


Abstract
This summary article presents an overview of the molecular relationships among the voltage-gated calcium channels and a standard nomenclature for them, which is derived from the IUPHAR Compendium of Voltage-Gated Ion Channels.1 The complete Compendium, including data tables for each member of the calcium channel family can be found at http://www.iuphar-db.org/iuphar-ic/.
Calcium Channel Subunits
Voltage-gated calcium channels mediate calcium influx in response to membrane depolarization and regulate intracellular processes such as contraction, secretion, neurotransmission, and gene expression. Their activity is essential to couple electrical signals in the cell surface to physiological events in cells. They are members of a gene superfamily of transmembrane ion channel proteins that includes voltage-gated potassium and sodium channels (Catterall, 1995).
The calcium channels that have been characterized biochemically are complex proteins composed of four or five distinct subunits, which are encoded by multiple genes (Fig. 1) (Catterall, 2000). The α1 subunit of 190 to 250 kDa is the largest subunit, and it incorporates the conduction pore, the voltage sensor and gating apparatus, and the known sites of channel regulation by second messengers, drugs, and toxins. Like the α subunits of sodium channels, the α1 subunit of voltage-gated calcium channels is organized in four homologous domains (I–IV) with six transmembrane segments (S1–S6) in each. The S4 segment serves as the voltage sensor. The pore loop between transmembrane segments S5 and S6 in each domain determines ion conductance and selectivity, and changes of only three amino acids (aa) in the pore loops in domains I, III, and IV will convert a sodium channel to calcium selectivity. An intracellular β subunit and a transmembrane, disulfide-linked α2δ subunit complex are components of most types of calcium channels. A γ subunit has also been found in skeletal muscle calcium channels and related subunits are expressed in heart and brain. Although these auxiliary subunits modulate the properties of the channel complex, the pharmacological and electrophysiological diversity of calcium channels arises primarily from the existence of multiple α1 subunits (Hofmann et al., 1994).

Subunits of CaV1 channels. The subunit composition and structure of calcium channels purified from skeletal muscle are illustrated. The model is updated from the original description of the subunit structure of skeletal muscle calcium channels. This model also fits available biochemical and molecular biological results for other CaV1 channels and for CaV2 channels. P, sites of phosphorylation by cAMP-dependent protein kinase; Ψ, sites of N-linked glycosylation. Predicted alpha helices are depicted as cylinders. The lengths of lines correspond approximately to the lengths of the polypeptide segments represented.
Calcium Currents
Calcium currents recorded in different cell types have diverse physiological and pharmacological properties, and an alphabetical nomenclature has evolved for the distinct classes of calcium currents (Tsein et al., 1995). L-type calcium currents require a strong depolarization for activation, are long-lasting, and are blocked by the organic L-type calcium channel antagonists, including dihydropyridines, phenylalkylamines, and benzothiazepines. They are the main calcium currents recorded in muscle and endocrine cells, where they initiate contraction and secretion. N-type, P/Q-type, and R-type calcium currents also require strong depolarization for activation. They are relatively unaffected by L-type calcium channel antagonist drugs but are blocked by specific polypeptide toxins from snail and spider venoms. They are expressed primarily in neurons, where they initiate neurotransmission at most fast synapses and also mediate calcium entry into cell bodies and dendrites. T-type calcium currents are activated by weak depolarization and are transient. They are resistant to both organic antagonists and to the snake and spider toxins used to define the N- and P/Q-type calcium currents. They are expressed in a wide variety of cell types, where they are involved in shaping the action potential and controlling patterns of repetitive firing.
Calcium Channel Genes
Mammalian α1 subunits are encoded by at least ten distinct genes. Historically, various names had been given to the corresponding gene products, giving rise to distinct and sometimes confusing nomenclatures. In 1994, a unified but arbitrary nomenclature was adopted in which α1 subunits were referred to as α1S for the original skeletal muscle isoform and α1A through α1E for those discovered subsequently (Birnbaumer et al., 1994). In 2000, a rational nomenclature was adopted based on the well defined potassium channel nomenclature (Chandy and Gutman, 1993; Ertel et al., 2000). Calcium channels were named using the chemical symbol of the principal permeating ion (Ca) with the principal physiological regulator (voltage) indicated as a subscript (CaV). The numerical identifier corresponds to the CaV channel α1 subunit gene subfamily (1 to 3 at present) and the order of discovery of the α1 subunit within that subfamily (1 through m). According to this nomenclature, the CaV1 subfamily (CaV1.1 to CaV1.4) includes channels containing α1S, α1C, α1D, and α1F, which mediate L-type Ca2+ currents (Table 1). The CaV2 subfamily (CaV2.1 to CaV2.3) includes channels containing α1A, α1B, and α1E, which mediate P/Q-, N-, and R-type Ca2+ currents, respectively (Table 1). The CaV3 subfamily (CaV3.1 to CaV3.3) includes channels containing α1G, α1H, and α1I, which mediate T-type Ca2+ currents.
