Article Text

Abstract
Rationale Chronic obstructive pulmonary disease (COPD) is a common lung disease leading to progressive decline in lung function. Inhibition of release of inflammatory mediators by p38 inhibitors may be a useful treatment for chronic inflammation of the airways thought to underlie the pathogenesis of the disease.
Objectives To evaluate the efficacy and safety of PH-797804, a potent and selective p38 inhibitor, in adults with moderate to severe COPD (Global Initiative for Chronic Obstructive Lung Disease stage II/III).
Methods This was a randomised, adaptive design, double-blind, placebo-controlled, parallel-group, multicentre trial. Patients were initially randomised to placebo, 0.5, 3, 6 or 10 mg PH-797804 once daily and treated for 6 weeks following a 2-week run-in.
Measurements and main results The primary endpoint was change from baseline in trough forced expiratory volume in 1 s (FEV1) compared with placebo after 6 weeks of treatment. Secondary endpoints included other spirometric parameters, transition dyspnoea index, rescue mediation use, high sensitivity C-reactive protein and symptoms. A total of 230 patients were assigned to treatment; placebo (n=45), 0.5 mg (n=20), 3 mg (n=47), 6 mg (n=70) and 10 mg (n=48). PH-797804 showed a statistically significant improvement in trough FEV1 at week 6 compared with placebo of 0.086 litre (95% Bayesian CI 0.008 to 0.164) and 0.093 litre (95% CI 0.018 to 0·166) at 3 and 6 mg PH-797804, respectively. PH-797804 3 mg and 6 mg showed an improvement in the baseline dyspnoea index/transition dyspnoea index total focal score at week 6. PH-797804 was well tolerated at all doses studied.
Conclusions PH-797804 demonstrated improvements over placebo in lung function parameters and dyspnoea in patients with moderate to severe COPD.
TrialRegNo NCT00559910.
- COPD Pharmacology
Statistics from Altmetric.com
Key messages
What is the key question?
-
What is the efficacy and safety of 6 weeks of treatment with PH-797804, an oral p38 inhibitor, in patients with moderate to severe chronic obstructive pulmonary disease (COPD)?
What is the bottom line?
-
Six weeks of treatment with PH-797804 led to improvements over placebo in lung function and dyspnoea in patients with moderate to severe COPD with few adverse events observed.
Why read on?
-
This study supports further investigation of PH-797804 as a potential treatment for patients with COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a common disease leading to a progressive decline in lung function. COPD is associated with high morbidity and death, and the direct and indirect socioeconomic costs of COPD are high.1 COPD is currently ranked as the fourth leading cause of death worldwide and is predicted to rise.2 ,3
Treatments are limited, providing inadequate symptomatic relief and reduction in exacerbations. No therapeutic class has consistently shown anti-inflammatory properties across the spectrum of disease severity. Hence, there is still a need for treatments with demonstrable anti-inflammatory activity and potential clinical benefit.4
Signalling through p38 mitogen-activated protein kinase (p38-MAPK) is required for the expression of a range of inflammatory mediators associated with the chronic lung inflammation characteristic of COPD, such as tumour necrosis factor α, interleukin-1 (IL-1), IL-6 and IL-8.5 p38-MAPK is expressed on inflammatory cells associated with COPD, and there is increased lung expression and activation of p38-MAPK in patients with COPD compared with smoking and non-smoking controls.6 p38-MAPK inhibition, but not corticosteroids, attenuated oxidative stress-induced cytokine release in an in vitro assay modelling some aspects of COPD-associated inflammation.7 Therefore, p38-MAPK inhibition is an attractive target for COPD.
