Article Text

Abstract
A four generation family is described in which some men of normal intelligence have epilepsy and others have various combinations of epilepsy, learning difficulties, macrocephaly, and aggressive behaviour. As the phenotype in this family is distinct from other X linked recessive disorders linkage studies were carried out. Linkage analysis was done using X chromosome microsatellite polymorphisms to define the interval containing the causative gene. Genes from within the region were considered possible candidates and one of these, SYN1, was screened for mutations by direct DNA sequencing of amplified products. Microsatellite analysis showed that the region between MAOB (Xp11.3) and DXS1275 (Xq12) segregated with the disease. Two point linkage analysis demonstrated linkage with DXS1039, lod score 4·06 at θ = 0, and DXS991, 3·63 at θ = 0. Candidate gene analysis led to identification of a nonsense mutation in the gene encoding synapsin I that was present in all affected family members and female carriers and was not present in 287 control chromosomes. Synapsin I is a synaptic vesicle associated protein involved in the regulation of synaptogenesis and neurotransmitter release. The SYN1 nonsense mutation that was identified is the likely cause of the phenotype in this family.
- synapsin I
- epilepsy
- mental handicap
- X linkage
Statistics from Altmetric.com
We were referred a young man with moderate learning difficulties and episodic aggressive outbursts. On taking the family history there were two male relatives with a similar history but there were also male relatives of normal intelligence with epilepsy. Although the features in the family were variable, the pedigree was compatible with an X linked inheritance pattern, we postulated a common genetic mechanism and proceeded to linkage studies.
METHODS
Family report
A simplified pedigree is shown in fig 1 and affected males are described below. The obligate carrier females are of normal intelligence and have no record of fits or aggressive behaviour. None of the affected individuals are dysmorphic. Chromosome analysis in the affected males was normal and they did not have an expansion of the FMR1 trinucleotide repeat. There are no medical records for I-3 (deceased) but the family report that he had tonic-clonic seizures and was of normal intelligence.

The pedigree. Genotypes for MAOA, MAOB, DXS8054, DXS1039, DXS991, and DXS1275 are shown against the individuals who were included in the linkage analysis. The region we deduce was inherited from the obligate carrier I-2 is boxed. The common haplotype is between MAOB and DXS1275.
II-5 is a 64 year old man of normal intelligence. He has had tonic-clonic seizures from the age of 16 years. Seizures occurred frequently, often daily, until a few years ago. His head circumference is on the 90th centile. Magnetic resonance imaging (MRI) showed mild generalised cerebral and cerebellar atrophy in keeping with his age. Both hippocampal bodies are moderately atrophic but without signal intensities.
III-2 is a 64 year old man who has had a cerebrovascular accident but was previously of normal intelligence. He had tonic-clonic seizures until aged 7. His head circumference is normal.
III-3 is a 53 year old man with a full scale IQ that has been formally assessed as 72 (with the recognised error in such assessments this places him within the borderline to mild learning difficulties range). His head circumference is normal. As a teenager, he showed extreme physical aggression (he was held in a high security psychiatric hospital between the ages of 11 and 18). Medical notes at the time described these episodes as epileptic in nature, though some episodes occurred after confrontations with staff. Exploratory brain surgery (cerebral corticography) was undertaken when he was 17 years old, without any significant findings. He also had generalised tonic-clonic seizures between the ages of 11 years and 18 years. An EEG report described “some evidence of spike focus” in the left temporal region. He currently lives alone, does not have seizures, and is in supported employment.
III-7 had his first fit at 18 years of age and has continued to have occasional seizures. He is of normal intelligence.
III-10 is a 47 year old man who presents clinically as functioning at the lower end of the mild learning difficulties range. He requires prompting with most activities of self care, and lives in a supported group home. He is macrocephalic (head circumference above the 97th centile for height) and has episodic aggressive outbursts. He has had a normal EEG and no history of seizures.
IV-1 is a 42 year old man of normal intelligence and with normal head circumference. He began to have partial and complex-partial seizures at the age of 16. His epileptic seizures have taken a number of forms. They have all occurred while falling asleep or during sleep. His wife describes repeated episodes when he would walk around the house during the night, engaging in repetitive stereotyped behaviours. He also has fits that consist of episodes of jerking of his left leg; these sometimes develop into jerking of his left leg and arm followed by unconsciousness. He does not have a history of abnormally aggressive behaviour. His EEG and MRI are normal.
