evidence for endogenous activators of these pathways. We hope our discussion can lead to a more nuanced and integrated understanding of how neurotransmitter transporters are controlled and may contribute to disorders that feature perturbed monoamine signaling, with an ultimate goal of developing better therapeutic strategies.
The mammalian nervous system is an incredibly complex system of neural circuits that communicates with both precision and flexibility. Key to ensuring this duality of signaling characteristics is synaptic modulation provided by the monoamine (MA) neurotransmitters serotonin (5-HT), dopamine (DA), and norepinephrine (NE) (Cooper et al., 2003). Although these molecules exhibit overlap in projections and synaptic control mechanisms, several key functions are generally ascribed to each. Thus, 5-HT signaling is most typically associated with mood, anxiety, aggression, and appetite, with 5-HT signaling dysregulation linked to disorders such as depression, obsessive-compulsive disorder (OCD), anxiety disorders, and autism spectrum disorder (ASD) (for review, see Olivier, 2015). DA has received prominent attention for its role in circuits supporting reward, attention, and movement, with perturbed DA signaling associated with addiction, attention-deficit hyperactivity disorder (ADHD), schizophrenia, and Parkinson’s disease (Viggiano et al., 2004b; Segura-Aguilar et al., 2014; Howes et al., 2015; Nutt et al., 2015 ). NE plays a prominent role in arousal, attention, executive function, and stress responses (Harley, 2004; Viggiano et al., 2004a; Morilak et al., 2005), with disorders such as ADHD, posttraumatic stress disorder. and depression often linked to disrupted central nervous system NE signaling (Southwick et al., 1999; Kim et al., 2008; Goddard et al., 2010). Prominent peripheral roles of 5-HT and NE are also known, as for example the role of the former in gut and platelet function (Mercado and Kilic, 2010; Mawe and Hoffman, 2013), and of the latter in broad control of autonomic function including heart rate, vasoconstriction, and lipolysis (Goldstein et al., 1983).
As with other signaling pathways, control mechanisms are in place to ensure the extent and temporal dynamics of MA effects. Prominent in the control of MA signaling is the clearance of released neurotransmitter, afforded by presynaptic transporter proteins (Gainetdinov and Caron, 2003; Blakely and Edwards, 2011; Kristensen et al., 2011). MA reuptake catalyzed by these transporters also provides a recycling pathway of neurotransmitter replenishment, augmenting levels achieved by de novo synthesis. Although important nuances exist [e.g., clearance of DA by the NE transporter (NET)] in cortex (Gresch et al., 1995; Siuta et al., 2010), uptake of 5-HT by organic cation transporters and/or DAT when SERT activity is genetically eliminated or blocked (Zhou et al., 2005; Baganz et al., 2008), each MA is cleared by the product of a single gene of the SLC6 transporter gene family: SLC6A2, (NET), SLC6A3 (DAT), SLC6A4 (SERT). Promoter, intronic, and exonic polymorphisms in these genes have been associated with multiple disorders, including, but not limited to, orthostatic intolerance and ADHD (NET), bipolar disorder, ADHD, and juvenile dystonia (DAT), depression, OCD, and ASD (SERT) (Hahn and Blakely, 2007; Kurian et al., 2011; Murphy and Moya, 2011). MA transporter connections to disease processes are also evident with respect to the actions of drugs that block their function, such as the 5-HT- (SSRI) and NE-selective reuptake inhibitors and cocaine, or those that lead to transport reversal, typified by d-amphetamine and methylenedioxymethamphetamine (Ecstasy) (Kristensen et al., 2011; Sitte and Freissmuth, 2015).
SLC6 family transporters energize substrate uptake via cotransport with Na+, with the MA transporters also exhibiting dependence on extracellular Cl−, and, for SERT, intracellular K+ (Blakely and Edwards, 2011). Structural features of ion coupling and substrate/antagonist binding have begun to be revealed through high-resolution structures and molecular modeling activities (Forrest et al., 2007; Tavoulari et al., 2009; Henry et al., 2011; Shan et al., 2011; Penmatsa et al., 2013). Although elegant and transformative, current structural studies have their limitations with respect to mechanisms of transporter regulation due to the limited homology in cytoplasmic regions (Torres et al., 2003b), the loss or manipulation of these domains in the course of crystallization (Penmatsa et al., 2013), or the relatively unstructured nature of these domains (Fenollar-Ferrer et al., 2014). This is a critical issue as these regions support the binding of a growing class of interacting proteins that dictate transporter localization, stability, and activity. Cytoplasmic domains also contain sites of posttranslational modifications, including lipidation (Foster and Vaughan, 2011) and phosphorylation (Ramamoorthy et al., 2011). Together, these modes of posttranslational regulation amply demonstrate that the MA reuptake process itself is under the control of signaling proteins whose actions provide additional points of control for MA signaling.
Over the past two decades, researchers have established multiple levels of MA transporter regulation, including modulation by gene transcription (Sacchetti et al., 2001; Tsao et al., 2008; Baudry et al., 2010; Harikrishnan et al., 2010), the control of protein trafficking to terminals (Torres et al., 2003a; Bjerggaard et al., 2004; Sucic et al., 2011), recruitment of transporters to the plasma membrane (Carvelli et al., 2002; Zhu et al., 2004a), localization of transporters to and mobility within membrane microdomains (Foster et al., 2008; Navaroli et al., 2011; Sakrikar et al., 2012), and internalization/recycling (Huff et al., 1997; Loder and Melikian, 2003; Jayanthi et al., 2004b; Samuvel et al., 2005), along with modulation of substrate recognition and transport capacity (Zhu et al., 2005; Foster et al., 2008; Steiner et al., 2009). These studies have been pursued with the conviction that a better understanding of transporter regulatory mechanisms may elucidate a broader network of genes and proteins that support risk for MA-associated brain disorders, as well as novel opportunities to modulate MA clearance without directly targeting the transporters themselves. In many cases, MA transporter regulatory processes involve the actions of protein kinases, which may act directly via phosphorylation of transporter cytoplasmic domains or indirectly by targeting transporter-associated proteins. Here we review our current understanding of kinase-dependent MA transporter regulation, focusing on posttranscriptional regulatory influences. After a brief introduction to the approaches common to the field of transporter regulation, we review their use for the study of kinase-modulated control of NET, DAT, and SERT. Although we limit ourselves to reports of MA transporter regulation, we expect that the findings we review will have, in many cases, their parallels with other transporters of the SLC6 family (for example, see Quick et al., 2004; Morioka et al., 2008; Cristovao-Ferreira et al., 2009; de Juan-Sanz et al., 2011).
I. Approaches for the Study of Kinase-Mediated Transporter Regulation
A. Monitoring Transporter Function
The most common technique for monitoring transporter function is the radiolabeled substrate uptake assay (for commentary on many assays, see Amara, 1998). Uptake assays involve incubating cells or tissue expressing the transporter of interest with a radiolabeled (most often [3H]) substrate of the transporter being studied, generally 5-HT, DA, or NE, and measuring the amount of substrate taken up by these cells as a function of time using a scintillation counter. Researchers often report use of this assay at a single concentration of substrate, although more informative data are obtained via a range of concentrations. Care must be taken to insure that properties of the assay in addition to that conferred by the transporter do not impact the rates determined, such as excess transporter protein relative to substrate concentrations, which can countermand assumptions of linearity inherent in drawing conclusions as to transport rates and their modulation. This saturation kinetic approach yields information regarding the substrate KM (a lumped constant containing any substrate-dependent steps in the transport cycle, including binding affinity) and the maximal velocity (Vmax) of substrate uptake that can aid researchers in determining the basis of changes in transport function when modulatory pathways are stimulated or inhibited. In recent years, nonisotopic approaches, such as using fluorescent transporter substrates, have been introduced to query neurotransmitter transport function (Mason et al., 2005). Such techniques are sometimes favored in avoiding the activation of receptors by the transporter substrate [e.g., study of DA interactions using ASP+ when endogenous D2 receptors could be active (Bolan et al., 2007)] or in avoiding potential intracellular actions of the neurotransmitter [e.g., ability of intracellular 5-HT to bind rab proteins (Walther et al., 2003)], interactions that may confound or limit assessments of modulation. It should also be recognized that because substrates can exert direct effects on transporter modulation, including that affected by protein kinases (Ramamoorthy and Blakely, 1999), the nature of the substrate chosen may impact conclusions drawn from such studies.
In addition to these approaches, electrophysiological measurement of substrate-induced currents can provide a relatively sensitive measure of surface transporter activity (Risso et al., 1996; Qian et al., 1997; Galli et al., 1998; Adams and DeFelice, 2002; Erreger et al., 2008; Schicker et al., 2012; Fraser et al., 2014). This has been performed in voltage-clamped oocytes and transfected cells, where inward currents are measured upon treatment with transporter substrate, and the amplitude of this current can be used to calculate the approximate amount of substrate moved, providing a measure of transporter activity. This technique can be used to compare transporter activity before and after pharmacological manipulations. Care must be taken when interpreting these results, however, because phosphorylation of the transporter may affect current movement, as with the phosphomimetic T62D mutation in hDAT, where substrate-induced currents are not observed in cells transfected with this mutant, despite its ability to transport DA, possibly due to equal bidirectional ion flow through this mutant transporter (Fraser et al., 2014). If a pharmacological manipulation similarly impacts the balance of inward and outward ion flow, one might mistake changes in substrate-induced currents for changes in uptake, even if no change in uptake occurs.
Electrochemical techniques are also used to measure extracellular monoamine levels and infer rates of uptake/MA clearance (Bunin and Wightman, 1998; Reed et al., 2003; Daws and Toney, 2007). These techniques involve the oxidation of monoamines by a carbon fiber probe (fiber or rotating disk) with substrate concentration at the electrode surface determined as a function of time from the oxidation current. One version of this technique termed chronoamperometry involves holding the potential of the electrode at a constant voltage, typically just above the peak of the oxidation potential for the MA under study, with either the evoked release and clearance of the MA assessed or the clearance rate from exogenously applied MA determined. This technique can be used both in vitro with cultured cells and in the brain in vivo to measure monoamine levels and transporter-mediated clearance over time. Most often the rate of decay of the oxidation current is taken a proxy measure of MA uptake, with transport KM and Vmax derivable provided substrate concentrations can be reliably estimated based on prior calibrations and titrated across the full dose-response range. In another version of the technique, termed fast-scan cyclic voltammetry, oxidation currents are measured across a range of potentials that ramped across a range of potentials generating repetitive redox cycles as a function of time. One advantage of this approach is that the chemical species being oxidized can be defined with greater certainty due to its unique redox “fingerprint." Because this approach also measures MA levels over time, it can be used to determine rates of monoamine uptake.
B. Assessment of Transporter Surface Expression—Steady-State and Kinetic Measures
Many studies reporting changes in transporter surface expression have used techniques that give measures of steady-state surface levels of transporter protein. In some cases, investigators have monitored the extent of radiolabeled antagonist binding to intact cells, defining the fraction of label bound to surface protein by displacing binding with a more hydrophilic, relatively membrane impermeant agent (such as the MA itself). This approach can be quite sensitive and highly quantitative and can be used when levels of the target transporter are too low for detection with conventional antibody-based approaches or when sensitive and specific antibodies are not available. It has the caveat that if the binding site becomes occluded due to conformational changes, a false conclusion as to the relocation of the transporter from the cell surface can be made. A common, antibody-based approach involves the biotinylation of surface proteins using a membrane-impermeant biotinylating reagent, typically tagging transporter lysine residues that are exposed at the cell surface. After cell lysis and detergent extraction, biotinylated transporters are captured on avidin or streptavidin-conjugated beads, eluted, and resolved using SDS-PAGE followed by Western blotting approaches. As with all antibody-based approaches, one should be mindful of the possibility that a change in the calculated fraction of surface versus total transporter protein could be influenced by posttranslational modifications (e.g., phosphorylation or lipidation) that could influence antibody binding. Although important confirmation of surface changes arises when one can show reciprocal changes in the nonbiotinylated fraction of proteins, this pool of transporter proteins may not be totally comprised of a free recycling pool of cytosolic proteins, because biotinylation may not label all surface transporters or a large, relatively immobile pool of transporters may be present. Caveats aside, this approach has been the workhorse for researchers in the transporter field to assess steady-state surface protein levels under basal and regulated conditions.
In addition to the use of biotinylation approaches to assess steady-state, surface transporter levels, researchers have also used reversible biotinylation methods to monitor the rates of endocytosis, recycling, and exocytosis (Loder and Melikian, 2003; Melikian, 2004; Sakrikar et al., 2012). For example, to determine endocytosis rates, biotin labeling of surface transporters tagged by a disulfide-linked biotinylating reagent can be removed as a function of time using a membrane impermeant reducing agent and endocytosis rates calculated because transporters that moved inside the cell retain their biotin tag and accumulate (assuming they do not rapidly recycle). Conversely, endocytosed protein can be monitored for reappearance on the cell surface as a function of time (transporter recycling) after stripping away biotin from residual surface transporters.
Although the covalent and reversible biotinylation of MA transporters has become a standard approach in the field for the study of steady-state and kinetic assessment of surface membrane trafficking, other approaches have been introduced that afford an assessment of the degree of surface labeling. In some cases, extracellular epitope antibodies have been developed, either based on native transporter sequence (Savchenko et al., 2003) or engineered epitope tags (Sorkina et al., 2009; Navaroli and Melikian, 2010; Rahbek-Clemmensen et al., 2014), that can be used in nonpermeabilized cells to assess transporter surface localization. These approaches have the advantage of being able to track the subcellular location of transporters in living cells (e.g., endocytosis), although here the caveat is that antibody binding may perturb transporter structure, affecting rates or extent of trafficking events. Thus, a better use for this method may be the labeling of transporters in fixed cells, where labeling has been obtained before and after a given stimulus (Savchenko et al., 2003).
A biophysical approach for the detection of transporter surface expression that can be used on single living cells involves the detection of transporter-dependent transient charge movements that occur with rapid changes in membrane potential (Kahlig et al., 2004). These charges arise from movement of free ions (e.g., Na+, Kl+) on and off the transporter as a consequence of membrane polarization and are proportional to the number of transporters at the cell surface. One caveat to this method is that unknown contributions are made to the density of transporter-bound charge by alterations in transporter conformations.
C. Determination of Transporter Membrane Microdomain Targeting
MA transporters, like other membrane receptors and channels, have been found to localize to cholesterol-rich, membrane microdomains, sometimes noted as “lipid rafts” (Jayanthi et al., 2004b; Foster et al., 2008; Sorkina et al., 2013). These domains are thought to serve as a site of concentrated assembly of membrane and membrane-associated proteins that serve to provide for higher order spatial and regulatory control of membrane protein function. To determine whether signaling pathways that regulate MA transporters involve changes in membrane microdomain localization (or requires such localization), investigators have generally used two approaches. One involves density gradient fractionation (e.g., sucrose gradients) of isolated membranes or membrane extracts, based on the idea that the different lipid/protein/cholesterol compositions of microdomains result in altered densities relative to non-raft membrane elements. Gradient fractions are then blotted with transporter antibodies to assess relative abundance in fractions colabeled with markers of membrane microdomains of interest (e.g., GM1 ganglioside, flottilin-1). To visualize the localization of MA transporters in membrane microdomains of intact cells, investigators have turned to fluorescent colocalization studies, targeting in particular the location of transporters in membrane microdomains that are enriched for GM1 ganglioside, which can be labeled using fluorescently tagged cholera toxin B subunit (Navaroli et al., 2011; Sakrikar et al., 2012). Transporters are most often visualized using antibodies-based detection strategies, although in some cases transporters have been labeled with fluorescently tagged small molecule transporter ligands (Chang et al., 2012; Eriksen et al., 2009; Kovtun et al., 2011).
D. Evaluation of Transporter Phosphorylation
Over the past two decades, it has become amply clear that MA transporters are posttranslationally modified by phosphorylation under basal conditions and in response to activation of multiple signaling pathways (Vrindavanam et al., 1996; Huff et al., 1997; Vaughan et al., 1997; Ramamoorthy et al., 1998; Ramamoorthy et al., 2011; Vaughan and Foster, 2013). Figures 1–3 show serine, threonine, and tyrosine residues on DAT, NET, and SERT that can act as potential phosphorylation sites that could be targeted by kinases in these signaling pathways. Both isotopic and nonisotopic methods are used to detect and monitor transporter phosphorylation. Isotopic methods are very sensitive and rely on the specificity of antibodies or other purification methods to isolate phosphorylate proteins. To monitor phosphorylation in situ (e.g., intact cells, synaptosomes), investigators isolate transporters from preparations incubated with [32P]-labeled sodium orthophosphate containing medium. The orthophosphate is taken up by the preparation and enters biosynthetic compartments, eventually labeling the cellular pool of ATP whose gamma phosphare will ultimately be transferred to the transporter as a consequence of kinase activity. [32P]-labeled proteins are immunoprecipitated, resolved by SDS-PAGE, and then visualized using autoradiographic or direct isotope imaging approaches. Radiolabeled ATP itself can be used to initiate phosphorylation of transporters or transporter-derived peptides in vitro. In this approach, purified kinases can be used to drive the specificity of labeling. Sites of labeling can be narrowed down by phosphoamino acid analysis whereby labeled protein/peptide is subjected to acid hydrolysis to reduce material to individual amino acids that are then resolved on thin layer chromatography (Sickmann and Meyer, 2001). Here the researcher can gain insight into whether phosphorylation is occurring on serine, threonine, and/or tyrosine residues. Exact sites of labeling are then inferred through mutagenesis studies. A nonisotopic variation on this approach involves the use of mass spectrometry to detect mass shifts of input or protease-derived peptides. The isotopic approach is very sensitive and can lead to detection of labeling at low stoichiometry and in cases of low transporter abundance. Mass spectrometry approaches typically require higher concentrations of transporter protein or purified peptides but can lead to direct identification of sites of phosphorylation and provide for precise stoichiometric assessments of transporter labeling. Finally, in some cases, investigators make use of phosphospecific antibodies that afford detection of site-specific modifications. A potential strength of this approach is that it can be used to monitor phosphorylation of transporters in intact cells and tissues using immunocytochemical approaches, although to date such an approach has yet to be implemented, largely due to a lack of suitable antibodies.

Potential phosphorylation sites in DAT. Model shows intracellular serine (yellow), threonine (green), and tyrosine (cyan) residues in the human DAT protein. Residue names in red are supported by the literature to be phosphorylated based on in vitro kinase assays with purified proteins, mutagenesis studies, phospho-specific antibodies, and/or mass spectrometry analysis (see text for discussion). Sites that show species variation between human, mouse, and rat are marked with an asterisk (*).
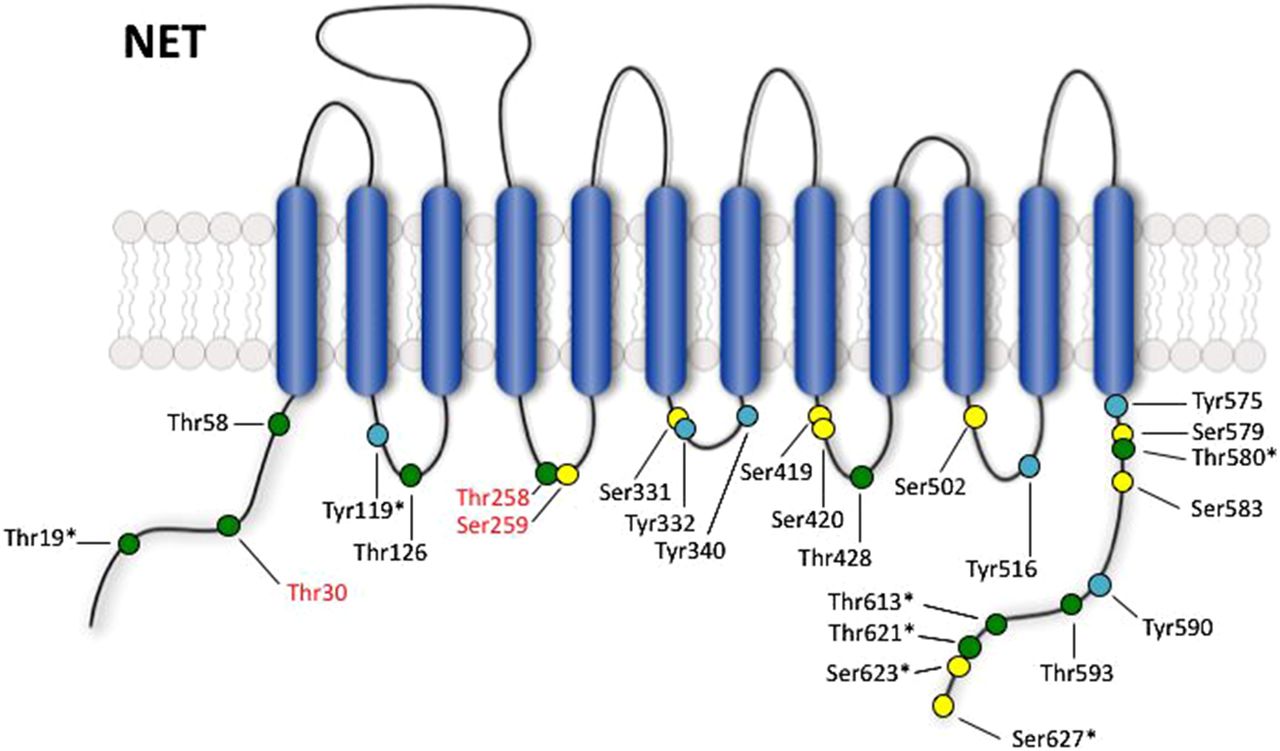
Potential phosphorylation sites in NET. Model shows intracellular serine (yellow), threonine (green), and tyrosine (cyan) residues in the human NET protein. Residue names in red are supported by the literature to be phosphorylated based on in vitro kinase assays with purified proteins, mutagenesis studies, phospho-specific antibodies, and/or mass spectrometry analysis (see text for discussion). Sites that show species variation between human, mouse, and rat are marked with an asterisk (*).

Potential phosphorylation sites in SERT. Model shows intracellular serine (yellow), threonine (green), and tyrosine (cyan) residues in the human SERT protein. Residue names in red are supported by the literature to be phosphorylated based on in vitro kinase assays with purified proteins, mutagenesis studies, phospho-specific antibodies, and/or mass spectrometry analysis (see text for discussion). Sites that show species variation between human, mouse, and rat are marked with an asterisk (*).
E. Detection of Transporter Protein-Protein Interactions
MA transporters interact with a vast array of proteins that can regulate their function in various ways (for review, see Sager and Torres, 2011). Much of the work that has been done to reveal these interactions has involved the use of biochemical techniques such as coimmunoprecipitations and GST-pulldown assays. In most instances, the proteins pulled down by these methods are then subjected to Western blotting for candidate interacting proteins, but some researchers have also used methods such as mass spectrometry-based approaches to probe for novel protein interactions. Other techniques that researchers have employed to investigate MA transporter-interacting proteins include yeast two-hybrid assays using transporter fragments (e.g., N and C termini) as bait to look for interactions in specific domains, fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer to assess direct interactions between proteins in intact preparations, and colocalization of immunofluorescence or fluorescent tags using microscopy to investigate compartmental interactions. In many cases, a combination of these techniques is used and repeated both in vitro and ex vivo from native preps to verify the authenticity and biologic relevance of the interactions. In this review, we do not provide a comprehensive review of MA transporter protein-protein interactions, but only note those known to us from publications to be regulated by kinase-linked signaling pathways.
II. Protein Kinase C—Overview
The most well extensively investigated kinases with respect to the regulation of MA transporters are the protein kinase C (PKCs), a family of Ser/Thr kinases so designated for the activation of classic isoforms (cPKCs) by Ca2+, in addition to diacylglycerol (DAG) (Zeng et al., 2012). The conventional PKC isoforms (cPKCs) include PKCα, PKCβ1, PKCβ2, and PKCγ (Dempsey et al., 2000). These kinases share two conserved domains, C1 and C2, that specify their interactions with DAG and Ca2+, respectively. Involvement of DAG in the action of cPKCs links their activation to the activity of phospholipase C (PLC), as well as to the receptors that target these enzymes as effectors. A second class of DAG-dependent PKCs, termed novel PKCs (nPKCs) although activated by DAG, lack sensitivity to Ca2+, a feature conferred by the absence of a C2 domain. The nPKCs include PKCδ, PKCδ1-3, PKCε, PKCη, and PKCθ. PKCs of the third, and less studied, class are designated as atypical PKCs (Dempsey et al., 2000). These enzymes (PKCι, PKCζ, and PKN1-3) are structurally related to cPKCs and nPKCs through their catalytic domain (C4), but they lack functional C1 and C2 domains and require neither DAG nor Ca2+ for activation. To our knowledge, nPKCs have yet to be implicated in MA transporter regulation and will not be discussed further.
Much of what has been reported concerning the role of PKCs in the regulation of MA transporters derives from studies of cells and tissues treated with phorbol esters, primarily phorbol 12-myristate 13-acetate (β-PMA, also called TPA) or phorbol 12,13 dibutyrate (PDBu), potent and membrane permeant mimics of DAG, and hence activators of both cPKCs and atypical PKCs. For both activators, analogs lacking PKC activity are available (4α-PMA, 4α−PDBu) and have been used to document PKC involvement in effects. To achieve cPKC versus nPKC selectivity for phorbol ester-mediated effects, subtype selective inhibitors or isoform-specific genetic manipulations can and should be pursued. Such efforts have supported regulation of MA transporters by specific and, in some cases, multiple PKC isoforms, sometimes appearing to work in opposition. An important challenge that remains for researchers in the field, one that relates as much to other kinases as well as PKCs, is the elucidation of endogenous pathways linked to specific receptors that capitalize on the potential revealed with kinase activators.
A. Regulation of Dopamine Transporter by Protein Kinase C
1. Regulation of Dopamine Transporter Activity by Protein Kinase C.
PKCs have been extensively investigated in the regulation of DAT, including studies using DAT cDNA transfected cells and rodent brain preparations (Table 1). Across these preparations, β-PMA treatments has been found consistently to result in a significant decrease in DA transport capacity, detected in kinetic studies as a decrease in the maximal velocity of DA transport (Vmax). These effects have been found to be sensitive to PKC inhibitors such as staurosporine and bisindolylmaleimide I (BIM-1) (Kitayama et al., 1994; Copeland et al., 1996; Huff et al., 1997; Chang et al., 2001; Granas et al., 2003). With regard to isoform specificity for PKC regulation of DAT, work in Xenopus laevis oocytes measuring tyramine-induced DAT-mediated currents demonstrated blockade of β-PMA-effects on DAT activity by the cPKC specific inhibitor Go6979 (5,6,7,13-Tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile), as well as by the Ca2+ chelator EGTA (Doolen and Zahniser, 2002). Although these findings suggest that Ca2+-dependent cPKCs likely mediate the effects of β-PMA, rottlerin, a δPKC inhibitor, also blocked β-PMA effects on DAT (Doolen and Zahniser, 2002). This contribution of δPKC has not been further investigated and may derive from the heterologous system these researchers used, and as such the biologic relevance of this PKC isoform in DAT regulation remains to be validated. However, further work exploring PKC isoform specificity for DAT regulation points to a greater complexity of enzymes and actions. Thus, studies conducted with the PKCβ specific inhibitor LY379196 [8((dimethylamino)methyl)-6,7,8,9,10,11-hexahydro-5,21:12,17-dimetheneo-18H-dibenzo(i,o)pyrrolo(3,4-1)(1,8)diazacyclohexandecine-18,10(19H)dione] as well as kinase knockout mice support a role for PKCβ in DAT regulation, although distinct from that observed in studies with phorbol ester stimulation, Chen et al. (2013) observed that treatment of mouse striatal synaptosomes with LY279196 blocked the ability of the D2 agonist quinpirole to increase DAT surface levels and DA uptake, suggesting a role for PKCβ in supporting elevated surface expression. LY279196 also blocked quinpirole-induced increases in p-extracellular signal-related kinase 1/2 (ERK1/2). In striatal synaptosomes from PKCβ knockout mice, reduced basal DA uptake and DAT surface levels were observed, as well as a lack of trafficking sensitivity to quinpirole. These observations argue that PKCβ may lie upstream in a signaling cascade connecting D2 receptor activation to ERK1/2, ultimately positively supporting DAT surface expression. In contrast to these findings, however, Zestos et al. (2016) found that perfusion of PKCβ inhibitors into the nucleus accumbens of rats had no effect on DA uptake in striatal synaptosomes, despite effects on AMPH-evoked DA efflux (discussed below). This lack of an effect on DA uptake in these studies may be due to the relatively short time course of PKCβ inhibition (30 minutes), which may not be sufficient to observe effects on basal DA uptake like those seen in PKCβ knockout mice. The findings of Zestos et al. on AMPH actions suggest that PKCβ inhibition has effects on mechanisms that support conformations of DAT linked to altered transporter states and/or trafficking, such as those supporting quinpirole-induced increases in DAT activity, as observed in the studies of Chen et al.
Regulation of DAT by protein kinases
Importantly, DA itself, acting through DAT, can induce a PKC-dependent downregulation of DAT. In rDAT LLC-PK1 cells, pretreatment with DA led to a decrease in subsequence DA uptake, an effect that was nonadditive with β−PMA and was blocked by BIM-1 (Gorentla and Vaughan, 2005). This downregulation was also blocked by cocaine pretreatment, suggesting DA acts through the transporter itself, and not other targets such as DA receptors, to cause this PKC activation and DAT downregulation. Other DAT substrates, AMPH and METH, also cause a cocaine-sensitive downregulation of DAT, although they appear to have differential dependence on PKC. Cervinski et al. (2005) demonstrated that METH-induced reduction of DA uptake in rDAT LLC-PK1 was blocked by BIM-1 treatment. In contrast to this, AMPH downregulation of DAT surface levels in transfected PC-12 cells was not blocked by BIM-1 and did not depend on a C-terminal endocytic sequence required for PKC-stimulated DAT endocytosis (discussed further in Regulation of Dopamine Transporter Membrane Compartmentalization and Trafficking by Protein Kinase C below) (Boudanova et al., 2008).