PH-797804 is a potent, selective p38-MAPK inhibitor,8 ,9 being evaluated as a potential oral anti-inflammatory COPD treatment. The primary objective of this trial was to assess the efficacy and safety of PH-797804 in adults with moderate to severe COPD defined by the guidelines applicable at the time (Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages II/III).10 Secondary objectives included understanding the dose–response relationship and the time course of response to PH-797804. These data were first presented at the European Respiratory Society 2010 Annual Congress in Barcelona, Spain.11
Methods
Patients
This randomised, double-blind, placebo-controlled, parallel-group, phase II trial was conducted between February 2008 and December 2009 in patients (40–80 years) with moderate to severe COPD in 38 centres across 13 countries. Patients had a diagnosis for at least 6 months and had a post-bronchodilator forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) ratio <0.7 with a post-bronchodilator FEV1 of 30–80% of predicted. Patients with stable disease (no exacerbation in the preceding month) and stable FEV1 during the run-in phase were eligible for enrolment. Key exclusion criteria included significant comorbidities or laboratory abnormalities, indicating significant concomitant disease. Long-acting bronchodilators and inhaled corticosteroids were not allowed 2 or 4 weeks prior to screening, respectively. To achieve this, withdrawal of treatment was necessary in some patients. Throughout the trial, patients received ipratropium bromide metered dose inhalers (MDIs; 40 μg four times a day) as maintenance therapy and salbutamol MDIs (100–200 μg as required) as rescue medication. Inclusion/exclusion criteria are described in the online data supplement. Independent ethics committees approved the trial and patients provided written informed consent. The trial was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines.
Trial design
The trial involved a screening visit, a run-in phase (weeks –2 and –1 to ensure stability of lung function), a baseline/randomisation visit (week 0), 6 weeks of double-blind treatment with PH-797804 or placebo (visits at weeks 1, 2, 3, 4 and 6), a 2-week run-out phase and a final follow-up visit (week 8). Patients withheld ipratropium bromide and salbutamol for at least 8 h before clinic visits.
The primary endpoint was the change from baseline in trough FEV1 compared with placebo after 6 weeks of treatment. Secondary efficacy endpoints included forced expiratory volume in 6 s (FEV6), FVC and inspiratory capacity (IC). Trough (prior to trial medication) measurements for these spirometry parameters were recorded at all visits. Measurements were also recorded post trial medication (weeks 0 and 6) and post salbutamol (screening and weeks –1, –2, 0 and 6). Baseline dyspnoea index12 ,13 was evaluated at week 0, and transition dyspnoea index (TDI) was evaluated at weeks 1, 2, 3, 4 and 6. Patients maintained a diary throughout the trial to record COPD symptoms, maintenance and rescue bronchodilator use and peak expiratory flow rate. Spirometry was performed using standardised equipment with centralised over-read/interpretation in accordance with guidelines.14
Safety endpoints included adverse events, laboratory data, ECGs and vital signs. High sensitivity C-reactive protein (hsCRP) was measured at each visit. Exploratory biomarkers Clara cell protein-16 (CC16), IL-6, surfactant protein D (SPD) and fibrinogen were measured at weeks 0 and 6.
Randomisation and masking
Patients were initially randomised (according to a computer-generated randomisation code) to one of five treatment groups in the ratio 1:1:1:2:1 for placebo, 0.5 mg, 3 mg, 6 mg, 10 mg once daily PH-797804, respectively, until the interim analysis.
An independent Data Monitoring Committee (DMC) conducted an unblinded interim analysis once 96 patients had completed 6 weeks of treatment. The DMC could drop a dose(s) in the event of statistical evidence of predefined futility or if unacceptable adverse events occurred, and could adapt the randomisation to increase the sample size for the 10 mg dose.
Trial treatments were supplied as PH-797804 capsules and matching placebo. Patients, investigator staff and the sponsor's project team were masked to treatment assignment throughout the trial until the database was locked. The randomisation code was provided to the DMC for the interim analysis. During the trial, the DMC could review unblinded data, at any time, in the event of safety concerns.
Statistical analysis
The analysis of the primary endpoint used Bayesian statistics. This approach has advantages over classical hypothesis testing because direct probabilistic statements about the effect size could be made (not addressed by p values) and information on placebo could be utilised from previous trials, reducing the number of placebo patients required.15 The Bayesian estimation of the dose–response relationship also means that the sample size is reduced compared with pairwise testing because information is used from all doses. Bayesian spline (normal dynamic linear model analyses)16 was applied that adjusted for baseline covariate.
It was of interest to characterise the dose–response relationship and calculate the probability of any dose achieving statistical significance and ascertaining whether the average treatment effect was minimally clinically relevant, defined as >=75 ml improvement over placebo in FEV1.17 A statistically significant result was defined if there was ≥95% probability that the treatment effect over placebo was positive. An additional hurdle required that the average effect over placebo was >75 ml. This hurdle was for internal decision-making purposes to give confidence that the magnitude of effect was sufficient to warrant further investigation.