IV-3 died in the bath aged 27 years. Unfortunately no neuropathological examination was undertaken. There was a recent history of “blackouts”, all occurring while in the bath; during these episodes he bit his tongue and also sustained a burn from hot running water. His EEG was normal and he was of normal intelligence; head circumference not recorded.
IV-4, now aged 25 years, was diagnosed as having nocturnal epilepsy at the age of six, though his mother had suspected the diagnosis from the age of three. He is of normal intelligence. His EEGs have shown bilateral non-specific changes and MRI was normal.
IV-5 is a 20 year old man with moderate learning difficulties. He has a diagnosis of autism manifested by characteristic impairments in reciprocal social interaction, social use of language, and restricted, repetitive behaviour, interests, and activities. He has outbursts of severe aggression that are described as of rapid onset, lacking a clear precipitant, with an intense rage of short duration. He has had two normal EEGs and no other evidence of seizures. His head circumference is above the 97th centile for his height.
Laboratory analysis
We extracted genomic DNA from EDTA coagulated blood by standard methods. In our initial linkage search we used 15 microsatellite fluorescent markers (multiplex screening set 8, Research Genetics Inc) spanning the X chromosome. We chose additional markers (DXS8054, DXS1039, DXS991, and DXS1275) using Entrez Genome Viewer (http://www.ncbi.nlm.nih.gov/cgi-bin/Entrez/) and dinucleotide repeats for MAOA and MAOB, following analysis of the first set of markers.1,2 We amplified 30 ng of template DNA using 1.25 units of Taq polymerase in 2 mM MgCl2, 200 μM dNTPs (deoxynucleotide triphosphates), and 0.1 μM of forward and reverse primers in a final volume of 25 μl. We used 30 cycles of 94°C for one minute, 58°C for one minute, and 72°C for one minute, with a final cycle of 72°C for 10 minutes. We electrophoresed samples through a polyacrylamide gel (6 M urea) on a ABI Prism 377 sequencer, obtaining the results using gene scan analysis software version 2.1 (PE Applied Biosystems) and analysing them using Genotyper 1.0 DNA fragment analysis software (PE Applied Biosystems).
We undertook two point linkage analyses using the MLINK program of the LINKAGE package,3 implemented in FASTLINK (version 2.2), accessed through the human genome project (HGP) (http://www.hgmp.mrc.ac.uk). For these analyses, we used an X linked recessive mode of inheritance and a disease allele frequency of 0.001.
We screened for synapsin I mutations by direct sequencing of genomic DNA from patients II-5 and III-2. We determined flanking intronic sequences for SYN1 exons by aligning cDNA (GenBank accession number NM_006950) with genomic sequences (exons 1–5 accession number AL009172 and exons 6–13 accession number Z84466).