In addition to alterations in DA uptake activity, PKC isoforms also appear to play critical roles in regulating DAT-dependent DA efflux. Johnson et al. (2005) reported that treatment of rat striatal synaptosomes and slices with β-PMA induces a Ca2+-independent efflux of DA through DAT, effects that could be blocked by the PKCβI/II inhibitor LY379196. AMPH-evoked DA efflux can also be blocked by the cPKC-specific inhibitor Go6976 as well as LY379196 (Johnson et al., 2005), suggesting that the contribution to AMPH actions to trigger reverse DA transport, which are known to involve elevations in intracellular Ca2+, rely on signaling through PKCβ isoforms. Consistent with this idea, PKCβ knockout mice demonstrate reduced, though notably not completely eliminated, AMPH-evoked DA efflux (Chen et al., 2009). Similar results were recently observed by the Gnegy group, who found that perfusion of PKCβ inhibitors into the nucleus accumbens of rats reduced AMPH-evoked DA efflux by approximately 50% (Zestos et al., 2016). These authors also observed a decrease in AMPH-induced stimulation of locomotor behavior. Interestingly, AMPH-evoked DA efflux in transfected HEK-293 cells as monitored by amperometry was found to be insensitive to PKCβ antagonism, although these inhibitors restored AMPH evoked efflux lost in the ADHD-associated DAT coding variant Val559 (Bowton et al., 2014).
The target of PKC isoforms in their regulation of AMPH-evoked DA efflux has not been determined, but work from Sucic et al. (2010) supports a role for the juxtamembrane N-terminal Thr62 residue, because Thr62Ala and Thr62Asp mutations significantly blunted AMPH-evoked efflux of MPP+ from HEK-293. Thr62 lies within a PKC phosphorylation consensus site, and homologous residues exist in both NET and SERT. Further work on the importance of this threonine residue has been performed on SERT, and will be discussed further in Regulation of Serotonin Transporter Activity by Protein Kinase C Isoforms.
2. Regulation of Dopamine Transporter Membrane Compartmentalization and Trafficking by Protein Kinase C.
When surface expression of DAT has been measured, phorbol ester-induced transport capacity changes are paralleled in dose and time by a decrease in steady-state surface DAT levels (Pristupa et al., 1998; Daniels and Amara, 1999; Melikian and Buckley, 1999; Holton et al., 2005; Boudanova et al., 2008). Kinetic trafficking studies indicate that these reductions in DAT surface expression derive from increased rates of endocytosis (Loder and Melikian, 2003). PKC-dependent DAT endocytosis has been reported to be both dynamin and clathrin dependent, with pharmacological inhibition of clathrin-mediated endocytosis, as well as siRNA knockdown of dynamin or clathrin light/heavy chains preventing β-PMA-induced DAT internalization (Sorkina et al., 2005). Gabriel et al. (2013) further supported the dynamin dependence of β-PMA-induced DAT internalization in mouse striatal slices and showed that this endocytosis arose exclusively from lipid raft domains, as opposed to constitutively endocytosed DAT that was dynamin independent and arose from both raft and non-raft compartments. β-PMA-induced DAT endocytosis requires a C-terminal FREKL motif, which is immediately upstream of the AYAIA motif that drives constitutive endocytosis (Holton et al., 2005). The fate of β-PMA-stimulated internalized DAT also appears to differ from DAT that is constitutively endocytosed. In both hDAT-HEK-293 cells and rat primary DA neurons, treatment with β-PMA leads to colocalization of DAT with markers of late endosomes/lysosomes, whereas DAT that is constitutively endocytosed colocalizes mostly with markers of early/recycling endosomes (Hong and Amara, 2013). It should be noted that a conflicting study also looking at rat primary DA neurons found no effect of PMA on DAT internalization (Eriksen et al., 2009), although at present the basis for these contradicting findings is unclear. β-PMA treatment also induces a marked increase in DAT ubiquitination, an effect that was absent with AMPH stimulation. As will be discussed in Protein Kinase C Regulation of Dopamine Transporter Protein-Protein Interactions, this ubiquitination is likely mediated by Nedd4-2 and may serve as a signal to route DAT to lysosomes for degradation. Indeed, in MDCK cells, β-PMA treatment again leads to colocalization of DAT with markers of late endosomes/lysosomes and degradation of the transporter within 2 hours (Daniels and Amara, 1999).
The role of PKC signaling in the effects of AMPH on DAT trafficking has been investigated by a number of studies with mixed results. As mentioned above, inhibition of PKC by BIM-1 did not block AMPH-stimulated DAT endocytosis. Additionally, this AMPH effect did not require the C-terminal FREKL motif necessary for PKC-stimulated DAT trafficking (Boudanova et al., 2008). In the work by Hong and Amara (2013) in rat primary DA neurons mentioned above, AMPH treatment led to colocalization of DAT with early/recycling endosomes and not late-endosomes/lysosomes as was seen with PMA treatment. The Amara laboratory recently provided evidence that AMPH-triggered DAT endocytosis is clathrin-independent and requires the small GTPase Rho (Wheeler et al., 2015), which mediates another dynamin-dependent mode of endocytosis (Croise et al., 2014). These lines of evidence are consistent with a PKC-independent mode of DAT internalization by AMPH. AMPH-stimulated trafficking of DAT may be impacted by PKC signaling, however, because mouse striatal synaptosomes treated with the PKCβ inhibitor LY379196 before AMPH exposure displayed an early decrease in DAT surface levels, followed by a later increase, opposite to what is seen with AMPH alone (Chen et al., 2009). Further complicating the story, in cells expressing the DAT Val559 variant that does not show AMPH-induced trafficking, treatment with a PKCβ inhibitor restored the ability of DAT to internalize in response to AMPH treatment, suggesting that PKCβ may actually antagonize AMPH-stimulated DAT endocytosis, at least in the context of the Val559 mutation (Bowton et al., 2014). Altogether, these results paint a complicated picture of the interaction between AMPH-stimulated and PKC-stimulated DAT trafficking. Hopefully future work will further clarify the role PKC isoforms play in the regulation of DAT activity and trafficking by AMPH.
Recently, Cremona et al. (2011) presented evidence that compartmentalization in distinct membrane microdomains contributes to Go6850-sensitive, PMA-triggered DAT internalization. These investigators identified the membrane raft-enriched protein flotillin-1 as a DAT-associated protein that is required for internalization of DAT upon PKC activation in transfected EM4 cells (a version of HEK-293 cells with increased adherence). Additionally, phosphorylation of flotillin-1 at Ser315 was found to be required for this effect on DAT internalization (Cremona et al., 2011), although whether this site is directly or indirectly targeted by PKCs (and by which isoform) to ensure PKC-dependent trafficking has not been established. However, the researchers also observed that flotillin-1 was required to maintain DAT membrane raft association, suggesting that PKC-dependent DAT endocytosis arises from specialized microdomains that contain flotillin-1. A conflicting study, however, reported that in both HEK-293 and HeLa cells, siRNA knockdown of flotillin-1 failed to block PMA-induced DAT endocytosis and may even slightly enhance DAT internalization. Loss of flotillin-1 did, however, increase the lateral mobility of DAT in the membrane as measured by a fluorescence recovery after photobleaching (FRAP) assay, suggesting that flotillin-1 may play a role in anchoring DAT, within membrane raft microdomains (Sorkina et al., 2013). The role PKC plays in the interactions between DAT and flotillin-1/membrane rafts still remains unclear; however. Cremona et al. (2011) also reported that flotillin-1 is required for AMPH-triggered, DAT-dependent DA efflux, a process others have reported to depend on DAT localization to cholesterol-rich raft domains (Jones et al., 2012). Together, these studies point to the existence of PKC-dependent regulation of DAT protein associations as critical to the response of the transporter to display altered function and/or trafficking.
A recent finding highlights the potential translational importance of understanding membrane microdomain targeting in relation to PKC-dependent DAT endocytosis. Sakrikar et al. (2012) studied the functional properties and localization of the ADHD-associated variant in DAT Cys615 and found the variant to exhibit elevated rates of constitutive endocytosis and recycling, coinciding with an insensitivity to the endocytic effects of phorbol esters and AMPH. This insensitivity to both of these agents is interesting considering the evidence discussed earlier that the endocytic effects of AMPH are PKC independent (Boudanova et al., 2008). Perhaps this loss of AMPH- and PKC-stimulated DAT trafficking could be due to a difference in this variant’s localization that impacts DAT endocytosis triggered by both AMPH and PKC. Importantly, the variant displayed reduced association with flotillin-1 as well as reduced membrane raft localization, quantified on the basis of colocalization with fluorescent Ctxβ labeling of GM1 ganglioside. These findings echo the trafficking insensitivity to PKC activation observed by Cremona et al. (2011) with loss of flotillin-1 or its mutation, as well as the increase in basal endocytosis observed by Sorkina et al. (2013) with the end result being dependent, perhaps, on technical considerations such as cell hosts and levels of expression. Interestingly, the DAT Cys615 mutant still supports AMPH-evoked DA efflux (Sakrikar et al., 2012) and thus may be useful in further segregating PKC-dependent control of trafficking versus function.
A recent report by Wu et al. (2015) found that the increased rate of constitutive endocytosis of the Cys615 variant could be rescued by overexpression of a constitutively active version of the tyrosine kinase Ack1, which functions downstream of the Rho-family GTPase Cdc42. The actions of this kinase appear to interact with PKC regulation of DAT because Ack1 or Cdc42 inhibition increased clathrin-dependent DAT internalization and reduced DAT surface levels and activity in a manner that was nonadditive with PMA. Additionally, overexpression of a either a constitutively active or a kinase-dead Ack1 both abolished any effect of PMA on DAT internalization rate, suggesting that PMA-stimulated trafficking of DAT may require inactivation of Ack1, which itself acts as a brake on DAT endocytosis. Negative regulation of Ack1 by PKC is supported by the finding that PMA reduced active phosphorylated Ack1. Altogether, these results are consistent with the idea that PKC may act in part to trigger DAT endocytosis by antagonizing this Cdc42/Ack1 brake on constitutive DAT endocytosis, and the increased internalization and loss of PKC-stimulated endocytosis of the Cys615 DAT variant may result from a blunting of this braking mechanism, possibly due to altered membrane localization.
Whether PKC regulates lateral movement of DAT into or out of membrane rafts, as well as how the kinase drives internalization of transporters from these compartments, continues to be studied. Some have even reported trafficking-independent modes of DAT regulation produced by PMA. Foster et al. (2008), using transfected LLC-PK1 cells, found that PMA treatments reduced DAT activity, accompanied by PKCα recruitment to membrane rafts and preferential phosphorylation of DAT in raft fractions. Notably, however, these investigators generated evidence that changes in surface DAT levels are required to establish reductions in DA transport function after PMA treatments, Thus, inhibition of clathrin-mediated endocytosis with either the chemical inhibitor concanavalin A (Con A) or a dominant-negative dynamin was sufficient to prevent internalization of the protein, but only partially prevented PKC-induced downregulation of DAT activity. Also, using a cholesterol depletor, methyl-β-cyclodextrin, they observed a partial blockade PKC-induced functional downregulation, despite changes in DAT internalization equivalent to that seen with PMA treatments. Further support for physiologic trafficking-independent regulation of DAT after PKC activation has been obtained from studies of brain synaptosomes, where functional downregulation has been observed after phorbol ester treatment in the presence of high sucrose, which blocks endocytosis (Park et al., 1988). The Vaughan laboratory more recently provided evidence that the trafficking-independent effects of PKC may involve regulation of palmitoylation of DAT. Inhibition of DAT palmitoylation by the palmitoyl acyltransferase inhibitor 2-bromopalmitate in rat striatal synaptosomes led to decreases in the Vmax of DA uptake independent of changes in DAT surface levels, supporting a role for this modification in trafficking-independent regulation of DAT activity (Foster and Vaughan, 2011). Importantly, activation and inhibition of PKC by PMA/BIM-1 decreased and increased palmitoylation of DAT, respectively, and mutation of the PKC target Ser7 to alanine abolished these effects on palmitoylation (Moritz et al., 2015). Critically, unlike wild-type DAT, this mutant showed no downregulation of uptake in response to PMA when endocytosis was blocked by ConA, suggesting that the trafficking-independent effects of PMA are likely mediated through phosphorylation of S7A, potentially via its role in regulating palmitoylation of DAT. Altogether, these studies seem to reveal a capacity of DAT to enter into a state of reduced function in response to PKC activation that may be difficult to observe in many conditions because of rapid endocytosis of inactivated transporter. In terms of the potential for translational relevance of trafficking-independent regulation by PKC pathways, Mazei-Robison and Blakely (2005) demonstrated that the human DAT variant Ala382 demonstrates greater reductions in DA uptake than can be explained by changes in DAT surface expression, as measured by biotinylation. Because wild-type DAT demonstrated comparable losses of surface transporters and DA uptake, the Val382 variant may interfere with the normal coincidence of DAT functional changes and transporter internalization. In vivo, such behavior could result in alterations in the normal homeostatic control of DAT that link changes in DA release to DA clearance. Obviously, a key step in such models is the identification of endogenous mechanisms that make use of PKC-dependent regulatory control of DAT, trafficking dependent or independent.
3. Regulation of Dopamine Transporter Phosphorylation by Protein Kinase C.
Along with producing changes in DA transport and/or DAT internalization, PKC activation also leads to increased transporter phosphorylation (summarized in Table 1). Elevated DAT phosphorylation has been observed in heterologous (Huff et al., 1997; Chang et al., 2001; Granas et al., 2003; Gorentla and Vaughan, 2005; Foster et al., 2012) and native (Cowell et al., 2000; Foster et al., 2012) preparations, although evidence suggests that transporter phosphorylation is not required for changes in DAT trafficking or activity. Thus, truncation of the DAT N terminus, which results in a significant loss of DAT phosphorylation after phorbol ester treatment, did not affect β-PMA-induced reductions in DAT activity in transfected cells (Granas et al., 2003; Khoshbouei et al., 2004). Interestingly, N-terminal truncation also completely blocks PKC-dependent DAT phosphorylation induced by methamphetamine treatment, although the drug still reduced DAT activity (Cervinski et al., 2005). Additionally, Chang et al. (2001) demonstrated that mutation of PKC consensus sites on DAT that prevent β-PMA-induced increases in DAT phosphorylation do not prevent reduction in transport or DAT surface levels, effects similar to those observed by Granas et al. (2003). In contrast to these findings, however, Lin et al. (2003) also showed that mutation of the N-terminal serine Ser7 blocks the ability of β-PMA to reduce the Vmax of DA uptake in transfected COS cells (Lin et al., 2003). This apparent differential dependence on the N terminus in PKC regulation of DAT may reflect differences in cell lines used and potentially different contribution of PKC isoforms in regulating DAT in these different contexts.
Studies by Vargas-Medrano et al. (2011) showed that inhibition of PKCβ abolished β-PMA-induced increases in DAT phosphorylation in PAE cells, suggesting that this isoform is required for DAT phosphorylation upon PKC activation. Considering work by Chen et al. (2013) discussed above seems to indicate that PKCβ may act to elevate DAT surface levels, it is possible that phosphorylation of DAT by PKC activation actually supports DAT surface expression, despite the net decrease in surface levels upon nonspecific PKC activation by β-PMA. In support of this, β-PMA treatment increases phosphorylation of Thr53, a residue whose phosphorylation seems to promote DAT surface expression (Foster et al., 2012). Based on work from Chen et al. (2013) that showed that PKCβ appears to function upstream of ERK1/2, which are strong candidates for targeting Thr53 (Gorentla et al., 2009), it is possible that PKCβ may act through ERK1/2 to increase Thr53 phosphorylation and therefore positively regulate DAT, whereas other PKC isoforms act to downregulate DAT activity, potentially through direct phosphorylation of the transporter or other interacting proteins. Caution is needed with respect to interpretations from heterologous expression systems; however, because these contexts may not accurately reflect the endogenous environment in which DAT interacts with PKC signaling pathways, including DAT-associated proteins. Possibly, DAT phosphorylation induced by PKC can shift the disposition of inward and outward facing conformations of the transporter, leading to functional changes that are only permitted in the context of a mature synapse or when exposed by other challenges. In this regard, Fog et al. (2006) documented that both PKCα and calmodulin-dependent protein kinase IIα (CaMKIIα) can phosphorylate multiple Ser residues on the DAT N terminus, sites previously implicated in AMPH-induced DA efflux. These results were corroborated by Moritz et al. (2013), who demonstrated that PKCα targets Ser4, Ser7, and Ser13 and that one of these PKC target sites, Ser7, appears to regulate conformational equilibria of DAT, because Ser7Ala and Ser7Asp mutations reduce and increase binding of a cocaine analog CFT, respectively. Interestingly, cocaine is also capable of blocking β-PMA-induced DAT phosphorylation in striatal synaptosomes, possibly at these N-terminal serine residues (Cowell et al., 2000), and similar effects were seen with GBR12909 in rDAT LLC-PK1 cells (Gorentla and Vaughan, 2005). These results further support the importance of the conformational state of DAT in PKC-dependent phosphorylation of the transporter.
4. Protein Kinase C Regulation of Dopamine Transporter Protein-Protein Interactions.
The number of DAT interacting proteins continues to grow. Members of the “DAT interactome” include PICK-1 (Torres et al., 2001), PP2Ac (Bauman et al., 2000), α-synuclein (Lee et al., 2001), Hic-5 (Carneiro et al., 2002), syntaxin 1A (Lee et al., 2004), RACK (Lee et al., 2004), CaMKII (Fog et al., 2006), Nedd4-2 (Sorkina et al., 2006), D2 DA receptors (Bolan et al., 2007), flotillin-1 (Cremona et al., 2011), Rin (Navaroli et al., 2011), and the κ-opioid receptor (Kivell et al., 2014). A few of these proteins have been implicated in the PKC regulation of DAT, including as noted earlier flottilin-1, a membrane raft-associated protein linked to PKC-dependent trafficking and AMPH actions. Several DAT-associated proteins have historical associations to PKC pathways (e.g., RACK1, PICK-1) in other systems but have yet to be linked to PKC-dependent DAT regulation. Several others (e.g., Hic-5) have been implicated in PKC-dependent regulation of other MA transporters, as noted below, and thus are likely also to participate in PKC regulation of DAT.
Sorkina et al. (2006) used an siRNA screen of DAT transfected cells to identify regulators of PKC-dependent DAT internalization, identifying the E3 ubiquitin ligase Nedd4-2. DAT is ubiquitinated in response to PMA treatments (Miranda et al., 2005) but when Nedd4-2 expression was reduced by siRNA approaches, DAT ubiquitination was blocked and DAT surface levels decreased to a lesser extent after β-PMA treatment. Importantly, mutation of DAT N-terminal lysine residues Lys19, Lys27, and Lys35 blocked transporter ubiqutination, and also prevented PMA-induced DAT internalization (Miranda et al., 2007; Vina-Vilaseca and Sorkin, 2010). Whether PKC directly regulates Nedd4-2 activity or drives its recruitment to DAT has yet to be demonstrated. In addition to potentially triggering DAT internalization, PKC-dependent ubiquitination mediated by Nedd4-2 transporter ubiqutination may also be involved in targeting DAT to different recycling pathways that can result in either recycling or degradation (Daniels and Amara, 1999; Hong and Amara, 2013).
Recent investigations with the plasma membrane-associated GTPase Rin further support the importance of membrane microdomain localization in the PKC-dependent regulation of DAT trafficking and activity (Navaroli et al., 2011). Navaroli and colleagues demonstrated that Rin directly associates with the DAT C-terminal “FREKLAYAIA” endocytic motif and that expression and activity of Rin is required for PKC-triggered DAT endocytosis. They also showed that Rin and DAT interactions increased with PKC activation and that this PKC-stimulated interaction was lost in DAT with mutation of the amino acids FREK to AAAA. Because these amino acids were previously shown to be required for β-PMA-induced DAT internalization, this loss of PKC-stimulated Rin association with DAT when these amino acids are mutated strongly supports a role for Rin in driving this motif-dependent PKC-stimulated DAT endocytosis. Interestingly, the PKC-stimulated DAT/Rin interactions occurred preferentially in membrane rafts. Whether the interaction between Rin and DAT in rafts is due to Rin supporting the endocytosis from raft domains or whether Rin may act to move DAT into or out of rafts or plays some other role remains to be clarified.
5. Receptor-Initiated Protein Kinase C Regulation of Dopamine Transporters.
Much of the work described above involves activation of PKC by phorbol esters, unnatural stimuli that are pathophysiologically linked to tumor promotion but not to normal synaptic modulation. As noted earlier, progress in the study of PKC mechanisms that regulate DAT will likely depend on the identification of receptor-mediated signaling pathways that rely on PKC signaling. As noted above, PKCβ isoforms have been reported to play a role in D2 receptor-mediated trafficking of DAT (Chen et al., 2013), possibly connected to DAT via ERK1/2 (see below). Additionally, Page et al. (2001) reported that an mGluR agonist decreases DA uptake in rat striatal synaptosomes, an effect that can be blocked by the PKC inhibitor Ro-31-8220 (3-[3-[2,5-Dihydro-4-(1-methyl-1H-indol-3-yl)-2,5-dioxo-1H-pyrrol-3-yl]-1H-indol-1-yl]propyl carbamimidothioic acid ester). PKC may also be involved in substance P-induced downregulation of DAT, because substance P treatment of HEK-293 cells coexpressing hDAT and the substance P receptor hNK-1 results in a decrease in DA uptake that is partially blocked by staurosporine and only slightly additive with PMA (Granas et al., 2003). The NK1 isoform is expressed by human midbrain DA neurons (NK-3 is more abundant in rat) where it may act to support substance P modulation of DA signaling (Whitty et al., 1997). NK-1 is a Gαq-coupled G protein-coupled receptor, consistent with coupling to PKC signaling pathways, although these have not been studied in the context of DAT regulation. Interestingly, microdialysis studies using local infusion of an NK-1 antagonist points to a capacity for the receptor to modulate cocaine-induced DA clearance (Loonam et al., 2003). PKC may also regulate reverse transport of DA induced by mGluR signaling in the rat substantia nigra (Opazo et al., 2010), by the σ2-receptor activation in rat striatal synaptosomes (Derbez et al., 2002), and by estradiol activation of membrane estrogen receptors in NGF-differentiated PC12 cells (Alyea and Watson, 2009), consistent with the discussion above relating PKC activity to DA efflux induced by AMPH treatment.
The role PKCβ plays in DAT-dependent DA efflux is particularly interesting in light of studies that have established ectopic activation of D2 receptors as a critical determinant of the disrupted function of the disease associated DAT coding variant Val559 (Mazei-Robison et al., 2008; Bowton et al., 2010, 2014). In initial studies (Bowton et al., 2010), the group found that anomalous DA efflux (“leak”) requires ongoing D2 receptor signaling, because it can be blocked by treatments with either raclopride, a D2 receptor antagonist, or pertussis toxin, which blocks Gi/o signaling through which D2 receptors signal. These studies suggest that endogenous D2 receptors on DAT transfected cells drive or sustain DA leaking through DAT Val559, whereas they lack this activity with wild-type DAT, either because of differences in heteromultimer formation or an ability of D2 receptors to amplify and sustain the inherent leak of the Val559 variant. In more recent studies, Bowton et al. (2014) implicated ectopic PKCβ signaling in both basal DA efflux and the insensitivity of the Val559 transporter to AMPH. Here, inhibition of PKCβ, like raclopride in the prior study, restored the ability of DAT Val559 to support AMPH-mediated DA efflux. These findings indicate that DAT Val559 exists in a state where D2-dependent PKCβ action drives transporter-mediated DA export in a manner that is normally impeded with wild-type DAT, either because D2 receptors are not continually flooded by DA leak or because other inhibitory processes are in place (that must then also be lost in the variant). Recently, Mergy et al. (2014) reported studies with DAT Val559 mice, providing evidence of both excessive extracellular DA and tonic presynaptic D2 receptor stimulation. The availability of the DAT Val559 mouse model provides an unprecedented opportunity to define the significance of D2 receptor mediated PKCβ signaling for DAT modulation in vivo.
B. Regulation of Norepinephrine Transporter by Protein Kinase C
1. Regulation of Norepinephrine Transporter Activity by Protein Kinase C.
As with DAT, activation of PKC by β-PMA has been reported to modify NET activity in various cell lines and ex vivo preparations (Table 2) (Lu et al., 1996; Apparsundaram et al., 1998b; Bönisch et al., 1998). Studies by Lu et al. (1996) demonstrated that treatment of primary rat midbrain-brain stem cultures with β-PMA leads to a time-dependent increase in NET mRNA levels, with elevations apparent with as little as 1 hour post β-PMA treatment, although no corresponding NE transport measures were presented, and likely more sustained treatments would have been needed to elevate NET protein levels. Indeed, Apparsundaram et al. (1998b) demonstrated that HEK-293 cells transfected to express hNET displayed significant reductions in NE uptake after short (0–60 minutes) β-PMA treatments, with kinetic analyses revealing a decrease in NE transport Vmax that is insensitive to inhibitors of transcription or translation (actinomycin D and cyclohexamide, respectively), suggesting that changes arise from altered transporter function or surface transporter protein trafficking. NET activity reductions with β-PMA were found to be blocked by the nonselective PKC inhibitors staurosporine and BIM-1 and to be accompanied by changes in surface expression, as monitored by biotinylation studies and whole cell [3H]nisoxetine binding assays. That such NE uptake-reducing actions of short β-PMA treatments are not limited to heterologous preparations was shown by Bauman et al. (2000) who documented NET activity reductions in studies of rat vas deferens minces, by Lee et al., (2006) who documented reductions in NET activity in cultured bovine primary endothelial cells, and by Jayanthi et al. (2002) who demonstrated rapid reductions in NE transport in rat placental trophoblast cultures. At present, however, specific PKC subtypes responsible for rapid NET regulation after phorbol ester treatments remain undefined, as do subtype(s) responsible for longer term transcriptional effects. However, as noted below, biochemical studies support a role of PKCε in relation to phosphorylation-dependent regulation of NET, whereas PKCα may participate when NET regulation is activated via a receptor-linked (substance P, NK-1) pathway.
Regulation of NET by protein kinases
2. Regulation of Norepinephrine Transporter Membrane Compartmentalization and Trafficking by Protein Kinase C.
As mentioned above, activation of PKC by β-PMA causes a reduction in the Vmax of NE uptake in cells expressing NET, and multiple groups have demonstrated that this decrease is paralleled by a reduction in NET surface levels. Jayanthi et al. (2004b) characterized this reduction in NET surface levels in rat placental trophoblast cultures and shown that it is due to an increase in internalization and not a change in recycling rates. Interestingly, the LWERLAYGIT sequence in the hNET C terminus, which matches the L(X)ERLAY(X)IT motif identified in studies of DAT, was shown by Melikian’s group to be required for efficient, constitutive endocytosis of NET (Holton et al., 2005). Whether these sequences (or the LWERL component, matching the aforementioned FREKL motif in DAT) are required for PKC-dependent endocytosis, has to our knowledge not been documented, although other mutagenesis studies support a role for C-terminal residues in NET plasma membrane trafficking (Bauman and Blakely, 2002). Jayanthi et al. (2004b) were the first to investigate membrane compartmentalization with respect to NET regulation in their studies of β-PMA induced NET internalization in cultured rat placental cells. The investigators found that blockade of clathrin-mediated endocytosis by ConA or dominant-negative dynamin I or II had no effect on β-PMA induced NET internalization, whereas treatment with filipin to block caveolae/lipid raft-mediated endocytosis blocked NET endocytosis. These findings are reminiscent of the findings noted above by Cremona et al. (2011) who implicated membrane rafts in β-PMA -induced DAT internalization. Additionally, using human trophoblast (HTR) cells that express hNET, β-PMA treatments were found to decrease NET presence in membrane raft fractions, defined biochemically (Jayanthi et al., 2006).
The translational significance of the previously noted observations remains ill-defined, although one can imagine that as with the ADHD-associated DAT mutation R615C, hNET mutations that result in improper targeting to membrane domains could yield anomalous regulatory behavior and compromised NE signaling fidelity. In this regard, Hahn et al. (2005) demonstrated altered basal and β-PMA regulated surface trafficking of multiple hNET nonsynonymous variants recovered in polymorphism screening projects. As with prior studies, these investigators documented downregulation of hNET activity in transfected (COS-7) cells after 30 minutes of β-PMA treatments. One variant studied, however, R121Q, exhibited an enhanced surface reduction, compared with wild-type hNET, after β-PMA treatment, whereas another, F528C, demonstrated β-PMA insensitivity. The locations of these variants in the hNET structure (R121Q, IL1; F528C, TM11), offer as yet few clues as to how such changes in regulation arise, nor are clinical implications readily inferred because of an absence of patient information in the studies where the variants were identified (Halushka et al., 1999; Iwasa et al., 2001). Now that phenotypes have been defined in humans (Kim et al., 2006; Shannon et al., 2000) and rodents (Shirey-Rice et al., 2013) arising from hNET genetic variation, clinically informative variants may be studied in the future, much as they have been in hDAT R615C and A559V, and in hSERT (see below).