Physiological function and pharmacology of calcium channels
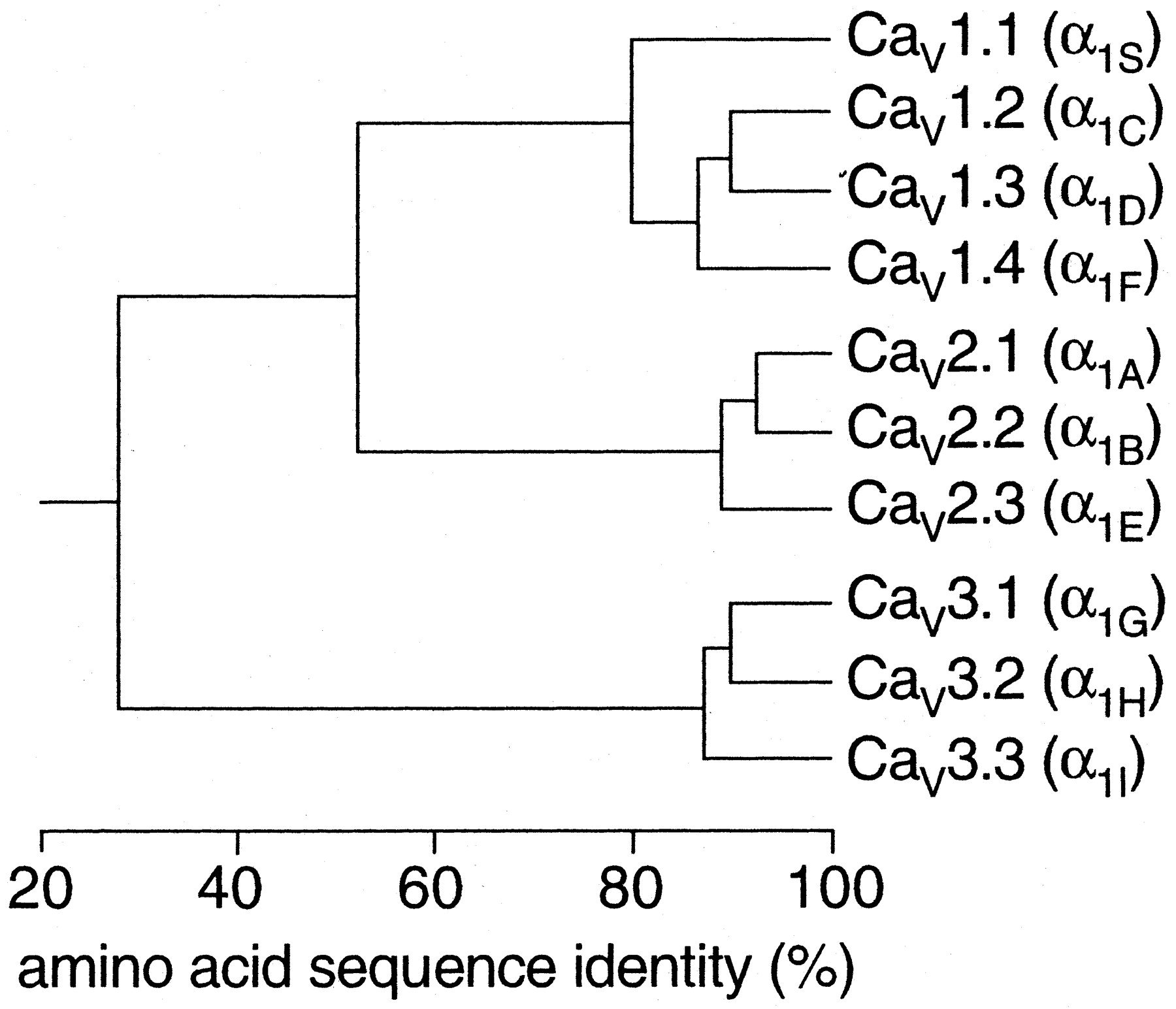
The complete amino acid sequences of these α1 subunits are more than 70% identical within a family but less than 40% identical among families. These family relationships are illustrated for the more conserved transmembrane and pore domains in Fig. 2. Division of calcium channels into these three families is phylogenetically ancient, as representatives of each are found in the Caenorhabditis elegans genome. Consequently, the genes for the different α1 subunits have become widely dispersed in the genome and even the most closely related members of the family are not clustered on single chromosomes.