The anticipated clinical dose was 6 mg. Simulations showed that with 48 patients per group and 64 for 6 mg, the power of the study was such that there was 94% probability of a dose achieving significance if the true effect size in FEV1 was 75 ml, and a 9% probability if the true effect size was only 20 ml (akin to type 1 error). The analysis of FEV1 used Bayesian statistics with an uninformative prior for placebo (WinBUGS18). No multiplicity adjustments were required because this issue was addressed implicitly in the Bayesian model fitted.
Secondary endpoints were analysed using a classical repeated measures analysis of covariance approach using SAS version 9.1.19 To adjust for multiplicity of testing several dose groups against placebo, for secondary endpoints, a controlled testing procedure was used to control the overall α level at 5%. The mixed-effect model includes all available information over time and provides appropriate, consistent estimates of model parameters under the assumption of ‘missing at random’.
Further details concerning the statistical methodology and models used can be found in the online data supplement.
All primary and secondary efficacy endpoints were analysed using the full analysis set, defined as all randomised patients with at least one valid FEV1 measurement in the double-blind phase. All safety analyses were conducted on the safety analysis set, defined as all patients who received at least one dose of PH-797804 or placebo.
Predefined futility rules
At the interim analysis, the dose–response relationship was estimated. The probability of a dose being futile was calculated. Doses could be stopped for futility if there was <10% chance of there being ≥75 ml improvement over placebo in FEV1 change from baseline. Discontinued doses still contributed information to the dose–response curve at the end-of-study analysis.
Results
A total of 230 patients were randomised to treatment and 201 completed double-blind treatment. Forty-five patients received placebo and 185 received PH-797804: 0.5 mg (n=20), 3 mg (n=47), 6 mg (n=70) or 10 mg (n=48). Figure 1 shows the trial profile. Demographics and baseline characteristics were similar among the five treatment groups (table 1).
Demographic and baseline characteristics

Trial profile. The full analysis set was used for all primary and secondary endpoints and was defined as all randomised patients who had at least one valid forced expiratory volume in 1 s measurement during the double-blind phase of the trial. The safety analysis set comprised all patients who received at least one dose of PH-797804 or placebo. *DBT=double-blind treatment.
Following the interim analysis, the 0.5 mg treatment group was dropped for futility and the randomisation ratio was modified as planned with post-interim patients randomised in the ratio 1:0:1:1:1 for placebo, 0.5 mg, 3 mg, 6 mg, 10 mg PH-797804, respectively.
The Bayesian analyses show that in this trial there is 99.2% probability that the true effect of 6 mg PH-797804 over placebo is greater than zero (ie, statistically significant). Additionally, the analysis shows that the probability of this effect being clinically relevant (>75 ml improvement) is 67.3%. The Bayesian CIs, called credible intervals (CrI), are wide and show that there is a probability of 0.95 that the true effect over placebo for 6 mg lies between 18 and 166 ml. The results are similar for 3 mg PH-797804 (table 2).
Trough FEV1: treatment comparisons versus placebo at week 6
There was only one outlier; a subject in the 3 mg PH-797804 group had an improvement in trough FEV1 of approximately 1 litre, influencing the mean effect; when removed from the analyses the mean effect at 3 mg was 61 ml (CrI=–9 to 132 ml).
The probability of 10 mg PH-797804 being better than placebo was 94.5% (almost classed as statistically significant), but this trial also required that the magnitude of effect was large enough (the probability that the true effect is 75 ml was <50%).
By contrast, 0.5 mg showed only numerical improvements over placebo of 59 ml.