3. Regulation of Norepinephrine Transport Phosphorylation by Protein Kinase C.
Significant evidence links transporter phosphorylation to the regulatory effects of PKC on NET. First, Jayanthi et al. (2004b) demonstrated that rat NET is phosphorylated under basal conditions in cultured rat trophoblasts with phosphorylation level increased β-PMA treatments. Subsequently, the same group demonstrated by phosphoamino acid analysis that β-PMA induced hNET phosphorylation in HTR cells occurs on both Ser and Thr residues, with site-directed mutagenesis studies implicating adjacent residues in IL2, T258, and S259 as likely kinase targets. Moreover, the T258A/S259A double mutant hNET, when expressed in HTR cells, exhibited reduced β-PMA induced phosphorylation and fails to support β-PMA-induced downregulation of NE transport or transporter internalization (Jayanthi et al., 2006). One aspect to consider in these studies is that the T258A/S259A double mutant also elevates basal hNET phosphorylation. Additionally, the double mutant fails to redistribute away from membrane raft fractions like wild-type hNET. Interestingly, mutation of individual residues in the T258/S259 dyad fails to blunt β-PMA-induced regulation, suggesting a redundant requirement for phosphate attachment in this domain for functional NET downregulation, although phosphorylation was found to be exquisitely sensitive to the S259A substitution. This is consistent with earlier work from Bönisch et al. (1998), who previously showed that S259A hNET retains sensitivity to β-PMA-induced NET downregulation. Interestingly, the T258A/S259A double mutant also lacks sensitivity to AMPH-induced internalization, an effect that is PKC independent, suggesting that these residues may play a more global role in regulating DAT endocytosis through multiple mechanisms (Annamalai et al., 2010). In toto, these findings provide significant evidence that phosphorylation of NET proteins is a prerequisite for the regulatory steps that culminate in PKC-dependent transporter downregulation, although some of these phosphorylation sites may play roles in PKC-independent modes of DAT internalization as well.
Finally, potential insights into PKC isoforms involved in these actions has been gleaned. After the group’s prior findings of Ca2+-independence of β-PMA effects on NET (Jayanthi et al., 2004b; Sung and Blakely, 2007), the novel PKC isoform PKCε was examined for its ability to trigger hNET phosphorylation in vitro using isolated membranes (Jayanthi et al., 2006). Indeed, PKCε elevated hNET phosphorylation (negligible in the absence of added kinase) and the impact of T258A and S259A mutations on phosphorylation was strikingly similar to that shown with intact cell studies. Although direct demonstration (as via mass spectrometry or phosphospecific antibody studies) of the targeting of the T258/S259 sites is needed, studies that manipulate these sites in vivo are anticipated and project an exciting pathway to further elucidating the synaptic and behavioral significance of NET regulation by PKC. An additional question arising from the NET IL2 phosphorylation studies is how they can be connected to the essential nature of residues 2–42 in the hNET N terminus for β-PMA effects on NE uptake and NET trafficking (Sung and Blakely, 2007). A similar issue arises with respect to C terminal involvement, particularly if the LWERL motif in this domain is required for PKC-dependent NET endocytosis, as predicted from DAT studies. The recent high-resolution structure of Drosophila melanogaster DAT (Penmatsa et al., 2013) reveals that the intracellular loops lie much closer to each other and to the cytoplasmic N and C termini that one would conclude from simple topology models, providing opportunities for regulatory events to arise from noncontiguous sequences. One might imagine for example that phosphorylation of T258/S259 could lead in changes in N- and C-terminus conformations, either directly or through protein interactions, one or more of which is sufficient to recruit key endocytic proteins that drive transporter internalization.
4. Protein Kinase C Regulation of Norepinephrine Transporter Protein-Protein Interactions.
As with DAT, NET interacts with a number of cytosolic and membrane proteins, including PP2A-Ar and c (Bauman et al., 2000; Sung et al., 2005), PICK-1 (Torres et al., 2001), Hic-5 (Carneiro et al., 2002), a-synuclein (Wersinger et al., 2006), and syntaxin 1A (Sung et al., 2003) as well as with the NK1R receptor (Arapulisamy et al., 2013). To our knowledge, the interactions of NET with PP2A subunits, as well as PICK-1, Hic-5, and a-synuclein, have not been evaluated for sensitivity to PKC activation, although a number of these proteins demonstrate published structural or functional interactions with one or more PKC isoforms. Syntaxin 1A/NET interactions have been shown to be sensitive to PKC activation (Sung et al., 2003). In cultured mouse sympathetic neurons, surface NET proteins were found to colocalize with syntaxin 1A, with colocalization most evident at varicosities. A similar pattern was observed in rat vas deferens, suggesting preferential targeting of the transporter near sites of NE release and syntaxin 1A pools involved in vesicular fusion (Sung et al., 2003). With the vas deferens preparation, these authors demonstrated coimmunoprecipitation of NET/syntaxin 1A complexes that was diminished after acute treatments with β-PMA. Additionally, cleavage of syntaxin 1A with BoNT/C1, which reduced NET activity on its own, abolished the ability of β-PMA to reduce NE uptake. Follow-up studies in transfected CHO and CAD cells revealed that β-PMA treatments also reduced the recovery of surface NET/syntaxin 1A complexes. In vitro studies support a direct interaction of the syntaxin 1A cytoplasmic domain with the NET N terminus, and deletion of residues 2–42 in intact NET precluded effects of β-PMA in reducing surface transporter levels, suggesting that either syntaxin 1A associations are required to localize NET to domains competent for PKC-dependent NET endocytosis or the physical association of the two proteins is an intrinsic requirement for recruitment of endocytic machinery. Interestingly, although syntaxin 1A overexpression increased NET surface levels in these studies, it did not increase NE uptake, suggesting that the additional NET at the surface was nonfunctional. Further studies revealed that overexpression of syntaxin 1A in HEK 293 cells expressing hNET results in a reduction in NE-activated NET single channel currents, supporting the idea that this interaction between syntaxin 1A and NET causes a reduction in NET activity, despite driving enhanced NET at the surface. This suggests that the regulated interaction between these two proteins by PKC likely has trafficking-dependent and -independent components. Although it is important to reflect seriously on the heterologous nature of these experiments, consideration should also be given to a dynamic interplay of the NE transport and release machinery, mediated through syntaxin 1A interactions, and the possibility that NET membrane insertion may have properties in common with, or coordinated with, NE release. Interestingly, AMPH also triggers hNET endocytosis in transfected CAD cells (Dipace et al., 2007); however, these effects arise in the context of enhanced, rather than reduced syntaxin 1A/NET associations, as seen with β-PMA treatments. As we note below, both trafficking and changes in NET/syntaxin 1A associations after AMPH treatments derive not from PKC activation, although they are Ca2+ dependent, but from CaMKII-linked pathways. These findings are reminiscent of the PKC independence of DAT trafficking after AMPH treatments (Boudanova et al., 2008). Additional studies are needed to understand whether distinct microdomains support both PKC and AMPH-induced NET downregulation or whether distinct signaling pathways converge on a common, transporter endocytosis-competent compartment.
5. Receptor-Initiated Protein Kinase C Regulation of Norepinephrine Transporter.
The potential for regulation of NET by endogenous signaling pathways dates to early findings of the acute effects of intraventricular injections of angiotensin to diminish CNS NE uptake (Palaic and Khairallah, 1967), although tools at the time did not afford further analysis of NE clearance. Although angiotensin II acting through the AT1 receptor induces PKC-dependent regulation of NET transcription in hypothalamus-brain stem neurons (Lu et al., 1996) more rapidly, the receptor induces PKC-independent increases in NE uptake that have been found in other studies to arise from elevations in NET surface expression (Savchenko et al., 2003) These effects are consistent with the general inhibitory effects of PKC activation on transporter activity and trafficking.
Rapid NET regulation by acetylcholine (ACh) has been linked to PKC-linked pathways. Early studies by Role and Perlman (1983) uncovered evidence for ACh downregulation of NE uptake Vmax by adrenal chromaffin cells, although the mechanism proposed involved a change in membrane potential, certainly reasonable given the electrogenic nature of the transporter (Galli et al., 1995, 1998). Later work by the Blakely laboratory (Apparsundaram et al., 1998a) demonstrated that muscarinic ACh receptors (likely M3 subtype) reduce the activity (Vmax) and surface expression of NET (surface [3H]nisoxetine binding) in the noradrenergic neuroblastoma SK-N-SH, effects blocked by coincubation with staurosporine and BIM-I, paralleling effects seen with β-PMA (Apparsundaram et al., 1998b). This regulation of NET by muscarinic ACh receptors may involve regulating NET/syntaxin 1A interactions, because treatment of M3-CHO cells with muscarinic agonists methacholine and carachol decreased the interaction between NET and syntaxin 1A, effects that are also seen with β-PMA (Sung et al., 2003). Although these studies are not informative with respect to subtypes of PKC involved in muscarinic receptor effects, NET alterations were abolished in cells treated with the intracellular Ca2+ chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), suggesting an important contribution of a cPKC isoform. Importantly, NET downregulation was insensitive to CaMKII-, NOS-, or PKA-linked pathways. Interestingly, chronic treatment of cells with β-PMA to downregulate PKCs eliminated the acute ability of β-PMA to diminish NET activity, whereas a portion of muscarinic receptor regulation of NET was retained, suggesting more complexity in receptor-dependent mechanisms than seen with phorbol ester treatments.
A role for PKC may also be involved in the regulation of NE uptake by the endothelins ET-1 and ET-3, with evidence arising through studies of rat hypothalamic ETB receptors (Hope et al., 2010). These investigators found that activation of ETB receptors by these peptides decreased NE uptake effects blocked by the PKC inhibitor GF-109203X [2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide] (Hope et al., 2008). However, the PKA inhibitor H-89, as well as the NOS inhibitors 7-Nitroindazole and L-NG-Nitroarginine methyl ester also blocked these effects, suggesting the involvement multiple pathways in the control of NET activity. PKC may also mediate the inhibitory effects of IFN-α on NET activity (Toyohira et al., 1998). In bovine adrenal medullary cells, IFN-α decreases the Vmax of NE uptake, and 48-hour treatment of these cells with β-PMA to induce downregulation of PKC leads to a blunting of these effects. Interestingly, IFN-α also induces a translocation of PKC from soluble to particulate fractions in a manner similar to β-PMA treatment. Unfortunately, more conventional methods of PKC inhibition have not been used to further study the actions of IFN-α on NET, so the true nature of the contribution of PKC to these effects is unclear.
A comprehensive study of NET regulation by substance P provides strong evidence for PKC-dependent alterations in transporter membrane raft localization (Arapulisamy et al., 2013). This work revealed that when HTR cells expressing hNET and the NK1 receptor were treated with substance P, both NET and NK1R surface levels decreased. Additionally, NET and NK1R could be captured in a complex at the cell surface basally, but in intracellular compartments after substance P treatment. Biochemical studies indicated that surface complexes were enriched in non-raft fractions but moved to raft fractions after substance P treatment. Importantly, PKCα was found to associate with NET in raft fractions after substance P treatment, where NET phosphorylation and subsequent internalization may be initiated. Why NET seems to move into rafts after substance P treatment but seems to do the opposite after β-PMA may be due to the combined effects of multiple PKC isoforms activated by β-PMA, whereas substance P may have more selective effects. Additionally, substance P may activate other signaling pathways that interact with PKC signaling to give rise to its effects. Regardless, these studies provide the best evidence that raft and non-raft membrane elements provide differential contexts for PKC-dependent modulation of NET by cell surface receptors. Importantly, the NET S258A/T259A double mutant that precludes PKC- and substance P-triggered NET endocytosis neither moves into rafts nor interacts with PKCα after substance P treatment. These findings provide critical evidence that PKC signaling and phosphorylation of NET, leading to transporter mobilization and functional downregulation, operate under physiologic conditions. Possibly these studies have more general relevance to reports of NET regulation by other PKC-linked receptors [e.g., H3 histamine (Imamura et al., 1996)] and a useful example as receptor control of other presynaptic transporters is evaluated.
C. Regulation of Serotonin Transporter by Protein Kinase C
1. Regulation of Serotonin Transporter Activity by Protein Kinase C Isoforms.
Research using native preparations and cell cultures concur that, as with DAT and NET, acute treatments with phorbol esters decrease 5-HT uptake, with a consistent reduction in 5-HT transport Vmax observed (Table 3). With the cloning of SERT cDNAs and development of SERT antibodies, it became possible to move beyond studies of radiotracer flux measurements of PKC regulation of SERT. Thus, using human SERT-transfected HEK-293 cells and SERT antibody-based biotinylation approaches, Qian et al. (1997) were able to provide direct evidence that surface expression of SERT protein was reduced in parallel with reduced 5-HT uptake and 5-HT induced inward currents after β-PMA treatments. In studies of rat platelet SERT regulation after PKC activation, Jayanthi et al. (2005) obtained evidence that SERT displays trafficking-independent modes of regulation. These authors found that when platelets were treated with β-PMA, 5-HT transport activity displays an early (5 minutes) increase in the 5-HT KM and no change in Vmax. These findings, suggesting a shift of surface transporters to less active conformations, concurred with biotinylation studies that revealed no change in surface SERT. With longer treatments (30 minutes or longer), a change in 5-HT transport Vmax predominates, and this alteration is accompanied by cell surface redistribution of transporters. Interestingly, decreases in the affinity for 5-HT have been reported for human platelets from OCD subjects treated with TPA, an effect that was not observed in platelets from control individuals (Marazziti et al., 2000). Although saturation kinetic studies were not performed, Prasad and colleagues reported that multiple naturally occurring, rare human SERT coding variants display greater sensitivity for uptake inhibition after treatment with β-PMA (Prasad et al., 2005). Two of these variants, Gly56Ala and Lys605Asn, located in SERT cytoplasmic domains, were subsequently uncovered in a screen of SERT in subjects with ASD (Sutcliffe et al., 2005). These findings suggest that SERT may reside in conformations more sensitive to PKC regulation (and other kinases; see below) in these disorders, the physical basis of which requires further examination.
Regulation of SERT by protein kinases
In contrast to efforts taken to explore PKC isoform selectivity in the regulation of DAT and NET, this issue has received little attention with respect to SERT regulation and remains an important area for future investigations. Ramamoorthy's group (Samuvel et al., 2005) reported that calcium chelation eliminated β-PMA-induced downregulation of SERT in midbrain synaptosomes, suggesting effects are mediated through one or more cPKC isoforms. Regulation of SERT by PKC also appears to be dependent on SERT conformation or activity state, because concurrent incubation with 5-HT (or other SERT substrates) blunts phorbol ester-induced 5-HT uptake reductions and transporter internalization in transfected HEK-293 cells (Ramamoorthy and Blakely, 1999). Because this action of 5-HT appears to require transport coincident with PKC activation, the mechanisms engaged after kinase activation to downregulate SERT appear to engage distinct transporter conformations, presumably driven by activity-dependent exposure of phosphorylation sites or binding sites for regulatory proteins.
As mentioned earlier, PKC may also play a role in regulating AMPH-evoked reverse transport through MA transporters. With regards to SERT, Sucic et al. (2010) showed that the more SERT-selective AMPH para-chloroamphetamine (PCA) induced significant 5-HT efflux in HEK-293 cells stably expressing SERT, but that mutation of the PKC consensus site Thr81 to either Ala or Asp abolished this efflux. Whether this residue is in fact a target for PKC in vivo remains to be determined, but its importance for SERT (and likely DAT and NET) conformational equilibrium was supported by molecular dynamics simulations that suggest that Thr81 mutants favor inward facing conformations. This hypothesis was further supported by an increased affinity of Thr81Ala SERT for ibogaine, which binds preferably to the inward facing conformation, and a decreased affinity for imipramine, which prefers the outward facing conformation. Additionally, FRET between the N and C termini in Thr81Ala SERT (using fluorophores attached to each terminus) was significantly reduced compared with wild-type SERT, consistent with this mutant favoring an inward conformation. Why this favoring of the inward facing conformation would abolish PCA-evoked 5-HT efflux is unclear, but the authors suggest another important role for the N terminus that might be regulated by phosphorylation of Thr81 or other N-terminal residues by PKC or other kinases. The authors fused the SERT N terminus to the membrane by attaching a Tac peptide to the N terminus and showed that PCA-evoked 5-HT efflux was abolished despite normal 5-HT uptake. This led the authors to conclude that flexibility of the N terminus is essential for AMPH actions on SERT (and likely DAT and NET) to induce reverse transport of substrate. Interestingly, interaction of the N terminus with IL4 of the related GABA transporter GAT1 has been shown to regulate both forward and reverse transport of GABA, supporting a role for flexibility of the N terminus in regulating the activity of SLC6 transporters (Hansra et al., 2004). Whether this flexibility is regulated through phosphorylation of residues such as Thr81 will be important to explore.
2. Regulation of Serotonin Transporter Membrane Compartmentalization and Trafficking by Protein Kinase C.
As noted above, studies with native and transfected preparations demonstrate a redistribution of cell-surface SERT after PKC activation, particularly after prolonged stimulation (Qian et al., 1997; Jayanthi et al., 2005). Kinetic trafficking analysis of SERT mobilization in transfected HEK-293 cells revealed that net relocation in this model arises from an increased rate of transporter internalization and not through a decrease in recycling of SERT to the surface (Samuvel et al., 2005). In the same report, this group reported that SERT in midbrain synaptosomes is localized to lipid raft microdomains and that phorbol ester treatment (30 minutes) results in a redistribution of transporters to these compartments, as assessed through sucrose gradient fractionation. Blakely’s group (Carneiro and Blakely, 2006) studied SERT membrane compartmentalization and regulation by PKC in human platelets. In resting platelets, SERT was found to be predominantly localized to Triton-soluble, intracellular membranes, although upon plating on collagen, SERT is recruited to plasma membrane focal adhesions labeled by phalloidin (Steiner et al., 2008). Surface-resident, platelet SERT proteins are largely present in a “membrane skeleton” (MS) fraction that is rich in focal adhesion proteins. Upon treatment with β-PMA, SERT translocates out of the MS to a distinct membrane fraction that is enriched for F-actin and nonbiotinylateable, suggesting an internalized compartment enmeshed in cytoskeletal elements driving endocytosis. These investigators also found that exogenous 5-HT elevates SERT abundance in the MS fraction. The SSRI citalopram, although not impacting SERT distribution on its own, completely blocked the ability of 5-HT to elevate SERT levels in the MS. Additionally, 5-HT blunted the ability of β-PMA to induce SERT relocation away from the MS. Together with the previously mentioned ability of 5-HT to attenuate PKC-dependent SERT internalization in transfected cells (Ramamoorthy and Blakely, 1999), these findings raise the possibility of changes in protein associations that are reciprocally influenced by SERT activity and PKC activation and that ultimately negotiate the surface residence time of these transporters.
3. Regulation of Serotonin Transporter Phosphorylation by Protein Kinase C.
Our group was the first to report the ability of protein kinase activators, including β-PMA, to result in SERT phosphorylation (Ramamoorthy et al., 1998; Jayanthi et al., 2005; Samuvel et al., 2005). Using orthophosphate metabolic labeling and immunoprecipitation methods with human SERT stably transfected HEK-293 cells, we detected a time- and dose-dependent elevation of basal SERT phosphorylation after β-PMA treatments that could be blocked by cPKC inhibitors. Similar effects have since been obtained in native preparations including platelets (Jayanthi et al., 2005) and synaptosomes (Samuvel et al., 2005). Using somewhat different approaches, Aguilera’s group also reported the ability of phorbol esters to induce SERT phosphorylation in synaptosomes (Najib et al., 2000; Gil et al., 2003). As with SERT trafficking and uptake modulation, PKC-dependent SERT phosphorylation in transfected cells was found to be sensitive to external 5-HT application. The IC50 for 5-HT attenuation of SERT phosphorylation induced by β-PMA was lower (∼70 nM) than the 5-HT transport KM, suggesting that although the 5-HT effect is mediated via interactions with SERT, 5-HT binding may stabilize a conformation with higher affinity than the ensemble of 5-HT bound states subserved by the 5-HT KM. We hypothesize that 5-HT may bias the transporter toward an open-out, high-affinity conformation, whereas PKC phosphorylation of SERT moves the transporter to a low affinity conformation, consistent with the elevated KM observed by Jayanthi et al. (2004a) with short β-PMA treatments.
Although there are strong correlations between SERT phosphorylation, loss of SERT activity and internalization, with respect to β-PMA dose and time dependence (Ramamoorthy et al., 1998; Jayanthi et al., 2005; Samuvel et al., 2005), and their sensitivity to 5-HT attenuation (Ramamoorthy and Blakely, 1999), there is still no direct evidence to support a requirement of SERT phosphorylation by PKC-induced changes in activity or trafficking. To date, no one has reported evidence that verifies a specific site(s) as responsible for in situ transporter phosphorylation by PKC. We reported initial evidence for the ability of GST fusion proteins of rat SERT N and C termini to be phosphorylated by purified PKCα in vitro, although specific sites were never thoroughly interrogated (Qian et al., 1995). Recently, Sorensen et al. (2014) used liquid chromatography tandem mass spectrometry approaches to identify and quantify in vitro phosphorylation of SERT-derived peptides incubated with purified kinases. These investigators reported labeling of Ser149, Ser277, and Thr603 by PKC in vitro. Although no evidence was provided in this study that these sites were phosphorylated on the intact SERT protein, the investigators found that a T276A/S277A double mutant prevented the decrease in SERT activity after a short (5 minutes) β-PMA treatment. Given its location on a short intracellular loop between TM4 and 5, PKC phosphorylation of the Ser277 residue could mediate the early, trafficking-independent effects of PKC. It should be pointed out that the equivalent residues in NET (T258/S259) are required for PKC-mediated internalization of this transporter (Jayanthi et al., 2006) as discussed earlier, suggesting a conserved role of these residues in mediating PKC-dependent regulation of monoamine transporters. Importantly, in the Sorensen study, mutation of any of the three PKC target SERT residues alone or in combination had no effect on the long-term effects of PKC on SERT, suggesting that the trafficking-dependent regulation of SERT may involve phosphorylation at other sites or be phosphorylation independent. That the former possibility may be involved can perhaps be gleaned from the studies of Jayanthi and colleagues who found a change in the SERT phosphoamino acid pattern over time after β-PMA treatments, with brief time points (5 minutes) that coincide with a KM change associated with Ser labeling versus longer time points (30 minutes) that coincide with a Vmax change associated with Thr labeling. With respect to a phosphorylation-independent impact of PKC on SERT endocytosis, Sakai et al. (1997) were unable to preclude SERT Vmax reductions by phorbol ester treatments of transfected COS-7 cells via mutation of canonical PKC sites, Ser8, Ser13, Ser277, Thr603, or Thr613, individually and in various combinations. Because phosphorylation per se was not assessed, we do not know whether these argue strongly against a connection of transporter phosphorylation to trafficking after PKC activation, although this certainly seems a possibility. If so, the Thr phosphorylation observed by Jayanthi et al. in platelets with longer β-PMA treatments could reflect a postendocytosis modification involved in cytoplasmic transporter routing or recycling.
4. Protein Kinase C Regulation of Serotonin Transporter Protein-Protein Interactions.
In considering the possible PKC-dependent mechanisms that may support SERT trafficking and/or activity, a consistent theme that emerges is the likelihood that targets of PKC activation influencing transporter distribution or activity may do so by altering interactions of SERT with the cytoskeleton. Thus a number of PKC-sensitive SERT protein associations involve proteins that interact with the actin cytoskeleton, with a prominent role suggested for SERT C-terminal interactions. The C terminus is intriguing because its expression in HEK-293 cells results in localization with cortical actin and can drive a reduction in SERT activity (Mochizuki et al., 2005). For example, Jess et al. (2002), via a yeast two-hybrid screen with the rat SERT C terminus, identified the protein MacMARCKS. MARCKS family proteins reversibly cycle between plasma membrane and endosomal membranes and are bona fide PKC substrates (Allen and Aderem, 1995). Although the authors could not validate a physical association of SERT with MacMARKS biochemically, they were able to demonstrate colocalization via confocal imaging approaches. Moreover, coexpression of MacMARCKS with SERT resulted in a ∼30% decrease in the Vmax of 5-HT uptake, an effect that was nonadditive with β-PMA effects, and staurosporine reversed this reduction up to control levels. Might MacMARCKS therefore be an intermediary in PKC-dependent SERT regulation? Some effects of overexpression of MacMARKS could be indirectly driven by changes in the actin cytoskeleton. However, although pharmacological disruption of actin polymerization with actinomycin D can reduce SERT Vmax and this loss of activity is reversed by actin stabilizing agents (Sakai et al., 2000), such a manipulation does not attenuate phorbol ester-induced SERT activity reductions. Additional studies are needed to determine whether MacMARCKS interactions with SERT are sensitive to PKC activation, whether MacMARCKS mutants disabled for PKC phosphorylation differentially impact SERT trafficking and function, and whether SERT C-terminal mutations can be developed to identify that selectively disrupt MacMARCKS interactions.
The SERT C terminus has also been implicated in the interactions platelet SERT makes with the focal adhesion protein Hic-5 in human platelets (Carneiro and Blakely, 2006). Hic-5 interactions with DAT were identified via a yeast two-hybrid screen and were subsequently found to extend to NET and SERT as well (Carneiro et al., 2002). Importantly, SERT:Hic-5 interactions are increased upon PKC activation by β-PMA over a time course that coincides with the time course of internalization of SERT, with both SERT and Hic-5 moving to F-actin enriched CS fractions. Interestingly, resealing platelet plasma membranes with in vitro transcribed Hic-5 resulted in a decrease in 5-HT uptake, suggesting a SERT-Hic-5 interaction may participate in both trafficking-dependent and -independent modes of PKC-dependent SERT regulation. Additional studies are needed to explore the significance of PKC-sensitive SERT/Hic-5 interactions in a neuronal context—Hic-5 is expressed in brain and localizes to synaptic terminals—and to consider whether aspects of PKC regulation of DAT/NET also involve modulation of Hic-5 associations.
The SERT C terminus was recently found to recruit the catalytic and regulatory subunits of the Ser/Thr phosphatase calcineurin, also known as PP2b, by studies initiated through GST-C terminal fusion pull down of brain extracts and subsequently confirmed in coimmunoprecipitation studies (Seimandi et al., 2013). Studies mapping the binding domain for calcineurin are consistent with a juxtamembrane area in proximity to or overlapping that likely to interact with Hic-5 (Carneiro et al., 2002). Importantly, phorbol ester-induced reductions in SERT activity are abrogated in SERT expressing HEK-293 cells transfected to also overexpress calcineurin. Interestingly, these investigators identified a peptide sequence from the N terminus (residues 30–60) that displayed phorbol ester-induced phosphorylation that was reduced in calcineurin transfected cells. Unfortunately, the exact site(s) of labeling were not identified and multiple potential Ser/Thr phosphorylation sites are present. Nonetheless, the investigators were able to capitalize on an inducible transgenic mouse model that results in adult-specific overexpression of calcineurin A. Although the transgenic manipulation was designed to deliver excess calcineuron (or an inhibitor) to forebrain neurons and not 5-HT neurons, and thus indirect effects may account for the findings, SERT activity was found to be elevated and animals displayed less depressive behavior. Together, these very interesting studies draw further attention to the physiologic importance of the mechanisms that control SERT regulation by PKC and other kinases. Given that experimental elevations in intracellular calcium increase SERT/calcineurin interactions, it would be interesting to evaluate whether PKC activation influences phosphatase interactions and whether external 5-HT modulates calcineurin associations. Calcineurin is not the only phosphatase reported to associate with SERT. Bauman et al. (2000) reported an association of SERT with the catalytic subunit of PP2A, finding that the interaction was stabilized by external 5-HT and reduced by β-PMA treatments.
The SERT N terminus was also reported to interact with multiple proteins. Of these, only one to date, syntaxin 1A, has been reported to be influenced by PKC activation (Samuvel et al., 2005). PKC-dependent interactions of syntaxin 1A are shared features of all three monoamine transporters. Quick (2003) demonstrated that presence or absence of syntaxin 1A dictated whether the 5-HT transport cycle is electrogenic or electroneutral, suggesting that PKC modulation of SERT/syntaxin 1A interactions could influence nerve terminal excitability or the degree to which membrane potential influences the 5-HT transport process.
III. cAMP-Dependent Protein Kinase/Protein Kinase A—Overview
Phosphorylation of proteins, including transporters, by PKA is common and important consequence of cAMP production by adenylyl cyclase, commonly activated by Gα-coupled G-protein receptors (GPCR) (Tasken and Aandahl, 2004). cAMP binds to the regulatory subunits of PKA, freeing the catalytic subunits to phosphorylate their targets (Sunahara and Taussig, 2002). Some adenylyl cyclases are activated by a rise in intracellular calcium, affording an indirect mechanism by which intracellular calcium elevations can lead to PKA activation (MacNeil et al., 1985). GPCRs that couple to Gαi inhibit the production of cAMP and thereby can reduce the capacity for PKA activation (Taylor et al., 2005). Ultimately, phosphodiesterase (PDE) proteins hydrolyze cAMP to restore the capacity of PKA regulatory proteins to sequester catalytic subunits. Together, these influences provide multiple opportunities by to regulate PKA and link its activity to external signals and cell activation state.