We designed seven primer sets to amplify the 13 coding exons and splice junctions. We sequenced the products using the DYEnamic ET dye terminator kit (Amersham Biosciences) and analysed reactions on a MegaBACE sequencer (Amersham Biosciences). We investigated the change identified in II-5 and III-2 in all individuals included in the linkage analysis, in III-5, and in 287 control chromosomes (90 female and 107 male controls).
RESULTS
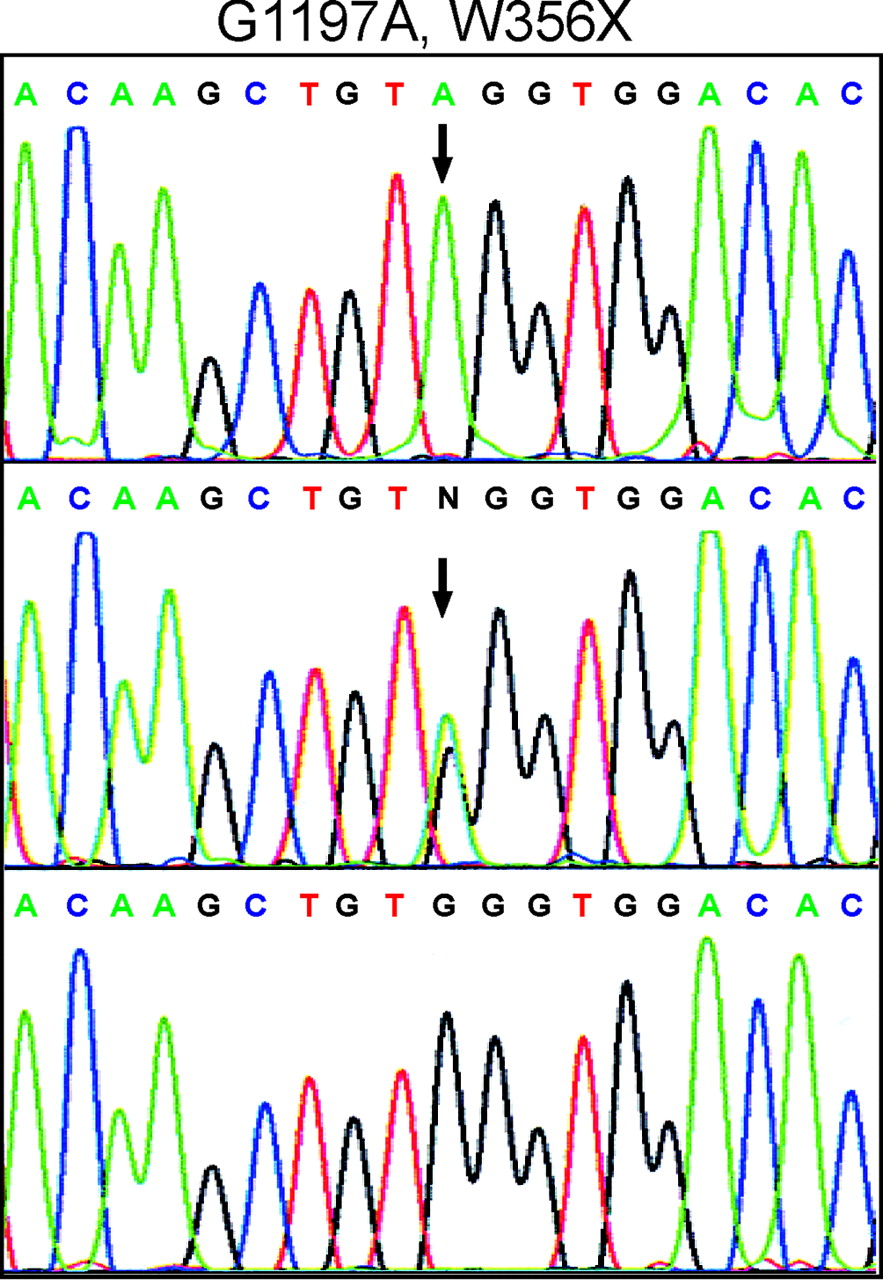
Haplotypes for polymorphic microsatellites at MAOA, MAOB (37 Mb from the tip of the short arm, http://www.ncbi.nlm.nih.gov/Entrez/), DXS8054 (39 Mb), DXS991 (48 Mb), and DXS1275 (62 Mb) are shown on fig 1. DXS8054, DXS1039, and DXS991 identified a common region in all affected individuals. Linkage analysis gave a two point lod score of 3.65 with DXS8054 at θ = 0, of 4.06 with DXS1039 at θ = 0, and of 3.63 with DXS991 at θ = 0. Recombinations between phenotype and the MAOA and MAOB markers were seen in the meioses leading to III3 and III7, effectively excluding these genes. Further evidence for this was that an unaffected man, III8, had the inferred grandmaternal alleles at the MAOA and MAOB loci. On the long arm, recombinations occurred between the phenotype and DXS1275 in the meioses leading to II3 and III10. Thus recombinations place the locus between MAOB and DXS1275, a region of approximately 25 Mb. We considered the candidate genes in this region and prioritised SYN1, the gene encoding synapsin I, for sequencing. We identified a transition (G1197A) in SYN1 exon 9 (fig 2) resulting in a nonsense codon (W356X). This mutation was present in all affected individuals and all carrier females included in the linkage analysis and in III-5, and was not detected in 90 female and 107 male controls.

{kind=link}
{kind=link}
Identification of SYN1 mutation. The top panel shows the genomic sequence from an affected male, the middle panel from a female obligate carrier, and the lower panel corresponding wild type sequence. The 1197G>A transition that produces a nonsense codon (W356X) is arrowed.
DISCUSSION
There is marked phenotypic variation in this X linked pedigree, to the extent that before starting linkage analysis and ultimately identifying the causative mutation we debated whether a single genetic disorder could account for all the problems diagnosed in the male members of the family. Seven of the 10 affected family members are of normal intelligence and have epilepsy. The natural history of the seizures has been variable—for example III-2 had seizures until he was seven years old, while IV-3 had his first seizure at the age of 27. IV-3 was unusual in that his fits only occurred while he was in the bath and his death was almost certainly related to a seizure. IV-1 predominantly has nocturnal partial seizures. None of these men with normal intelligence and epilepsy have a history of aggressive outbursts and, where recorded, they have a normal head circumference.
The scans in the three men who have had MRI have been unremarkable. Two of the 10 affected family members have moderate learning difficulties and are macrocephalic, and one, not macrocephalic, has mild learning difficulties. These three men have had episodic rages during which they are extremely aggressive. One of them, III-3, was an inpatient in a high security psychiatric unit from the age of 11 to 18 because of this, and III-10 was an inpatient in a learning disability hospital from his early teens until he recently moved to a staffed community home for people with learning disabilities. The aggressive outbursts that occur in three of the men could be epileptic in nature as they are described as being episodic, of rapid onset, often with no obvious precipitant, and the intense rage is of short duration.
Our mapping studies showed that all the affected male members of the family had inherited a common region of the X chromosome and placed the disorder centromeric to the MAOB locus (Xp11.23) and DXS1275 (Xq12), a region of 25Mb spanning the centromere. This region contains many genes but we focused our attention on synapsin, SYN1, because of the observations that electrical stimulation of the amygdala at intensities that induce short duration EEG changes in wild type mice cause electrographic changes of longer duration and overt seizures in SYN1 deficient mice; and there is an increased incidence of seizures in SYN1 deficient mice following vestibular stimulation.4,5 There are two SYN1 isoforms resulting from the use of different splice acceptors for the final exon (exon 13) and encoding proteins of 669 and 705 amino acids. We identified a nonsense change at codon 356. This mutation is in the ninth of the 13 SYN1 exons. As premature termination codons that are 5′ of the last intron by more than 50 nucleotides generally lead to nonsense mediated mRNA decay, this transcript is more likely to be degraded than to produce a truncated protein.6 Unfortunately as synapsin I is a neuronal specific protein and as there was no neuropathological examination in the only deceased affected individual, we have not been able to study protein expression in relevant samples from affected individuals.
Given that mice lacking SYN1 have a lower seizure threshold, it is very likely that this nonsense mutation is responsible for the phenotype in this family. Synapsins are a family of related phosphoproteins associated with the cytoplasmic surface of synaptic vesicles. Vertebrates have three synapsin genes (I, II, and III) that can generate five distinct isoforms.7SYN2 and SYN3 map to human chromosomes 3p25 and 22q12.3, respectively. The family members and their isoforms are differentially expressed in subsets of neurones, but the functional significance of this is unknown.8 A high level of expression of synapsin II relative to synapsin I has been reported in excitatory synapses, while a relatively high level of synapsin I has been observed in inhibitory synapses.9
Before synapse formation, synaptic vesicles are uniformly distributed along the axons. After synapse formation neurotransmitter vesicles redistribute into two pools, one in the active zone ready for release, and a reserve pool that can be recruited in response to increased activity. The density of synaptic vesicles in presynaptic terminals is decreased in SYN1 deficient mice and this is more marked in the reserve pool than in the active zone.4,10 Synaptic vesicles in the reserve pool are thought to be reversibly tethered to the actin based cytoskeleton by synapsin in a phosphorylation dependent manner.11 Upon polarisation, synapsin I is phosphorylated at two C-terminal sites by CaM kinase II11,12 and the synaptic vesicles are liberated from the reserve pool to enter the release pool where they undergo docking and priming at the active zone plasma membrane to become competent for fusion and subsequent neurotransmitter release. It is also postulated that synapsins regulate the kinetics of neurotransmitter release during priming of synaptic vesicles at the plasma membrane.13
A growing body of evidence suggests that synapsins play a role during neuronal development and synapse formation. When hippocampal neurones from SYN1 deficient mice are cultured, the axons are shorter and have fewer branches than in controls, and synapse formation is delayed.14 Thus synapsin I plays a developmental role in axon elongation, branching, and synaptogenesis, and an ongoing role in the distribution of neurotransmitter vesicles and neurotransmitter release. It is intriguing that in this family some individuals have learning difficulties while others, of normal intelligence, have varying types of seizure. We have no explanation for this phenotypic variation. The normal MRI findings in the three men who have had imaging are in keeping with the normal brain size and structure in SYN1 deficient mice.4,10,15
Considerable progress has been made in recent years in the investigation of familial epilepsies, with causative mutations being identified in subunits of a number of voltage gated and ligand gated ion channels.16 This is the first report of a family with a defect in a synaptic vesicle associated protein. Further studies are needed to address whether synapsin abnormalities play a role in other patients with epilepsy or learning disability.