Sequence similarity of voltage-gated calcium channel α1 subunits. Phylogenetic representation of the primary sequences of the calcium channels. Only the membrane-spanning segments and the pore loops (∼350 amino acids) are compared. First, all sequence pairs were compared, which clearly defines three families with intra-family sequence identities above 80% (CaV1, CaV2, CaV3). Then, a consensus sequence was defined for each family and these three sequences were compared with one another, with inter-family sequence identities of ∼52% (CaV1 versus CaV2) and 28% (CaV3 versus and CaV1 or CaV2).
Calcium Channel Molecular Pharmacology
The pharmacology of the three families of calcium channel is quite distinct. The CaV1 channels are the molecular targets of the organic calcium channel blockers used widely in the therapy of cardiovascular diseases. These drugs are thought to act at three separate, but allosterically coupled, receptor sites (Table 1). Phenylalkylamines are intracellular pore blockers, which are thought to enter the pore from the cytoplasmic side of the channel and block it. Their receptor site is formed by amino acid residues in the S6 segments in domains III and IV, in close analogy to the local anesthetic receptor site on sodium channels (Hockerman et al., 1997; Hofmann et al., 1999; Striessnig, 1999). Dihydropyridines can be channel activators or inhibitors and therefore are thought to act allosterically to shift the channel toward the open or closed state, rather than by occluding the pore. Their receptor site includes amino acid residues in the S6 segments of domains III and IV and the S5 segment of domain III. The dihydropyridine receptor site is closely apposed to the phenylalkylamine receptor site and shares some common amino acid residues. Diltiazem and related benzothiazepines are thought to bind to a third receptor site, but the amino acid residues that are required for their binding overlap extensively with those required for phenylalkylamine binding.
The CaV2 family of calcium channels is relatively insensitive to dihydropyridine calcium channel blockers, but these calcium channels are specifically blocked with high affinity by peptide toxins from spiders and marine snails (Miljanich and Ramachandran, 1995). The CaV2.1 channels are blocked specifically by ω-agatoxin IVA from funnel web spider venom. The CaV2.2 channels are blocked specifically by ω-conotoxin GVIA and related cone snail toxins. The CaV2.3 channels are blocked specifically by the synthetic peptide toxin SNX-482 derived from tarantula venom. These peptide toxins are potent blockers of synaptic transmission because of their specific effects on the CaV2 family of calcium channels.
The CaV3 family of calcium channels is insensitive to both the dihydropyridines that block CaV1 channels and the spider and cone snail toxins that block the CaV2 channels, and there are no widely useful pharmacological agents that block T-type calcium currrents (Heady et al., 2001). The organic calcium channel blockers mibefradil is somewhat specific for T-type versus L-type calcium currents (3- to 5-fold). The peptide kurtoxin inhibits the activation gating of CaV3.1 and CaV3.2 channels. Development of more specific and high affinity blockers of the CaV3 family of calcium channels would be useful for therapy and for more detailed analysis of the physiological roles of these channels.
This section of the compendium summarizes the major molecular, physiological, and pharmacological properties for each of the ten calcium channels that have been functionally expressed. Quantitative data are included for voltage dependence of activation and inactivation, single-channel conductance, and binding of drugs and neurotoxins, focusing on those agents that are widely used and are diagnostic of channel identity and function.
Footnotes
-
↵1 This work was previously published in Catterall WA, Chandy KG, and Gutman GA, eds. (2002) The IUPHAR Compendium of Voltage-Gated Ion Channels, International Union of Pharmacology Media, Leeds, UK.
-
DOI: 10.1124/pr.55.4.8.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}