A. Regulation of Dopamine Transporter by Protein Kinase A
1. Regulation of Dopamine Transporter Activity by Protein Kinase A.
Far fewer studies have evaluated the role played by PKA in regulation of monoamine transporters, including DAT, than for PKC isoforms, and the results have been rather mixed. With respect to DAT activity, PKA has been suggested to have no effect, stimulatory or inhibitory. Initially, Tian et al. (1994) reported that that, in contrast to GABA uptake in rat striatal synaptosomes, DA uptake is insensitive to adenylyl cyclase or PKA activating agents forskolin or 8-Bromo-cAMP (8-Br-cAMP). Similar conclusions were reached by Copeland et al. (1996) as well as Zhu et al. (1997) and Daniels and Amara (1999) using transfected MDCK cells. By using rotating disk voltammetry to achieve higher time resolution for potential DAT modulation, Batchelor and Schenk (1998) reported that DA uptake in rat striatal suspensions can be stimulated by 8-Br-cAMP and forskolin at 1 minute but not at 12 minutes, with effects blocked by a PKA inhibitor and attributed to a Vmax elevation. Adding further complication, studies with DAT-transfected Sf9 and COS-7 cells revealed in these models a simulation of DA transport capacity (Vmax) with PKA inhibition by Rp-cAMPS [(R)-Adenosine, cyclic 3′,5′-(hydrogenphosphorothioate)] but no effect of PKA stimulatory agents—Sp-cAMPS [(S)-Adenosine, cyclic 3′,5′-(hydrogenphosphorothioate)] or 8-Br-cAMP (Pristupa et al., 1998)—consistent with tonic, PKA-dependent inhibitory control of DAT in these models. However, Page et al. (2004) found that activation of PKA by 8-Br-cAMP stimulated DA uptake in rat striatal synaptosomes. This increase in uptake was blocked by inhibitors of both PKA and CaMKII, which may function downstream of PKA in this regulation (further discussion of CaMKII below). These authors also used very brief treatments of pharmacological agents, suggesting effects may as suggested by others be highly time dependent. Overall though, these issues have yet to be resolved and may in part derive from species differences, tissue and cell preparation variations, and mode of drug application. Also, for native preparation studies, consideration should be given for possible heterogeneity in DA terminal populations, particularly dorsal versus ventral striatum, as well as trauma induced with tissue harvest/fractionation.
2. Regulation of Dopamine Transporter Membrane Compartmentalization and Trafficking by Protein Kinase A.
Few studies have provided evidence of trafficking pathways connected to DAT regulation by PKA signaling. Pristupa et al. (1998) monitored the localization of human DAT expressed in Sf9 cells after acute treatment with the PKA antagonist Rp-cAMPS, observing an increase in plasma membrane associated labeling, consistent with their finding of elevated DA transport activity with this agent and tonic PKA-dependent inhibitory tone on DAT trafficking. No effects were seen with the PKA activator Sp-cAMPS, however, suggesting a high basal PKA activity in this model that could preclude further DAT regulation. Recent work from the Amara laboratory has implicated PKA signaling in the regulation of Rho-mediated DAT internalization, specifically in response to AMPH treatment (Wheeler et al., 2015). In both SK-N-SH cells as well as rat primary DA neurons, dibutyryl-cAMP and the PKA inhibitor KT5720 [(9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid hexyl ester] blocked and enhanced AMPH-stimulated reduction in DA uptake/DAT surface levels, respectively. Additionally, agonism of the Gαs-coupled D1/D5 and β2 receptors, and thereby activation of PKA, also blocked the effects of AMPH on DAT activity/surface levels. Whether this PKA regulation of DAT plays a role in regulating Rho-mediated internalization of DAT apart from these AMPH effects remains to be seen. No studies have as yet reported an influence of PKA pathways on DAT localization to membrane microdomains.
3. Regulation of Dopamine Transporter Phosphorylation by Protein Kinase A.
Vaughan et al. (1997) found no ability of forskolin or 8-Br-cAMP to elevate basal DAT phosphorylation in metabolically labeled rat striatal synaptosomes (or to modulate DA uptake), whereas as noted above, β-PMA treatments caused a robust elevation in phosphorylation. Similarly, the PKA inhibitor H89 (N-[2-[[3-(4-Bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide) had no impact of DA uptake or on basal phosphorylation of rat DAT in metabolically labeled LLC-PK1 cells (Cervinski et al., 2005). Although these studies support a general lack of regulation of DAT by PKA-linked pathways, Vaughan’s group reported the ability of the purified DAT N terminus to be phosphorylated by PKA in vitro (Gorentla et al., 2009) and for phosphopeptides to match peptides labeled in synaptosomes labeled by combined phorbol ester/okadaic acid treatments, with mutation studies consistent with targeting of Ser7 (Moritz et al., 2013). The evidence presented by the authors that Ser7 can influence conformational equilibria means that only if DAT is assessed with measures sensitive to such changes of structure can the influence of this site be detected. These studies leave open the possibility that PKA, directly or indirectly, may modulate DAT, which could include transporter phosphorylation, although the physiologically relevant context may not yet have been identified. Given the possibility of conformational state modulation, further insights into potential significance of PKA pathways may come from studies of signaling modulation of DAT protein complexes, particularly interactions mediated by the DAT N-terminus.
4. Receptor-Initiated Protein Kinase A Regulation of Dopamine Transporter.
The most intimate connection between a cell surface receptor and DAT involves the presynaptic DA D2 receptor (D2R), which has reported both to interact physically with DAT (Lee et al., 2007) and to modulate DAT trafficking and function (Meiergerd et al., 1993). D2Rs are Gαi-coupled receptors, and their ability to attenuate cAMP production and PKA activation could provide a mechanism to initiate PKA-dependent regulation of DAT. In this regard, Schenk’s group (Meiergerd et al., 1993; Batchelor and Schenk, 1998), using rotating disk voltammetry reported a rapid (30 seconds) ability of the D2R agonist quinpirole to enhance DAT-mediated DA clearance in rat striatal minces, an effect blocked by the D2R antagonists sulpiride as well as the PKA inhibitor H89. The authors also showed that systemic D2 antagonist haloperidol could slow the clearance of DA evoked by K+ stimulation in vivo (Meiergerd et al., 1993). Cass and Gerhardt (1994), using chronoamperometry techniques, also provided evidence for in vivo D2R regulation of DAT activity. The opposing effects of H89 on D2R activation in the in vitro Schenk studies are puzzling if one assumes the effects are via PKA antagonism, given that D2Rs should act to diminish PKA activation. H89 also appears to have antagonistic activity on DAT activity, in these studies, on its own. As such, the D2R connection to DAT may involve other effectors. Using voltage-clamped Xenopus laevis oocytes expressing DAT and D2Rs, Mayfield and Zahniser (2001) demonstrated that D2R regulation of DAT is voltage independent, ruling out an effect mediated indirectly by modulation of ion gradients. Moreover, these authors demonstrated that the D2R-mediated increase in DAT activity was pertussis toxin sensitive (hence Gαi dependent) and supported by elevated DAT surface expression. Because G proteins are heterotrimers, pertussis toxin sensitivity could mean as much a role for signaling by the βγ subunits as the a subunit. Indeed, evidence has emerged to implicate a PKCβ-ERK pathway, activated by bg after D2R activation, as opposed to attenuation of PKA signaling (Moron et al., 2003; Bolan et al., 2007; Chen et al., 2013). MAPK pathways regulating DAT are further discussed below. Whether attenuation of PKA signaling is a parallel determinant, acting in concert with the PKCβ-ERK pathway, remains worth considering. DA D3 receptors (D3R), also Gαi coupled, are expressed by DA projections to the nucleus accumbens and have been reported to regulate DAT (Zapata and Shippenberg, 2002; Zapata et al., 2007), suggested to be through pathways like that of presynaptic D2Rs. In summary, although evidence has been advanced over the years in consideration of PKA-dependent DAT modulation, such a pathway, if it exists, remains poorly understood.
B. Regulation of Norepinephrine Transport by Protein Kinase A
1. Regulation of Norepinephrine Transport Activity by Protein Kinase A.
Bunn et al. (1992), following up observations of reduction of NE transport by treatment in bovine cultured adrenal chromaffin cells with overnight pertussis toxin, examined the more immediate impact (15 minutes) of cell treatments with 8-Br-cAMP, observing a reduction in uptake activity in both control and pertussis toxin-treated cells. Forskolin treatments, regardless of pertussis toxin treatment, yielded biphasic effects, with stimulation of uptake at lower concentrations. Apparsundaram et al. (1998a) examined the sensitivity of human NET expressed by SK-N-SH cells to acute pharmacological manipulations of PKA signaling (e.g., forskolin, 8-BrcGMP, Rp-cAMPS) and found no alterations in NE transport activity. Similar findings were obtained using the same cell model by Bönisch et al. (1998), although they observed a robust decrease in uptake in the adrenal chromaffin cell-derived PC-12 cells with these treatments. Whether the presence of cAMP sensitivity in the bovine chromaffin and PC-12 cells versus SK-N-SH cells reflects a species difference or a difference between adrenal chromaffin cells and more “neuron-like” cells is unclear. Owing to the paucity of studies supporting a consistent role for PKA contributions to NET regulation (or a lack of effects), studies examining the kinase in modulation of NET phosphorylation or regulation of NET protein interactions are lacking.
2. Receptor-Initiated Protein Kinase A Regulation of Norepinephrine Transporter.
α2 Adrenergic receptors act as NE autoreceptors on noradrenergic terminals to regulate feedback control of NE release and are Gαi-coupled (Starke, 2001). There is an expectation, therefore, that these receptors should regulate NET, by analogy with D2 and D3 DA autoreceptors that regulate DAT trafficking and function. However, Callado and Stamford (2000) were unable to detect an influence on endogenous NE clearance assessed by fast cyclic voltammetry of the α2 antagonist BRL 4408 (2-[2H-(1-methyl-1,3-dihydroisoindole) methyl]-4,5-dihydroimidazole) was applied to locus ceruleus brain slices. With respect to heteroreceptors, Hope and colleagues obtained evidence for cAMP-dependent modulation of NE uptake in hypothalamic preparations downstream of endothelin 1 and 3 receptors (Hope et al., 2008, 2010; Vatta et al., 2015), effects likely mediated by changes in NET trafficking. These receptors however do not couple directly to cAMP production, and thus the cAMP dependence of effects are likely indirect. Altogether, studies with pharmacological manipulations have hinted at a potential role of cAMP/PKA signaling in acute NET regulation, although more detailed and precise measures are needed, particularly with respect to potential PKA targeting of NET or NET-associated proteins, to have confidence of an important role of this pathway. No in vivo studies to examine a contribution of PKA-dependent mechanisms in NET-mediated NE clearance have been reported.
C. Regulation of Serotonin Transporter by Protein Kinase A
1. Regulation of Serotonin Transporter Activity by Protein Kinase A.
As with DAT and NET, investigations of PKA-dependent modulation of SERT are few in number. Cool et al. (1991) showed that SERT activity in human placental JAR cells is elevated by the cAMP-elevating agents cholera toxin, db-cAMP, and forskolin. Although the effect of cholera toxin could be blocked by the PKA inhibitor H-9, SERT activity elevations require chronic drug exposure with the first effects evident at ∼8–16 h after treatment. Later work demonstrated that these treatments elevate SERT steady-state mRNA levels (Ramamoorthy et al., 1993), suggesting a transcriptional regulation of SERT downstream of PKA activation, findings confirmed in later transcriptional reporter studies (Heils et al., 1995). Others have sense detected cAMP stimulation of SERT mRNA expression in 5-HT neurons (Rumajogee et al., 2002). PKA-dependent changes in gene expression explain the elevated 5-HT Vmax seen in the Cool et al. study after treatment of JAR cells with cAMP elevating agents, as well as the increased density of [125I]RTI-55 binding sites in the Ramamoorthy et al. study. Interestingly, although of unknown significance, Cools et al. also detected a significant reduction in the 5-HT KM. Possibly, other PKA-dependent changes in SERT that impact transporter conformational states such as alterations in SERT protein associations or the JAR membrane environment occur as a result of PKA activation, but are largely obscured by the pronounced changes seen in elevated SERT mRNA and protein expression. Evidence of such a nontranscriptional effect of chronic cAMP elevations comes from studies of Yammamoto et al. (2013) of transfected SERT in serotonergic RN46A cells who subjected cells to chronic treatments with dbcAMP or forskolin and generated evidence for an increase in SERT protein levels that arose from a reduced rate of transporter degradation, possibly as a result of a change in SERT protein or its associations/localization. SERT is found in multiple cell types in the intestine, and SERT activity, RNA, and protein can be detected in human Caco-2 cells, an intestinal epithelial cell model (Martel et al., 2003; Iceta et al., 2006). Studies in this model have yielded evidence of rapid (30 minutes) inhibitory effects of db-cAMP or forskolin treatments (Iceta et al., 2006), although PKA dependence was not assessed. With respect to brain preparations, Awtry et al. (2006) came to opposite conclusions, reporting that 15-minute treatment of rat prefrontal cortical synaptosomes with the PKA activator Sp-cAMPS and the PKA inhibitor Rp-cAMPS robustly increased and decreased 5-HT uptake, respectively. Together, these studies with second messenger modulation and pharmacological manipulation of PKA signaling suggest that SERT is likely under regulation by PKA with mechanisms dependent on cell context and that occur at a number of levels, including changes in both transcription and post-transcriptional mechanisms.
2. Regulation of Serotonin Transporter Membrane Compartmentalization and Trafficking by Protein Kinase A.
As noted above, SERT protein levels, surface expression, and activity have been reported to be elevated by chronic PKA activation, but as of yet, influences of acute cAMP elevations on SERT trafficking or compartmentalization are unknown and represent an interesting direction for future studies.
3. Regulation of Serotonin Transporter Phosphorylation by Protein Kinase A.
In studies of metabolically labeled HEK-293 cells transfected with human SERT, Ramamoorthy et al. (1998) reported that SERT basal phosphorylation is insensitive to PKA inhibition but can be elevated by PKA activation. Thus treatments (2 hours) with forskolin or cholera toxin robustly elevated SERT phosphorylation, effects blocked by coincubation of these agents with the PKA inhibitor KT5720. Although labeling was found to be PKA dependent, these studies do not allow one to know whether PKA directly phosphorylates SERT or whether labeling occurs through an intermediate kinase. By using PKC antagonists, however, this kinase was ruled out as a contributor to PKA-dependent SERT phosphorylation. Despite clear evidence of SERT phosphorylation induced by PKA activation, no functional effects on SERT activity were detected. Possibly, the lack of functional effects may derive from the heterologous nature of SERT expression, because potential phospho-SERT binding proteins may not be expressed, or alternatively functional effects of PKA phosphorylation may be contingent on the presence of other signals (e.g., coincident PKC or PKG activation). Interestingly, SERT phosphorylation elevated by the PP1/2A inhibitor okadaic acid was insensitive to PKA inhibition. This suggests that, presuming PKA-induced SERT phosphorylation occurs in vivo, another phosphatase may target sites phosphorylated by PKA-linked pathways. In their studies of the ability of purified kinases to phosphorylate human SERT cytoplasmic domain peptides, Sorensen et al. (2014) detected modest labeling of segments of both the N and C termini, although exact sites were not identified and no canonical PKA sites exist in these regions. In light of the evidence presented by Ramamoorthy et al. for phosphorylation of intact SERT in cells, the Sorensen et al. findings may indicate that PKA phosphorylation sites must be presented in the context of a fully-folded protein or may derive from the action of a PKA-modulated kinase that ultimately targets SERT. In this regard, two of the three SERT peptides labeled by PKA in the Sorensen et al. study are much more efficiently phosphorylated by either CaMKII or p38 MAPK.
4. Receptor-Initiated Protein Kinase A Regulation of Serotonin Transporter.
In their studies of Caco-2 modulation by PKA-linked pathways, Iceta et al. (2009) found that a 5-HT7 receptor agonist inhibited, in a PKA-dependent manner, SERT activity, whereas a 5-HT1A agonist increased SERT activity. Because 5-HT7 and 5-HT1A receptors are predicted to elevate versus inhibit PKA activity, these findings represent important support for the physiologic use of PKA signaling in the regulation of intestinal SERT, although as yet native intestinal preparations have not been similarly queried. With respect to SERT in the CNS, Awtry et al. (2006) found that brief nicotinic acetylcholine receptor agonist treatments elevated, whereas antagonists inhibited, SERT activity in rat prefrontal cortical synaptosomes. Importantly, nicotinic stimulation of SERT activity was blocked by coincident antagonism of PKA. These interesting studies have not been pursued, leaving unanswered whether they might relate to changes in SERT phosphorylation, protein interactions, or transporter trafficking. Because altered serotonergic signaling continues to be a focus for the study of addiction plasticity mechanisms (Muller and Homberg, 2015), further research in this area may be of clinical relevance.
IV. cGMP-Dependent Protein Kinase/ Protein Kinase G—Overview
PKG proteins are serine/threonine kinases that are activated by cGMP (Francis et al., 2010). In contrast to PKA proteins, where a kinase complex is formed from catalytic and regulatory units, PKG proteins feature internal inhibitory domains that upon binding cGMP are displaced from the catalytic domains to permit phosphorylation of target proteins. There are two genes that encode PKG proteins, PKGI and PKGII (Hofmann et al., 1992). PKGII is N-myristoylated, permitting anchoring of the kinase to the plasma membrane. Additionally, PKGI is alternatively spliced into PKG1α and PKG1β. PKG proteins are activated by increases in cGMP generated by guanylyl cyclase (GC). One of the major regulators of PKG signaling is nitric oxide synthase (NOS), which is regulated by intracellular Ca2+/calmodulin to generate nitric oxide (NO). NO binds to GC proteins to stimulate cGMP production, leading then to PKG activation (Potter, 2011). NO has other actions in cells, and thus NOS activation alone cannot be taken as evidence for a role of PKG in transporter regulation. cGMP production and hydrolysis as well as PKG can be targeted by various agents whose use has helped uncover roles for the kinase in monoamine transporter regulation.
A. Regulation of Dopamine Transporter by Protein Kinase G
Two papers in 1994, one from Kuhar’s group (Pogun et al., 1994) and one from Lonart and Johnson (1994), established a sensitivity of rat brain DAT to agents that elevate NO production, with both reports finding suppression of transport activity via a reduction in DA transport Vmax. Lonart and Johnson also observed an increase in prelabeled DA efflux, findings also observed by Buyukuysal (1997). In the latter study as well as in a more recent study by Schenk’s group (Volz and Schenk, 2004), a GC inhibitor did not prevent DA efflux or DAT modulation, suggesting that these NO effects are likely neither cGMP nor PKG dependent. Other studies have continued to report effects of endogenous (Chaparro-Huerta et al., 1997) or pharmacological (Cao and Reith, 2002; Mike et al., 2003; Kiss et al., 2004) NO donors or NOS inhibitors on DAT activity in vitro or in vivo, but to date the available data point to activities independent of PKG modulation of DAT, such as through nitrosylation of DAT (Park et al., 2002) or DAT regulatory proteins. Similarly, as of this date, no reports of cell surface receptors linked to DAT modulation that signal through PKG have appeared, nor is their work linking signaling through cGMP/PKG pathways to changes in DAT trafficking or membrane localization. One study (Gorentla et al., 2009) reported the ability of purified PKG to phosphorylate a rat DAT-N terminal peptide in vitro, although as of yet the site supporting labeling has not been described nor is their evidence of PKG-dependent phosphorylation of intact DAT protein either after heterologous expression or from native tissue preparations. Altogether, little evidence is presently available to consider PKG-linked signaling pathways as key to the regulation of DAT proteins.
B. Regulation of Norepinephrine Transporter by Protein Kinase G
As with DAT, studies have reported the ability of NO donors to regulate NET activity. Thus, Kaye et al. (1997) found that a 1-hour S-Nitroso-N-acetyl-DL-penicillamine (SNAP) treatment could reduce NE uptake activity of PC12 cells as well as in cultured superior cervical ganglion neurons. Studies with PC12 cells cocultured with inducible NOS expressing endothelial cells led to a reduction in NE uptake that could be blocked by a NOS inhibitor. Although SNAP treatments significantly elevated cGMP levels, the reduction in NE uptake was not mimicked by cGMP exposure, suggesting that NO may regulate NET through direct modification of the transporter. Indeed, subsequent studies by this group found that SNAP effects were lost in a NET mutant bearing a Cys351Ser mutation, consistent with nitrosylation versus actions through a cGMP/PKG pathway (Kaye et al., 2000). There are several studies that implicate receptors signaling through NO-independent, cGMP pathways to regulate NET activity. Specifically, members of the atrial natriuretic peptide family (ANP, BNP, CNP) signal through a transmembrane receptor guanylate cyclase and have been reported to stimulate NET activity in brain and adrenal preparations (Fernandez et al., 1990; Porzionato et al., 2010). How the cGMP signal propagates to regulate NET activity has unfortunately not been further pursued, although the second messenger has been reported to induce release of NE in sympathetic terminals via a PKG-dependent phosphorylation and inactivation of PDE3, leading to an elevation of cAMP and activation of PKA (Chan et al., 2012). These studies remind us of the interconnectedness of protein kinase-mediated signaling mechanisms that pathways regulating transporter trafficking and function may seldom (or never) rely on a single kinase and that following the trail from receptor to transporter may yield mechanistic insights as to transporter regulation not accessible through single kinase-oriented pharmacological paradigms. Our review of the literature indicates that whether PKG signaling directly or indirectly influences NET trafficking or membrane localization or regulates NET phosphorylation state or the composition of NET protein complexes is unknown.
C. Regulation of Serotonin Transporter by Protein Kinase G
1. Regulation of Serotonin Transporter Activity by Protein Kinase G.
In contrast to the limited evidence for a significant role for PKG signaling in DAT or NET regulation, a significant body of research has accumulated to implicate the kinase in modulation of SERT activity. NO donors have been found to both inhibit (Asano et al., 1997; Bryan-Lluka et al., 2004) and elevate (Gespach et al., 1986; Miller and Hoffman, 1994; Kilic et al., 2003; Zhu et al., 2004a) SERT activity, with differences here likely arising from the reagents used for NO generation and time/concentration of application. The findings of a stimulatory action appear to be more physiologically relevant, because treatment of many preparations with cGMP elevating or mimicking agents, including platelets (Launay et al., 1994), cell lines (Miller and Hoffman, 1994), synaptosomes (Ramamoorthy et al., 2007), and SERT transfected cells (Prasad et al., 2005) has been shown to increase SERT activity. Consistent with this surmise, Kilic et al. (2003) found that stimulation of SERT in transfected cells by a NO generator was blocked by GC inhibition, whereas Zhu et al. (2004a) found stimulation of both an NO donor and 8-Br-cGMP to be blocked by a PKG inhibitor. Moreover, Zhu et al. (2004b) found that pharmacological inhibition of the cGMP degrading enzyme PDE5 with sildenafil or zaprinast elevated SERT activity in cultured cells and these effects were precluded by PKG inhibition. Notably, the actions of these agents was found to be specific for SERT among the monoamine transporters, consistent with the general lack of significance of this pathway in DAT and NET regulation, as noted above. By using rat midbrain synaptosomes, Ramamoorthy et al. (2007) also reported loss of 8-Br-cGMP stimulation of SERT activity with PKG antagonist cotreatment, whereas PKC and PKA inhibitors were ineffective.
2. Regulation of Serotonin Transporter Membrane Compartmentalization and Trafficking by Protein Kinase G.
As will be further amplified in studies of receptor-dependent PKG modulation of SERT discussed below, PKG-dependent pathways appear to support both a trafficking-dependent and -independent mode of SERT regulation, with an elevation in surface expression arising after PKG activation. Thus, Zhu et al. (2004a) reported an elevation in surface expression of rat SERT in RBL-2H3 cells after sildenafil treatments, assessed by surface radioligand binding, as well as in human SERT transfected CHO cells, assessed by radioligand binding and biotinylation. These effects were blocked by a PKG inhibitor. Prasad et al. (2005) also found elevated surface density by cell surface binding and biotinylation methods after treatments of human SERT-transfected HeLa cells with 8-Br-cGMP. With respect to trafficking-independence, Ramamoorthy et al. (2007) detected no elevated surface expression after 8-Br-cGMP treatments of rat midbrain synaptosomes, as determined by cell surface biotinylation techniques, despite findings of an elevation in 5-HT transport Vmax and not in KM. These findings may relate to the ability of PKG signaling to induce a trafficking-independent mode of SERT regulation, mediated by p38 MAPK activation (Zhu et al., 2004a; Chang et al., 2012), as discussed later in this review. In this regard, Chang et al. (2012) found that surface labeled SERT proteins in serotonergic RN46A cells exist largely in a confined state within lipid raft-like membrane microdomains. Treatment of cells with 8-Br-cGMP mobilized SERT, increasing the diffusion rate of single particle labeled transporters, although SERT proteins remained confined to rafts. These findings, along with studies with cytoplasmic domain peptides and actin cytoskeleton disrupting agents, indicate that one consequence of PKG activation may be to release SERT from cytoskeletal tethers that constrain transport conformation and function.
3. Regulation of Serotonin Transporter Phosphorylation by Protein Kinase G.
Initial studies of SERT phosphorylation in transfected HEK-293 and HeLa cells revealed significant elevations in SERT phosphorylation induced by 8-Br-cGMP (Ramamoorthy et al., 1998, 2007; Prasad et al., 2005), although a PKG antagonist did not attenuate SERT phosphorylation triggered by okadaic acid, suggesting surveillance of PKG-dependent SERT phosphorylation by phosphatases other than PP1 or PP2A (Ramamoorthy et al., 1998). Using phosphoamino acid analysis of 8-Br-cGMP treated synaptosomes, Ramamoorthy et al. identified specific phosphorylation of Thr residues. Through systematic evaluation of SERT, Thr mutants expressed in CHO cells, Thr276 was identified as key both to phosphorylation and uptake stimulation. Thus a Thr276Ala mutant, possessing no alterations in 5-HT transport activity, lost phosphorylation and uptake stimulation after 8-Br-cGMP treatments, whereas a Thr276Asp mutation showed constitutively elevated transport activity and no further sensitivity to 8-Br-cGMP. Consistent with the findings by Ramamoorthy's group of a contribution made by PKG-dependent regulation to trafficking-independent SERT modulation, the constitutively elevated transport upregulation of the Thr276Asp mutant did not arise in the context of an elevation in SERT surface protein. Although the identity of the site phosphorylated after PKG activation in native preparations is unknown, these results provide strong evidence that phosphorylation of Thr276 is a key for PKG regulation of SERT.
The Thr276 site lies in a small cytoplasmic loop between TMs4 and 5 and would seem more likely to influence transport kinetics by altering the structure of the permeation pathway for 5-HT than in altering the interactions needed to change SERT trafficking. This idea has been reinforced by studies by Zhang et al. (2007) who found that the OCD associated, TM5 SERT coding variant Ile425Val exhibits elevated transport capacity in transfected HeLa cells, without an apparent change in surface expression, is not further responsive to a NO generator or 8-Br-cGMP and has hyperactivity quelled by treatment of cells with a GC or PKG inhibitor. Prasad et al. (2009), also examining human SERT in human SERT in transfected HeLa cells, although agreeing with the above studies that the Ile425Val mutant is insensitive to 8-Br-cGMP, found Ile425Val to cause an increase in surface transporter expression both by radioligand binding and biotinylation methods. Similarly, a different SERT variant at the same position, Ile425Leu, also shows elevated cell surface density. The basis for these differences remains unresolved but likely results from technical differences in heterologous expression methods used by the two laboratories. Regardless, these studies draw a compelling connection between SERT phosphorylation at Thr276 and elevated trafficking and/or activity as a contributor to risk for neuropsychiatric disorders. Elevated SERT activity in association with OCD risk is consistent with the common use of SERT-antagonizing SSRIs for treatment. The idea of psychiatric disease risk conferred by SERT hyperfunction has been further strengthened by the finding that the autism-associated SERT Ala56 variant confers constitutively elevated SERT activity in parallel with elevated basal phosphorylation where neither uptake activity or phosphorylation can be further elevated by cGMP signaling (Prasad et al., 2005; Sutcliffe et al., 2005).
Despite clear evidence from multiple groups that SERT phosphorylation is elevated after activation of cGMP-PKG signaling, other research indicates that direct phosphorylation of SERT by PKG is not necessary for this regulation. First, sequences containing the critical Thr276 residue do not conform to a classic PKG consensus sequence. Second, when Wong et al. (2012) in the Rudnick laboratory examined the regulation of SERT by a mutated PKGIα (M438G) that can use an exogenously supplied ATP analog for phosphorylation of its substrate, these investigators found that although SERT activity was increased by 8-Br-cGMP, no incorporation of a [33P]labeled form of the analog was observed, suggesting another kinase acts downstream of PKG to regulate SERT, likely by phosphorylating Thr276. Sorensen et al. (2014) also found no activity of purified PKG to phosphorylate purified cytoplasmic domain peptides in vitro, although they did identify activity, as noted above, for PKC at Ser277. The reader will recall from our prior discussion that PKC activation has been implicated in NET phosphorylation at the analogous position to Ser277. Thus, given the NET findings, one may consider whether PKG signaling might act to regulate SERT through a PKC isoform. Arguing against this idea are the findings that the PKC inhibitor BIM-1 does not attenuate 8-Br-cGMP stimulation of SERT activity or SERT phosphorylation (Ramamoorthy et al., 2007). Together, these studies call for further studies oriented to identifying the downstream targets of PKG that support phosphorylation and regulation of SERT and a characterization of the use of Thr276 for SERT regulation in vivo.