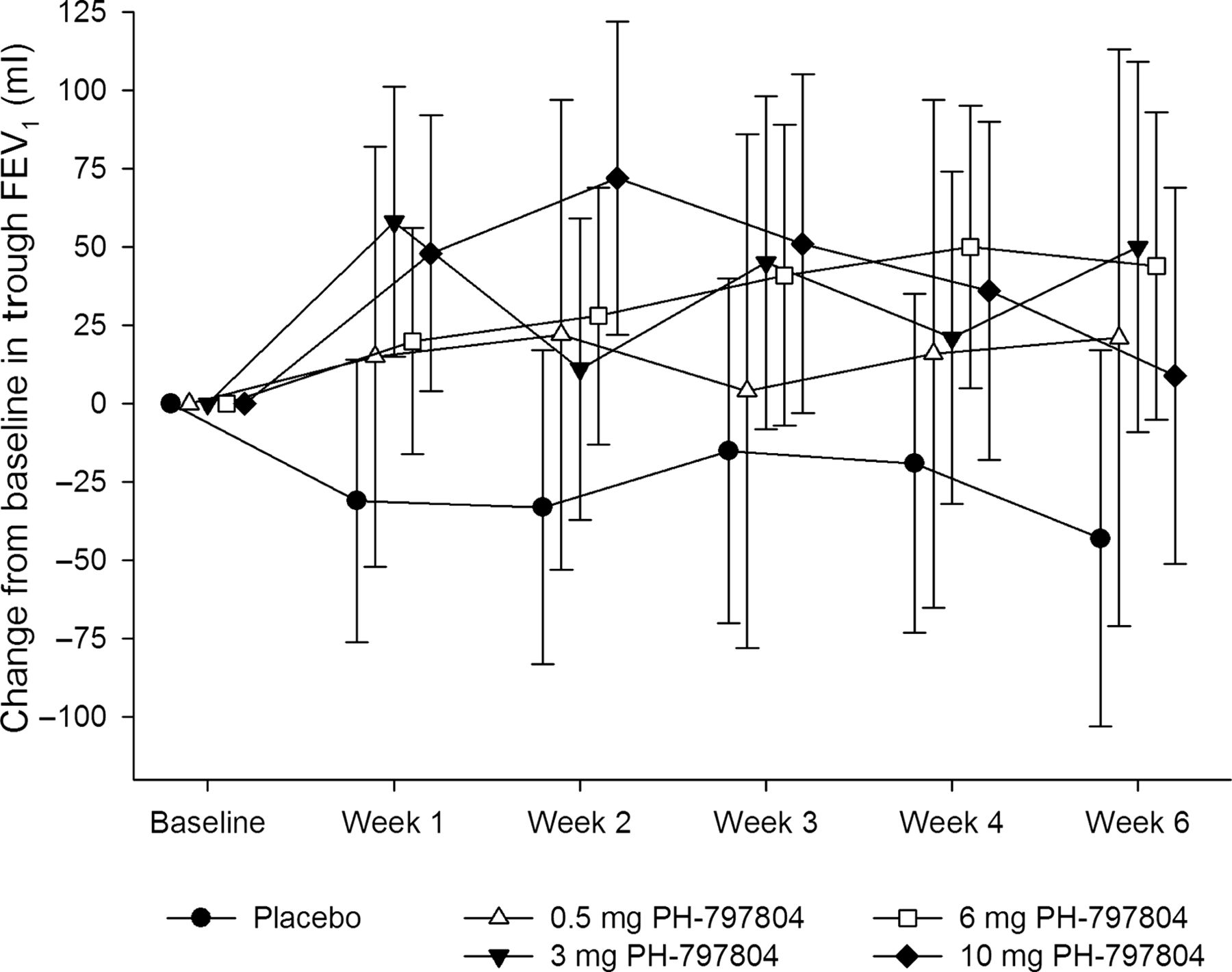
Figure 2 shows the mean change from baseline in trough FEV1 for each treatment group over the 6 weeks of treatment. FEV1 declined by 52 ml in the placebo group, while it increased in each of the PH-797804 groups. There was an improvement in FEV1 over placebo as a function of time with clear evidence of a treatment effect given the separation of all active treatments from placebo. The observed maximum estimated effect was in the 10 mg group at week 2, this difference then reduced over time. The results of the statistical analysis of trough FEV6, FVC and IC at week 6 are presented in table 3.
Trough FEV6, FVC and IC: treatment comparisons versus placebo at week 6

Mean change from baseline in trough forced expiratory volume in 1 s (FEV1) (ml) during treatment. The symbols and bars represent the means and 95% CIs.
PH-797804 at doses of 3, 6 and 10 mg produced a statistically significant improvement in trough FEV6 at week 6 versus placebo, with mean changes from placebo of 115 ml (SE=54), 87 ml (SE=50) and 94 ml (SE=55), respectively. PH-797804 at doses of 6 and 10 mg produced a statistically significant improvement in trough FVC at week 6 versus placebo, with mean changes from placebo of 111 ml (SE=54) and 128 ml (SE=59), respectively. PH-797804 at doses of 3, 6 and 10 mg produced a statistically significant improvement in trough IC at week 6 versus placebo, with mean changes from placebo of 119 ml (SE=57), 119 ml (SE=53) and 96 ml (SE=58), respectively.
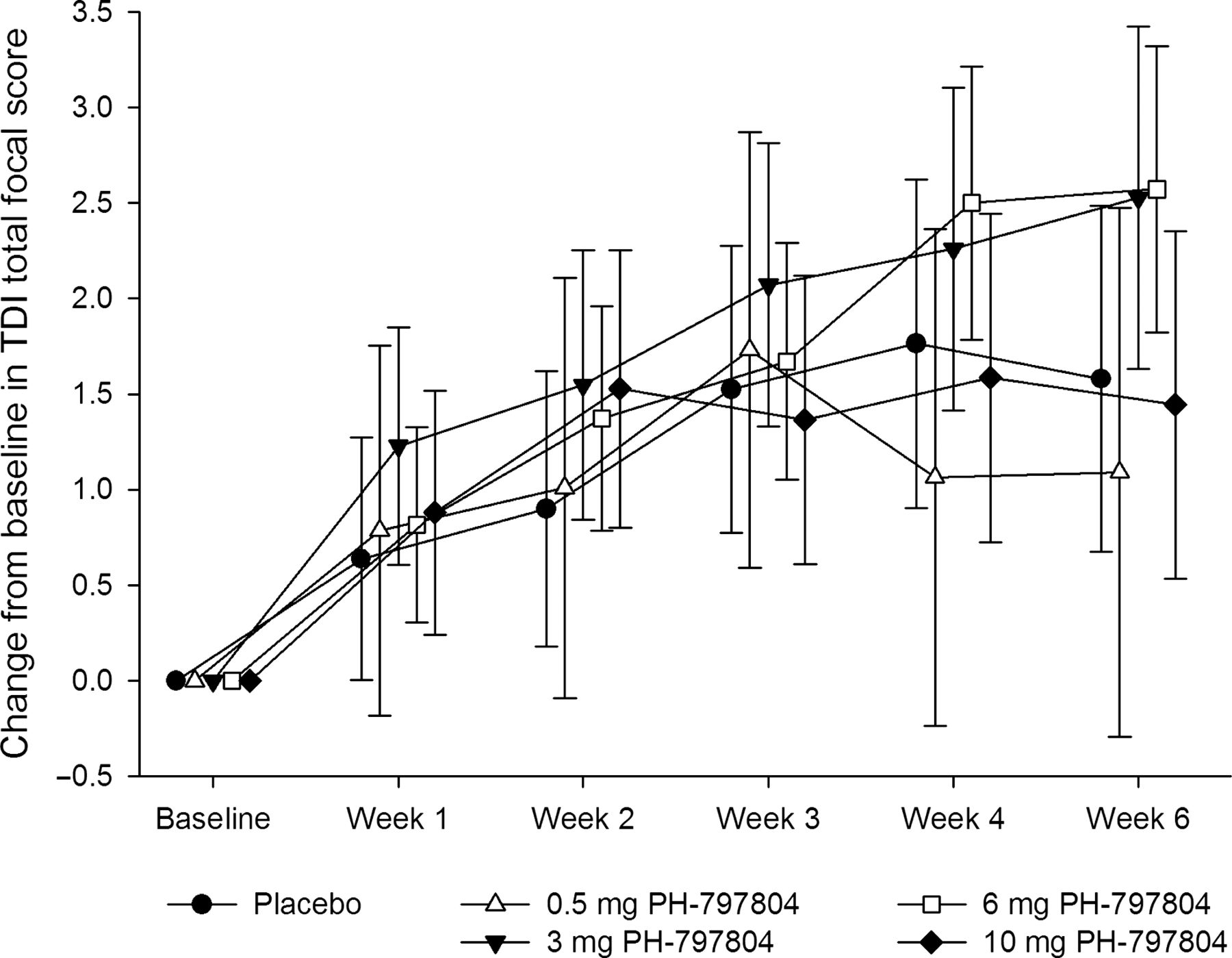
TDI total focal score: treatment comparisons versus placebo for change from baseline at week 6
No statistically significant differences from placebo in FEV1, FEV6, FVC or IC were observed when post-dose (PH-797804 or bronchodilator) measurements were compared with pre-dose measurements.
PH-797804 at doses of 3 and 6 mg showed an improvement in the TDI total focal score at week 6 at the 10% and 5% significance level, with a 0.95 point (SE=0.646) and 0.99 point (SE=0.596) improvement, respectively (table 4). This improvement was driven by the magnitude of effort component as opposed to the magnitude of task or functional impairment components. Mean TDI for each treatment over time is presented in figure 3.
{kind=link}
{kind=link}
{kind=link}
Mean change from baseline in transitional dyspnoea index (TDI) total focal score during treatment. The symbols and bars represent the means and 95% CIs.
Table 5 summarises treatment comparisons with placebo for the change from baseline in rescue medication (salbutamol) usage. A statistically significant decrease in rescue medication use was observed with 6 mg PH-797804 with a mean daily use of 0.66 less actuations of salbutamol at week 6.
Treatment comparisons versus placebo for change from baseline in mean rescue medication usage at week 6
Log hsCRP values were analysed and adjusted for log baselines. A decrease in hsCRP compared with placebo occurred with the 3, 6 and 10 mg doses of PH-797804, with statistically significant differences from placebo observed in the ratio of the means of 0.633 (p=0.033), 0.588 (p=0.011) and 0.594 (p=0.021), respectively after 6 weeks treatment. There were no statistically significant differences from placebo for CC16, IL-6, SPD or fibrinogen.
COPD symptoms were assessed using an internal unvalidated daily paper diary. Individual item analyses found no significant differences over placebo at the end of treatment compared with baseline. A total dyspnoea incidence score was calculated to determine the number of dyspnoea-experienced and dyspnoea-free days. Trends were seen for 6 mg to affect dyspnoea days.
The most frequently reported adverse events were COPD exacerbation, rash and nasopharyngitis (table 6). Rash was the most frequently reported treatment-related adverse event. All reported rashes resolved on cessation of treatment. Three patients (6 mg PH-797804) experienced serious adverse events considered to be treatment related by the investigator; two had gastrointestinal (GI) haemorrhages (one with a history of GI complications including past GI bleed, partial gastrectomy and heavy alcohol intake; the event occurred on day 2 of the trial). The other had been self-medicating with over-the-counter aspirin. One patient had left and right bundle branch block (also present on pretrial ECGs). No significant trends were noted in any laboratory safety parameters or vital signs. There were no significant trends in mean ECG parameters, although there was an increase in the percentage of patients with changes from baseline in QT interval corrected for heart rate using Fridericia's formula (QTcF) >30 ms. No patients had a QTcF of >500 ms.
All-cause treatment-emergent adverse events in at least two patients in any treatment group
Discussion
To the authors’ knowledge, this is the first clinical trial to demonstrate a positive effect of a p38 inhibitor on a range of clinical outcomes in COPD. Six weeks of treatment with PH-797804 in patients with COPD (GOLD II/III) maintained on a background of short-acting bronchodilators led to statistically significant improvements in lung function associated with improvements in dyspnoea and rescue medication usage.
By week 6, the 6 mg PH-797804 group showed an improvement over placebo in prebronchodilator FEV1 of 93 ml and a 0.99-point improvement in the TDI (one point change is considered clinically meaningful).12 ,13 The minimum target effect on FEV1 was predefined as 75 ml over placebo, based on data obtained with the phosphodiesterase 4 inhibitor roflumilast, which showed placebo-corrected increases of 49 ml and 80 ml in prebronchodilator FEV1 in two 6-month studies.17 This magnitude of the change in FEV1 is below the defined minimally clinically important difference of 100 ml derived from the improvement in FEV1 bronchodilator trials.20 It is recognised that FEV1 improvements may be less with drugs having an anti-inflammatory rather than a bronchodilator effect. However, the change in TDI and decrease in rescue medication alongside the other changes in lung function suggest that the effects may be of clinical relevance.
PH-797804 at doses of 3 and 6 mg consistently showed an improvement in FEV1 over placebo throughout the trial. In addition, FEV1 improved compared with baseline throughout the trial in all PH-797804 groups, whereas the placebo group showed a decline in lung function over the duration of the trial. This decline is greater than expected in a 6-week trial and may have been contributed to by the fact that subjects were maintained on short-acting bronchodilators alone.
Other lung function parameters, such as FVC, FEV6 and IC, showed improvements consistent with the FEV1 improvement; this was maintained in post-bronchodilator measures, indicating an additive effect of PH-797804 on top of salbutamol. There was no acute effect of PH-797804 on lung function, supporting its mode of action as anti-inflammatory rather than a direct bronchodilator.
PH-797804 reduced hsCRP at doses above 0.5 mg, indicating a systemic anti-inflammatory effect. This effect was maintained throughout the 6-week dosing period. Statistically significant effects were not observed on other systemic biomarkers measured in this study. A 12-week study in patients with COPD, with another p38 inhibitor, losmapimod, examined the effect of p38 inhibition on sputum neutrophils. No statistically significant effect was seen on this endpoint. However, a statistically significant reduction in plasma fibrinogen and trends towards an effect on systemic IL-6, IL-8 and CRP were observed but no statistically significant changes in lung function.21
The most consistently efficacious dose of PH-797804 was 6 mg, not the highest dose of 10 mg; the reason for this is unknown. The SEs of the mean data are large, therefore this may be due to variability. An alternative explanation could be tachyphylaxis, although the mechanism of this is unknown. This phenomenon will be investigated in longer-term studies. Potential differences in baseline severity of COPD, systemic exposure compared with other dose groups, and preponderance of adverse events potentially leading to a higher dropout rate or functional unblinding do not explain the apparent lower effect size of the 10 mg group.
This study was on a background of short-acting bronchodilators alone and it recognised that these are not the mainstay of treatment in COPD. It’s anticipated that PH-797804 would also show additive effects to long-acting bronchodilators. This is being studied in ongoing trials (clintrials.gov NCT0153919 and NCT01321463).
A further confounding feature in this study is the effect of withdrawal of treatment with long-acting β agonists in some patients in the run-in period, which was considered to have influenced the results of many pharmacological trials.22 In our study a similar number of subjects (<50%) in each of the arms of the study had treatment withdrawn, which makes it less likely that differences between placebo and treatment groups resulted from differences in treatment withdrawal between the groups.
Data from trials of agents such as roflumilast, tiopropium, salmeterol and fluticasone suggest that, although the changes from baseline in FEV1 may decrease with time in patients with COPD, the difference between active treatment and placebo generally appears to be maintained across the duration of the studies.23–27 The duration of this trial (6 weeks of dosing) was relatively short compared with the likely long-term use of efficacious COPD medications and the long-term effects of PH-797804 will be explored in longer studies.
An effect on COPD exacerbations could not be adequately assessed during a 6-week trial, however we propose that the anti-inflammatory properties of PH-797804 which led to the improvements in lung function and dyspnoea measures may translate to reductions in COPD exacerbations over a longer period.
The overall safety profile of PH-797804 in this trial was good, with most patients assigned to PH-797804 treatment groups completing the double-blind treatment phase. The most commonly reported treatment-related adverse event was acneform rash, which resolved upon cessation of treatment. This has been reported with other p38 inhibitors.28 There were no significant effects on liver function, unlike with other p38 inhibitors, when increases in alanine transaminase and aspartate transaminase have been reported.29
In conclusion, PH-797804 showed improvements in lung function and dyspnoea scores in patients with moderate to severe COPD treated with short-acting bronchodilators. These data are encouraging and support further investigation of this compound in patients with COPD.
Acknowledgments
The authors would like to thank the investigators and patients who participated in the trial. Medical writing support was provided by Samantha Abel, Valley Writing Solutions Ltd. This support included drafting the initial manuscript based on discussions with the authors and subsequent updates based on author comments. This assistance was funded by Pfizer Global Research and Development.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Funding This trial and all trial-related activities including medical writing support for the manuscript were funded by Pfizer Global Research and Development, Sandwich, UK.
-
Competing interests R Allan, L Tan and I Jones are or were employees of Pfizer and own or owned a limited amount of Pfizer stock. W MacNee has received funding for patient recruitment and participation in this trial and has received funding for research projects unrelated to this study from Pfizer. W MacNee has also received funding for work unrelated to this study from Almirall, Boehringer-Ingelheim, GlaxoSmithKline, Grifols, Novartis and Janssen. MC De Salvo has received funding for patient recruitment and participation in this trial.
-
Ethics approval This was a multicentre study and was approved by multiple independent ethics committees as appropriate for each centre/country.
-
Provenance and peer review Not commissioned; externally peer reviewed.