4. Protein Kinase G Regulation of Serotonin Transporter Protein-Protein Interactions.
In considering SERT associations with other proteins, we begin with a discussion of PKG itself. Steiner et al. (2009) studied interactions of PKG isoforms with SERT using the RN46A rat serotonergic neuroblastoma model. Immunocytochemical methods revealed colocalization of SERT with PKGI but not PKGII. They also found that siRNA targeted to the PKGI isoform blocked 8-Br-cGMP stimulation of SERT. Finally, using coimmunoprecipitation methods, a complex of SERT and PKGIa could be identified. These findings are consistent with findings by Zhang and Rudnick (2011) who, using PKG-depleted HeLa cells, found that PKGI but not PKGII transfection supports 8-Br-cGMP-stimulated SERT activity and that PKGI coimmunoprecipitates with SERT. These investigators went on to show that a PKGII mutant lacking the capacity for myristoylation was competent for SERT association and phosphorylation as well as supporting SERT stimulation, indicating that the membrane anchor afforded by myristoylation precludes localization of PKGII in SERT complexes where regulation is initiated. Although no evidence exists to support a direct PKGIa/SERT interaction, these studies indicate that if SERT is not a direct target of PKG, the kinase may target another SERT-associated protein as a key determinant of transporter regulation. Possibly, SERT-associated PKG could lead to the phosphorylation and inactivation of a SERT-associated phosphatase, with the result being an indirect elevation of SERT phosphorylation by a different kinase. Whereas PMA reduces recovery of PP2Ac/SERT complexes, no change in PP2Ac/SERT associations was detected after 8-Br-cGMP treatments (Zhang et al., 2007). The influence of cGMP/PKG signaling on the association with SERT of other phosphatases [e.g., calcineurin (Seimandi et al., 2013)] and with PKGIα, remains to be pursued.
Findings of SERT/PKGIα associations are all the more striking given evidence of an association of neuronal NOS (nNOS) with SERT (and a GC/NOS-coupled GPCR, see below), suggesting the assembly of a much larger signaling complex capable of compartmentalizing the influence of cGMP on the transporter (Chanrion et al., 2007). Whereas physical determinants of PKG interaction with SERT remain unknown, the type II C-terminal PDZ recognition motif is required for SERT/nNOS interactions. nNOS interactions appear to reduce SERT activity, with evidence indicating a reduction in surface expression. Conversely, 5-HT uptake stimulated NOS activity and cGMP production, suggesting a novel mechanism by which SERT activity could have broader influences. How SERT activity influences NOS function is unknown, although a mutant SERT unable to physically interact with nNOS does not elevate cGMP production, suggesting some form of communication between these partners regarding SERT conformations. Possibly, these findings may relate to demonstrations of elevated, PKG-dependent transporter phosphorylation associated with the gain of function SERT mutation Ala56 (Prasad et al., 2005) and by the ability of 5-HT uptake to antagonize PKC-dependent SERT phosphorylation and trafficking (Ramamoorthy and Blakely, 1999). Although the stimulatory influence of PKG signaling on SERT are difficult to reconcile with nNOS-mediated inhibition of SERT, the latter effects may relate to an influence on basal versus stimulated SERT activity or may relate to chemical versus natural stimulation of these pathways. Notably, in these studies, SERT activity was not influenced by NOS activation or inhibition.
5. Receptor-Initiated Protein Kinase G Regulation of Serotonin Transporter.
The first indications of a connection between PKG and SERT arose in studies of histamine modulation of 5-HT uptake activity in human platelets (Gespach et al., 1986). In these early studies, the rapid stimulatory actions of histamine were found to be mimicked by the NO-generating agent sodium nitroprusside. Further studies in this system (Launay et al., 1994) revealed the actions of a novel, H2 type histamine receptor that can elevate cGMP, produce an increase in SERT Vmax with no change in 5-HT KM and induce SERT stimulation blocked by a GC inhibitor. Although PKG signaling plays important roles in platelet activation and secretion downstream of cGMP elevations (Eigenthaler et al., 1992; Li et al., 2003), studies of histamine modulated SERT function have yet to progress to implicate PKG in this pathway. Launay et al. (2006) also described a NO-dependent pathway emanating from 5-HT2B receptor activation in IC11 cells. Previously, this group found that 5-HT2B receptors associate with NOS via PDZ domain interactions and that 5-HT2 agonist induces cGMP production (Manivet et al., 2000). In their later study, pharmacological PKG inhibition was used to demonstrate a role for the kinase in basal SERT phosphorylation and that induced by 5-HT2B agonism. Lastly, the group moved to embryonic raphe cultures to demonstrate 1) basal phosphorylation dependent on tonic 5-HT2B receptor activation and 2) elevated SERT phosphorylation triggered by either 8-Br-cGMP or 5-HT2B agonism. These effects were not examined, however, in the presence of a PKG antagonist. The relevance of these findings in vivo has yet to be established. Recently, Ye et al. (2014) found that although 5-HT2B receptor mRNA is not highly expressed in the adult mouse midbrain, mRNA levels were significantly negatively correlated with midbrain 5-HT levels, suggesting an as yet underappreciated autoregulatory role in adult 5-HT homeostasis.
Further support for receptor-linked PKG pathways in the service of SERT modulation comes from studies of A3 adenosine receptors (A3AR). Miller and Hoffman (1994) provided the first evidence for an A3AR-PKG linked pathway in their studies of SERT in RBL-2H3 cells. These authors found that the general adenosine receptor agonist NECA rapidly (minutes) increased 5-HT uptake, an effect blocked by NOS and GC inhibition. The effects of NECA were mimicked by 8-Br-cGMP and the PKG inhibitor H8 blocked SERT stimulation. Blakely’s group pursued further studies in the RBL-2H3 model, confirming a role for the A3AR subtype in SERT stimulation via the use of selective agonists and antagonists and finding that NECA-induced SERT stimulation could be blocked by the PLC inhibitor U73122 [1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione], as well as by Ca2+ chelation by BAPTA-AM (Zhu et al., 2004a). BAPTA-AM was unable to block stimulation by NO generators or 8-Br-cGMP, suggesting Ca2+ signaling activates this pathway upstream of NO generation, likely through activating nitric oxide synthase (NOS). By using cell surface radioligand binding methods, NECA effects were found to derive from an increase in SERT surface levels, with activity and trafficking effects blocked by PKG inhibition. Further evidence for a trafficking effect of A3AR-PKG pathways came from studies with the membrane impermeant cysteine modifying reagent MTSET, which can inactivate SERT by modifying key cysteine residues involved in the translocation mechanism. Although MTSET could reduce basal 5-HT uptake, SERT stimulation was still evident, consistent with recruitment of intracellular transporters after PKG activation. These studies also revealed a trafficking-independent mode of SERT regulation mediated by PKG-dependent activation of p38 MAPK, to be discussed further below. In these studies, whereas p38 inhibition blocked NECA-induced increases in 5-HT uptake, it failed to block the increase in SERT surface levels, suggesting that trafficking-dependent and -independent pathways collaborate to elevate SERT activity after PKG activation. Subsequently, Zhu et al. (2007) established the presence of an A3AR-PKG-SERT pathway in serotonergic terminals, finding that the A3AR agonist IB-MECA (N6-(3-Iodobenzyl)adenosine-5′-N-methyluronamide) stimulated SERT activity in midbrain and hippocampal, but not striatal synaptosomes, effects observed in wild-type but not A3AR knockout mice. As with cell line studies, 5-HT uptake regulation arose from a change in transport Vmax as opposed to 5-HT KM. IB-MECA-induced SERT stimulation was also found to be blocked by PKG and p38 MAPK antagonists. Finally, A3AR activation was found to enhance SERT-mediated 5-HT clearance in vivo as assessed by chronoamperometry. In more recent studies, Zhu et al. (2011) used coimmunoprecipitation methods and A3AR/SERT cotransfected CHO cells to show that A3ARs can be trapped in a physical complex with SERT and that recovery of the complex is enhanced with A3AR stimulation, effects blocked by PKG inhibition. Whether A3ARs and SERT assemble and are stabilized at the cell surface, versus in trafficking compartments before insertion, is unknown. That receptor/transporter interactions may be of physiologic and translational significance was suggested by studies of the A3AR coding variant Leu90Val identified in subjects with autism (Campbell et al., 2013). The variant was found to induce elevated basal and agonist stimulated cGMP production and SERT stimulation after IB-MECA stimulation and to remain associated SERT beyond the time when wild-type A3AR/SERT associations have returned to basal levels (Zhu et al., 2011). These studies indicate that A3AR/SERT associations are a consequence of receptor conformations associated with agonist occupancy, possibly stabilized by PKG-dependent phosphorylation of the transporter, and provide support for ectopic elevation of SERT expression, trafficking, or function as a risk factor in autism spectrum disorders.
V. Ca2+/Calmodulin-Dependent Protein Kinase II—Overview
CaMKII is a major effector of signaling pathways that result in elevations in cytosolic Ca2+ and is a critical regulator of many cellular processes. CaMKII exists as multiple isoforms encoded by four genes (α, β, γ, and δ), although the α and β isoforms are expressed mostly in the brain (Ma et al., 2015). CaMKII holoenzymes exist as multimers of 12 subunits, and different isoforms of CaMKII can mix to form heteromeric CaMKII holoenzymes made up of different relative compositions of these various isoforms (Shonesy et al., 2014). The activity of the enzyme is regulated by autoinhibition, which is relieved upon binding to calmodulin in its Ca2+-bound state, a process that underlies the Ca2+-dependent stimulation of CaMKII activity. As the release of DA, NE, and 5-HT is triggered by elevations in cytosolic Ca2+, a coupling of CaMKII activation to transporter trafficking and function offers a potential mechanism for linking neurotransmitter clearance capacity to activity-dependent neurotransmitter release. As we will see below, significant evidence supports an important role for CaMKII isoforms in multiple aspects of biogenic amine transport regulation. Where the evidence is drawn from the use of CaMKII inhibitors that do not distinguish between isoforms, we will simply use the general notation of “CaMKII” to describe the available evidence and note specific isoforms where the use of subtype-specific molecular approaches permits a finer designation.
A. Regulation of Dopamine Transporter by Calmodulin-Dependent Protein Kinase II
1. Regulation of Dopamine Transporter Activity by Calmodulin-Dependent Protein Kinase II.
At present, limited evidence supports a role for CaMKII in regulating DA uptake by DAT. Uchikawa et al. (1995) reported DA uptake in rat striatal synaptosomes exhibits a biphasic sensitivity to the addition of external Ca2+, with 1 mM Ca2+ significantly elevating DA uptake over Ca2+ free buffer, followed by a reduction as Ca2+ concentrations were further increased. Because an absence of external Ca2+ is a nonphysiologic state, the stimulatory effect of 1 mM Ca2+, which elevated DA transport Vmax with no effect on IM, should best be seen as revealing a requirement to support basal transport capacity and could be indirect (e.g., impact on Na+ gradient via Na/Ca2+ exchange). Regardless, these investigators found that the stimulatory effect of 1 mM Ca2+ could be attenuated by either the calmodulin antagonist W-7 (N-(6-Aminohexyl)-5-chloro-1-naphthalenesulfonamide) or the CaMKII antagonist KN-62 (4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl] phenyl isoquinolinesulfonic acid ester). Earlier in our presentation, we noted we noted that Page et al. (2004) had reported that radiolabeled DA uptake activity in rat striatal synaptosomes could be enhanced by treatments with a membrane permeant cAMP analog. These effects were rapid and transient (evident with 5-minute treatment, lost within a few minutes thereafter), which changes impacting DAT Vmax but not DA KM. These stimulatory effects could be blocked by KN-62, suggesting convergence on a CaMKII pathway that can positively regulate DA uptake capacity. Further investigations of mechanism supporting these effects have not appeared. Lin et al. (2003) found that treatment of COS cells transfected with rDAT with KN-62 had no effect on DA uptake, suggesting that the kinase plays no essential role in sustaining basal DA uptake. Consistent with this, in studies where CaMKIIα knockout (KO) and knock-in mice have been used to assess contributions of CaMKIIα expression/activity to AMPH evoked DA release (see below), no alterations in basal DA uptake, expression, or surface trafficking were evident (Steinkellner et al., 2012), although the opportunity for compensations in the context of life-long kinase manipulations must be taken into account. Repeating the Uchikawa and Page experiments noted above with these genetic models would be helpful in assessing potential CaMKII engagement in DAT regulation.
Whereas little support for CaMKII regulation of DA uptake exists, substantial evidence supports a role for the kinase in DAT-dependent DA efflux triggered by AMPH or DAT mutations. Historically, vesicular and nonvesicular, transporter-mediated neurotransmitter release have been considered distinguishable on the basis of Ca2+ dependence, with vesicular release being Ca2+ dependent and transporter reversal being Ca2+ independent. Such distinctions, however, have been drawn in relation to efflux that arises from changes in the ion gradients (e.g., Na+/Cl−) needed to drive concentrative uptake, as for example when the Na+/K+ ATPase is poisoned with ouabain, when extracellular Na+ is lowered experimentally, or during ischemia (Kim et al., 1995; Elverfors et al., 1997; Oliva et al., 2013). As previously noted, DAT, NET, and SERT support another form of efflux that occurs upon AMPH stimulation and here significant evidence supports an important role for Ca2+ as well as CaMKII. By using whole cell patch-clamp and amperometric methods, Gnegy et al. (2004) demonstrated that AMPH-induced currents and AMPH-induced DA release could be blocked by the Ca2+ chelator BAPTA-AM. These investigators also reported that BAPTA blocked AMPH-evoked DA release from striatal synapsotomes. Importantly, AMPH treatment of DAT transfected cells produced a rise in intracellular Ca2+ that could be blocked by thapsigargin or cocaine, supporting a model whereby AMPH is first transported into cells where it can then produce release of endoplasmic reticulum Ca2+ stores. Subsequently, AMPH was shown to activate CaMKII in DAT transfected cells (Wei et al., 2007). Multiple studies have found that CaMKII inhibition can attenuate AMPH-evoked DA release (Weatherspoon and Werling, 1999; Fog et al., 2006; Steinkellner et al., 2012), although in at least one study this sensitivity emerged only after repeated treatment of animals with AMPH (Kantor et al., 1999), suggesting that CaMKII action on DAT may play a key role in AMPH sensitization. Fog et al. (2006) also found that intracellular perfusion of activated CaMKIIα enhanced AMPH-induced DA efflux. Most recently, Steinkellner et al. (2014) reported a reduction in AMPH-evoked DA release in vivo via microdialysis in CaMKIIa knockout mice as well as a blunting of AMPH-induced locomotor activation. Consistent with these findings, transgenic expression of a CaMKII inhibitory peptide blocks the locomotor stimulating actions of AMPH in Drosophila melanogaster (Pizzo et al., 2014).
An important role for CaMKII in sustaining conformations that trigger DAT-mediated DA efflux has further advanced in studies of the human DAT variant Val559. This variant was first identified by Grunhage et al. (2000) in a female subject with bipolar disorder, although the rarity of the mutation and a lack of segregation with the disorder in the initial kindred limited further analyses. Subsequently, Blakely’s group reidentified the variant in two male siblings with ADHD (Mazei-Robison et al., 2005, 2008), and subsequently Galli and Sutcliffe’s groups found the variant in two unrelated males with ASD (Bowton et al., 2014). Strikingly, the DAT Val559 variant supports DAT-dependent DA efflux that can be reversed by CaMKII inhibition (Mazei-Robison et al., 2008; Bowton et al., 2010), mimicking the state of DAT in the presence of AMPH. Indeed, AMPH lacks the ability to trigger DA efflux in the DAT Val559 mutant, serving rather to attenuate elevated basal efflux (Mazei-Robison et al., 2008). Recently, Mergy et al. (2014) provided in vivo evidence via microdialysis studies of knock-in mice that the DAT Val559 variant supports a tonic elevation in basal extracellular DA, consistent with chronic DAT-mediated DA efflux, although whether CaMKII sustains DAT Val559 efflux activity in vivo has yet to be addressed.
2. Regulation of Dopamine Transporter Membrane Compartmentalization and Trafficking by Calmodulin-Dependent Protein Kinase II.
To date, only modest evidence supports a role for CaMKII in basal or regulated DAT trafficking. In studies of the actions of insulin to support DAT surface expression, CaMKII appeared to oppose the actions of insulin signaling pathways on DAT and was found to be required for AMPH-induced reduction in Akt activity and DAT surface levels (Wei et al., 2007). Recently, Sakrikar et al. (2012) reported the identification of the DAT coding substitution Arg615Cys in a subject with ADHD. In this work, the Cys615 variant was found to redirect DAT to a novel plasma membrane compartment in transfected HEK-293 cells where it recycles constitutively and lacks sensitivity to PKC and AMPH-modulated trafficking. Interestingly, the variant also results in the emergence of functional inhibition by the CaMKII inhibitor KN-93 [N-[2-[N-(4-chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide] that is dictated by a reduction in DAT Vmax and that is absent in wild-type DAT. Biotinylation studies revealed that KN-93 inhibition of DA uptake capacity was not a result of a change in surface DAT density, but rather indicates a change in DAT conformation leading to functional inactivation. Additionally, the engagement of CaMKII pathways for AMPH-induced DAT trafficking was lost in the DAT Cys615 mutant. Whereas wild-type DAT displays a CaMKII-dependent AMPH redistribution from the cell surface, this effect is lost in the mutant. As noted above, AMPH can inhibit Akt via a KN-93 sensitive CaMKII pathway (Wei et al., 2007), and the surface microdomain or trafficking vesicles within which DAT Cys615 resides appear to lack regulation by CaMKII/Akt pathways. Together, these findings reveal a role for CaMKII activation in surface DAT trafficking, mediated by Akt-linked pathways, as well as conformations impacting intrinsic DAT functional activity, although at present the latter contribution is most evident in considering a role for CaMKII in DA efflux and only becomes evident with respect to DA transport in the context of DAT mutations, likely in modulating key DAT protein associations, including CaMKII itself (see below).
3. Calmodulin-Dependent Protein Kinase II Regulation of Dopamine Transporter Phosphorylation.
As noted above, an important role for CaMKII activity in AMPH-evoked DA efflux has been defined through the use of organic and peptide CaMKII inhibitors, intracellular kinase perfusion and the use of CaMKII KO/knock-in mouse models. The question naturally arises as to whether this contribution arises from direct, CaMKII-mediated DAT phosphorylation. Khoshbouei et al. (2004) identified five Ser residues in the DAT N terminus that through mutation studies appear to support AMPH-induced DA efflux in transfected cells. Khoshbouei et al. was able to restore significant AMPH-induced DA efflux in HEK-293 cells by restoring Ser7 and Ser12 to a human DAT mutant lacking the five N-terminal Ser residues, whereas other substitutions were without effect, suggesting one or more of these as likely key to AMPH action. Although none of these residues match canonical motifs for known protein kinases, Fog et al. (2006) demonstrated N-terminal human DAT peptides to be efficiently phosphorylated by CaMKIIα. Consistent with these findings, Gorentla et al. (2009) demonstrated that that the purified rat DAT N terminus acquires Ser phosphorylation upon incubation with CaMKII. At present, information is lacking as to the site(s) that support CaMKII phosphorylation of DAT in vivo, although Javitch’s group provided support for a role of the N-terminal Ser residues in AMPH behavioral activation using transgenic expression of wild-type and mutant human DAT constructs in Drosophila melanogaster (Pizzo et al., 2014). The Galli laboratory recently reported an importance for nearby Lys residues Lys3 and Lys5 and PIP2 interactions with DAT on AMPH-evoked DA release (Hamilton et al., 2014), and this interaction may also be related to CaMKII phosphorylation of N-terminal serines. However, the phosphomimetic mutations S2D and S4D were not able rescue the defect in AMPH-stimulated DA efflux induced by alanine substitutions of these lysine residues, suggesting that the effects of these Lys mutations are independent of phosphorylation of these serine residues. Importantly, phosphorylation was not directly measured in the K/A mutant DAT, and there are other nearby N-terminal serines whose phosphorylation may be impacted by this phosphatidylinositol (4,5)-bisphosphate (PIP2) interaction, perhaps through regulation of CaMKII binding. Further work is necessary to determine if there is an interaction between these modes of regulation of AMPH-evoked DA efflux. Finally, both of the ADHD-associated DAT variants identified by the Blakely laboratory, Val559 and Cys615, display elevated transporter phosphorylation in transfected cells (Bowton et al., 2010; Sakrikar et al., 2012), and although the in vivo significance of these findings or their reliance on CaMKII pathways have yet to be determined, both mutations disrupt aspects of DAT function and trafficking that we have noted as under the influence of CaMKII.
4. Calmodulin-Dependent Protein Kinase II Regulation of Dopamine Transporter Protein-Protein Interactions.
Using the yeast 2-hybrid methodologies, Fog et al. (2006) identified CaMKIIα as a potential interacting partner with the DAT C terminus and appears to play a role in regulating DAT activity. Fusion protein pull-down studies and coimmunoprecipitation experiments confirmed an association of the DAT C terminus with CaMKIIα in both transfected cells and brain extracts as well as interactions with the full-length transporter. The site of interaction between kinase and transporter was mapped to the distal C terminus, including residues 612–617. Perfusion of cells with a C-terminal GST fusion protein containing the last 24 amino acids (C24) blocked AMPH-induced DA efflux. Finally, a mutant DAT with Ala substitutions of residues 612–614 failed to support enhanced AMPH-induced DA efflux by CaMKIIα overexpression. Subsequent studies by the Gether laboratory demonstrated the ability of the DAT C24 peptide to block DAT-CaMKIIα interactions in vivo as well as AMPH-evoked DA efflux and AMPH-induced locomotor hyperactivity (Rickhag et al., 2013). Together, these studies make a compelling case that CaMKIIα both associates with DAT and regulated the functional states of the transporter, as manipulated by AMPH. Moreover, they suggest that disrupting DAT protein-protein interactions could prove of therapeutic benefit for disorders linked to elevated CaMKII/DAT interactions and/or anomalous DA efflux. Steinkellner et al. (2012) evaluated the impact of a mouse model of Angelman syndrome that features a mutation in the UBE3A gene. This mutation renders reduced activity of CaMKII due to increased phosphorylation at inactivating Thr605/606 sites. Angelman Ube3a knock-in mice displayed elevated basal MPP+ efflux, whereas CaMKIIα mutant mice displayed reduced basal efflux. However, as expected, Angelman mice demonstrated blunted AMPH-evoked DA release, despite normal levels of DAT/CaMKII expression and interaction. Further efforts that exploit these observations to evaluate contributions of DAT to altered behaviors in these mice are needed to more fully appreciate the connection between altered DAT and DA signaling and the disorder. Finally, both of the ADHD-associated DAT variants identified to date demonstrate elevated CaMKII interactions and elevated DAT phosphorylation (Mazei-Robison et al., 2008; Bowton et al., 2010; Sakrikar et al., 2012), with the DAT Val559 variant demonstrating excess DA efflux. The recent generation of the DAT Val559 model offers an opportunity to bring the idea of the therapeutic utility of manipulating CaMKII/DAT interactions to a test.
The current model for how CaMKII participates in AMPH-triggered DA efflux involves binding of the kinase to the transporter C terminus followed by phosphorylation of one or more Ser residues in the transporter N terminus. This phosphorylation is then thought to facilitate conformational changes that place the transporter in a “ DA efflux-willing” conformation. Likely, phosphorylation of distal residues in a cytosolic domain is insufficient to stabilize conformations that must engage multiple transmembrane helices involved in the transport mechanisms. Rather, phosphorylation may perturb the interactions of DAT-associated proteins that impact conformational stability. The best candidate to date for such a protein is syntaxin 1A. Syntaxin 1A is a plasma membrane SNARE protein that is conventionally understood to support the fusion of synaptic vesicles via regulated coiled-coil interactions with its vesicle and plasma membrane SNARE partners (Sudhof and Rothman, 2009). Quick and colleagues initially described the ability of syntaxin 1A to regulate the trafficking and activity of GAT1 GABA transporters (Horton and Quick, 2001; Quick, 2002), work subsequently extended to other neurotransmitter transporters, including SERT (Haase et al., 2001), NET (Sung et al., 2003), and DAT (Lee et al., 2004). Importantly, the interaction of syntaxin 1A with SLC6 transporters appears to occur through a direct partnership with acidic residues of the N terminus, providing an opportunity for CaMKII-dependent N-terminal phosphorylation to modulate interactions. Indeed, Binda and colleagues (2008) demonstrated that AMPH treatment increases DAT/syntaxin 1A interactions in transfected cells and mouse synaptosomes and that overexpression of syntaxin 1A elevates AMPH-evoked DA efflux. Inhibition of CaMKII was found to both block the ability of syntaxin 1A to interact with DAT and to promote AMPH-evoked DA efflux. Together, these findings support a model whereby AMPH-dependent phosphorylation of DAT by CaMKII stabilizes syntaxin 1A interactions with DAT, and this complex promotes a DA efflux willing transporter conformation. Because this interaction would normally lead to a leaching of cytosolic DA pools, were it to happen under normal conditions, it is likely that the syntaxin 1A/DAT complex is either transient or inhibited by other mechanisms or occurs in the context of other changes in DAT or DAT-associated proteins that preclude DA efflux. Cervinski et al. (2010) detected a reduction in metabolic phosphorylation of DAT in rat striatal tissue with cleavage of syntaxin 1A with botulinum neurotoxin C (BoNT/C). Although the pathway through which this change in phosphorylation occurs has not been established and the effects of BoNT/C may be indirect, they suggest that a reciprocal relationship may exist between DAT phosphorylation states and DAT/syntaxin 1A interactions. Interestingly, a recent study from the Galli laboratory reveals coding mutations in the human syntaxin 1A and DAT genes, found in subjects with autism, that individually reduce DAT/syntaxin 1A associations and diminish AMPH-evoked DA efflux (Cartier et al., 2015). Although studies are needed to associate synaptic changes with these effects, they remind us that basal interactions between DAT and syntaxin 1A are readily detectible in vivo and thus are likely to play key roles in the normal functional modulation of DAT. The possibility also remains open that DAT-mediated DA efflux arises as a consequence of normal signaling (Falkenburger et al., 2001), and if so, CaMKII/syntaxin 1A pathways would be of prime consideration as a conduit for such signals.
5. Receptor-Initiated Regulation of Dopamine Transporter by Calmodulin-Dependent Protein Kinase II.
To date, the contributions made by CaMKII pathways to DAT regulation have been elicited largely through pharmacological or genetic manipulations that target the kinase versus neural signaling-linked pathways. Possibly, CaMKII may participate in DAT regulation as a consequence of intracellular Ca2+ elevations at synaptic terminals after neuronal depolarization. DAT proteins are believe to be perisynaptic (Nirenberg et al., 1996, 1997) and thus CaMKII modulation of DAT could arise in the context of repeated activation, as occurs with burst firing states of DA neurons. Presynaptic D2 DA receptors, which we previously discussed as important regulators of DAT, are known to signal to other efforts through CaMKII-linked pathways, although to date whether D2 receptor-mediated control of DAT in vivo is mediated by CaMKII is unclear. One suggestive study that this may be the case, and be of disease relevance, involves the anomalous DA efflux produced by the ADHD-associated DAT Val559 variant. Bowton et al. (2010) showed that this spontaneous efflux, when studied in transfected HEK-293 cells or cultured DA neurons, is supported by a D2 DA receptor mediated mechanism linked to Gi/Go signaling, CaMKII activation, and DAT N-terminal phosphorylation (specifically at Ser7, 12, and 13). Studies with the DAT Val559 mouse support constitutive activation of presynaptic DA D2 receptors (Mergy et al., 2014), consistent with a positive feedback model whereby initial DA leak produced by the DAT Val559 variant can stimulate further DA efflux via D2 receptor activation, although CaMKII engagement was not evaluated. The question remains however as to when, with wild-type DAT, a D2-CaMKII pathway would be engaged. Activation of presynaptic D2 DA receptors inhibits vesicular DA release and stimulates DAT trafficking, thereby enhancing DA clearance capacity. Possibly, DAT mutations that induce anomalous DA efflux subvert this D2-mediated DAT regulatory pathway. Studies are needed that more closely delineate the control of vesicular fusion and DAT trafficking by D2 receptors and whether CaMKII plays an important role in the functional modulation of the latter process.
B. Regulation of Norepinephrine Transporter by Calmodulin-Dependent Protein Kinase II
1. Regulation of Norepinephrine Transporter Activity by Calmodulin-Dependent Protein Kinase II.
Several studies indicate that NET-mediated NE transport activity is positively regulated by CaMKII-linked pathways. Thus, Apparsundaram et al. (1998a) found that treatment of noradrenergic SK-N-H cells with KN-93 induced a rapid reduction in NET activity, effects reproduced in other models (Mandela and Ordway, 2006; Sung and Blakely, 2007; Hope et al., 2010). Conversely, Uchida et al. (1998), studying rat PC-12 cells found that elevation of extracellular Ca2+ enhanced NET activity, relative to Ca2+ free medium, in a calmodulin and CaMKII inhibitor-sensitive manner, effects mediated by an increase in NE uptake Vmax and a reduction in NE KM. The treatment of cells with CaMKII inhibitor in the former studies and the nonphysiologic, Ca2+ free reference point for the findings in the latter studies make a case for CaMKII in supporting basal NET activity. However, in PC-12 cells, in Ca2+ containing medium, Mandela and Ordway (2006) found that treatment of cells with short pulses of KCl increased NE uptake Vmax, an effect dependent on extracellular Ca2+ and lost when cells were treated in the presence of the CaMKII inhibitor KN-93. These findings suggest that CaMKII can contribute to both basal and stimulated NET activity. Interestingly, myosin light chain kinase inhibitors, which are targets of CaMKII, also reduced NE uptake and blocked the KCl-stimulated increase in NE uptake, suggesting these kinases may act downstream of CaMKII in this mode of NET regulation. More recently, using SK-N-SH cells, Chung et al. (2013) detected a rapid KCl-stimulated enhancement of NET function, as measured by [131I]-metaiodobenzylguanidine that, as found with PC-12 cell stimulation, arose from an elevation in NE transport Vmax and was blocked both by CaMKII and myosin light chain kinase inhibitors.
CaMKII also appears to play a role in NET substrate efflux triggered by AMPH, although this has been far less well investigated than DAT-mediated DA efflux, and the available study indicates that this contribution becomes apparent only after repeated, intermittent AMPH exposure. Thus, Kantor et al. (2004) described enhancement of DA efflux by PC-12 cells that was dependent on external Ca2+ and is blocked by the voltage-gated Ca2+ channel (VGCC) inhibitors ω-conotoxin and nifedipine. The reader will recall that evidence suggests that DAT-mediated DA efflux after AMPH treatment relies more on intracellular Ca2+ stores than extracellular Ca2+, suggesting differences in either NET versus DAT induction of efflux or the model systems used (e.g., presence of VGCCs). Importantly, the effects of repeated (but not single dose) AMPH on PC-12 cells to trigger DA efflux were found to be blocked by KN-93. (Kantor et al., 2004). Interestingly, AMPH also elicited a greater increase in Ca2+ elevations after repeated treatment, suggesting possible changes in expression/activity of Ca2+ channels as well. The actions of AMPH to elevate Ca2+ levels were blocked by desipramine, suggesting that AMPH-induced depolarization may be responsible for Ca2+ channel activation. Consistent with this idea, Cameron et al. (2015) recently reported an ability of AMPH to activate VGCCs via transporter-mediated depolarization. Although these studies were of DAT expressing cells, NET is also electrogenic and generates significant currents in response to substrate interactions (Galli et al., 1998).
2. Regulation of Norepinephrine Transporter Membrane Compartmentalization and Trafficking by Calmodulin-Dependent Protein Kinase II.
The studies described above support a role for CaMKII in the control of basal NE uptake capacity. In the studies by Uchida et al. (1998), where significant elevations in NET Vmax were identified after addition of Ca2+ to PC-12 cells incubated in Ca2+ free medium, these authors also identified a significant elevation in [3H]desipramine binding sites in isolated membranes. Possibly this measure reflects a movement of NET to the cell surface in this model, although no explicit definition of the membranes used as cell surface versus intracellular membranes was provided. Sung and Blakely (2007), however, demonstrated using biotinylation approaches that restoration of extracellular Ca2+ elevates surface NET density in transfected CHO cells. The increased density of [3H]desipramine binding sites observed by Uchida et al. with external Ca2+ restoration could also reflect a CaMKII-dependent change in conformation that exposes binding sites that were previously “hidden.” In support of trafficking-independent regulation, Mandela and Ordway (2006) found no change in binding of [3H]nisoxetine after depolarizations that elicited CaMKII-dependent elevations in NET activity using whole cell binding assays. Savchenko et al. (2003), using a surface-epitope antibody approach, reported a significant elevation of NET surface levels in brain stem and superior cervical ganglion (SCG) cultures after KCl-induced depolarizations, although no evaluation of Ca2+ or CaMKII-dependence was explored.
A role for CaMKII in NET surface trafficking has been proposed in relation to the effects of AMPH. Thus, Dipace et al. (2007) reported that AMPH treatment of NET-transfected CAD cells induces transporter internalization that can be prevented by CaMKII inhibition. AMPH treatments are also accompanied in the CAD model by an elevation in intracellular Ca2+ and CAMKII activation as detected through immunoblotting of CaMKII Thr286. Additionally, both BAPTA-AM and blockade of Ca2+ channels using Cd2+ attenuate AMPH effects. In contrast, AMPH is capable of reducing surface expression of NET and NET activity in stably-transfected trophoblast cells (HTR) via a reduced recycling mechanism that is insensitive to intracellular Ca2+ chelation with BAPTA-AM or CaMKII inhibition (Annamalai et al., 2010). These studies underscore the differential capacities of neuronal and nonneuronal hosts to support seemingly equivalent changes in transporter function through different mechanisms. In this regard, Matthies et al. (2010) demonstrated reduced surface expression of NET in mouse brain slices and single varicosities of cultured SCG neurons after AMPH treatment. Future studies that extend this paradigm to an evaluation of the role of Ca2+/CaMKII mechanisms in basal and AMPH-modulated NET trafficking should yield important insights.
3. Calmodulin-Dependent Protein Kinase II Regulation of Norepinephrine Transporter Phosphorylation.
Uchida and colleagues, seeking possible sites for phosphorylation that might be relevant to the CaMKII-dependent changes in PC-12 NET activity seen with extracellular Ca2+ restoration, tested synthetic rat NET peptides for their ability to support phosphorylation by purified CaMKII. Only one peptide, from the NET C terminus that contains Ser579 and Ser583 supported phosphorylation, with a KM (6.6 µM) comparable to a synthetic CaMKII peptide substrate (14.7 µM), although at ∼20% the efficiency. To date, however, no evidence of CaMKII-dependent phosphorylation of full-length NET, either under basal or stimulated conditions, has been reported.
4. Calmodulin-Dependent Protein Kinase II Regulation of Norepinephrine Transporter Protein-Protein Interactions.
To date, exploration of effects of CaMKII linked pathways on NET protein associations has involved studies of syntaxin 1A interactions. Sung and Blakely (2007) extracellular Ca2+-dependent changes in the interaction of mouse NET with syntaxin 1A in mouse brain synaptosomes as well as in human NET transfected CHO cells. Although KN-93 reduced basal NET activity in both brain and transfected cell preparations, this treatment did not influence NET/syntaxin 1A interactions, nor was the impact of KN-93 sensitive to botulinum toxin C, which cleaves syntaxin 1A, whereas they eliminated PKC-dependent NET regulation. Studies with AMPH modulation of NET trafficking, however, have provided evidence for a CaMKII-sensitive pathway that can influence transporter/syntaxin 1A interactions. Thus, treatments with AMPH that reduce NET surface expression in transfected CAD cells, increase cell surface NET/syntaxin 1A interactions (Dipace et al., 2007). These effects were magnified in a mutant NET lacking N-terminal sequences (28–47), a deletion that also leads to greater AMPH-induced elevations in cytosolic Ca2+ and CaMKII activation. Treatments of cells with KN-93 that as noted above blocks AMPH induced internalization in these cells also attenuates the increase in NET surface syntaxin 1A. These studies highlight differences in studies of Ca2+/CaMKII-dependent NET trafficking and protein associations that likely relate to the mechanisms by which Ca2+ elevations arise, namely via direct manipulation or cell depolarization versus AMPH treatments, with the former manipulation driving elevated surface expression and enhanced activity and reduced syntaxin 1A interactions and the latter manipulation supporting DA efflux and internalization. Also, the studies of NET/syntaxin 1A interactions in the Binda et al. (2008) study examined surface NET as opposed to the total pool of transporters. Possibly, NET/syntaxin 1A interactions become enhanced at the cell surface as a prelude or requirement for transporter internalization by AMPH but then upon internalization, these interactions are reversed.
5. Receptor-Initiated Regulation of Norepinephrine Transporter by Calmodulin-Dependent Protein Kinase II.
At present, no reports have linked presynaptic receptor activation to CaMKII-dependent changes in NET function, trafficking, or protein associations. By using inhibitors, Hope and colleagues found no contributions of CaMKII pathways to endothelin regulation of NET activity in rat hypothalamic preparations, although KN-62 had basal inhibitory effects (Hope et al., 2010). Lu et al. (1996) found rapid, stimulatory actions of angiotensin II on hypothalamic NET activity mediated by AT1 receptors, effects consistent with elevations in NET surface expression found by Savchenko et al. (2003) after angiotensin II treatments. Neither group evaluated the role of CaMKII in angiotensin II effects, although the AT1 receptor is known to couple through intracellular Ca2+-linked pathways. Lu et al. provided evidence that a PKC-linked pathway is involved in chronic angiotensin II regulation of NET mRNA expression. Because PKC activation leads to NET internalization (see above) in most systems, another Ca2+ linked pathway, possibly CaMKII, may support the elevated surface expression evident with acute angiotensin II treatments. Indeed, multiple studies support the ability of AT1 receptors to regulate cell signaling via CaMKII-linked pathways (Du et al., 2004; Palomeque et al., 2009; Zhang et al., 2010).
C. Regulation of Serotonin Transporter by Calmodulin-Dependent Protein Kinase II
1. Regulation of Serotonin Transporter Activity by Calmodulin-Dependent Protein Kinase II.
Evidence for a role of CaMKII in regulating SERT has been little to nonexistent until recently. In placental cells (BeWo, JAR), Jayanthi et al. (1994) demonstrated that inhibitors of calmodulin (CGS93-47, W7, camidazolium) reduced 5-HT uptake via a reduction in transport Vmax and an increase in 5-HT KM. No evidence was presented to suggest that these effects were mediated via CaMKII pathways. Indeed, the inhibitory effects of CGS could be seen in resealed membrane vesicles lacking ATP, suggesting they are kinase activity independent as well as derived from trafficking-independent mechanisms. Quick’s group provided the first direct evidence for CaMKII regulation of SERT in studies of the transporter coexpressed with syntaxin 1A in Xenopus oocytes (Ciccone et al., 2008). Prior work by Quick demonstrated that the SERT transport cycle is electroneutral when coexpressed with syntaxin 1A. Treatment of oocytes expressing SERT/syntaxin 1A with KN-93, but not the CaMKII inactive analog KN-92, restored electrogenicity to SERT. No influence in electrogenicity was seen with KN-93 treatments in the absence of syntaxin 1A, indicating a key role for the SNARE protein in mediating coupling stoichiometry regulation. Regardless of coupling stoichiometry effects, no effect of KN-93 was seen on 5-HT uptake, indicating a specific role of CaMKII/syntaxin 1A in modulating SERT electrogenicity, presumably by dictating the amount of nonstoichiometric ion flow. Syntaxin 1A interactions with NET also reduce uncoupled ion flow in this transporter (Sung et al., 2003), suggesting a broader relevance to the changes in electrogenicity seen with SERT. Importantly, the SERT findings were replicated in patch-clamped rat thalamocortical neurons that have been shown to express SERT during development. The model that emerges from these studies is that CaMKII phosphorylation events could serve to modulate SERT conformation, stabilizing the transporter in a conformation that precludes nonstoichiometric ion flow. This model of course raises the important question of when and how the cell uses the electrogenic capacity accorded by SERT when CaMKII is inhibited
Very recently, a role for CaMKII in supporting PCA-triggered 5-HT efflux has emerged, complementing studies previously noted that implicate the kinase in AMPH actions on DAT (Steinkellner et al., 2015). Steinkellner and colleagues reported that the efflux of [3H]MPP+ through SERT in transfected HEK-293 cells is reduced in a dose-dependent manner by KN-93 as well as another CaMKII antagonist, autocamptide-2 related inhibitor peptide. Interestingly, a concentration of KN-93 (10 µM) that elicited a strong reduction in efflux capacity has no effect on 5-HT uptake, pointing to a selective contribution to conformations supporting reverse transport triggered by the AMPH analog. Consistent with these data, efflux of [3H]5-HT by cortical synaptosomes was blunted by KN-93 and autocamptide-2 related inhibitor peptide. Even more compelling was the finding of a loss of SERT-dependent efflux activity in synaptosomes and slices from CaMKIIα KO mice despite normal levels of SERT and 5-HT uptake. Finally, the ability of MDMA to increase locomotion, which is known to be SERT dependent (Bengel et al., 1998), was found to be diminished in the CaMKIIα KO mice, whereas cocaine-induced hyperactivity is normal.
2. Regulation of Serotonin Transporter Membrane Compartmentalization and Trafficking by Calmodulin-Dependent Protein Kinase II.
At present, no evidence supports a contribution of CaMKII signaling to SERT surface trafficking or membrane microdomain localization. Given recent findings, however, that SERT mobility within membrane microdomains can be affected by a peptide derived from the SERT C terminus (Chang et al., 2012), a domain that supports SERT/CaMKII interactions (Steinkellner et al., 2015), higher resolution approaches are needed. The Ca2+ activated phosphatase calcineurin has been found to interact with the SERT C terminus in a Ca2+-dependent manner and to promote SERT surface expression and 5-HT uptake (Seimandi et al., 2013), although the relationship of this phosphatase to CaMKII interactions with SERT or functional modulation (e.g., efflux) has not been established. The binding site mapped for calcineurin is near the plasma membrane, whereas the expected interaction for CaMKII, based on DAT studies, is more distal; the work suggests that important insights related to SERT modulation are to be gained by further study of these Ca2+-dependent regulatory mechanisms.
3. Calmodulin-Dependent Protein Kinase II Regulation of Serotonin Transporter Phosphorylation.
Ramamoorthy et al. (1998) first explored the possibility of CaMKII regulation of SERT phosphorylation in examining the ability of KN-93 to inhibit SERT phosphoprylation triggered by incubation of transfected HEK-293 cells with okadaic acid, where no sensitivity was observed. Certainly the heterologous expression system used in these and the mode of phosphorylation activation, through phosphatase inhibition, do not allow one to rule out contributions in other contexts or with more physiologic stimuli. Studies examining SERT phosphorylation after calcineurin inhibition of brain preparations, for example, could reveal contributions of CaMKII not detected with okadaic acid treatments of transfected cells. Suspecting that SERT phosphorylation might contribute to the ability of CaMKII to modulate SERT electrogenicity, Ciccone et al. (2008) mutated a putative phosphorylation site in the SERT N terminus, Ser13, and found the mutant to elevate SERT-dependent Na+ flux without a change in 5-HT uptake and to lose sensitivity to KN-93. Importantly, recent work form Sorensen et al. (2014) demonstrated that CaMKII can phosphorylate Ser13 in the N terminus of SERT in vitro, adding evidence that the effects observed by Ciccone and colleagues with the Ser13Ala mutant may have derived from prevention of CaMKII phosphorylation at that site. Studies are now needed to extend these findings to an evaluation of the ability of CaMKII to support phosphorylation of SERT in native brain preparations and to understand the functional consequences of such modifications in vivo.
4. Calmodulin-Dependent Protein Kinase II Regulation of Serotonin Transporter Protein-Protein Interactions.
The principal protein-protein interaction linked to the regulation of SERT to date is that of syntaxin 1A. In their studies of SERT/syntaxin 1A interactions, Ciccone et al. (2008) provided evidence that the ability of KN-93 to induce SERT electrogenicity arises from a CaMKII-dependent inhibition of SERT/syntaxin 1A interactions that normally maintain 5-HT transport as an electroneutral event. Thus, KN-93 inhibited SERT charge flux only when syntaxin 1A is coexpressed with SERT and KN-93 diminishes recovery of syntaxin 1A in SERT coimmunoprecipitation experiments. Moreover, the SERT Ser13Ala mutant that results in KN-93 insensitivity also reduces syntaxin 1A interactions with the transporter. Together with the later findings of Steinkellner et al. (2015) who provided evidence of an interaction of CaMKII with the SERT C terminus, these studies lead to a model whereby CaMKII interactions with SERT support phosphorylation of the N terminus at Ser13 to support syntaxin 1A interactions. Thus CaMKII could serve to shift the transporter to a state supporting heightened membrane depolarization through 5-HT-dependent SERT currents. Because CaMKII interactions also support AMPH analog induced 5-HT efflux, further study of this process is likely to yield insights not only into the molecular details of presynaptic 5-HT homeostasis but also give insights into their subversion by psychotropic agents.
5. Receptor-Initiated Regulation of Serotonin by Calmodulin-Dependent Protein Kinase II.
At present, the evidence supporting CaMKII modulation of SERT function and protein interactions arises from studies where CaMKII is pharmacologically inhibited or lost through genetic ablation, and as such we lack an understanding of whether these features are under dynamic control as could be triggered through neural activity or receptor-linked mechanisms. Certainly membrane depolarization, leading to Ca2+ influx and CaMKII activation at nerve terminals, could engage these mechanisms, although evidence as yet to support such a contention is lacking. Ansah et al. (2003) found that presynaptic alpha2 adrenergic receptors, acting in a Ca2+-channel-dependent manner, could diminish 5-HT uptake in synaptosomes and diminish 5-HT clearance in vivo. A specific role for CaMKII was not investigated. However, kinetic studies indicated that the reduction in SERT activity was due to a regulation of 5-HT affinity, suggesting the engagement of a pathway linked to the alteration of transporter structure. Additionally, the alpha2 agonist UK14304 (brimonidine) significantly attenuated efflux of 5-HT from brain slices triggered by the AMPH analog fenfluramine, harkening back to data demonstrating a requirement for CaMKII in pCA-induced 5-HT release. Given the findings of others, one possible explanation for the Ansah et al. findings is that the alpha2 receptor, by inhibiting Ca2+ channel activation, reduces CaMKII activity, leading to changes in SERT conformation that attenuate fenfluramine-induced 5-HT efflux (and in this case, conformations that reduce 5-HT uptake and clearance).
For the kinases noted below, less comprehensive evaluations across all three monoamine transporters have been pursued and as such, we will not maintain subsection structures to discuss impact on function, trafficking, and associated proteins, etc.
VI. ERK1/2—Overview
ERK1 and ERK2 [sometimes noted as p44 and p42 mitogen activated protein kinase (MAPK), respectively] are two highly similar members of the MAPK family (85% sequence identity) that also includes c-Jun N-terminal kinase and p38 MAPK. Signaling by receptor tyrosine kinases and GPCRs through the small GTPase Ras, the Ser/Thr kinase Raf, and the dual specificity kinase MEK1/2 (MAPK kinase) leads to phosphorylation and activation of ERK1/2 (Roskoski, 2012). Inhibition of MEK1/2 is sufficient to block activation of ERK1/2 and is a strategy that is commonly used to implicate ERK1/2 in regulatory mechanisms. ERK1/2 inhibitors, such as SL327 (α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzeneacetonitrile], can also be used to study these kinases, although it should be noted that neither this strategy nor MEK1/2 inhibition, selectively targets one kinase over the other, so distinguishing the roles of the individual proteins is not possible with these tools. ERK1/2 are proline-directed kinases that prefer serines and threonines that are immediately followed by proline residues. These kinases are involved in activating a number of transcription factors and can also activate other kinases. As will be discussed below, ERK1/2 are also involved in the regulation of monoamine transporters.
A. Regulation of Dopamine Transporter by ERK1/2
Much of the work concerning ERK1/2 regulation of DAT relies on preventing their activation through inhibition of the upstream activators MEK1/2. Thus, Rothman et al. (2002) first noted the ability of the MEK1/2 inhibitor PD98059 (2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) to reduce DAT binding, as assessed by radioligand binding ([125I]RTI-55) in rat striatal synaptosomes. Consistent with these findings, Moron et al. (2003) demonstrated that MEK1/2 inhibition with U0126 (1,4-Diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene), as well as PD98059, reduced DA uptake in rat striatal synaptosomes, effects attributed to a reduction in DA uptake Vmax. Similarly, Lin et al. (2003) found U0126 to reduce DA uptake Vmax by heterologously expressed rat DAT. Both Moron et al. and Lin et al. provided evidence that DA uptake reductions after MEK1/2 inhibition derive from reduced DAT cell surface expression. This inhibition also increased colocalization between hDAT and clathrin light chain in HEK293 cells, and the reduction in DAT surface level was blocked by the clathrin inhibitor ConA (Moron et al., 2003). These results suggest that ERK1/2 activation downstream of MEK1/2 acts to stabilize DAT at the surface and antagonize internalization signals that trigger clathrin-mediated endocytosis of DAT. Providing evidence that ERK1/2 regulation could be bidirectional, Moron et al. (2003) observed that transfection of a constitutively-active MEK into human DAT-expressing HEK cells increased DA uptake Vmax. Finally, Owens et al. (2012) observed that in vivo administration of the ERK1/2 inhibitor SL327 to rat striatum reduced the rate of exogenous DA clearance as measured by high-speed chronoamperometry. Together, these results reveal MEK1/2 signaling, likely through ERK1/2, acts to sustain or elevate DAT surface expression and DA clearance capacity.
The target of ERK1/2 in regulating DAT is unknown at this point, although evidence has been provided to indicate that DAT phosphorylation may contribute. Lin and colleagues, using rat DAT transfected LLC-PK1 cells, observed a reduction in transporter phosphorylation after treatment of cells with U0126, suggesting that one or more phosphorylation sites on DAT may contribute positively to ERK1/2-dependent surface DAT trafficking. In an exploration of potential phosphorylation sites that could support changes in DA uptake produced by U0216 treatments, Ala substitutions yielded complex effects, with mutations noted that either augmented, inverted, or reversed uptake changes brought about by MEK inhibition. Although not all mutations were evaluated, substitutions at Ser12, Ser13, Ser62, Ser581, Thr591, and Thr612 were found to both block the reduction in basal phosphorylation arising from U0126 treatments as well as to diminish the inhibition seen on DA uptake. Gorentla et al. (2009), using purified N and C termini from rat DAT, observed significant phosphorylation of the N terminus with both ERK1 and 2 in vitro, with phosphoamino acid analysis indicating phosphorylation by ERK1 on Thr residues. Studies with mutant N termini revealed Thr53 as essential for phosphorylation by ERK1, consistent with the location of the residue in a motif for proline-directed protein kinases (PPQTP) that is conserved in human DAT. Importantly, further studies in this and a later report (Foster et al., 2012) provided evidence for the use of Thr53 as a phosphoacceptor site in rat striatum, leading to the development of a P-Thr53 specific antibody that reveals labeling after ERK1 incubations. Interestingly, PMA treatment of rDAT-LLCPK1 cells increased Thr53 phosphorylation, despite the fact that PKC was unable to phosphorylate this residue in vitro. This is consistent with the observation described earlier that PKCβ likely activates ERK1/2, and this may be the route through which PMA increases Thr53 phosphorylation. Additionally, mutation of this residue to alanine or aspartate resulted in a significant decrease in surface DAT and DA uptake in rDAT-LLCPK1 cells. Total DAT levels were also reduced to a similar extent as surface DAT, suggesting that this residue is necessary for stabilizing DAT protein levels. These mutations also abolished AMPH-induced MPP+ efflux in transfected cells, suggesting this residue is also critical to establish DA efflux conformations. As an equivalent loss of AMPH evoked efflux capacity was observed for both Ala and Asp substitutions, the precise role of phosphorylation at Thr53 in AMPH-induced DA efflux remains to be established.
The biologic relevance of ERK1/2 regulation of DAT is supported by findings that ERK1/2 signaling appears to be necessary for upregulation of DAT by D2/D3 receptors. Stimulation of DAT activity by activation of these receptors has been demonstrated both in vivo and in vitro (Cass and Gerhardt, 1994; Dickinson et al., 1999) and the dependence on ERK1/2 signaling was demonstrated by coexpressing either D2 or D3 with hDAT in EM4 cells and treating with the D2/3 agonists quinpirole or PD128907 [(4aR,10bR)-3,4a,4,10b-Tetrahydro-4-propyl-2H,5H-[1]benzopyrano-[4,3-b]-1,4-oxazin-9-ol hydrochloride] (Bolan et al., 2007; Zapata et al., 2007). These treatments increased uptake of the fluorescent DAT substrate ASP+ (which does not activate DA receptors), and this increase was blocked by PD98059, a MEK1/2 inhibitor. Importantly, inhibition of PI3K by LY294002 [2-(4-morpholino)-8-phenyl-4H-1-benzopyran-4-one] also blocked the increase in cells expressing the D3 receptor, suggesting perhaps some crosstalk between these signaling pathways occurs when D3 is activated. Indeed, it has been suggested that activation of ERK1/2 signaling by D3 activation depends on PI3K activation because of its utilization of the βγ pathway of Gi to signal through RTKs such as EGFR, as opposed to D2 receptor signaling that seems to act primarily through the alpha subunit of the Gi protein to activate ERK1/2 signaling (Beom et al., 2004). D3 activation is also different in that DA uptake is initially increased 5 minutes after quinpirole treatment, but actually decreased at a later 30-minute time point, possibly also due to activation of other signaling pathways as discussed above. ERK1/2 signaling downstream of D2 receptor activation after repeated AMPH exposure also appears able to compensate for reduced insulin/PI3K/Akt signaling in hypoinsulinemic rats and restore normal DAT activity (Owens et al., 2012). The researchers also showed that this repeated AMPH exposure increased pERK1/2, as would be expected based on the in vitro results discussed above, but they unfortunately did not address the dependence of the rescue of DAT function on ERK1/2 signaling. The authors did, however, demonstrate that inhibition of ERK1/2 alone decreased the rate of clearance of intrastriatally applied DA as measured by high-speed chronoamperometry, which is consistent with the in vitro and ex vivo results described above.
Another GPCR that signals through ERK, the kappa opiate receptor (KOR) has been found to modulate DAT function in an ERK1/2-dependent mechanism (Kivell et al., 2014). The KOR agonist salvinorin A has been found to elevate DAT uptake capacity in a pertussis toxin-sensitive and MEK1/2-dependent manner, measured in KOR/DAT cotransfected EM4 cells using ASP+ accumulation. Salvinorin A also increases DA clearance in striatal minces as assessed by rotating disk amperometry, effects blocked by PD98059, as well as DA uptake in rat striata synaptosomes. As seen with D2/D3 agonists, salvinorin also elevates surface expression of DAT, a likely mechanism supporting the increased DA transport Vmax. Lastly, coimmunoprecipitation and bioluminescence resonance energy transfer/FRET studies support the formation of a KOR/DAT protein complex. Whether this complex also harbors D2/D3 receptors or these receptor/transporter complexes are distinct remains to be determined. Although D2/D3 association with DAT is believe to be agonist independent, Kivell and colleagues found elevated associations with salvanorin exposure. Altogether, these studies provide significant evidence that the ERK1/2 pathway regulates DAT in native preparations downstream of GPCR activation, although the mechanism by which ERK1/2 acts to control DAT trafficking following receptor activation is ill-defined.
B. Regulation of Norepinephrine Transporter by ERK1/2
As of this writing, evidence of a role for ERK in rapid NET regulation is lacking. Although evidence supports signaling by AT1 receptors via ERK1/2 linked pathways (Yang and Raizada, 1999), and activation of these receptors rapidly elevate NET activity and surface expression (Lu et al., 1996), the involvement of MAPKs in these effects remains to be explored. However, Kivell et al. (2014) found that in cells transfected to express the KOR and human NET, no change in NET activity results from salvinorin A application, whereas as noted above a significant elevation of DA uptake occurs with the same agonist when the cells were transfected with DAT.
C. Regulation of Serotonin Transporter by ERK1/2
As with NET, little evidence exists to implicate ERK1/2-linked pathways in SERT regulation. Benmansour et al. (2014) observed that inhibition of MEK1/2 by PD98059 or U0126 blocked the slowing of 5-HT clearance induced by acute estradiol administration in rat hippocampus, providing evidence for a role of this pathway in rapidly downregulating SERT in vivo downstream of estrogen signaling. ERK1/2 may be involved in the regulation of SERT by 5-HT1B receptors. These receptors have been shown to increase 5-HT uptake in vitro as well as in vivo in rat hippocampus (Daws et al., 1999, 2000; Hagan et al., 2012; Montanez et al., 2014), and although it has not been demonstrated which signaling cascade exerts these effects, researchers have shown that 5-HT1B robustly activates ERK2 in CHO cells, and this activation is significantly greater than that of 5-HT1A receptors, which do not seem to impact SERT activity (Mendez et al., 1999). In studies examining the transporter specificity and ERK1/2 dependence of salvinorin A modulation of DAT via KORs, Kivell et al. (2014) found that cells cotransfected with KOR and SERT cDNAs supported salvinorin modulation of 5-HT uptake, but in a direction (inhibition) opposite to that seen with DAT (stimulation). The signaling cascade downstream of KOR activation leading to SERT downregulation has yet to be defined and as will be noted below, published work suggests a role for a different MAPK, p38 MAPK, in SERT regulation rather than ERK1/2 (Bruchas et al., 2011).
VII. p38 Mitogen-Activated Protein Kinase
p38 MAP kinases exist as four isoforms (α,β γ, and δ), although the α isoform is the most well-characterized and widely expressed (Cuadrado and Nebreda, 2010). p38 MAPK can be activated downstream of MKK3/6 (distinct from the MEKs for ERK1/2 and c-Jun N-terminal kinase) by multiple dimensions of cellular stress, including reactive oxygen species and inflammatory cytokines. Cytokine activation of p38 has received great attention by immunologists because of its critical role in mediating the effects of proinflammatory cytokines in multiple aspects of immune system function (Huang et al., 2009). The recent appreciation for the role of immune molecules in regulating brain function, however, has underscored the importance for p38 signaling in the CNS, including through regulation of MA transporter function. Studies looking at the role of p38 in regulating these transporters generally rely on the use of nonisoform specific inhibitors of p38 such as SB203580 [4-(4′-fluorophenyl)-2-(4′-methylsulfinylphenyl)-5- (4′-pyridyl)-imidazole], or activators such as anisomycin. Importantly, anisomycin also suppresses protein synthesis (Barros et al., 1997), and although the concentrations used to inhibit p38 are sufficiently low to prevent this effect, the dual actions of this drug require follow-up evaluation with p38 MAPK inhibitors for validation of any anisomycin effects. As with ERK1/2, p38 isoforms are also proline-directed, preferring serine and threonine residues followed by a proline residue.
A. Regulation of Dopamine Transporter by p38 Mitogen-Activated Protein Kinase
Whereas significant evidence implicates ERK1/2 in DAT regulation, little evidence supports a role for p38 MAPK linked pathways. Gorentla et al. (2009) did find that a rat DAT N-terminal peptide containing Thr53 can be phosphorylated in vitro by purified p38α MAPK; however, no evidence exists that this site is targeted by p38 MAPK in vivo. Indeed, p38 MAPK inhibition is without effect on KOR-mediated regulation of DAT, which as noted above is likely mediated by an ERK1/2 pathway. Zhu et al. (2005) found that the p38 MAPK activator anisomycin reduced DA uptake by human DAT in transfected CHO cells, effects suppressed by the p38 MAPK inhibitor SB203580. Interestingly, this effect is in the opposite direction seen for NET or DAT in the same cell host where uptake stimulation is seen (see below). Lin et al. (2003) found that the p38 MAPK inhibitor SB202190 failed to alter basal DA uptake of rat DAT transfected COS cells. However, several N-terminal Ala substitutions (Val14, Ser2, Ser4, Ser12, Ser13, Ser21, Ser45) exposed a capacity for SB202190-induced uptake stimulation, driven by an elevation in transport Vmax, suggesting that a DAT-inhibitory p38 MAPK regulatory pathway may be engaged by changes in DAT structure induced by other signals. Interestingly, these substitutions enhanced uptake inhibition by PI3K inhibition. These findings are reminiscent of studies by Prasad et al. (2005), who found that SERT coding variation can impart opposite effects on p38 MAPK and PKC-dependent 5-HT uptake modulation, suggesting a dynamic interplay between transporter regulatory pathways. Such an idea gains additional support from studies of Quick et al. (2004) on GAT1 GABA transporters where tyrosine kinase modulation of GAT1 is influenced by the state of PKC activation. Together, these findings warrant further investigation of a role for p38 MAPK in a more physiologic context and in relation to other regulatory signals. That latent regulation by a p38 MAPK pathway may explain the findings of van Heesch et al. (2014), who found that low dose (133 µg/kg) of lipopolysaccharide (LPS), an inflammatory agent, could rapidly induce changes in DA turnover suggestive of elevated DAT activity. Possibly related to these findings, Wu et al. (2014) found that injection of the proinflammatory cytokine TNF-α increased DAT levels. At a higher dose of LPS (1 mg/kg), Lai et al. (2009) observed a reduction in DAT protein levels in mouse striatum without an effect on VMAT2 levels. Although the signaling pathways, explanations for dose dependency, and mechanisms for these effects of LPS have not been worked out, Zhu et al. (2010) have implicated a p38 MAPK pathway in SERT modulation by peripheral LPS (see below).
B. Regulation of Norepinephrine Transporter by p38 Mitogen-Activated Protein Kinase
The first indications of a role for p38 MAPK-linked pathways in the modulation of NET arose from studies of the ability of insulin to elevate [3H]NE uptake in SK-N-SH cells. Apparsundaram et al. (2001) found that insulin inducted a time (detected by 30 minutes)- and dose-dependent elevation in NET activity, kinetically resolved as an increase in NE uptake Vmax. In keeping with the rapid action of insulin in this model, NET stimulation was insensitive to inhibitors of transcription or protein synthesis and was not accompanied by changes in transporter surface expression, as measured by whole cell [3H]nisoxetine binding or cell-surface biotinylation. Studies from the Klip laboratory indicated a role for p38 MAPK in insulin regulation of GLUT4 activity, where the kinase was found to further augment glucose uptake beyond what can be accounted for by surface trafficking, implying p38 MAPK-dependent catalytic activation (Sweeney et al., 1999). In the SK-N-SH model, insulin was found to activate p38 MAPK, effects blocked by SB203580. This same inhibitor also effectively blocked insulin stimulation of NET activity, whereas the inactive analog SB202474 was without effect. Tests with inhibitors of other ERK1/2-linked pathways also failed to reduce insulin effects. Activation of p38 MAPK has been found to lead to activation of PP2A activity (Westermarck et al., 2001), and the phosphatase has been shown to associate with NET (Bauman et al., 2000). Consistent with a model whereby activation of PP2A by p38 MAPK supports insulin-stimulated NET activity, okadaic acid and calyculin A both blocked NET stimulation, whereas the PP1 inhibitor tautomycin and the calcineurin (PP2B) inhibitor cyclosporine lacked activity. Further studies in the model have not been pursued, and thus we do not know whether these effects are mediated through changes in NET phosphorylation, although one can speculate that activation of PP2A by p38 MAPK may dephosphorylate one or more sites critical for shifting NET to an inactive conformation. Zhu and colleagues, in their exploration of the ability of inflammatory cytokines to elevate SERT activity via p38 MAPK pathways, tested CHO cells transfected with human NET for sensitivity to anisomycin, observing a rapid, SB203580-sensitive elevation of transport activity, consistent with the Apparsundaram et al. studies and demonstrating that the regulatory control of NET by p38 MAPK pathways could be reproduced after heterologous expression. More recently, Mannangatti et al. (2011) reported that p38 MAPK-linked pathways are involved in rapid upregulation of NET activity in rat prefrontal cortex synaptosomes induced by intraperitoneal cocaine. NET activity stimulation was paralleled by elevated synaptosomal p38 MAPK phosphorylation, which may involve a broader effect of the psychostimulant given that noradrenergic terminal contributions to these synaptosomes is likely modest. In vitro treatment of synaptosomes with cocaine reproduced the effects of intraperitoneal cocaine on NET activity, with additional evidence indicating an elevation in transporter surface expression mediated by reduced constitutive endocytosis as well as an elevation of NET phosphorylation. Further studies by the group in transfected human trophoblast cells led to the detection of a requirement for Thr30 in the ability of cocaine to both elevate NET activity and phosphorylation and reduce endocytosis. Most recently, this group demonstrated that p38 inhibition by SB203580 blocked cocaine-induced NET stimulation and phosphorylation in mouse prefrontal cortex synaptosomes, and intraperitoneal injection of SB203580 also blocked cocaine-induced NET upregulation and reduced cocaine sensitization and CPP (Mannangatti et al., 2015). Finally, intravenous injection of a TAT peptide containing a NET N-terminal fragment including Thr30, but not one with a T30A substitution, completely blocked cocaine-mediated NET upregulation and phosphorylation and significantly blunted cocaine CPP. These comprehensive studies identify an important role for p38 MAPK-linked pathways in the trafficking of NET induced by cocaine and suggest that posttranslational modifications of the NET N terminus can impede interactions of proteins required to drive transporter endocytosis. How cocaine triggers NET phosphorylation remains to be defined, but may involve cocaine stabilization of transporter conformations more readily phosphorylated by basal p38 MAPK activity or conformations that destabilize PP2A/NET interactions, reducing transporter dephosphorylation, with p38 MAPK dependence arising indirectly due to a requirement for kinase activation to elevate PP2A activity. Perhaps even more curious is the ability of cocaine to elevate p38 MAPK phosphorylation, though it is not clear as of yet whether this effect derives from NET interactions or through an independent mechanism. In summary, evidence exists to support both trafficking-dependent and -independent regulation of NET by p38 MAPK-linked pathways, depending on the system under study and the mode of activation of p38 MAPK.
C. Regulation of Serotonin Transporter by p38 Mitogen-Activated Protein Kinase
As with NET, studies have linked p38 MAPK pathways to both trafficking-dependent and -independent modes of SERT regulation. The first evidence of a connection between kinase and transporter arose from studies by Zhu et al. (2004a) who, in the course of their investigations of A3AR modulation of SERT via PKG-linked pathways in cultured RBL-2H3 cells, obtained evidence that receptor activation led to elevated levels of activated p38 MAPK, whereas p38 MAPK inhibitor blocked stimulation of SERT activity. In a later paper, the group found that A3ARs in mouse midbrain, hippocampal, and cortical synaptosomes could elevate SERT activity in a p38 MAPK-dependent manner, adding evidence to the physiologic relevance of the findings from RBL-2H3 cells (Zhu et al., 2007). Because p38 MAPK inhibition blocked NECA stimulation of 5-HT uptake but did not block NECA-induced elevation of surface SERT, this group proposed a model whereby A3AR activation signals through both PKG and p38 MAPK, with activation of the PKG pathway tied to transporter trafficking and catalytic activation, with PKG activation of p38 MAPK responsible for the latter, activity-modulating step. Consistent with these data, direct activation of p38 MAPK with anisomycin has been found to elevate 5-HT uptake in RBL-2H3 cells (Zhu et al., 2005), in serotonergic RN46A cells (Zhu et al., 2005), in human SERT transfected HeLa cells (Prasad et al., 2005, 2009), in mouse synaptosomes (Zhu et al., 2006), in human platelets (Zhu et al., 2005), and in human lymphoblastoid cells (Sutcliffe et al., 2005). We note, however, that another group failed to replicate these effects using rat synaptosomes or rat cDNA transfected cells (Andreetta et al., 2013). Species differences would not appear to be involved in this discrepancy, because RBL-2H3 and RN46A cells are both rat derived. We found that in vitro studies of p38 MAPK regulation of SERT are very sensitive to sample handling, cell host, and expression level. In both RBL-2H3 and transfected HeLa cells, the stimulatory activity of anisomycin was found to arise from a reduction in 5-HT KM with no change in 5-HT Vmax nor a change in SERT surface expression. These findings support a model where p38 MAPK activation leads to an increase in the probability that SERT adopts a conformation with high 5-HT affinity (Steiner et al., 2008). Consistent with this idea, anisomycin treatment of RN46A cells leads to enhanced potency of 5-HT to antagonize [125I]RTI-55 binding (Zhu et al., 2005).
Discovery of a regulation by SERT by p38 MAPK as a component of PKG-linked transporter regulation raises the question as to whether cell surface receptors can bypass PKG activation to activate p38 MAPK signaling and thereby influence SERT without attendant changes in surface expression. Inflammatory cytokines are well known to signal through receptor-activated p38 MAPK signaling (Huang et al., 2009). Using RN46A cells, Zhu et al. (2006) established that both IL-1β and TNFα could replicate the effects of anisomycin in inducing a rapid (peak at 20 minutes) increase in SERT activity in parallel with p38 MAPK activation. The increase in SERT activity produced by IL-1β can be blocked by SB203580 (effects are partially antagonized) and is nonadditive with anisomycin effects, consistent with action through a common pathway. Saturation analysis of IL-1β effects also revealed a shift in the 5-HT KM with no change in transport Vmax. Interestingly, TNFα induced a change in both KM and Vmax, denoting a more complex mechanism of SERT regulation, part of which is p38 MAPK dependent. Importantly, both agents exerted SERT stimulatory actions in mouse midbrain and striatal synaptosomes similar to those seen in RN46A cells, with effects blocked (or blunted in the case of TNFα) by SB203580. The IL-1R peptide inhibitor (IL-1Ra) fully antagonized IL-1β effects on SERT in synaptosomes, whereas anisomycin effects were insensitive to this agent, consistent with a more direct mode of action of p38 MAPK activation by anisomycin. Importantly, IL-1β stimulation of SERT activity is absent in synaptosomes prepared from IL-1R KO mice (Zhu et al., 2010). Moreover, peripheral injection of lipopolysaccharide (LPS) was found to elevate CNS SERT activity, effects lost with intraperitoneal SB203580 coinjection or using IL-1R KO mice and paralleled by an increase 5-HT clearance rates in vivo (Zhu et al., 2010). Finally, several behavioral effects that attend peripheral LPS administration (immobility in the tail suspension and forced swim tests) were absent in lost in both IL-1R and SERT KO mice or with SB203580 injections, suggesting that inflammatory regulation of SERT via p38 MAPK pathways may induce depression-like behavior. In this regard, Baganz et al. (2015) recently demonstrated that p38α MAPK is required in 5-HT neurons to support both activation of brain SERT and anxiety and depression-like behaviors after an intraperitoneal LPS injection. These studies provide definitive evidence that the α isoform of p38 MAPK is engaged in SERT regulation by immune stimuli in vivo and identify mechanisms by which peripheral inflammatory insults can change behavior through alterations in 5-HT signaling (see also Baganz and Blakely, 2013).
Evidence for trafficking-independent control of SERT activity downstream of IL-1R/p38 MAPK activation has been provided at the single molecule level by Chang et al. (2012), who used SERT antagonist-conjugated quantum dots (Qdots) to monitor SERT mobility on the surface of RN46A cells. These investigators found that either 8-Br-cGMP or IL-1β treatment increased the lateral diffusion of SERT on the cell surface, effects blocked by p38 MAPK inhibitor. A similar effect was observed after actin destabilization by CytoD, as well as after treatment with a SERT C-terminal peptide, suggesting this domain is required for interactions with the actin cytoskeleton. Importantly, this peptide also led to an increase in 5-HT uptake. The authors proposed a model involving p38 MAPK signaling as regulating the tethering of SERT to the actin cytoskeleton with untethering allowing SERT to change conformations and more readily enter a state of enhanced activity. Whether these effects derive from direct phosphorylation of SERT by p38 MAPK is not yet clear. Recently, Sorensen et al. (2014) reported that purified p38 MAPK can phosphorylate a peptide containing SERT Thr616, a canonical site for praline-directed kinases like p38 MAPK, in vitro. Curiously, mutation of this residue in full-length human SERT to either Ala or Asp decreased the Vmax of 5-HT uptake to ∼60–70% and reduced transporter surface expression, as determined by whole cell binding experiments. Although these studies are difficult to interpret given the equivalent impact of substitutions designed to obviate or mimic phosphorylation, they suggest that further studies of the Thr616 site in more native settings may shed light on modulation of SERT by p38 MAPK.
Whereas significant evidence supports a trafficking-independent mode of SERT regulation downstream of IL-1R/p38 MAPK activation, other studies have provided evidence that the kinase can also impact mechanisms that dictate SERT surface density. Thus, Samuvel et al. (2005) found that treatment of rat midbrain synaptosomes with the p38 MAPK inhibitor PD169316 (as well as SB203580) leads to a rapid (2.5–10 minutes) reduction in 5-HT uptake that is supported kinetically by a transport Vmax reduction and molecularly through a reduction of surface transporter protein. The pathway through which the reduction in surface expression arises was found to be distinct from that supporting phorbol ester-induced SERT endocytosis, although both PD169316 and phorbol ester treatments result in reductions in PP2Ac and syntaxin 1A associations. Interestingly, whereas phorbol ester elevates SERT phosphorylation, p38 MAPK inhibitor treatment led to a reduction in basal SERT phosphorylation. Indeed, p38 MAPK would appear to be the major determinant of basal SERT phosphorylation in synaptosomes. Moreover, p38 MAPK inhibition reduced AMPH-induced SERT phosphorylation. These findings indicate that basal p38 MAPK is required to sustain steady-state levels of SERT protein on the surface and the mechanisms involved are not equivalent to those acting to modulate SERT trafficking by PKC. Consistent with this idea, these authors found no change in membrane microdomain localization with p38 MAPK inhibitor treatment and transporter endocytosis was not enhanced, as it is with phorbol ester treatment. The latter finding indicates that basal levels of p38 MAPK activity are needed to support transporter trafficking to the cell surface. Lau et al. (2009) reached the same conclusion in their study of surface SERT expression in mouse stem cell-derived serotonergic neurons.
Bruchas et al. (2011) have provided evidence that p38 MAPK-dependent changes in SERT trafficking support long-term behavioral changes that arise from social defeat stress or repeated, forced swim stress. Thus, these investigators detected elevated SERT activity and surface levels in whole brain and midbrain synaptosomes after social defeat stress or repeated, forced swim stress, along with a decrease in social interaction scores, increased forced swim immobility time, and increased cocaine conditioned place preference (Schindler et al., 2012). Notably, when mice with 5-HT neuron-specific deletion of p38α MAPK were exposed to these stressors, the changes in uptake and SERT trafficking as well as behavior were lost. An inhibitor of the kappa opioid receptor, norBNI, also blocked these effects and the kappa opioid receptor agonist U50,488 (2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide) increased SERT activity and surface levels, an effect that absent in 5-HT neuron-specific p38α MAPK knockout mice. These authors propose that stress induces dynorphin release to trigger behavioral changes through a kappa receptor/p38α MAPK pathway that elevates SERT trafficking and reduces extracellular 5-HT availability. These studies, as well as those linking LPS-induced anxiety and depressive behaviors to p38 regulation of catalytic SERT modulation, suggest that targeting p38α MAPK may be a useful strategy in treating disorders normally treated through the use of SERT antagonists.
Additional evidence for the disease relevance of p38 MAPK linked SERT regulation arises from studies of SERT mutations found in subjects with autism spectrum disorder (ASD). Sutcliffe et al. (2005) identified five coding variants in subjects diagnosed with ASD. A common finding with these variants is a constitutive increase in 5-HT uptake activity when expressed in HeLa cells (Prasad et al., 2009; Ye and Blakely, 2011). Moreover, for several of these variants, their activity cannot be further increased by p38 MAPK activation (Prasad et al., 2005, 2009), suggesting these variants may be constitutively poised in the state normally produced after p38 MAPK activation. Expression of SERT molecules with constitutively elevated 5-HT clearance could drive developmental onset disorders linked to 5-HT such as ASD owing to the early embryonic expression of 5-HT and SERT (Daws and Gould, 2011). Recently, a mouse with a knock-in mutation of the most common of these variants, Ala56, was generated and found to exhibit behavioral perturbations reminiscent of ASD (Veenstra-VanderWeele et al., 2012). Importantly, the Ala56 variant not only conferred elevated SERT activity in vivo but also the SERT protein was found to exhibit elevated p38 MAPK dependent SERT phosphorylation. These studies raise the possibility that attenuation of the activity of p38α MAPK or intervention in the pathway through which it regulates SERT may reduce ASD risk or symptoms.
VIII. Phosphatidylinositol 3-Kinase (PI3K)/Akt—Overview
PI3K is a lipid kinase composed of kinase (p110) and anchoring subunits (p85) that generates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) from phosphatidylinositol (4,5)-bisphosphate (PIP2), and this PIP3 recruits Akt [also known as protein kinase B (PKB)] to the membrane via binding through the Akt pleckstrin homology domain (Reckamp, 2012). These actions of PI3K are opposed by the membrane bound lipid phosphatase PTEN, which catalyzes the reverse reaction (PIP3 to PIP2) and thus reduces Akt membrane recruitment. Upon membrane binding of Akt, this kinase is then activated through dual-phosphorylation by the mTORC2 complex at Ser473 and by the Ser/Thr kinase PDK1 at Thr308 (Hemmings and Restuccia, 2012). Akt exists in three isoforms in mammals, Akt1, Akt2, and Akt3, each of which have many known downstream targets that regulate cell growth and protein synthesis. The PI3K/Akt signaling pathway is activated by a number of receptors, including receptor tyrosine kinases (RTKs) such as the insulin receptor through activation of PI3K by IRS-1 and Ras and GPCRs through activation by Gβγ subunits. As will be described below, these signaling pathways appear to play critical roles in regulating monoamine transporters.
A. Phosphatidylinositol 3-Kinase/Akt Regulation of Dopamine Transporter
With respect to pathways that appear to sustain normal DAT activity, signaling through PI3K/Akt is one of the best characterized, including studies in both transfected cells and native preparations, using both inhibitors and activators. For instance, Carvelli et al. (2002) observed that acute application of the PI3K inhibitor LY294002 led to a decreased DA uptake Vmax in rat striatal synaptosomes and in human DAT transfected HEK-293 cells, effects accompanied by a reduction in DAT cell surface expression. Conversely, overexpression of a constitutively active PI3K in HEK-293 cells increased both DA uptake and DAT surface levels. In their studies of kinase inhibitors and activators, Lin et al. (2003) also observed inhibitory actions of LY294002 on rat DAT activity and surface expression in transfected cells, findings accompanied by a reduction in DAT phosphorylation. This decrease was absent in T62A, S581A, and T612A mutants, however, suggesting that one or more of these sites may be the target of a kinase downstream of PI3K signaling. Importantly, these mutations also blocked the effect of LY294002 on reducing the Vmax of DA uptake, suggesting that phosphorylation of one or more of these residues may be required for the effects of PI3K signaling on DAT activity, although indirect structural explanations cannot as yet be ruled out. As noted above, activation of PI3K and production of PIP3 leads to recruitment and activation of Akt. Studies in multiple preparations support a role for Akt in PI3K-dependent support of basal DAT surface expression and activity. Garcia et al. (2005) observed that both a pharmacological inhibitor of Akt (ML9) and transfection of a dominant-negative Akt (K179R) reduced DAT surface expression and DA uptake in DAT transfected HEK-293 cells. In vivo, viral overexpression of the Akt activator IRS2 in the substantia nigra of rats that had been maintained on high-fat diets alleviated some DAT-related deficits seen in these rats (Speed et al., 2011). These included restoration of reduced striatal DAT surface levels and DA uptake. Importantly, this treatment also rescued reduced pAkt levels seen in these rats. Although an explicit mechanism by which PI3K/Akt leads supports DAT surface expression remains to be defined, the initial studies by Carvelli et al. on the effects of LY294002 revealed protection against transporter surface reductions with either ConA to inhibit clathrin-mediated endocytosis or a dynamin dominant negative construct, suggesting that PI3K/Akt promotes enhanced surface delivery of DAT that is normally balanced by basal, dynamin-dependent transporter endocytosis. Gorentla et al. (2009) demonstrated that Akt1 was unable to phosphorylate the DAT N terminus in vitro, suggesting that either another kinase may act downstream of Akt or the N terminus is not the target for Akt phosphorylation of DAT. Another possible explanation is that Akt2, rather than Akt1, may function downstream of PI3K to exert its effects on DAT activity and phosphorylation. This is supported by the findings of Speed et al. (2010) that showed that inhibition of Akt2, and not Akt1, reduced DAT surface levels in rat striatal tissue.
Support for DAT surface trafficking by the PI3K/Akt pathway would also be expected to be seen in studies of AMPH-induced DA efflux. Lute et al. (2008) demonstrated that LY294002 reduced the capacity of cultured DA neurons to support AMPH-induced DA efflux. Biotinylation studies and measurements of DAT transient currents that correlate with cell surface transporter expression yielded evidence that these effects were supported by transporter redistribution. Consistent with these studies, inhibition of either PI3K or Akt in vivo in mice via direct application of inhibitors to the striatum has been found to reduce AMPH-evoked DA release as measured by high-speed chronoamperometry (Williams et al., 2007; Speed et al., 2011). Similarly, viral overexpression of the Akt activator IRS2 in the substantia nigra of high fat-fed rats alleviated the reduction in AMPH-induced hyperlocomotion. AMPH-induced reductions in cell surface expression also appear to be mediated via the Akt pathway. Thus, Wei et al. (2007) found that AMPH produced a time-dependent reduction in activated Akt, effects blocked by cocaine and thus DAT mediated. Evidence presented indicates that AMPH induced an increase in CaMKII activity that in turn reduced Akt activity, leading to reduced DAT surface expression that could be offset with the CaMKII inhibitor KN-93.
Whereas the studies noted above indicating PI3K/Akt support for DAT surface expression are important from a fundamental perspective, they gain in importance from the evidence that PI3K/Akt pathways appear to regulate DAT as a consequence of insulin receptor activation and underlie effects of the hormone on various aspects of appetitive behavior and psychostimulant action (Daws et al., 2011; Niswender et al., 2011). Early studies by Figlewicz et al. (1994) demonstrated that DAT mRNA levels in the substantia nigra and ventral tegmental area could be increased by intracerebroventricular injection of insulin, findings supported by detection of insulin receptor expression in these nuclei (Figlewicz et al., 2003) and elevations of DAT mRNA in the hyperinsulinemic Zucker rat (Figlewicz et al., 1998). A rapid action of insulin on DAT was first noted using ex vivo analysis of DA clearance in brain preparations with rotating disk voltammetry, where the hormone was found to normalize reductions in DAT-mediated DA clearance observed with fasting (hypoinsulinemia) (Patterson et al., 1998). In their studies of PI3K-dependent regulation of DAT in transfected cells, Carvelli et al. (2002) found that insulin exposure increased DAT-mediated DA uptake and blocked AMPH-induced DAT internalization. Subsequent studies with diabetic rodents or animals subjected to diet-induced obesity have provided compelling data that alterations of peripheral insulin production or action are monitored centrally and transduced by CNS insulin receptor/PI3K/Akt pathways in DA neurons, with a major effector being DAT. Thus animals made diabetic (e.g., with streptozotocin) demonstrate decreased CNS DA uptake and amphetamine self-administration (Galici et al., 2003), reduced DAT-mediated DA clearance and locomotion (Owens et al., 2005; Sevak et al., 2007), and reduced fMRI bold responses to AMPH (Williams et al., 2007). Speed et al. (2010) showed that raising rats on high-fat diets reduces Akt activation and DAT surface expression, reducing the locomotor actions of AMPH. Although most studies to date have concentrated on a role for insulin modulation of DAT in presynaptic terminals, Mebel et al. (2012) provided evidence that a component of insulin’s ability to regulate behavior is via VTA somatodendritic regulation of DAT-mediated DA clearance. Together, these studies paint a compelling picture that peripheral metabolic state and feeding behavior, as well as other DA-linked processes, are coordinated significantly through a CNS insulin/PI3K/Akt/DAT pathway.
A potential contribution of PI3K signaling to DAT regulation in the realm of functional activity modulation has recently come to light by way of findings of a physical and functional interaction of PIP2 with the transporter. Hamilton et al. (2014) provided the first evidence for interactions of PIP2 with DAT via coimmunoprecipitation studies using either transfected cells or brain extracts. Consistent with findings of PIP2 interactions with the SERT N terminus (see below), these authors found that the DAT N terminus interacts with PIP2, mediated by electrostatic interactions involving residues K3 and K5. Using a PIP2 sequestration approach, the group provided evidence in transfected cells that PIP2-DAT interactions are critical for AMPH-induced DA efflux, whereas inward DA transport was unaffected. Finally, using an elegant DAT restoration approach in Drosophila melanogaster lacking DAT, these investigators observed that the K/A mutant DAT demonstrated reduced recovery of AMPH-induced locomotion compared with wild-type DAT. These findings indicate that mechanisms that can deplete or augment PIP2 levels in vivo could influence DAT functional states, as read out via AMPH responsiveness. Although the PI3K pathway was not evaluated in these studies, studies with a Gq-coupled M1 acetylcholine receptor that can deplete PIP2 levels provide some support for this idea. Regardless, these studies, along with recent efforts to model PIP2/DAT interactions (Khelashvili et al., 2015), support an increasingly complex and dynamic contribution by which the N terminus coordinates both lipid and protein interactions to regulate the transporter.
B. Phosphatidylinositol 3-Kinase Regulation of Norepinephrine Transporter
To our knowledge, the first implication of a role for PI3K-linked pathways in the regulation of NET comes from studies of Apparsundaram et al. (2001) who observed, using cultured SK-N-SH cells, that the structurally distinct PI-3K inhibitors wortmannin and LY294002 inhibited NE uptake activity in a dose- and time-dependent manner, with effects seen within 20–30 minutes of drug treatment. These effects were accompanied by reduced whole cell [3H]nixoxetine binding (but not total membrane binding), suggesting support for basal NET trafficking. Previously, Uchida et al. (1998) showed that wortmannin could suppress the enhancement of NE uptake that occurs with restoration of Ca2+ to PC12 cells, although this effect was attributed to an effect on myosin light chain kinase.
Contrasting with these studies using pharmacological modulators, studies with insulin signaling through PI-3K/Akt paint a more complex picture. Apparsundaram et al. (2001) found that insulin could rapidly stimulate NE uptake in SK-N-SH cells, effects blocked by PI-3K inhibitors (wortmannin and LY294002) as well as by the p38 MAPK inhibitor SB203580, and as previously noted, this effect did not occur with evidence of an increase in overt changes in transporter surface expression, implicating catalytic activation. The physiologic significance of these findings remains unexplored and they stand in contrast to earlier studies by Boyd et al. (1985), who found that insulin rapidly (10 minutes) reduced NE uptake activity in rat whole brain primary cultures. The authors argued in subsequent studies that the NE uptake monitored in these cultures derived from NET due to sensitivity to Na+ depletion and the uptake inhibitors maprotiline and desipramine (Boyd et al., 1986). Although a dissection of signaling pathways supporting these insulin actions was not explored, the group later reported that insulin stimulates PI3K activity in this preparation (Patel et al., 1993). Consistent with these findings, Figlewicz et al. (1993) reported that insulin could rapidly (20 minutes) reduce NE uptake in PC-12 cells, whereas the related peptide IGF-1 did not. This group went on to show that intracerebroventricular insulin reduced NET mRNA levels in contrast to the elevation observed for DAT mRNA expression (Figlewicz et al., 1994). More recently, Robertson et al. (2010) demonstrated that insulin applied to hippocampal slices rapidly reduces NET activity, effects supported by a reduction in surface expression measured by biotinylation. The group also used SCG cultures where NET trafficking could be studied with optical approaches in individual boutons, finding again that insulin reduced transporter density at the plasma membrane and increased colocalization with the endosomal marker rab11a. Similar studies were seen with hNET transfected CHO cells. Although PI-3K was not investigated, the group employed pharmacological and kinase dead dominant negative construct to determine a requirement for Akt signaling to support insulin effects in the CHO model. Both Akt1 and Akt2 could be colocalized with NET in hippocampal slices and insulin was shown to activate Akt in the preparation through phospo-kinase blots. Importantly, specific inhibitors of either Akt1 or Akt2 blocked insulin effects and even appeared to lead to elevated NET surface expression, attesting to a key role of this pathway and likely redundancy of kinase isoforms. Finally, the group used in vivo chronoamperometry to show that streptozotocin-induced diabetes results in an enhanced rate of NE clearance in vivo, effects that can be restored by local insulin application. Hippocampal NE levels were found to be elevated without a change in TH or DBH levels, suggesting that changes in NET may drive augmented recovery and storage of NE. An intriguing linkage of the findings of Akt/NET interactions to psychiatric disorders was pursued based on the ability of the antipsychotic clozapine to activate Akt. As predicted by the more direct studies, the group found that clozapine reduced surface NET levels.
The Galli group has gone on to pursue pathways through which Akt may modulate NET, implicating the mTORC2 complex (Siuta et al., 2010). In these studies, the mTORC2 regulatory protein rictor was eliminated in neurons and found to impair Akt activation. These mice have deficits in prepulse inhibition, a feature of subjects with schizophrenia. Moreover, they demonstrate elevated NET expression and cortical hypodopaminergia, consistent with NET as a major clearance mechanism for DA in the cortex. Importantly, the NET inhibitor nisoxetine reversed PPI deficits. Altogether, these studies point to the possibility that changes in NE and DA signaling seen in schizophrenia (and other neuropsychiatric disorders) may have an important component driven by altered Akt modulation of both NET and DAT, respectively.
C. Phosphatidylinositol 3-Kinase /Akt Regulation of Serotonin Transporter
Investigations of the role of PI3K linked pathways in the rapid regulation of SERT are far less numerous than for DAT or NET. One way in which PI3K may regulate SERT activity is through control of PIP2 levels, which, as with DAT, interacts with the SERT N terminus and regulates PCA-induced 5-HT efflux (Buchmayer et al., 2013). Manipulation of this interaction did not appear to affect 5-HT uptake, however. A recent in vivo study using chronoamperometry to monitor SERT activity in the hippocampus of ovariectomized rats provided evidence that estradiol via the alpha isoform of the estrogen receptor can rapidly diminish the ability of fluoxetine to inhibit 5-HT clearance (Benmansour et al., 2014), effects that can be reversed by local application of wortmannin or LY294002. These drugs did not impact clearance rates per se, only sensitivity to fluoxetine, suggesting an effect on SERT conformations required to bind antagonist or induction of fluoxetine-insensitive transporters. Although a mechanism by which these effects arise is as yet unclear, the ability of fluvoxamine to decrease 5-HT clearance after estradiol treatment was also restored by blockade of IGF-1 and mGluR1, suggesting these receptors may act upstream of PI3K. Ramamoorthy’s group recently reported that Akt X, an antagonist of Akt activation, reduces 5-HT transport activity in a time- and dose-dependent manner, with effects seen as early as 5 minutes (Rajamanickam et al., 2015). siRNA knockdown of Akt (isoform not specified) also reduced SERT activity, as did small molecule inhibitors of both Akt1 and Akt2. Kinetic studies indicate that the effects on SERT activity of Akt inhibition derive from a reduced transport Vmax, with biotinylation studies supporting diminished SERT surface expression that is established by reduced trafficking of transporters to the cell surface. These findings are reminiscent of the positive support for basal DAT surface expression by PI3K/Akt pathways. The effects of Akt inhibition on basal SERT activity and trafficking may arise through GSK3β activation as GSK antagonists lead to augmented SERT activity. Interestingly, Akt X also causes a significant reduction in basal SERT phosphorylation. Whether Akt regulation of SERT occurs in native preparations, whether it is controlled by cell surface receptors, and the nature of the sites supporting Akt-sensitive SERT phosphorylation are unknown. As reported above, p38 MAPK pathways contribute significantly to the control of basal SERT phosphorylation, and thus the possibility of convergence of Akt and p38 MAPK pathways regulating SERT may be fruitful to explore in the coming years.
IV. Tyrosine Kinases
Tyrosine kinases (TKs) fall into two classes: receptor tyrosine kinases (RTKs) and intracellular tyrosine kinases, which can be either cytosolic or membrane anchored. RTKs can be activated by many different types of ligands such as neurotrophic factors such as BDNF for TrkB and GDNF for Ret, or hormones such as insulin, and this ligand binding results in dimerization of receptor subunits that induces activating autophosphorylation (Huang and Reichardt, 2003; Hemmati et al., 2014). This initiates a complex signaling cascade involving small GTPases and both Ser/Thr kinases as well as intracellular tyrosine kinases such as Src-family kinases that ultimately can impact transporter expression, trafficking, or activity. Research demonstrating a role for RTKs kinases in regulating MA transporters has generally depended on activation of RTKs with their respective endogenous ligands, although studies have also appeared where TKs, either RTKs or intracellular tyrosine kinases, have been implicated via the impact of TK inhibitors such as genistein and tyrphostin 23/25 or exposure to Tyr-phosphatase inhibitors such as vanadate. Inhibitor-based approaches are much less specific, because they do not generally reveal the identity of the kinase mediating observed effects. As mentioned in previous sections, RTK activation could drive Ser/Thr phosphorylation of transporters through the activation of transporter-targeting Ser/Thr kinases such as PI3K/Akt via insulin signaling, although actions through intracellular tyrosine kinases such as Src-family kinases are also supported by the literature. Although clear examples of transporter regulation initiated by tyrosine kinases exist—some that have been already discussed—this area has received much more limited attention than transporter regulation by Ser/Thr directed protein kinases but likely will expand in coming years, triggered by findings we will allude to below. Because of the small number of studies in this area, we will review these efforts more generally.
A. Pharmacological Evidence for Tyrosine Kinase Modulation of Monoamine Transporter Activity
The first studies exploring a potential role for TK in MA transporter regulation arose from the studies of Helmeste and Tang (1995) who examined the actions of TK inhibitors on human platelet SERT activity and antagonist binding. These studies revealed rapid inhibition of 5-HT transport after in vitro treatments with genistein and methyl 2,5-dihydroxysuccinamate without comparable changes in inhibitor binding, consistent with either activity or trafficking effects. Aguilera's group pursued findings that treatment of rat brain synaptosomes with tetanus toxin (TeTx) or its HC fragment leads to noncompetitive reductions in 5-HT uptake (Inserte et al., 1999), effects suggested to be independent of the toxin’s well know inhibition of vesicular secretion (Najib et al., 1999). In cultured neuron studies, the same group reported that genistein inhibited 5-HT uptake, effects that were not additive with effects of TeTx, whereas vanadate elevated transport function, effects that were sensitive to TeTx inhibition (Pelliccioni et al., 2001). Unfortunately, although the toxin is thought to enter cells via interactions with cell surface gangliosides, possibly associated with synaptotagmin proteins (Rummel et al., 2007), the specific target for these SERT regulatory effects remains elusive, limiting further progress (Calvo et al., 2012), although evidence of SERT P-Tyr labeling has been reported (see below). With respect to intracellular Tyr kinases, Zarpellon et al. (2008) reported that the Src inhibitors SU6656 [(3Z)-N,N-Dimethyl-2-oxo-3-(4,5,6,7-tetrahydro-1H-indol-2-ylmethylidene)-2,3-dihydro-1H-indole-5-sulfonamide] and PP2 rapidly (minutes) inhibit 5-HT uptake in human platelet suspensions, whereas vanadate increased uptake. These effects were retained in reserpine-treated platelets, suggesting they derived from SERT modulation versus an effect on vesicular uptake or storage. Annamalai et al. (2012) also observed PP2-induced inhibition of SERT in rat platelets, although these effects were only evident with 24-hour treatments compared with 30-minute treatments, whereas vanadate-induced elevations in SERT activity were evident at both time points. These authors also reported dose- and time-dependent inhibition of SERT activity in transfected HTR cells, with effects evident at 6 hours posttreatment. Kinetic studies indicated Vmax reductions, consistent with protein stability reductions, detected as an increase loss over time of the total recycling pool of biotinylated SERT proteins. How these findings in rat platelets and transfected cells relate to the more rapid effects detected in human platelets is as yet unclear and should be further pursued. Rapid (minutes) inhibitory effects on 5-HT uptake have also been reported after treatment of human platelets with inhibitors of the intracellular TK Syk (Pavanetto et al., 2011), suggesting that SERT proteins, at least in platelets, may be the target of multiple cytosolic TKs. A reduced 5-HT uptake Vmax was reported after Syk inhibition in the latter study, which the authors attribute to stabilization of the transporter in an inward-facing conformation, due to a loss of imipramine binding sites, although the whole cell nature of the binding studies precludes elimination of SERT internalization as a possible explanation.
With respect to NET, only two studies to our knowledge have reported on effects of TK inhibitors on NET activity, with conflicting findings. Apparsundaram et al. (2001) reported reductions of basal and insulin-stimulated NE transport inhibition after acute (minutes) TK inhibition by genistein evident in SK-N-SH cells, whereas, over the same time frame, Toyohira et al. (2010) reported elevations of NET activity in the same cell model, as well as in transfected COS-7 cells, where increased density of membrane binding sites was found. Possibly, differences in culture conditions explain the differences in findings of the two groups, although as yet they remain unexplained.
Simon et al. (1997) first reported pharmacological evidence of a role for TK signaling with respect to DAT regulation, with findings of reduced DA uptake in mouse striatal homogenates achieved through a reduction in DA transport Vmax. Similar, although less potent effects were observed with the TK inhibitor tyrphostin 23. Consistent with these studies, Doolen and Zahniser (2001), studying DAT-expressing Xenopus laevis oocytes, observed that genistein, lavendustin A, and tyrphostin 25 reduced both DA uptake, substrate (tyramine)-induced currents, and DAT leak currents that were paralleled by reduced surface hDAT as measured by [3H]WIN35,428 binding. Interestingly, the Src inhibitor PP2 did not replicate these findings, suggesting a role for other, as yet undefined TK-linked pathways. Zahniser’s group replicated these findings in rat striatal synaptosomes and primary mesencephalic cultures, again demonstrating a loss of DA uptake and DAT surface density (Hoover et al., 2007). Perhaps the effects observed here are mediated by the Cdc42-activated tyrosine kinase Ack1, which was discussed previously in the discussion of PKC regulation of DAT (Wu et al., 2015). Again, this pathway appeared to promote DAT surface expression because inhibition of either Ack1 or Cdc42 resulted in increased clathrin-dependent internalization and reduced surface DAT.
Altogether, these studies provide important, albeit indirect, evidence for a role of TK signaling in the regulation of MA transporters, raising questions as to the physiologic triggers for these pathways, which we turn to in the next section.
B. Evidence for Endogenous Pathways Triggering Regulation of Monoamine Transporters by Tyrosine Kinases
Beyond the role of insulin receptor signaling in control of NET and DAT noted earlier in this review, multiple observations across each of the MA transporters provides compelling evidence that RTK activation by endogenous ligands impact one or more facets of MA transporter biology. In rat brain neuronal cultures, Pelliccioni et al. (2001) observed a stimulation of 5-HT uptake after basic fibroblast growth factor (bFGF) treatments that could be attenuated by TeTx treatment. Specific FGF receptors involved in these effects have yet to be elucidated, although further studies by this group pointed to similar a similar stimulatory activity on 5-HT uptake by bFGF, nerve growth factor (NGF) and epidermal growth factor (EGF) in rat brain synaptosomes (Gil et al., 2003). In the latter studies, whereas TeTx inhibited basal 5-HT uptake, it did not preclude elevations of transport activity, suggesting different pathways engaged by TeTx and these RTK-linked growth factors in regulating SERT activity in this preparation. To date, however, the signaling pathways responsible for these findings have not been further clarified. Benmansour et al. (2008) provided evidence that intracerebroventricular or intrahippocampal injections in anesthetized rats of brain-derived neurotrophic factor (BDNF) reduced 5-HT clearance rates in vivo as assessed by chronamperometric recordings, effects suppressed by the TrkB receptor antagonist K252a [9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3',2',1'-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester]. In unanesthetized animals, however, using microdialysis, intrahippocampal injections of BDNF were found to decreased extracellular 5-HT levels, effects attributed to reductions in 5-HT release. Although the effects of intracerebroventricular BDNF on 5-HT clearance arose 3 days after growth factor injection, intrahippocampal injections led to reductions in clearance within 30 minutes, suggesting engagement of rapid, posttranslational mechanisms. Interestingly, Daws et al. (2007) reported reduced rates of 5-HT clearance in vivo using amperometric recordings of aged mice genetically deficient for BDNF (BDNF+/− animals) that did not correlate with changes in SERT density (although see Guiard et al., 2008, where density changes were observed), suggestive of functional modulatory pathways connecting neurotrophin and transporter, although perhaps indirect. Although clearly complexities remain in terms of how BDNF modulates SERT over time scales of minutes to months, the relationship of the BDNF/TrkB pathway and SERT function to depression and antidepressant mechanisms points to the translational potential of further studies of this interconnection (Haase and Brown, 2015). Keeping up with the sex of animals in further studies may also be beneficial given findings by Benmansour et al. (2014) that estradiol slows SERT-mediated 5-HT clearance in a manner sensitive to TrkB inhibition, as produced using K252a, and findings by Ren-Patterson et al. (2006) of sex specificity and estrogen effects in biochemical and behavioral measures from BDNF+/− and SERT +/− mice.
Aside from studies of insulin receptor-linked pathways, NET has received much less attention than SERT with respect to RTK-mediated regulatory pathways, aside from studies related to long-term modulation of noradrenergic neuron differentiation and survival (Wakade et al., 1996; Ren et al., 2001; Kreusser et al., 2006; Traver et al., 2006). One example is the observation of Toyohira et al. (2010) that the EGF receptor inhibitor tyrphostin A can increase NE transport activity in transfected COS-7 cells, although by a mechanism of action that presumably does not involve transcriptional control mechanisms but that has yet to be elucidated. Additionally, Rodriguez Fermepin et al. (2009) observed that BDNF and NT-4 can rapidly enhance NET activity in hypothalamic preparations via a TrkB- and PLC-dependent pathway.
With respect to DAT regulation by RTK-linked pathways, in the studies of Hoover et al. (2007) from Zahniser’s group where genistein was found to rapidly reduction DAT Vmax and surface expression in rat brain synaptosomes, these authors also reported that DAT activity could be rapidly (30 minutes) stimulated in primary cultures by BDNF in a TrkB-, MAPK-, and PI3K-dependent manner. Kinetic studies indicated enhancement of DA transport Vmax accompanied by an elevation in DA KM. These studies were conducted with serum-starved preparations, and thus the stimulation may reflect restoration of basal activity/trafficking. Other RTKs have also been shown to play a role in regulating DAT, including Ret, a receptor tyrosine kinase that is activated by GDNF and that has been shown to be essential for DA neuron development and survival. Li et al. (2006) demonstrated that Ret transfection into rat embryonic mesencephalic cultures induced transcription of DAT as measured by reverse-transcription polymerase chain reaction. Also, mice expressing a constitutively active form of Ret, MEN2B, have elevated levels of DAT and display increased rates of DA uptake as measured by in vivo voltammetry (Mijatovic et al., 2008). Interestingly, overexpression of Src, a known downstream effector of Ret signaling, in human placental trophoblast cells increased DA uptake through transiently transfected human DAT (Annamalai et al., 2012), potentially identifying a downstream tyrosine kinase effector for mediating the effects described above. The heterologous nature of DAT expression here indicates that Src effects are not linked to changes in DAT transcription from its endogenous promoter, reminding us of the posttranscriptional mechanisms engaged by Src to modulate SERT in the same study. Recently, Zhu et al. (2015) identified a GDNF/Ret signaling pathway that influences DAT trafficking in the nucleus accumbens, mediated by the Rho family guanine exchange factor Vav2. GDNF stimulation of heterologously expressed DAT/Ret (N2A cells) resulted in reduced transporter surface expression, with no change in total DAT levels and decreased DA uptake. The actions of GDNF to regulate DAT were blocked by treatment of cells with Vav2 shRNA. By using a split ubiquitin system to monitor protein-protein interactions in yeast, evidence was also gained for a physical interaction between Ret and DAT, results confirmed by coimmunoprecipitation methods using striatal and nucleus accumbens extracts. Additionally, activation of Ret lead to enhanced recovery of DAT/Vav2 complexes. Finally, the authors demonstrate that Vav2 KO mice have deficits in cocaine effects on DAT trafficking, DA uptake, and locomotor sensitization. Together, these studies provide strong evidence for functional and structural interactions between DAT and components of the GNDF signaling pathway. Whether DAT is modified though through the actions of GDNF is unknown.
C. Evidence of Tyrosine Phosphorylation of Monoamine Transporters
Certainly whether SERT, NET, or DAT are subject to tyrosine phosphorylation as a facet of their regulation cannot be inferred from the ability of RTKs or cytosolic TKs to modulate transporter trafficking or activity, because such regulation may arise through intermediate, transporter-associated proteins. The strongest evidence to date for transporter Tyr phosphorylation derives from studies of SERT, although specific sites supporting phosphorylation have not been identified. Thus, immunoprecipitation of the transporter from human platelets, followed by immunoblotting with P-Tyr antibodies revealed labeled bands that comigrated with SERT immunoreactive species (Zarpellon et al., 2008). Future studies with platelets drawn from SERT KO mice would be helpful in assessing the specificity of these results. Nonetheless, the authors reported a significant elevation of P-Tyr labeled SERT after vanadate treatments, consistent with authentic Tyr phosphorylation. Moreover, treatment of platelets with PP2 and SU6656 resulted in diminished P-Tyr labeling, consistent with a role for Src in SERT phosphorylation. Interestingly, a reduction in imipramine binding sites was observed after PP2/SU6656 treatments, suggesting a change in SERT conformation may attend transporter Tyr phosphorylation. Pavanetto et al. (2011) also reported evidence of P-Tyr incorporation in human platelet SERT by immunoblotting of SERT immunoprecipitates, with evidence presented indicating a decrease in labeling from platelets treated with the Syk kinase inhibitor piceatannol, paralleling reductions in 5-HT uptake. Corroborative findings of piceatannol-induced reductions in SERT phosphorylation were also obtained using a metabolic labeling strategy. The drug also reduced the apparent density of imipramine binding sites, suggesting that phosphorylation can change access of the antagonist to both substrate and antagonist binding sites, reminiscent of studies demonstrating catalytic activation of SERT by p38 MAPK noted above. Evaluation of SERT trafficking after piceatannol treatment and examination of drug effects using platelets and other SERT-expressing preparations from Syk KO mice would be helpful going forward. Annamalai et al. (2012) also provided evidence for P-Tyr labeling of rat platelets using a metabolic labeling paradigm. In these studies, basal P-Tyr was minimal and vanadate elevated P-Tyr (defined as acid-resistant [32P] incorporation), an effect blocked by coincubation with genistein, consistent with a capacity for direct targeting of SERT by one or more TK. That Src might be one of these TKs was evaluated in transfected HTR cells, with evidence elaborated from alterations in 5-HT transport evident in assays conducted using PP2 treatments (uptake inhibition), Src overexpression (uptake stimulation), and Src siRNA treatments (uptake inhibition). The heterologous expression model allowed for a demonstration that cotransfection of SERT with Src results in elevated P-Tyr labeling as well the nomination, through mutant studies, of Y47 and Y142 as Tyr phosphorylation sites. Mutation of these sites individually precluded Src-mediated elevations of SERT protein levels and 5-HT uptake. Two other cytoplasmic facing Tyr residues (Y350, Y358) exist and may also be targets of Src-mediated phosphorylation. Studies of mutants at these sites were not possible, however, because of losses of basal protein expression and will need to be pursued with more direct strategies such as mass spectrometry. Regardless, these comprehensive studies provide significant evidence that SERT is subject to endogenous regulation via P-Tyr modification that, as noted earlier, appears to enhance SERT protein stability. Next steps of course involve elucidating how P-Tyr incorporation results in changes in SERT protein turnover.
No studies to our knowledge have evaluated NET for P-Tyr labeling under basal or stimulated conditions. Although in the Annamalai studies of Src-induced elevation of 5-HT uptake in transfected HTR cells similar findings were obtained with NET (and DAT, but not taurine transporter) transfected cells. With respect to DAT, Simon et al. (1997) failed to detect a phosphoprotein of the size of DAT, as probed by phosphotyrosine immunoblotting methods, after genistein treatment of mouse striatal homogenates, although immunoprecipitation to enrich for DAT was not pursued. Foster et al. (2003) also found no reduction in DAT basal phosphorylation when metabolically labeled striatal slices were treated with purified Tyr phosphatase in contrast to efficient dephosphorylation evident with the Ser phosphatase PP1. Given the evidence obtained to date in SERT studies and the growing evidence for DAT modulation by RTKs and intracellular TKs, we suspect that P-Tyr studies with endogenous activators of these pathways will provide evidence of DAT Tyr phosphorylation and possibly open a new era of exciting studies connecting growth factor signaling to the regulation of DA synapses.
X. Conclusions
The past few decades have yielded significant insights into how MA transporters are regulated, and the field is now beginning to appreciate the importance of disruoted transporter regulation in the pathophysiology of MA-associated diseases. As our review demonstrated, kinases and their associated signaling pathways are central to the regulation of MA transporter trafficking and activity, and understanding how these pathways act to impact MA transporter function remains of critical importance to generating an integrated picture of how synaptic MA signaling is dynamically controlled. Interactions between different kinase pathways have been demonstrated in various native and in vitro preparations, revealing an extensive and complex network of regulatory pathways that regulate MA transporter trafficking and function. When and how these various signaling pathways might be engaged in an in vivo context is in many cases unclear, but hopefully this review provided some level of clarity in its effort to integrate and compare studies of MA transporter regulation across different species and preparations. Future studies that use more specific pharmacological inhibitors and activators of kinases, genetic model systems, conditional genetic strategies, and disease-associated transporter mutations will hopefully lead to a clearer picture of the relevance of MA transporter regulation in the powerful control that kinase-linked pathways exert over MA transport as well as how perturbed control contributes to human disease risk.
Acknowledgments
We gratefully acknowledge Raajaram Gowrishankar for careful initial review of our manuscript.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Bermingham, Blakely.
Footnotes
The authors gratefully acknowledge the support of National Institutes of Health National Institute on Drug Abuse [Award DA35559 (D.P.B)], National Institute of Mental Health [Award MH096972, MH095044, MH105094, MH094527 (R.D.B.)], and a SFARI grant from the Simons Foundation.
Abbreviations
- ACh
- acetylcholine
- ADHD
- attention-deficit hyperactivity disorder
- ASD
- autism spectrum disorder
- BAPTA-AM
- 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- BDNF
- brain-derived neurotrophic factor
- bFGF
- basic fibroblast growth factor
- BIM-1
- bisindolylmaleimide I
- BoNT/C
- botulinun neurotoxin C
- 8-Br-cAMP
- 8-Bromo-cAMP; BRL 4408, 2-[2H-(1-methyl-1,3-dihydroisoindole) methyl]-4,5-dihydroimidazole
- CaMKII
- calmodulin-dependent protein kinase II
- Con A
- concanavalin A
- cPKC
- conventional PKC isoform
- DA
- dopamine
- DAG
- diacylglycerol
- D2R
- dopamine D2 receptor
- D3R
- dopamine D3 receptors
- DAT
- dopamine transporter
- EGF
- epidermal growth factor
- ERK1/2
- extracellular signal-related kinase 1/2
- GC
- guanylyl cyclase
- GF-109203
- 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide
- Go6979
- 5,6,7,13-Tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile
- GPCR
- Gα-coupled G-protein receptors
- HTR
- human trophoblast
- 5-HT
- serotonin
- KO
- knockout
- KOR
- kappa opiate receptor
- IB-MECA
- N6-(3-Iodobenzyl)adenosine-5′-N-methyluronamide
- LPS
- lipopolysaccharide
- LY379196
- 8((dimethylamino)methyl)-6,7,8,9,10,11-hexahydro-5,21:12,17-dimetheneo-18H-dibenzo(i,o)pyrrolo(3,4-1)(1,8)diazacyclohexandecine-18,10(19H)dione
- MA
- monoamine
- MAPK
- mitogen-activated protein kinase
- NE
- norepinephrine
- NET
- NE transporter
- NGF
- nerve growth factor
- NO
- nitric oxide
- NOS
- nitric oxide synthase
- nNOS
- neuronal nitric oxide synthase
- nPKC
- novel protein kinase C
- OCD
- obsessive-compulsive disorder
- PCA
- para-chloroamphetamine
- PDBu
- phorbol 12,13 dibutyrate
- PDE
- phosphodiesterase
- PD98059
- 2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one
- PKA
- protein kinase A
- PKB
- protein kinase B
- PKC
- protein kinase C
- PKG
- protein kinase G
- PI3K
- phosphatidylinositol 3-kinase
- PIP2
- phosphatidylinositol (4,5)-bisphosphate
- PIP3
- phosphatidylinositol (3,4,5)-trisphosphate
- PLC
- phospholipase C
- Ro-31-8220
- 3-[3-[2,5-Dihydro-4-(1-methyl-1H-indol-3-yl)-2,5-dioxo-1H-pyrrol-3-yl]-1H-indol-1-yl]propyl carbamimidothioic acid ester; Rp-cAMPS, (R)-Adenosine, cyclic 3′,5′-(hydrogenphosphorothioate)
- RTK
- receptor tyrosine kinase
- SERT
- serotonin transporter
- SNAP
- S-Nitroso-N-acetyl-DL-penicillamine
- Sp-cAMPS
- (S)-Adenosine, cyclic 3′,5′-(hydrogenphosphorothioate)
- SSRI
- 5-HT-selective reuptake inhibitor
- SU6656
- (3Z)-N,N-Dimethyl-2-oxo-3-(4,5,6,7-tetrahydro-1H-indol-2-ylmethylidene)-2,3-dihydro-1H-indole-5-sulfonamide
- TK
- tyrosine kinase
- β-PMA
- phorbol 12-myristate 13-acetate
- U0126
- 1,4-Diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene
- VGCC
- voltage-gated Ca2+ channel
- W-7
- N-(6-Aminohexyl)-5-chloro-1-naphthalenesulfonamide
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
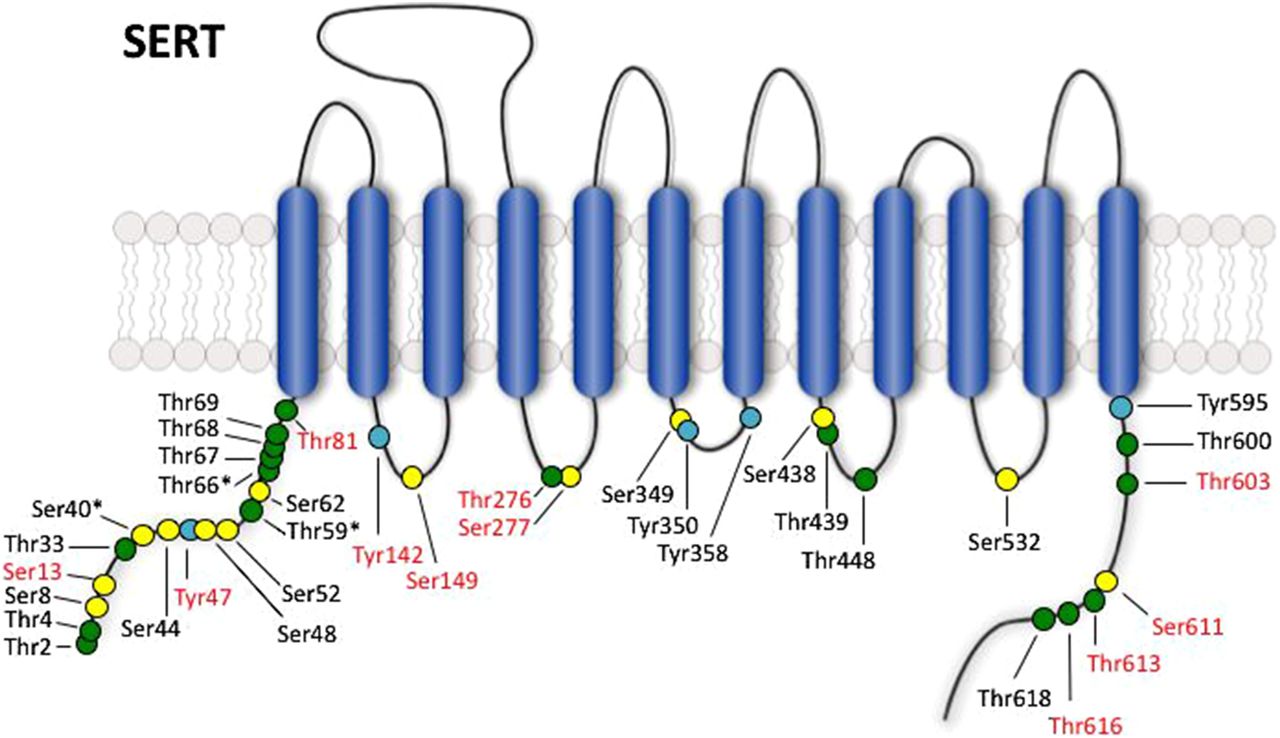{kind=link}
Jump to section
- Article
- Abstract
- I. Approaches for the Study of Kinase-Mediated Transporter Regulation
- II. Protein Kinase C—Overview
- III. cAMP-Dependent Protein Kinase/Protein Kinase A—Overview
- IV. cGMP-Dependent Protein Kinase/ Protein Kinase G—Overview
- V. Ca2+/Calmodulin-Dependent Protein Kinase II—Overview
- VI. ERK1/2—Overview
- VII. p38 Mitogen-Activated Protein Kinase
- VIII. Phosphatidylinositol 3-Kinase (PI3K)/Akt—Overview
- IV. Tyrosine Kinases
- X. Conclusions
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters