


Abstract
The mammalian bombesin receptor family comprises three G protein-coupled heptahelical receptors: the neuromedin B (NMB) receptor (BB1), the gastrin-releasing peptide (GRP) receptor (BB2), and the orphan receptor bombesin receptor subtype 3 (BRS-3) (BB3). Each receptor is widely distributed, especially in the gastrointestinal (GI) tract and central nervous system (CNS), and the receptors have a large range of effects in both normal physiology and pathophysiological conditions. The mammalian bombesin peptides, GRP and NMB, demonstrate a broad spectrum of pharmacological/biological responses. GRP stimulates smooth muscle contraction and GI motility, release of numerous GI hormones/neurotransmitters, and secretion and/or hormone release from the pancreas, stomach, colon, and numerous endocrine organs and has potent effects on immune cells, potent growth effects on both normal tissues and tumors, potent CNS effects, including regulation of circadian rhythm, thermoregulation; anxiety/fear responses, food intake, and numerous CNS effects on the GI tract as well as the spinal transmission of chronic pruritus. NMB causes contraction of smooth muscle, has growth effects in various tissues, has CNS effects, including effects on feeding and thermoregulation, regulates thyroid-stimulating hormone release, stimulates various CNS neurons, has behavioral effects, and has effects on spinal sensory transmission. GRP, and to a lesser extent NMB, affects growth and/or differentiation of various human tumors, including colon, prostate, lung, and some gynecologic cancers. Knockout studies show that BB3 has important effects in energy balance, glucose homeostasis, control of body weight, lung development and response to injury, tumor growth, and perhaps GI motility. This review summarizes advances in our understanding of the biology/pharmacology of these receptors, including their classification, structure, pharmacology, physiology, and role in pathophysiological conditions.
I. Introduction
The unusual name of this family of receptors, bombesin (Bn1), comes from the original terminology used by V. Erspamer and his colleagues to name the first natural ligand described, bombesin, which was an amidated tetradecapeptide isolated from the skin of the European frog Bombina bombina (Erspamer et al., 1970, 1972) (Fig. 1). They isolated many related peptides from other frog skins, and most were named after the genus of frog from which they were isolated (Erspamer and Melchiorri, 1973; Erspamer, 1988). In terms of their structural similarities they were originally divided into three general groups (Fig. 1): the bombesin group, which all had a carboxyl terminus of Gly-His-Leu-Met-NH2 (bombesin, alytesin, and [pGlu1]bombesin6–14); the ranatensin group, which had a carboxyl terminus of Gly-His-Phe-Met-NH2 (ranatensin, ranatensin R and C, litorin, rodhei-litorin, and [Glu (Ote)2 or (Ome)2]litorin); and the phyllolitorin group, which had a carboxyl-terminal Gly-Ser-Phe/Leu-Met-NH2 (phyllolitorin, [Leu8]phyllolitorin, and [Thr5,Leu8]phyllolitorin) (Erspamer, 1988; Falconieri Erspamer et al., 1988) (Fig. 1). Recent molecular studies show that the occurrence of these peptides in amphibian skins is more complicated than originally thought with both Leu and Phe penultimate forms present in the same frog species in many cases (Nagalla et al., 1996; Spindel, 2006). For example, in the skin of the frog, Bombina orientalis [Leu13]bombesin, [Phe13]bombesin, and [Ser3,Arg10,Phe13]bombesin (SAP bombesin) are found, and each of these three forms is derived from separate genes (Nagalla et al., 1996; Spindel, 2006).

Structures of GRP, NMB, and Bn-related agonists and antagonists. The entire structures of the different peptides are shown except for GRP, which has 27 amino acids, and only the COOH-terminal 14 amino acids are shown (the biologically active end). Both natural occurring agonists and some of the antagonists referred to in the text are shown. Ψ,–CONH peptide bond changed to–CH2NH–; pGlu, pyroglutamic acid; Cpa, chlorophenylalanine; NMC, neuromedin C; F5, pentafluoro-.
Subsequently, in mammals two Bn-like peptides were isolated, gastrin-releasing peptide (GRP) (McDonald et al., 1979) and neuromedin B (NMB) (Minamino et al., 1983). GRP, a 27-amino acid peptide was originally isolated from porcine stomach and shares the same seven carboxyl-terminal amino acids with bombesin (McDonald et al., 1979) accounting for similar biological activity (Fig. 1). The decapeptide of GRP was later isolated from porcine spinal cord and originally called neuromedin C (Minamino et al., 1984b), although it is recommended that a more appropriate name is either GRP-10 or GRP18–27 (Anonymous, 1988). The mammalian equivalent of ranatensin, NMB, was isolated from porcine spinal cord and shown to be a decapeptide (Minamino et al., 1983), which also occurs in precursor forms of 30 and 32 amino acids (Minamino et al., 1985). The carboxyl-terminal seven amino acids are identical in ranatensin, except for the replacement of threonine in NMB for valine in ranatensin at the fifth position from the carboxyl terminus (Fig. 1).
Studies of GRP and NMB immunoreactivity as well as mRNA studies have demonstrated that these peptides and their mRNA are widely distributed in mammals in both the nervous system and peripheral tissues, especially the gastrointestinal tract (Penman et al., 1983; Wada et al., 1990; Battey and Wada, 1991; Spindel et al., 1993; Moody and Merali, 2004). In the alimentary tract GRP-like IR is found primarily in neurons as well as in the submucosal and myenteric plexuses and not in endocrine cells (Penman et al., 1983). With Northern blots the highest levels of mRNA occur in the colon with lower amounts in the stomach and small intestine (Sunday et al., 1988). In the spinal cord GRP-IR was found in both the posterior and anterior horn, and in the CNS GRP-IR and mRNA are widely distributed in neurons with high levels in the hypothamic nuclei, forebrain, and medullary nuclei that participate in autonomic functions, as well as in sensory nuclei (Panula et al., 1982, 1988; Wada et al., 1990; Battey and Wada, 1991; Spindel et al., 1993). NMB-IR and mRNA are found throughout the GI tract, but generally at lower levels than GRP except in the esophagus (Spindel et al., 1993). In general, in the brain and spinal cord, NMB-IR is greater than GRP-IR (Minamino et al., 1984a), and NMB mRNA is most abundant in the olfactory bulb, dentate gyrus, and dorsal root ganglia, whereas GRP mRNA is highest in the forebrain and some hypothamic nuclei (Wada et al., 1990; Battey and Wada, 1991). In most brain regions the NMB mRNA distribution does not overlap with GRP (Wada et al., 1990; Battey and Wada, 1991; Moody and Merali, 2004; Ohki-Hamazaki et al., 2005).
The mammalian bombesin peptides, GRP and NMB, demonstrate a broad spectrum of pharmacological and biological responses. GRP stimulates smooth muscle contraction in both the gastrointestinal tract and urogenital system and has profound effects on GI motility, stimulates release of numerous gastrointestinal hormones/neurotransmitters, stimulates secretion and/or hormone release from the pancreas, stomach, colon, and numerous endocrine organs, has potent effects on immune cells (macrophages, dendritic cells, lymphocytes, and leukocytes) (Ruff et al., 1985; De la Fuente et al., 1991, 1993; van Tol et al., 1993; Del Rio and De la Fuente, 1994; Del Rio et al., 1994; Plaisanci é et al., 1998; Makarenkova et al., 2003), has potent growth effects on both normal tissues and tumors; has potent CNS effects, including regulation of circadian rhythm, thermoregulation; regulation of anxiety and the fear response, regulation of food intake, and behavioral effects and is involved in mediating numerous CNS effects on the GI tract (Tache et al., 1988; Bunnett, 1994; Martinez and Tache, 2000; Jensen et al., 2001; Jensen, 2003; Grider, 2004; Jensen and Moody, 2006). In many tissues the effects of NMB overlap with those of GRP; however, NMB has specific effects in some tissues such as contraction of smooth muscle, growth effects in various tissues (Moody et al., 2000; Matusiak et al., 2005), CNS effects including effects on feeding, thermoregulation; regulation of TSH release, stimulation of various CNS neurons, behavioral effects; and effects on spinal sensory transmission (von Schrenck et al., 1989; Rettori et al., 1992; Ladenheim et al., 1997b; Ohki-Hamazaki, 2000; Merali et al., 2006; Oliveira et al., 2006). GRP and to a lesser extent NMB affects the growth and/or differentiation of a number of important human tumors including colon, prostate, lung, and some gynecologic cancers (Cuttitta et al., 1985; Schally et al., 2000; Jensen et al., 2001; Glover et al., 2003; Jensen and Moody, 2006).
Early studies on the biologic effects of the different bombesin peptides isolated from frog skins, primarily examining their effects on contraction of isolated smooth muscle preparations from various tissues, demonstrated markedly varying potencies, which suggested that more than one subtype of bombesin receptor might exist (Falconieri Erspamer et al., 1988; Regoli et al., 1988; Severi et al., 1991). Binding studies and the development of highly selective antagonists established unequivocally the existence of two different classes of receptors in mammalian tissues mediating the actions of these peptides (Jensen et al., 1978; Moody et al., 1978; Jensen and Gardner, 1981; Coy et al., 1988; von Schrenck et al., 1989, 1990; Ladenheim et al., 1990; Jensen and Coy, 1991; Metz et al., 1992). One class had a high affinity for GRP and a lower affinity for NMB (termed GRP-R, GRP receptor, or GRP-preferring receptor) and the other class had a higher affinity for NMB than for GRP (termed NMB-R, NMB receptor, or NMB-preferring receptor) (Jensen and Gardner, 1981; Moody et al., 1988, 1992; von Schrenck et al., 1989, 1990; Ladenheim et al., 1990, 1992; Wang et al., 1992). Subsequently, two mammalian receptors with high affinity for GRP (Spindel et al., 1990; Battey et al., 1991) or NMB (Wada et al., 1991) have been cloned in addition to a closely related orphan receptor (Gorbulev et al., 1992; Fathi et al., 1993b) and one related receptor from amphibians (Nagalla et al., 1995), which will be discussed in more detail below (Table 1).
Current bombesin receptor nomenclature and general characteristics See text for references.
II. Molecular Basis for Nomenclature
Once the receptors were defined using binding studies, cross-linking studies, and studies of biological activity (Kris et al., 1987; Sinnett-Smith et al., 1988; Tache et al., 1988; von Schrenck et al., 1989; Huang et al., 1990; Ladenheim et al., 1990; Lebacq-Verheyden et al., 1990), an active effort to clone the GRP-R was undertaken by Dr. Eliot Spindel, Oregon Regional Primate Center, and Dr. James Battey, National Institutes of Health. In 1990 using electrophysiological and luminometric Xenopus oocyte expression assays, Spindel et al. (1990) succeeded in cloning the GRP-R from murine Swiss 3T3 cells, which express high levels of this receptor (Rozengurt, 1988). The cDNA for the same receptor was isolated and described by Battey et al. in 1991 by using an enriched library from Swiss 3T3 cells and specific oligonucleotide probes on the basis of information from a partial sequence of the GRP-R in these cells obtained after solubilization and purification using wheat germ agglutinin-agarose and ligand affinity chromatography (Feldman et al., 1990). Pharmacology studies demonstrated that the cloned receptor preferred GRP to NMB and its activation was blocked by specific GRP-preferring receptor antagonists (Rozengurt, 1988; Battey et al., 1991). Subsequently, using low stringency conditions with a mouse GRP-R cDNA probe (Wada et al., 1991), the NMB-R was cloned from a cDNA library made from the rat esophagus, a tissue that had been reported to have a high density of NMB-Rs (von Schrenck et al., 1989, 1990). The structure of the cDNA of the human GRP-R and NMB-R were described from a small cell lung cancer cell line in 1991 (Corjay et al., 1991).
In 1992 a novel receptor was cloned from guinea pig uterus (Gorbulev et al., 1992), which showed the highest amino acid identity to the GRP-R (52%) and the NMB-R (47%). This receptor bound GRP and NMB, but only with relatively low affinities (IC50 of 290 and 20,000 nM, respectively). The human analog of this novel receptor was cloned in 1993 (Fathi et al., 1993b), and expression studies showed that it was specifically activated by bombesin-related peptides but only with low affinity and thus was classified as an orphan receptor. It was termed BRS-3 for bombesin receptor subtype 3 (Fathi et al., 1993b). Subsequent binding studies and signaling studies using synthetic ligands of bombesin with high affinity for hBRS-3 (Mantey et al., 1997) demonstrated that it had low affinity not only for GRP and NMB but also for all known naturally occurring bombesin related peptides (Wu et al., 1996; Mantey et al., 1997; Pradhan et al., 1998; Ryan et al., 1998a,b) and therefore it remains an orphan receptor. Subsequently it was cloned from mouse (Ohki-Hamazaki et al., 1997a), rat (Liu et al., 2002), and sheep (Whitley et al., 1999).
In the search for receptors for bombesin-related peptides in amphibians (Nagalla et al., 1995), clones that had a sequence similar to the mammalian GRP-R and NMB-R were isolated. A clone that encoded for a novel bombesin receptor, which had 61, 56, and 70% amino acid identities to the human GRP-R, NMB-R, and BRS-3, respectively, was isolated (Nagalla et al., 1995). This receptor had the highest affinity for [Phe13]-bombesin, the form most prevalent in frog brain and had lower affinity for GRP and NMB. This receptor was called BB4 for bombesin receptor subtype 4 (Nagalla et al., 1995). Subsequent detailed binding studies and studies of cell signaling confirmed these findings and showed that this receptor had greater affinity for [Phe13]bombesin than any other naturally occurring bombesin-related peptide (Katsuno et al., 1999). At present no mammalian equivalent of this receptor has been described and therefore it is not included in the classification discussed in the following sections. Recently in chickens a receptor was cloned that had high amino acid identity to frog BB4 (fBB4) (70%) as well as to human BRS-3 (69%) and lower for human GRP-R (58%) and human NBR-R (52%) (Iwabuchi et al., 2003). When expressed this receptor had low affinity for GRP and NMB, but it retained high affinity for [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 (Iwabuchi et al., 2003), a synthetic analog which has high affinity for hBRS-3, GRP-R, NMBR, and fBB4 (Mantey et al., 1997; Pradhan et al., 1998). It was proposed that this receptor be termed chBRS-3.5 because of its resemblance to both fBB4 and BRS-3. No mammalian equivalent of this receptor has been described and therefore it is also not included in the following classification.
On the basis of the preceding molecular studies, three classes of mammalian bombesin receptors are proposed for which the nomenclature and a few features are summarized in Table 1. Although the usual International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC-IUPHAR) nomenclature uses the endogenous mammalian ligand, the substantial historical use of the frog peptide bombesin in the field to describe this system was retained. The BB1 through BB3 receptors will each be dealt with in more detail in the following sections, but a few important points will be covered briefly here. The BB1 receptor was previously referred to as the NMB receptor, NMB-R, or NMB-preferring receptor. This terminology is the same used for this bombesin receptor subclass in the Sigma-RBI Handbook of Receptor Classification and Signal Transduction (Watling, 2007) and is the same as the BB1 in the “BJP Guide to Receptors and Channels” (Alexander et al., 2006). The BB2 receptor was previously referred to as the GRP-R, GRP receptor, or GRP-preferring receptor (Table 1). This terminology is the same used for this bombesin receptor subclass in the Sigma-RBI Handbook of Receptor Classification and Signal Transduction (Watling, 2007) and is the same as the BB2 subclass in the “BJP Guide to Receptors and Channels” (Alexander et al., 2006). The BB3 receptor was previously referred to as the BRS-3 receptor, BRS-3, and bombesin receptor subtype 3 (Table 1). This terminology is the same used for this bombesin receptor classes in the Sigma-RBI Handbook of Receptor Classification and Signal Transduction (Watling, 2007) and is the same as the bb3 receptor in the “BJP Guide to Receptors and Channels” (Alexander et al., 2006). Finally, the amphibian BB4 receptor does not have a mammalian equivalent so is not included in this classification. This receptor was also not classified in the Sigma-RBI Handbook of Receptor Classification and Signal Transduction (Watling, 2007) or the “BJP Guide to Receptors and Channels” (Alexander et al., 2006).
III. BB1 Receptor
A. Early Studies of the BB1 Receptor
Before the identification of the BB1 in 1989 in rat esophageal muscle tissue sections by direct binding studies using 125I-Bolton-Hunter-labeled NMB and subsequent esophageal muscle strip contraction studies (von Schrenck et al., 1989), there were no early studies that unequivocally established the existence of BB1. Numerous previous studies had demonstrated that the frog peptides ranatensin and litorin, which closely resembled NMB (Minamino et al., 1983), had potent effects on various tissues and especially on smooth muscle contraction, which in some classes had differences from bombesin (Falconieri Erspamer et al., 1988; Regoli et al., 1988). However, these differences were not significant enough to clearly establish the existence of a separate class of BB1 receptors (Minamino et al., 1983; Falconieri Erspamer et al., 1988; Regoli et al., 1988). Although there had been many binding studies to numerous tissues from the late 1970s, in almost all cases 125I-[Tyr4] bombesin or another radiolabeled bombesin analog was used (Moody et al., 1978; Ladenheim et al., 1993b; Shapira et al., 1993). Unfortunately, bombesin has high affinity for both BB1 and BB2, making it more difficult to distinguish subtypes. Numerous classes of selective BB2 receptor antagonists were developed before the cloning of the BB1, and these also confirmed the presence of the BB1 on esophageal smooth muscle (von Schrenck et al., 1990). After the pharmacologic description of BB1 on esophageal muscle and before its cloning in 1991, by use of selective BB2 receptor antagonists or binding studies with radiolabeled NMB and selective agonists or BB2 receptor antagonists, BB1 receptors were demonstrated in the CNS (Ladenheim et al., 1990) and on gastric smooth muscle cells (Severi et al., 1991).
B. Cloned BB1 Receptor and Receptor Structure
The human BB1 receptor is a 390-amino acid protein, and it shows an 89% amino acid identity with the rat BB1 (Corjay et al., 1991). The human BB1 receptor has 55% amino acid identities with the human BB2 (Corjay et al., 1991) and 47% with the human BB3 receptor (Fathi et al., 1993b). The human BB1 receptor has two consensus sites for potential PKC phosphorylation and three potential N-linked glycosylation sites (Corjay et al., 1991). Hydropathy plots yielded results consistent with a seven-transmembrane structure typical for a G protein-coupled receptor (Corjay et al., 1991). The BB1 receptor has been cloned from rat (Wada et al., 1991) (Fig. 2), mouse (Ohki-Hamazaki et al., 1997a), and the frog, B. orientalis (Nagalla et al., 1995). Cross-linking studies demonstrate that the mature human BB1 receptor had a molecular mass of 72 ± 1 kDa and when deglycosylated 43 ± 1 kDa (Benya et al., 1995b). Detailed cross-linking and serial deglycosylation studies using enzymatic digestion in the rat BB1 receptor demonstrated a molecular mass of 63 kDa in the membrane and showed that there were no O-linked carbohydrates, but that the mature BB1 receptor was a sialoprotein (Kusui et al., 1994). However, each of the potential N-linked glycosylation sites was, in fact, glycosylated, with tri-antennary and/or tetra-antennary complex oligosaccharide chains (Kusui et al., 1994).
C. BB1 Receptor Genomic Organization
The human BB1 receptor gene is localized at human chromosome 6p21-qter and in the mouse on chromosome 10 (Table 1). Both the human, rat, and mouse genes contained three exons with two introns (Corjay et al., 1991; Wada et al., 1991; Ohki-Hamazaki et al., 1997a; Ohki-Hamazaki, 2000). In the mouse the gene for the BB1 receptor spanned more than 10 kb with exon 1 of the BB1 gene separated from exon 2 by 6 kb, and this in turn is separated from exon 3 by 3 kb (Ohki-Hamazaki et al., 1997a). In human and mouse the first intron of the BB1 gene was located between transmembrane domains 3 and 4 and the second between transmembrane domains 5 and 6 (Corjay et al., 1991; Ohki-Hamazaki et al., 1997a). The first intron interrupted a codon for arginine located immediately COOH terminal to the transmembrane domain 3, and the second intron was located between glutamine and methionine codons in both the mouse and human BB1 gene (Corjay et al., 1991; Ohki-Hamazaki et al., 1997a). The positions of the first and second introns were identical in the mouse and human BB1 receptor gene (Corjay et al., 1991; Ohki-Hamazaki et al., 1997a).
D. BB1 Receptor Expression
Expression levels of BB1 receptor mRNA have been reported in human, mouse, rat, and monkey (Corjay et al., 1991; Wada et al., 1991; Ohki-Hamazaki et al., 1997a; Sano et al., 2004). In the monkey, in which it was studied in detail, the highest levels of BB1 mRNA are found in the CNS and in the testis (Sano et al., 2004). In the CNS the BB1 receptor was expressed widely in different brain regions including the amygdala, caudate nucleus, hippocampus, hypothalamus, thalamus, brain-stem, spinal cord, and peripheral tissues in addition to the testis and the stomach, which is a similar distribution to that found in rats and mice (Wada et al., 1991; Ohki-Hamazaki et al., 1997a; Ohki-Hamazaki, 2000; Sano et al., 2004). In the rat and mouse, BB1 mRNA is present in high amounts in the olfactory region and esophagus (Wada et al., 1991; Ohki-Hamazaki et al., 1997a). Binding studies and studies of biological activity provide evidence for BB1 on both gastrointestinal and urogenital smooth muscle cells (von Schrenck et al., 1989; Severi et al., 1991; Bitar and Coy, 1992; Kim et al., 1993). Binding studies have confirmed the widespread distribution of BB1 in the brain showing especially high levels in the olfactory tract of the rat (Ladenheim et al., 1990, 1992, 1993a).
Using binding studies and/or assessment of BB1 mRNA, BB1 receptors have been shown to exist on a large number of different tumors (Reubi et al., 2002; Jensen and Moody, 2006) including CNS tumors (glioblastomas) (Wada et al., 1991; Wang et al., 1992), small cell and non-small cell lung cancers (Corjay et al., 1991; Moody et al., 1992, 2000; Toi-Scott et al., 1996; Siegfried et al., 1997; Jensen and Moody, 2006), carcinoids (intestinal, thymic, and bronchial) (Reubi et al., 2002), human ovarian epithelial cancers (Sun et al., 2000b), and pancreatic cancer cell lines (Jensen and Moody, 2006).
E. BB1 Receptor Pharmacology
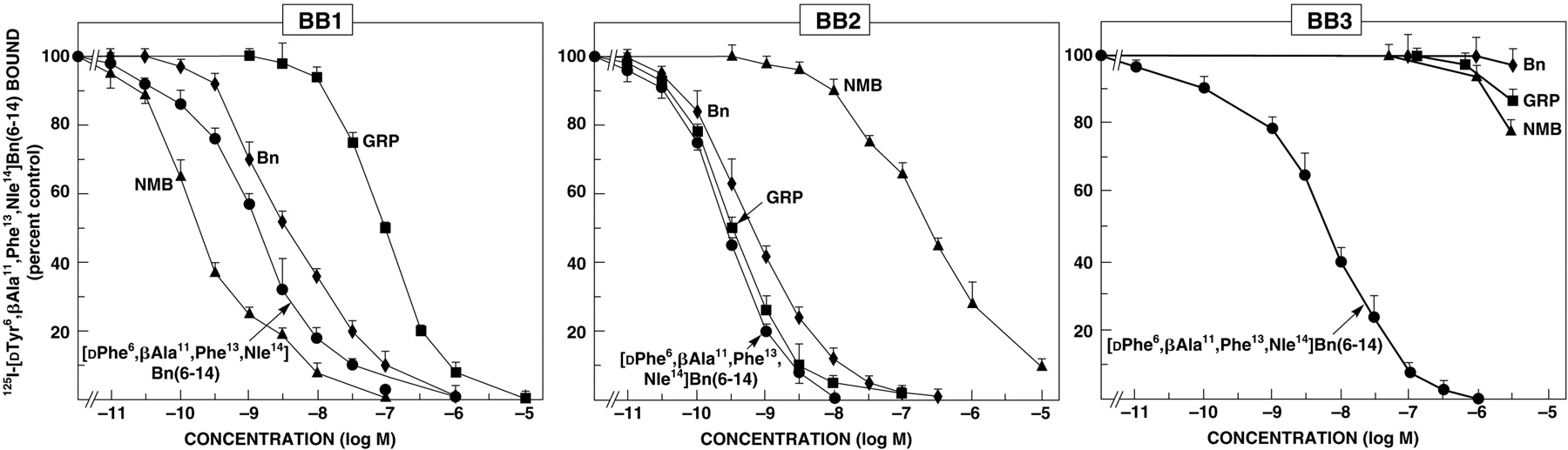
1. BB1 Receptor Agonists.
The human BB1 receptor (Moody et al., 1992; Benya et al., 1995b; Reubi et al., 2002) as well as the rat BB1 receptor (von Schrenck et al., 1989, 1990; Wang et al., 1992; Ladenheim et al., 1992, 1993a) has a >100-fold higher affinity for NMB than for GRP (Fig. 1, Table 1). Bombesin and the frog peptides, ranatensin and litorin, also had relatively high affinity for the BB1 receptor (affinities 1- to 10-fold less than those for NMB) (Wang et al., 1992; Mantey et al., 1997; Katsuno et al., 1999) (Tables 1 and 2). The synthetic bombesin analog [d-Phe6,β-Ala11,Phe13,Nle14]-bombesin6–14 (Mantey et al., 1997), which has high affinity for the human BB3 receptor also has a high affinity for the human BB1 receptor as well as the human BB2 receptor and fBB4 (Mantey et al., 1997; Pradhan et al., 1998) (Table 2).
Affinity of bombesin receptor subtypes for various agonist / antagonists See text for definitions of compound structures for each specific receptor.
2. BB1 Receptor Antagonists.
Whereas the search for high-affinity receptor antagonists for the BB2 receptor has been very successful (section IV.E.1.) (Jensen and Coy, 1991; Jensen et al., 1993; de Castiglione and Gozzini, 1996), results with the BB1 receptor have been much less successful and only a few high-affinity receptor antagonists are available. None of the strategies used for making high-affinity BB2 antagonists were successful with the BB1 receptor, including the synthesis of bombesin or NMB COOH-terminal pseudopeptide analogs, COOH-terminal truncated analogs or [des-Met10]-NMB amides, alkylamides, or esters (Lin et al., 1995). Subsequently, it was discovered that certain substituted somatostatin analogs selectively antagonized the BB1 receptor compared with the BB2 receptor (Orbuch et al., 1993). The most potent analog was cyclo-somatostatin-octa[d-Nal-Cys-Tyr-d-Trp-Lys-Val-Cys-Nal-NH2], which had a 100-fold higher affinity for the BB1 receptor than the BB2 receptor (Ki 230 versus 3000 nM) (Orbuch et al., 1993; Ryan et al., 1999) (Table 2). Unfortunately this analog also interacted with high affinity with somatostatin receptors (IC50 0.80 nM) and μ-opioid receptors (IC50 430 nM) (Orbuch et al., 1993). Substitution of an ornithine for Lys greatly reduced the affinity for somatostatin receptors, and a related analog (BIM-23127) inhibited NMB cell signaling in rat BB1 receptor transfected Rat-1 cells (Lach et al., 1995) and selectively reversed NMB feeding suppression, but had no effect on the action of GRP (Ladenheim et al., 1997b). However, a recent study reported that BIM-23127 also functions as a receptor antagonist of both human and rat urotensin-II receptors (Herold et al., 2003), limiting its utility. Peptoid antagonists of BB1 have been described, including PD 165929 (Eden et al., 1996) and PD 168368 (Ryan et al., 1999), which have high affinity and selectivity for BB1. In a detailed comparison of bombesin receptors from different species, PD 168368 was found to have a similar high affinity (Ki 15–45 nM) for BB1 receptors from each species, a 30- to 60-fold lower affinity for the BB2 receptor from different species, and a >300-fold lower affinity for the BB3 receptor or fBB4 (Ryan et al., 1999) (Table 2). It also inhibited NMB-stimulated cellular signaling in a competitive manner (Ryan et al., 1999) as well as inhibiting NMB-induced proliferation of rat C6 glioblastoma cells (Moody et al., 2000) and NMB stimulation of NCI-H1299 lung cancer cell proliferation (Moody et al., 2000).
F. BB1 Receptor Structural Basis of Receptor Binding/Activation
1. BB1 Receptor Agonist Binding/Activation.
Structure-function studies of NMB demonstrated that the COOH-terminal octapeptide is the minimal peptide length required for BB1 receptor activation and the full decapeptide was required for full affinity for the BB1 receptor (Lin et al., 1996). NMB differs from GRP in the COOH octapeptide, which is the biologically active end (Broccardo et al., 1975; Lin et al., 1996), at three residues: substitution of a leucine in NMB for a histidine in GRP at position 3, a threonine for valine at position 6, and a phenylalanine for leucine at position 9 of NMB from the amino terminus (Minamino et al., 1983; Lin et al., 1996) (Fig. 1). Structure-function studies of all naturally occurring bombesin-related peptides for BB1 and BB2 receptors suggested the presence of the phenylalanine instead of leucine, as the penultimate amino acid from the COOH terminus in NMB was not important for selectivity for the BB1 receptor, Single amino acid substitutions in NMB demonstrated the Leu for His substitution in position 3 was the most important for determining high affinity and selectivity for the BB1 receptor (Lin et al., 1996) (Fig. 1).
A chimeric receptor approach (Fathi et al., 1993a) and homology screening after computer alignment of bombesin receptor family members (Sainz et al., 1998), followed by site-directed mutagenesis studies, have been used to explore the molecular basis of NMB high affinity and selectivity for the BB1 receptor over the BB2 receptor (Fig. 3). A study of BB1/BB2 chimeric receptors (Fathi et al., 1993a) demonstrated that differences in the amino terminus of the two receptors were of minimal importance for high-affinity NMB interaction. High affinity and selectivity for the BB1 receptor were primarily determined by differences in transmembrane (TM) domain 5 (Fathi et al., 1993a) (Fig. 3). Site-directed mutagenesis of the amino acid differences between the BB1 receptor and the BB2 receptor in this region demonstrated that the substitution of an Ile216 instead of Ser in the comparable position of the TM5 of the BB2 receptor was the critical difference accounting for high-affinity NMB interaction with the BB1 and not the BB2 receptor (Fathi et al., 1993a). A second study (Sainz et al., 1998) used a different approach to select potentially important amino acids for NMB selectivity for the BB1 receptor and further study. Using amino acid sequence alignment of bombesin receptor family members and identifying conserved amino acids in members with similar peptide affinities (Akeson et al., 1997), four amino acids were identified that could be important for high-affinity bombesin binding to either the BB1 or BB2 receptor (Akeson et al., 1997) (i.e., in the BB1 receptor, Gln123, Pro200, Arg290, and Ala310, and in the BB2 receptor, Gln121, Pro199, Arg288, and Ala 308). Possible gain-of-affinity mutants were made in the BB3 receptor, which has a low affinity for NMB (Mantey et al., 1997; Ryan et al., 1998a,b), by substituting alone or in combination each of these four BB1 receptor amino acids for the comparable amino acid(s) of the BB3 receptor (Arg127, Ser205, His294, and Ser315) (Fig. 3). It was found that each of these four amino acids is important for determining NMB affinity because the affinities for NMB of the BB3 mutants with these BB1 receptor amino acids substituted one at a time were increased (Sainz et al., 1998). The substitution of all four amino acids for the comparable amino acids in the BB3 receptor, which has a very low affinity for NMB (i.e., Ki 3450 nM), increased the affinity and the potency for NMB, almost up to that seen with the native BB1 receptor (Sainz et al., 1998). This study helped to define the binding pocket for NMB by identifying four amino acids needed for high-affinity NMB interaction in markedly different BB1 regions [transmembrane domain 2 (Gln123), extracellular domain 2 (Pro200), extracellular domain 3 (Arg290), and transmembrane region 7 (Ala310)] (Fig. 3) (Sainz et al., 1998).
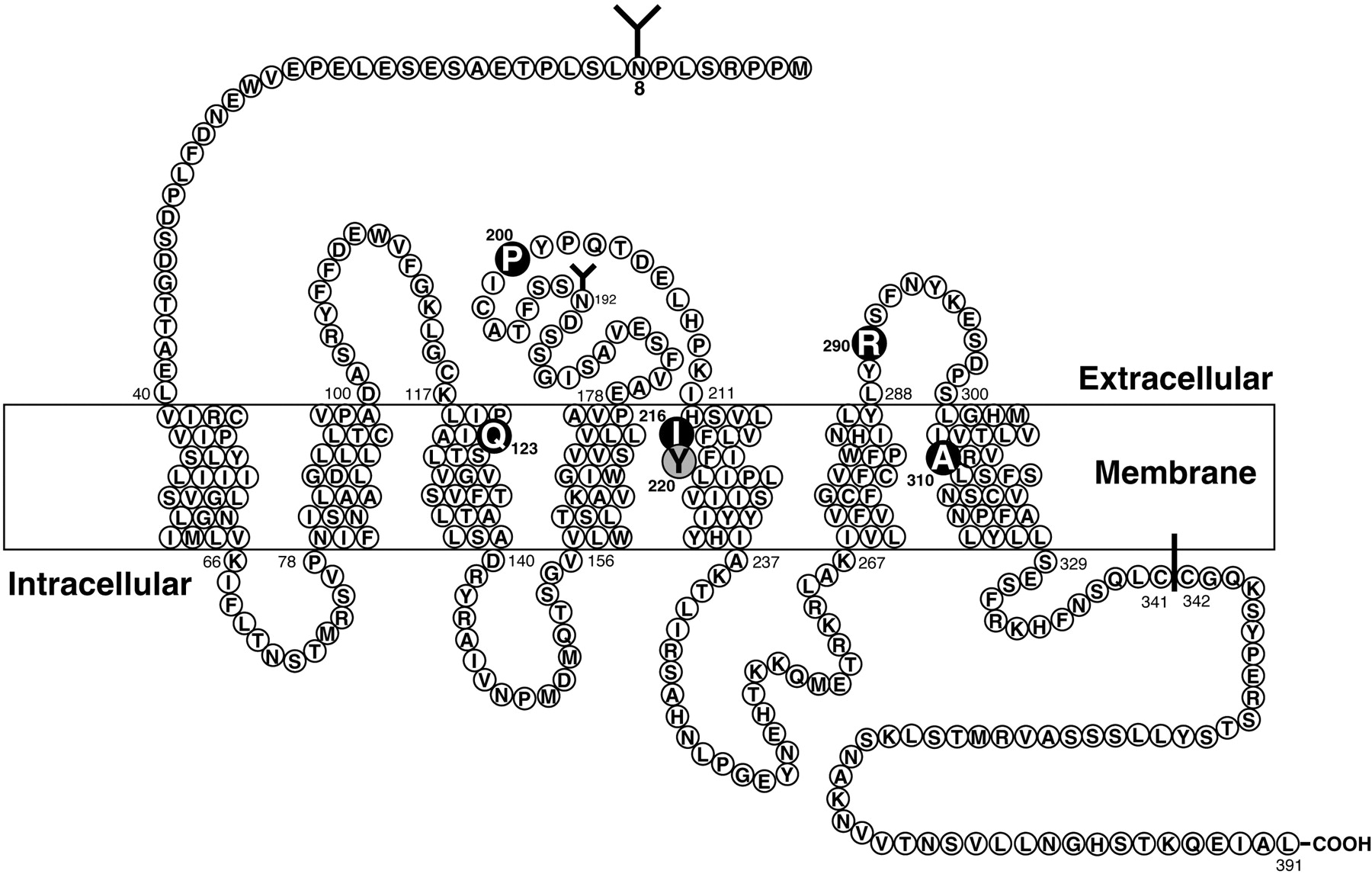
Schematic representation of the rat BB1 receptor showing the postulated transmembrane topology, sites for NH2-linked glycosylation, possible palmitoylated cysteines in the cytoplasmic tail, and the key amino acids for high-affinity NMB interaction (dark circles) or interaction with the BB1 receptor specific peptoid antagonist PD 168368 (shaded circles). Amino acid data are from Wada et al. (1991), data for NMB high-affinity sites are from Fathi et al. (1993a) and Sainz et al. (1998), and data for PD 168368 are from Tokita et al. (2001a).
2. BB1 Receptor Antagonist Binding.
Using a chimeric receptor approach combined with site-directed mutagenesis and receptor modeling, the molecular basis of selectivity of the BB1 receptor antagonist, PD 168368 was studied (Tokita et al., 2001a) (Fig. 3). PD 168368 is a new class of antagonists described as a peptoid, because this group of antagonists are nonpeptide ligands, which were designed using the chemical structure of the mammalian neuropeptide of interest as a stating point (Horwell et al., 1994; Horwell, 1995). This approach has yielded antagonists for cholecystokinin, somatostatin, tachykinins, and bombesin receptors (Boden et al., 1993; Boyle et al., 1994; Horwell et al., 1994; Horwell, 1995; Eden et al., 1996; Tran et al., 1998; Tokita et al., 2001a). However, little is known about the molecular basis of their affinity and whether they resemble peptide or other nonpeptide ligands in the basis of their selectivity and affinity (Tokita et al., 2001a). The receptor extracellular domains were shown not to be important for the selectivity of PD 168369 by studying both loss-of-affinity BB1 receptor chimeras in which the extracellular domains of the BB1 were replaced by those from BB2, one at a time or the reverse study performed by making PD 168368 gain-of-affinity chimeras in the BB2 receptor (Tokita et al., 2001a). Additional PD 168368 loss- and gain-of-affinity chimeric studies made by exchanging the upper transmembrane regions of BB1 and BB2 receptors showed that differences in the upper TM5 were the key determinants of selectivity of PD 168368 (Tokita et al., 2001a). Site-directed mutagenesis studies of the different amino acids between the BB1 receptor and the BB2 receptor in the upper TM5 region demonstrated that the substitution of Tyr at position 220 of BB1 for Phe in the comparable position in BB2 was the critical difference (Tokita et al., 2001a) (Fig. 3). Three-dimensional modeling studies showed the critical Tyr220 was facing the interior of a large binding pocket formed primarily by transmembrane domains 3 to 7 and minimum energy conformation of the ligand showed that it was dominated by a large hydrogen bond-accepting region around the nitrophenyl group (Tokita et al., 2001a). It was concluded that the Tyr220 hydroxyl group of the BB1 receptor was critical for interacting with the nitrophenyl group of PD 168368, probably primarily by hydrogen bonding. This result showed that the binding of this peptoid antagonist was similar to that reported with other nonpeptide antagonists, in that it was primarily dependent on interaction with transmembrane regions (Tokita et al., 2001a).
G. BB1 Receptor Signaling, Activation, and Modulatory Processes (Internalization, Down-Regulation, and Desensitization)
The human BB1 receptor (Moody et al., 1986, 1992, 1995a; Corjay et al., 1991; Benya et al., 1995b), as well as the rat BB1 receptor (Wada et al., 1991; Jones et al., 1992; Wang et al., 1992; Dobrzanski et al., 1993; Lach et al., 1995; Akeson et al., 1997; Tsuda et al., 1997b; Vigne et al., 1997; Hou et al., 1998) is coupled to phospholipase C, resulting in breakdown of phosphoinositides, mobilization of cellular calcium, and activation of protein kinase C. BB1 receptor activation also results in the stimulation phospholipase A2 (Moody et al., 1995a) and phospholipase D by a PKC-dependent and -independent mechanism (Tsuda et al., 1997b) but does not activate adenylate cyclase (Benya et al., 1992). BB1 receptor stimulation also results in activation of tyrosine kinases (Lach et al., 1995; Tsuda et al., 1997b) stimulating tyrosine phosphorylation of p125FAK by a phospholipase C-independent mechanism that requires p21rho and the integrity of the actin cytoskeleton (Tsuda et al., 1997b). BB1 receptor activation also stimulated tyrosine phosphorylation of paxillin and MAP kinase activation (Lach et al., 1995). The native and transfected rat BB1 receptor in BALB 3T3 cells have been shown to behave in a similar manner in their binding and signaling cascades (Benya et al., 1992), demonstrating the usefulness of this cell line for studying BB1 receptor interaction and signaling.
The BB1 receptor is coupled to heterotrimeric guanine-nucleotide binding proteins in both native and BALB 3T3-transfected cells (Benya et al., 1992; Wang et al., 1993). In an Xenopus oocyte assay with the injection of antisense oligonucleotides, Gαq was identified as a mediator of the BB1 receptor response (Shapira et al., 1994). With an in situ reconstitution assay with purified G protein α subunits, it was found that cells expressing the BB1 receptor activated Gαq, but not Gαt or Gαi/o (Jian et al., 1999). This activation was enhanced by βγ dimers with a relative potency of βγ > β1γ 2 >> β1γ1. In this study (Jian et al., 1999), these results were contrasted with those for the BB2 receptor, and differences were found in their kinetics of activation and preference for Gαq proteins from different sources and for βγ dimers, demonstrating distinct coupling mechanisms for these two closely related receptors (Jian et al., 1999).
In contrast with the BB2 receptor there have been few studies of BB1 receptor modulatory processes (internalization, down-regulation, or desensitization). Both the human (Benya et al., 1995b) and rat BB1 receptors (Benya et al., 1992, 1994c; Wang et al., 1993) are rapidly internalized with receptor activation of the BB1 receptor. The rat BB1 receptor internalized 60 to 80% of the bound ligand, and human BB1 receptors internalized 70% of the bound ligand. In addition to being rapidly internalized by BB1 receptor-bearing cells, the ligand is rapidly degraded by these cells (Benya et al., 1992; Wang et al., 1993). Protease inhibitors markedly decreased ligand degradation by either rat native or rat BB1 receptor-transfected BALB 3T3 cells (Benya et al., 1992; Wang et al., 1993) with the acid proteinase inhibitor, leupeptin being the most potent followed by bacitracin > chymostatin > phosphoramidon >> bestatin and amastatin. The BB1 receptor also undergoes desensitization, which is mediated by receptor down-regulation and internalization (Benya et al., 1994c). Preincubation for 3 h with 3 nM NMB markedly attenuated the ability of a maximally effective concentration of NMB (1 μM) to subsequently stimulate either native or BB1-transfected BALB 3T3 cells but did not alter the response to other stimulants (Benya et al., 1994c). This desensitization was associated with a rapid decrease in BB1 receptors due to internalization of the receptors. Restoration of receptor number and response recovered over a 6-h period, and it was not dependent on new protein synthesis but was due to receptor recycling, because it was inhibited by the recycling inhibitor, monesin, a monocarboxylic acid cation ionophore (Benya et al., 1994c).
H. BB1 Receptor Function in Various Tissues and in Vivo
One of the main difficulties in assessing the effects of BB1 receptor activation in the CNS as well as in peripheral tissues, especially in older studies, is that bombesin was frequently used as the agonist, and it interacts with both BB1 and BB2 receptor with relatively high affinity. Furthermore, many tissues possess both BB1 and BB2 receptors, and therefore it was difficult to assess whether a particular response was due to activation of the BB1 or BB2 receptors present.
Numerous effects of NMB in both in vivo and in vitro studies have been reported, but it is not clear in many cases which are physiological and which are pharmacological. Studies comparing the potencies of NMB to GRP as well as binding studies or antagonist studies provide evidence that the BB1 receptor can stimulate contraction of urogenital and gastrointestinal smooth muscle (esophageal, gastric, colon, and gallbladder) (Regoli et al., 1988; von Schrenck et al., 1989, 1990; Severi et al., 1991; Kilgore et al., 1993; Parkman et al., 1994; Milusheva et al., 1998), potently inhibit thyrotropin release from the pituitary gland by acting as an autocrine and paracrine regulator (Rettori et al., 1992; Pazos-Moura et al., 1996; Ortiga-Carvalho et al., 2003), and have potent CNS effects including inhibiting food intake independent of BB2 stimulation (Ladenheim et al., 1994, 1996b, 1997b; Merali et al., 1999; Ladenheim and Knipp, 2007) and mediating aspects of the stress and fear responses as well as various behaviors such as spontaneous activity (Merali et al., 2002, 2006).
BB1 receptor knockout mice are now available and have undergone a limited number of investigations for actions of NMB (Ohki-Hamazaki et al., 1999; Oeffner et al., 2000; Yamada et al., 2002b, 2003; Yamano et al., 2002) (Table 1). In these mice the hypothermic effect of NMB was reduced by 50% without a change in the GRP response, supporting a possible BB1 receptor-mediated role in thermoregulation: NMB-mediated gastric smooth muscle contraction was not affected, suggesting this is mediated not through BB1 receptors, and no effect on feeding could be confirmed, although NMB did not have an effect in the control animals (Ohki-Hamazaki et al., 1999). The satiety effects of the BB1 receptor are mediated through peripheral neural pathways different from those mediating the satiety effects of the BB2 receptor, because only the satiety effects of BB1 receptors are inhibited by capsaicin treatment, suggesting the involvement of primary sensory afferent neurons (Ladenheim and Knipp, 2007). Recently, NMB has found to be expressed in human and rodent adipose tissue and to be regulated by changes in energy balance. It was proposed that because of the known anorectic effects of NMB centrally, it may form part of a new adipose tissue-hypothalamic regulating system for food intake (Hoggard et al., 2007). In BB1 receptor knockout mice dysregulation of the thyroid occurred, suggesting that BB1 receptor pathways are significantly involved in both TSH gene regulation and function (Oliveira et al., 2006), dysfunction in response to stress was seen (Yamada et al., 2002b; Yamano et al., 2002), impairment in the modulation of the CNS 5-HT system in response to stress occurred (Yamano et al., 2002), and an impairment of learning and memory was seen (Yamada et al., 2003). The alterations in the CNS 5-HT and stress in these animals is particularly interesting, because the dorsal raphe nucleus is one of the brain regions that has a preponderance of BB1 receptors (Wada et al., 1990; Ladenheim et al., 1992; Pinnock et al., 1994; Merali et al., 2006), which are located on 5-HT neurons, and stimulation of this nucleus by NMB stimulates release of 5-HT, resulting in anxiogenesis (Merali et al., 2006). In a study in rats using BB1 and BB2 receptor agonists and antagonists (Bédard et al., 2007), data were provided to show that both GRP and NMB affect the stress response. NMB affected both anxiety and fear responses, whereas GRP affected only fear responses (Bédard et al., 2007).
Whereas the growth effects of the BB2 receptor in normal and especially in neoplastic tissues have received the most attention, stimulation of the BB1 receptor and/or administration of NMB has been shown to have growth-promoting effects in a number of neoplastic tissues. NMB is an autocrine growth factor for non-small cell lung cancer with 14 of 14 such cell lines possessing BB1 receptors in one study (Siegfried et al., 1997), and in four non-small cell lung cancer cell lines examined in detail NMB was synthesized and released into the media by the tumor cell in 7 to 15 times greater amounts than was GRP (Siegfried et al., 1997). Blockade of the BB2 receptor only partially blocked the proliferative effect of NMB on these cells, demonstrating the importance of BB1 receptor activation for the proliferative effects in these tumor cells (Siegfried et al., 1997). Furthermore, in human colon cancers NMB and the BB1 receptor are coexpressed, and they act in an autocrine growth fashion (Matusiak et al., 2005). Activation of BB1 receptors causes proliferation of rat C6 glioblastoma cells (Moody et al., 1995a), BB1 receptor transfected RAT-1 cells (Lach et al., 1995), small cell lung cancers (Moody et al., 1992), and adrenal zona fasciculata cells (Malendowicz et al., 1996).
I. BB1 Receptor in Diseases
At present, no disease has been shown to be caused specifically by alterations in the BB1 receptor. Activation of the BB1 receptor in various human cancers (particularly human small cell lung cancers, non-small cell lung cancers, colon, cancer, and various carcinoid tumors) due to an autocrine growth pathway may have an important effect on their growth (Moody et al., 1992; Moody and Jensen, 1996; Siegfried et al., 1999; Matusiak et al., 2005; Jensen and Moody, 2006). In various studies BB1 receptors were overexpressed by 55% of small cell lung cancers, 67% of non-small cell lung cancers, 46% of intestinal carcinoids, and a proportion of colon cancers, prostate cancers, and CNS tumors such as glioblastomas (Moody et al., 1995a; Reubi et al., 2002; Matusiak et al., 2005; Jensen and Moody, 2006).
Numerous studies (Rettori et al., 1992; Pazos-Moura et al., 1996; Ortiga-Carvalho et al., 2003) including BB1 receptor knockout studies (Oliveira et al., 2006) support the conclusion that NMB plays an important physiological role in the regulation of thyrotropin release, having primarily an inhibitory effect. NMB is produced in the pituitary (Jones et al., 1992), and it is proposed that NMB functions as a tonic inhibitor of TSH secretion, acting as an autocrine/paracrine regulator (Rettori et al., 1992; Oliveira et al., 2006) (Table 1). Conditions with increased TSH release such as hypothyroidism are associated with decreased pituitary NMB levels (Jones et al., 1992; Ortiga-Carvalho et al., 2003), whereas in hyperthyroidism in which the TSH levels are suppressed; there is an increased pituitary NMB level (Jones et al., 1992; Ortiga-Carvalho et al., 1997). These results suggest NMB could play an important role in human thyroid disorders causing hyperor hypofunction.
The role of NMB in human feeding disorders is unclear at present. Two genetic studies have suggested that the NMB gene is a possible candidate for eating disorders and predisposition to obesity (Oeffner et al., 2000; Bouchard et al., 2004).
IV. BB2 Receptor
A. Early Studies of the BB2 Receptor
Many of the early studies provided limited information on the BB2 receptor, as discussed in section III.A. for the BB1 receptor. This occurred because many of the tissues studied are now known to possess both BB2 and BB1 receptors and in most studies bombesin analogs were used, which have high affinity for both subclasses of receptors. This situation continued after the isolation of GRP in 1978 (McDonald et al., 1979), even though it had greater selectivity than bombesin analogs for the BB2 over the BB1 (von Schrenck et al., 1989; Lin et al., 1995; Benya et al., 1995b; Reubi et al., 2002), because of its limited availability. In vivo studies were even more difficult to interpret because numerous studies demonstrated that GRP-related peptides can have both a direct action on tissues as well as indirect action as they are potent for stimulating the release of many hormones (gastrin, insulin, somatostatin, CCK, pancreatic polypeptide, enteroglucagon, pancreatic glucagon, and gastric inhibitory peptide) (Greeley et al., 1986; McDonald et al., 1979, 1983; Modlin et al., 1981; Ghatei et al., 1982; Knuhtsen et al., 1987; Pettersson and Ahren, 1987; Kawai et al., 1988; Hermansen and Ahren, 1990). With the development of selective BB2 receptor antagonists (von Schrenck et al., 1990; Jensen and Coy, 1991; Benya et al., 1995b) and the increased use of BB2 selective ligands such as GRP, it became clear that a separate GRP-preferring receptor existed, even before the cloning of the mouse and human BB2 receptor in the early 1990s (Spindel et al., 1990; Battey et al., 1991; Corjay et al., 1991) (Table 2). It subsequently became clear that a number of the tissues that had been extensively used to characterize bombesin receptors/responses such as pancreatic acinar cells (Jensen et al., 1978; Jensen, 1994) and Swiss 3T3 cells (Rozengurt, 1988) possessed only BB2 receptors, whereas other tissues such as the CNS (Battey and Wada, 1991; Ladenheim et al., 1992) and smooth muscle preparations possessed both BB1 and BB2 receptors (Severi et al., 1991).
B. Cloned BB2 Receptor and Receptor Structure
The human BB2 receptor has 384 amino acids and shows high homology (90% amino acid identities) with the mouse BB2 receptor (Corjay et al., 1991) (Fig. 4). The human BB2 receptor has 55% amino acid identities with the human BB1 receptor (Corjay et al., 1991) and 51% with human BB3 receptor (Fathi et al., 1993b). Hydropathy analysis of the predicted human BB2 structure revealed seven regions of hydrophobic amino acids consistent with a seven-transmembrane structure typical for G protein-coupled receptors (Corjay et al., 1991). There were two consensus sites of potential PKC phosphorylation and two potential sites for N-linked glycosylation in the human BB2 receptor (Corjay et al., 1991). The BB2 receptor has been completely or partially cloned from 21 species (Baldwin et al., 2007) and the most highly conserved regions are in the transmembrane domains and the third intracellular domain (Baldwin et al., 2007). The presence of a likely disulfide bond between cysteines at the end of the extracellular domain 1 and middle of extracellular domain 2 (Cys113 and Cys196 in human BB2) is preserved in all noninsect species (Baldwin et al., 2007) (Fig. 4). Solubilization studies as well as cross-linking studies demonstrate that the mature human BB2 receptor has a molecular weight greater than that predicted from the structure (Kris et al., 1987; Rozengurt, 1988; Feldman et al., 1990; Huang et al., 1990; Staley et al., 1993; Benya et al., 1994b; Kusui et al., 1994; Williams and Schonbrunn, 1994; Benya et al., 1995b). Cross-linking studies demonstrate that the mature human BB2 receptor has a molecular mass of 60 ± 1 kDa and the mouse BB2 receptor has a molecular mass of 82 ± 2 kDa and when each is deglycosylated the molecular mass is 43 ± 1 kDa (Kris et al., 1987; Rozengurt, 1988; Huang et al., 1990; Benya et al., 1994b; Kusui et al., 1994; Williams and Schonbrunn, 1994; Benya et al., 1995b). These results demonstrate that 35% of the molecular mass of the mature human BB2 receptor is due to glycosylation, whereas in the mouse BB2 receptor it is 47%. This difference is probably due to the existence of two potential sites of N-linked glycosylation in the human BB2 receptor compared with four potential sites in the mouse BB2 receptor (Spindel et al., 1990; Battey et al., 1991; Corjay et al., 1991; Benya et al., 1995b) (Fig. 4). Using cross-linking studies with serial deglycosylation by enzymatic digestion (Kusui et al., 1994, 1995), and a molecular approach involving mutating the four potential N-linked glycosylation sites either alone or in combination in the murine BB2 receptor followed by receptor expression and cross-linking analysis (Benya et al., 1994d), the murine BB2 receptor was shown to be glycosylated at all four potential N-linked sites (Asn5, Asn20, Asn24, and Asn191) (Benya et al., 1994d; Kusui et al., 1994, 1995). The extent of glycosylation varied, however, with carbohydrate residues of 12 kDa on Asn5, 10 kDa on Asn20, 5 kDa on Asn24, and 9 kDa on Asn191 (Benya et al., 1994d). The presence of the glycosylation on Asn24 and Asn191 was especially important for sorting and expression of the murine BB2 receptor on the plasma membrane (Benya et al., 1994d). Digestion of the cross-linked receptor with different enzymes demonstrated that the murine BB2 receptor was not a sialoprotein, contained no O-linked glycosylation, and had four tri-antennary and or tetra-antennary complex oligosaccharide chains (Kusui et al., 1994). Studies using baculovirus expression of the BB2 receptor (Kusui et al., 1995) demonstrated that neither full glycosylation was needed for receptor expression on the cell surface nor did the glycosylation have to be tri- or tetra-antennary for expression, because in the baculovirus only 11 kDa of glycosylation was seen on different sites, and the glycosylation was entirely bi-antennary complex oligosaccharide chains (Kusui et al., 1995).

Schematic representation of the murine BB2 receptor showing the postulated transmembrane topology, sites for NH2-linked glycosylation, possible palmitoylated cysteines in the cytoplasmic tail, and the key amino acids for high-affinity GRP interaction or signaling (dark circles) or interaction with the BB2 selective antagonist statin analog JMV594 or the pseudopeptide analog JMV641 (shaded circles). Amino acid data are from Spindel et al. (1990) and Battey et al. (1991); GRP high-affinity sites are from Akeson et al. (1997), Donohue et al. (1999), Carroll et al. (2000b), Lin et al. (2000), Tokita et al. (2002), Glover et al. (2003), and Nakagawa et al. (2005); and data for JMV594 and JMV641 are from Tokita et al. (2001b).
C. BB2 Receptor Genomic Organization
The human BB2 receptor gene was localized to Xp22 (Maslen and Boyd, 1993; Xiao et al., 2001) and the murine BB2 receptor gene to X chromosome between the Pdha-1 and Amg loci (Maslen and Boyd, 1993). Both the human (Xiao et al., 2001) and murine (Weber et al., 2000) BB2 receptor gene organizations have been studied in detail. The human BB2 receptor gene has three exons (Corjay et al., 1991; Xiao et al., 2001) spanning more than 27 kb with intron 1 and intron 2 being 23 and 1.6 kb (Xiao et al., 2001). Exon one encodes the first three membrane-spanning domains of the BB2 receptor, and the splice site is located in the proximal second intracellular loop (residue 137). Exon 2 encodes for the transmembrane regions 4 and 5 and most of the third intracellular loop with the splice site located at residue 254. Exon 3 encodes for transmembrane domains 5 as well as the cytoplasmic carboxyl terminus of the BB2 receptor (Xiao et al., 2001). Two major transcription start sites for the human BB2 receptor gene were found in gastrointestinal and breast cancer cells located 43 and 36 bp downstream of a TTTAAA motif, which is identified 407 to 402 bp upstream of the ATG start codon (Xiao et al., 2001). Truncation studies of the transfected promoter region suggested that a cyclic AMP response element motif located 112 bp upstream of the major transcription start site is required to confer basal BB2 receptor promoter activity in duodenal cancer cells (Xiao et al., 2001).
D. BB2 Receptor Expression
Expression levels of BB2 receptor mRNA have been reported in human, mouse, and monkey (Spindel et al., 1990; Battey et al., 1991; Corjay et al., 1991; Ohki-Hamazaki et al., 1997a; Sano et al., 2004). BB2 receptor mRNA distribution was studied in detail in the monkey, in which it is found in the greatest amount in the pancreas and in lesser amounts in the stomach, prostate, skeletal muscle, and CNS (Sano et al., 2004). This result generally agrees with studies of location of the human BB2 receptor gene, in which a very strong signal was found in the normal pancreas with four specific transcripts of 9, 4.6, 3.1, and 2.1 kb sizes, a weaker signal in the stomach with two transcripts of 9 and 3.1 kb, and a very weak 9-kb transcript signal in whole brain and adrenal gland (Xiao et al., 2001). In the monkey CNS BB2 receptor mRNA was widely expressed with the highest amounts in hippocampus, hypothalamus, amygdala, and pons (Sano et al., 2004). In the mouse BB2 receptor mRNA was present in high amounts in the digestive tract and in the colon, but not in the stomach or small intestine (Battey et al., 1991). Detailing mapping in the rat brain was reported, which showed BB2 receptor expression in all brain regions, with the highest amounts of BB2 receptor mRNA in the hypothalamus, particularly the suprachiasmatic and supraoptic nuclei, and in the magnocellular preoptic nucleus in the basal ganglia and the nucleus of the lateral olfactory tract (Battey and Wada, 1991).
Detailed CNS location of the murine BB2 receptor has been reported using a specific BB2 receptor antibody (Kamichi et al., 2005). The BB2 receptor was widely distributed in the mouse brain in the isocortex, hippocampal formation, pyriform cortex, amygdala, hypothalamus, and brain stem (Kamichi et al., 2005). Strong BB2 immunoreactivity was observed in many nuclei of the amygdala and in the nucleus tractus solitarius (Kamichi et al., 2005). Double-labeling studies in the amygdala demonstrated subpopulations of BB2 receptors present in the GABAergic neurons, providing support for a possible role of BB2 receptors mediating memory by modulating neurotransmitter release in the local GABAergic network (Kamichi et al., 2005).
Binding studies have confirmed the widespread distribution of BB2 receptors in the brain, showing high levels in the cortex as well as the suprachiasmatic and supraoptic nuclei of the rat (Ladenheim et al., 1990, 1992, 1993a; Moody and Merali, 2004). Binding studies and studies of biological activity provide evidence for BB2 receptors on both gastrointestinal and urogenital smooth muscle cells (Severi et al., 1991; Kilgore et al., 1993; Ladenheim et al., 1997a; Milusheva et al., 1998; ter Beek et al., 2004; Fleischmann et al., 2005). BB2 receptors in the gastrointestinal tract are also found in gastric antral G cells (Giraud et al., 1987), other gastric mucosa cells (D cell, mucus cell, and parietal cell) (Nakamura et al., 1988), and pancreatic acinar cells (Jensen et al., 1978, 1988a; Jensen, 1994). In the epithelial cells lining the normal human gastrointestinal tract, BB2 receptor mRNA was only found in the antrum with the esophagus, jejunum, and ileum and not in the descending colon (Ferris et al., 1997).
BB2 receptors are present on a large number of different tumors using binding studies and immunohistochemical localization with specific receptor antibodies and/or assessment of BB2 receptor mRNA. BB2 receptors have been widely studied in prostate cancer (Reubi et al., 2002; Jensen and Moody, 2006; Patel et al., 2006), small cell lung cancer (Corjay et al., 1991; Toi-Scott et al., 1996; Jensen and Moody, 2006; Patel et al., 2006), non small cell lung cancer (Corjay et al., 1991; Toi-Scott et al., 1996; Siegfried et al., 1997; Jensen and Moody, 2006), breast cancer (Gugger and Reubi, 1999; Reubi et al., 2002; Jensen and Moody, 2006; Patel et al., 2006), head and neck squamous cell cancer (Lango et al., 2002; Jensen and Moody, 2006), colon cancer (Carroll et al., 1999b, 2000a; Jensen et al., 2001; Glover et al., 2003; Patel et al., 2006), uterine cancer (Fleischmann et al., 2005), various CNS/neural tumors (glioblastomas, neuroblastomas) (Jensen and Moody, 2006), ovarian cancer (Sun et al., 2000b), gastrointestinal carcinoid tumors (Reubi et al., 2002; Scott et al., 2004), and renal cell cancers (Reubi et al., 2002; Heuser et al., 2005).
E. BB2 Receptor Pharmacology
1. BB2 Receptor Agonists.
The human BB2 receptor (Frucht et al., 1992; Benya et al., 1995b; Reubi et al., 2002) and the rat (von Schrenck et al., 1990; Ladenheim et al., 1992, 1993a; Lin et al., 1996; Katsuno et al., 1999; Ryan et al., 1999), mouse (Huang et al., 1990; Ryan et al., 1999), and guinea pig BB2 receptors (Jensen and Gardner, 1981; Mantey et al., 1993) have >50-fold higher affinity for GRP than for NMB (Fig. 2). Bombesin and various frog peptides, including ranatensin, litorin, PG-L, and [Phe13]bombesin also have high affinities for the BB2 receptor, where as other frog peptides such as phyllolitorin, [Leu8]phyllolitorin, [Ser3,Arg10,Phe13]-bombesin and Xenopus NMB have low affinities for this receptor (Jensen and Gardner, 1981; Frucht et al., 1992; Mantey et al., 1997; Katsuno et al., 1999) (Fig. 1; Table 2). The synthetic bombesin analog, [d-Phe6,β-Ala11, Phe13,Nle14]bombesin6–14 (Iwabuchi et al., 2003), which has high affinity for human BB3 receptor, also has high affinity for the BB2 receptor as well as the BB1 receptor and fBB4 (Mantey et al., 1997; Pradhan et al., 1998; Ryan et al., 1998b).
2. BB2 Receptor Antagonists, Partial Agonists, and Biased Agonists.
a. BB2 receptor antagonists.
There have been a large number of different compounds reported to function as BB2 receptor antagonists (Jensen and Coy, 1991; Jensen et al., 1993; de Castiglione and Gozzini, 1996). They can be divided into six general classes of BB2 receptor antagonists (Jensen and Coy, 1991; Jensen et al., 1993; de Castiglione and Gozzini, 1996) (Table 2). All classes are peptides or peptoid antagonists, except for class 6, which are flavone derivatives, isolated from extracts of the mulberry tree Morus bombycis (Mihara et al., 1995). These six classes include substituted substance P analogs (class 1), [d-Phe12]bombesin analogs (class 2), modified position 13–14 bombesin or position 26–27 GRP analogs (class 3), desMet14 or GRP27 analogs (class 4), peptoids (class 5), and finally the nonpeptide analogs, kuwanon G and H (class 6) (Fig. 1).
Jensen and coworkers noted in 1984 that the d-amino acid-substituted substance P (SP) analog, [d-Arg1,d-Pro2,d-Trp7,9,Leu13]SP, not only functioned as a substance P receptor antagonist, but also inhibited both radiolabeled bombesin binding and bombesin-stimulated amylase release from guinea pig pancreatic acini, which are now known to possess only BB2 receptors. Later, they showed that various d-amino acid-substituted substance P analogs had broad inhibitory activity against a number of GPCR (Jensen et al., 1988b; Zhang et al., 1988). The inhibition of the action of bombesin by [d-Arg1,d-Pro2,d-Trp7,9,Leu13]SP was competitive in nature with a Schild plot having a slope of 0.996, and the inhibition was specific for the substance P and BB2 receptor, because it did not inhibit vasoactive intestinal peptide, secretin, or carbamylcholine-stimulated secretion (Jensen et al., 1984). Subsequent studies demonstrated that numerous d-amino acid substance P and SP4–11 analogs including [d-Arg1,d-Phe5,d-Trp7,9,Leu13] SP functioned as BB2 receptor antagonists (Jensen et al., 1988b; Woll and Rozengurt, 1988b; de Castiglione and Gozzini, 1996). These analogs were reported to inhibit bombesin-stimulated growth of lung cancer cells and Swiss 3T3 cells (Woll and Rozengurt, 1988a,b) as well as a number of other bombesin-stimulated changes in the CNS and peripheral tissues (Jensen and Coy, 1991). This class of BB2 receptor antagonists is now rarely used, not only because of their relatively low affinities for the BB2 receptor (1–40 μM) but also because of their lack of selectivity for the BB2 over the BB1 receptor. In addition, some show agonist activity in various tissues (von Schrenck et al., 1990; Jensen and Coy, 1991; Patel and Schrey, 1991; Lin et al., 1995; Mantey et al., 1997; Katsuno et al., 1999) (Table 2). These various d-amino acid-substituted SP analogs were reported not only to inhibit the action of bombesin but also to function as antagonists of substance P, cholecystokinin, vasopressin, and endothelin (Zhang et al., 1988; Langdon et al., 1992; Jarpe et al., 1998). Subsequent detailed studies of the mechanism of action of these substance P analogs provided evidence that they were functioning as biased agonists rather than antagonists. This will be discussed in the next section dealing with biased agonists.
Early bombesin structure-function studies demonstrated that Trp8 and His12 in the COOH terminus of bombesin were essential for biologic activity (Broccardo et al., 1975; Rivier and Brown, 1978; Märki et al., 1981). The substitution of a number of d-amino acids (d-Phe, d-chlorophenylalanine, and d-Tyr) for His12 in bombesin analogs produced antagonists (class 2) (Heinz-Erian et al., 1987; Saeed et al., 1989) (Fig. 1). These antagonists inhibited bombesin-stimulated amylase release from pancreatic acini (Heinz-Erian et al., 1987; Saeed et al., 1989) and the satiety effect of bombesin in rats (Flynn, 1997), which were both due to BB2 receptor activation. The use of these antagonists is limited by their relatively low affinities for the BB2 receptor (0.4–10 μM), their low aqueous solubility, and their low selectivity for BB2 over BB1 receptors (Lin et al., 1995; Mantey et al., 1997; Katsuno et al., 1999).
Numerous studies have demonstrated that the biologically active portion of GRP or bombesin is the COOH terminus (Broccardo et al., 1975; Rivier and Brown, 1978; Heimbrook et al., 1988; Lin et al., 1996). In 1988 Coy and coworkers reported a new class of BB2 receptor antagonists by substituting pseudopeptide bonds (Ψ bonds) (i.e., each CONH group one at a time replaced by CH2NH) into the COOH terminus of bombesin, a strategy that had been used successfully to make antagonists for gastrin, secretin, and substance P (Martinez et al., 1985; Rodriguez et al., 1986; Coy et al., 1988; Qian et al., 1989; Haffar et al., 1991) (Fig. 1; Table 2). Two of the pseudopeptides were antagonists with the Ψ 13–14 analogs having a higher affinity than the Ψ 9–10 bond analog. This Ψ13–14 bombesin analog was the first bombesin receptor antagonist described with an affinity <0.1 μM (Coy et al., 1988). Subsequent studies demonstrated that this analog had 50- to 100-fold higher selectivity for the BB2 receptor in human or rat than the BB1 receptor (Benya et al., 1995b; Ryan et al., 1999). This antagonist was shown to inhibit a number of BB2 receptor-stimulated processes including bombesin-stimulated enzyme secretion from isolated acini and growth of Swiss 3T3 cells as well as of various small cell lung cancer cell lines (Coy et al., 1988, 1989; Trepel et al., 1988; Liu et al., 2002). A subsequent study described short-chain pseudopeptide bombesin receptor antagonists (such as [d-Phe6,Cpa14,Ψ13–14]Bn6–14) that had fewer proteolytic sites and could be more easily synthesized (Coy et al., 1989, 1990, 1992a; Jensen and Coy, 1991) (Table 2). Furthermore, some of the Ψ13–14 analogs had partial agonist activity in some species (particularly the rat), which was not seen in a number of the newer, shortened substituted pseudopeptide analogs such as [d-Phe6,Cpa14,Ψ13–14]Bn6–14 (Dickinson et al., 1988; Coy et al., 1990, 1992a; Houben and Denef, 1991) (Fig. 1). A number of the shortened d-Phe substituted [Ψ13–14]Bn6–14 analogs are >100-fold more selective for the BB2 over the BB1 receptor (von Schrenck et al., 1990; Mantey et al., 1997; Katsuno et al., 1999). Subsequently, a particularly potent group of pseudopeptide antagonists, having a d-Pro-Ψ(CH2NH)-Phe-NH2 moiety at the COOH terminus of GRP, were described (Leban et al., 1993). One of the most potent and widely used analogs in this series is (3-PhPr)-His,Trp,Ala,Val,d-Ala,His,d-Pro-Ψ(CH2NH)-Phe-NH2 (BW2258U89) [(Ki 0.001 nM murine BB2) (Leban et al., 1993); 0.7 nM rat BB2 (Mantey et al., 1997), and 10 nM human BB2 (Moody et al., 1996a)]. BW2258U89 has >10,000 fold selectivity for the rat BB2 over the rat BB1 receptor (Mantey et al., 1997; Katsuno et al., 1999) (Table 2). BW2258U89 was reported to inhibit small cell lung cancer growth (Moody et al., 1995b) and to inhibit bombesin-stimulated gastrin release in vivo in dogs and rats (Singh et al., 1992) and blocked the satiety effect of bombesin in rats (Kirkham et al., 1994). An additional series of substituted pseudopeptide analogs with position 14 substitutions in addition to the Ψ13–14 bond have been described and widely used by Schally's group for inhibition of various tumor cell growth (Radulovic et al., 1991a; Cai et al., 1992, 1994; Qin et al., 1994, 1995; Jungwirth et al., 1998; Bajo et al., 2004). Two analogs with high potency in this group include [d-Phe6,Ψ13–14,Tac14]Bn6–14 (tac = thiazolidine-4-carboxylic acid) (RC-3950-II) (Cai et al., 1994) (Ki 0.078 nM, murine BB2 receptor) and [d-Tpi6,Ψ13–14]bombesin6–14 (RC-3095) (Ki 0.92 nM, murine BB2 receptor) (Reile et al., 1994; Qin et al., 1994, 1995). A final group of potent antagonists in this class were synthesized by J. Martínez's group, with the most potent being JMV641 and JMV594 (Azay et al., 1996; Lamharzi et al., 1998). JMV641 [H-d-Phe,Gln,Trp,Ala, Val,Gly,His-NH-*CH[CH2-CH(CH3)2]-**CHOH-(CH2)3-CH3 [where * is (S) and ** is 92% of (S isomer)], contains a pseudopeptide bond that mimics the transition state analog (Ki murine BB2 0.85 nM) (Azay et al., 1996) and has a >3000-fold selectivity for the BB2 over the BB1 receptor (Tokita et al., 2001b). JMV594 [d-Phe6, statine13]Bn6–14] (where statine = 4-amino-3-hydroxy-6-methylhepatanonoic acid) also has a high affinity for the murine BB2 receptor (Ki 0.60 nM) (Azay et al., 1998; Llinares et al., 1999) and has >5000-fold selectivity for the BB2 over the BB1 receptor (Tokita et al., 2001b) (Table 2).
The fourth class of BB2 receptor antagonists are all [desMet14]Bn or [desMet27]GRP analogs (Jensen and Coy, 1991; Jensen et al., 1993; de Castiglione and Gozzini, 1996), but vary widely in chemical groups attached, including desMet amides (Heimbrook et al., 1989; Wang et al., 1990a,b), alkylamides (Camble et al., 1989; Heimbrook et al., 1989; Wang et al., 1990a,b), esters (Heimbrook et al., 1989; Wang et al., 1990b; Coy et al., 1992b), hydrazides (Wang et al., 1990b), and with other COOH-terminal groups attached (Heimbrook et al., 1989, 1991) (Fig. 1; Table 2). A number of these analogs have high potency for the BB2 receptor in all species studied and have high selectivity for the BB2 over the BB1 receptor (Heimbrook et al., 1989; Jensen and Coy, 1991; Jensen et al., 1993; Benya et al., 1995b; de Castiglione and Gozzini, 1996; Mantey et al., 1997; Katsuno et al., 1999). Two widely used antagonists in this class are [d-Phe6]Bn6–13 methyl ester or its analogs (Wang et al., 1990b; Coy et al., 1992b) and Ac-[N-GRP20–26 ethyl ester (Heimbrook et al., 1989), with each having high affinity for the BB2 receptor (Ki 2–5 nM) (Heimbrook et al., 1989; Wang et al., 1990b; Coy et al., 1992b; Benya et al., 1995b; Mantey et al., 1997; Katsuno et al., 1999) and having >1000-fold selectivity for the BB2 over the BB1 receptor (von Schrenck et al., 1990; Katsuno et al., 1999). [d-Phe6]Bn6–13 methyl ester and/or Ac-N-GRP20–26 ethyl ester are reported to inhibit GRP-stimulated mitogenesis in 3T3 cells (Heimbrook et al., 1989) (Fig. 1), GRP-dependent acid secretion (Heimbrook et al., 1989), GRP-induced signaling in small cell lung cancer cells, GRP/Bn-induced smooth muscle contraction (Maggi et al., 1992), and BB2 receptor-mediated pancreatic enzyme secretion (Wang et al., 1990b) and in vivo to inhibit bombesin/GRP-stimulated pancreatic enzyme secretion (Varga et al., 1991; Coy et al., 1992b), satiety (Stratford et al., 1995; Ladenheim et al., 1996a), hypothermia (Cai et al., 1994), and acid secretion (Weigert et al., 1997). In vivo a number of these antagonist were found to have a short duration of action (Alptekin et al., 1991; Coy et al., 1992b), and it was found that by adding a d-Ala11 in place of Gly11 in bombesin, as well as lipophilic moieties to the amino terminus, the in vivo stability was improved, and analogs with long duration of action were obtained. [d-pentafluoro-Phe6,d-Ala11]Bn6–13 methyl ester not only retained high affinity for the BB2 receptor (Ki human BB2 0.9 nM; rat BB2 5 nM) but it also had >400- to 10,000-fold selectivity for the BB2 over the BB1 receptor in rat and human (Coy et al., 1992b; Benya et al., 1995b) and a 15-fold longer duration of action in vivo (Coy et al., 1992b) (Fig. 1). This analog was subsequently used in a number of human studies (Guex and Peitsch, 1997; Hildebrand et al., 2001), which will be reviewed in section IV.H.
In contrast to the BB1 receptor (Eden et al., 1996; Moody et al., 2000; Tokita et al., 2001a), there are no selective peptoid BB2 receptor antagonists (class 5). However, PD 176252 is a peptoid antagonist that has nanomolar affinity for both the BB2 (Ki 1 nM) and BB1 receptor (Ki 0.1 nM) (Ashwood et al., 1998; Moody et al., 2003b). Subsequent studies demonstrated that PD 176252 inhibited the growth of lung cancer cells, potentiated the growth inhibitory effects of histone deacetylase inhibitors (Moody et al., 2006a); inhibited GRP/Bn-stimulated signaling in lung cancer cells (Ca2+ and tyrosine phosphorylation of p125FAK) and the stimulation of increases in c-fos mRNA (Moody et al., 2000) and growth (Moody et al., 2000), and in rats had an anxiolytic effect in vivo (Merali et al., 2006).
The only nonpeptide, nonpeptoid antagonists of BB2 receptors reported are kuwanon G and kuwanon H, two closely related flavone compounds that were isolated from the Mulberry tree, M. bomcycis (Mihara et al., 1995). Only one study (Mihara et al., 1995) has examined their ability to interact with BB2 receptors on Swiss 3T3 cells. Kuwanon G and kuwanon H had affinities of 290 and 470 nM, respectively for the murine BB2 receptor and kuwanon H had a 22-fold higher affinity for the murine BB2 receptor than for the rat BB1 receptor (Mihara et al., 1995). Kuwanon H inhibited both bombesin-stimulated changes in cytosolic calcium and growth in Swiss 3T3 cells, which are both mediated by BB2 receptors (Mihara et al., 1995).
b. BB2 receptor partial agonists.
None of the naturally occurring mammalian or frog bombesin-related peptides is a partial agonist for the BB2 receptor (Jensen et al., 1978, 1988a; von Schrenck et al., 1989; Lin et al., 1996). However, one of the main difficulties found with the various classes of peptide antagonists is that in some species or some cellular systems they demonstrated partial agonist activity or even full agonist activity, whereas they are antagonists in other species or cell systems (Coy et al., 1991b, 1992a; Jensen and Coy, 1991). This fact was reported for both class 3 pseudopeptide analogs as well as for class 4 potent desMet14 bombesin analogs in a number of studies (Dickinson et al., 1988; Coy et al., 1990, 1992a; Wang et al., 1990b; Houben and Denef, 1991; Wu et al., 1995). Furthermore, some BB2 receptor antagonists functioned as partial agonists for BB1 receptors (Ryan et al., 1996). Detailed studies with both bombesin pseudopeptide and desMet14 analogs, which functioned as pure BB2 receptor antagonists in the guinea pig or mouse, demonstrated that many showed partial agonist activity in the rat BB2 receptor (Coy et al., 1990, 1991b; Wang et al., 1990b; Jensen and Coy, 1991). The conclusion from these studies was that there exist important differences in the ability of the same ligand to activate the BB2 receptor from different species with the rat having less stringent peptide structural requirements for BB2 receptor activation than the guinea pig or mouse. The expression level of the BB2 receptor can have a marked effect on the magnitude of various agonist responses such as phospholipase C activation with stimulation of phosphoinositide breakdown (Tsuda et al., 1997a) and calcium mobilization (Wu et al., 1995) or stimulation of mitogenesis (Wu et al., 1995). This receptor density may contribute to the presence or magnitude of the partial agonist activity of some of these compounds in different tissues.
c. BB2 receptor-biased agonists.
As discussed in section IV.E.2.a. after the initial description of the ability of d-amino acid substituted analogs of substance P to function as bombesin receptor antagonists by Jensen et al. in 1984, the same group reported that some of these analogs could function as broad-spectrum antagonists inhibiting the activation of a number of peptide hormone GPCRs (Jensen et al., 1988b; Zhang et al., 1988). It is now clear that these compounds can inhibit activation of a wide range of different G protein-coupled receptors (i.e., substance P, cholecystokinin, vasopressin, and endothelin) (Zhang et al., 1988; Langdon et al., 1992; Jarpe et al., 1998). A number of subsequent studies have proposed different mechanisms for the ability for the substituted SP analogs to function as broad-spectrum GPCR antagonists, with some studies, but not others, suggesting that they function as biased agonists at the BB2 receptor (Jarpe et al., 1998; Sinnett-Smith et al., 2000; MacKinnon et al., 2001; Djanani et al., 2003). Initially it was shown (Jarpe et al., 1998) that the substance P analog, [d-Arg1,d-Phe5,d-Trp7,9,Leu11]SP, at concentrations that inhibited bombesin-stimulated calcium mobilization at the BB2 receptor, stimulated c-Jun kinase activation and cytoskeletal changes. To explain this unexpected result it was proposed (Jarpe et al., 1998) that the substance P analog functions as a biased agonist in that it causes the BB2 receptor to preferentially activate Gα12 over Gαq, and this results in activation of the Gα12-stimulated events (i.e., c-Jun kinase activation and changes in cytoskeletal events) and inhibition of the Gαq-stimulated events (i.e., calcium mobilization). A later study (Sinnett-Smith et al., 2000) challenged this hypothesis by providing evidence that d-amino acid-substituted SP analogs prevented BB2, bradykinin, and vasopressin receptor activation of both Gα12 and Gαq. A more recent study (MacKinnon et al., 2001) provided evidence that [d-Arg1,d-Phe5,d-Trp7,9, Leu11]SP differentially modulates the activation of the G proteins Gα12, Gαi, and Gαq. This unique ability allows BB2 receptor activation to couple to Gαi and at the same time to block Gαq, supporting the proposal that [d-Arg1,d-Phe5,d-Trp7,9,Leu11] SP is functioning as a biased agonist at the BB2 receptor.
F. BB2 Receptor Structural Basis of Receptor Binding/Activation
1. BB2 Receptor Agonist Binding/Activation.
Structure-function studies of GRP or bombesin demonstrate that the COOH-terminal heptapeptide is the minimal peptide length required for BB2 receptor activation and the COOH-terminal nonapeptide is the minimal fragment required for full affinity for BB2 (Mazzanti et al., 1982; Heimbrook et al., 1988; Lin et al., 1996). GRP differs from NMB in three residues in the biologically active COOH decapeptide: a histidine in GRP eight amino acids from the COOH terminus instead of Leu in NMB, at a valine five amino acids from the COOH terminus in GRP instead of a threonine, and a leucine at the penultimate position of GRP instead of phenylalanine in NMB (Minamino et al., 1983; Lin et al., 1996). Structure-function studies of all natural occurring bombesin-related peptides for BB2 and BB1 receptors suggested that primarily the presence of His for Leu and to a lesser extent the presence of Leu for Phe were the most important differences in GRP from NMB determining high affinity and selectivity for the BB1 receptor (Lin et al., 1996). Correlating biological activity with binding affinity, especially of antagonists, demonstrated that the presence of a COOH-terminal amino acid in position 14 of bombesin is not essential for high affinity for the BB2 receptor, but it is essential for biologic activity (Coy et al., 1988; Wang et al., 1990a, 1992).
From studies correlating binding results with biological activity, especially for COOH-terminal pseudopeptides, a model was proposed for the biologically active conformation of GRP/Bn at the BB2 receptor (Coy et al., 1988, 1991b; Wang et al., 1990a). In a study (Coy et al., 1988) of the effects on the affinity and potency of bombesin for the BB2 receptor of substitution of a Ψ bond (i.e., CH2NH2 instead of CONH) between each amino acid pair at the COOH terminus, it was found only Ψ13–14 and Ψ9–10 substitutions resulted in peptides that retained affinity for the BB2 receptor but did not activate it and thus functioned as antagonists. Because previous studies of somatostatin analogs had shown that hydrogen bonding was the prime factor in stabilizing the conformation of the peptide (Sasaki et al., 1987), the loss of efficacy with retention of affinity in these two bombesin pseudopeptides suggested that the elimination of these CO groups was probably having an effect on the conformation of the peptide owing to both loss of a potential intramolecular hydrogen-bonding point and increased rotation about the C–N bond (Coy et al., 1988). The model proposed (Coy et al., 1988) was based on the known solution conformation of somatostatin in which the COOH terminus of bombesin had a β-bend beginning at Val10 and the rest of the amino acid chains arranged in an antiparallel β-pleated sheet. In this model the hydrogen bonding between Leu13-Leu14 CO groups and Ala9-Val10-CO groups is important, and their destruction by a pseudopeptide bond would lead to a conformational shift and loss of efficacy. Support for this conformation has come from studies of both agonists and antagonists (Kull et al., 1992; Wang et al., 1990a; Coy et al., 1991a). Only the agonist results will be discussed here with the antagonist result in the next section. The proposed folded conformation of the COOH terminus of GRP/bombesin was supported by findings from a study of various covalently cyclized analogs of the COOH terminus of bombesin (Coy et al., 1991a). By using such an approach both agonists and antagonists were identified, supporting the proposal that both BB2 receptor agonists and antagonists probably adopted a folded conformation. A subsequent study (Lin et al., 1996) demonstrated that one cyclized analog, [d-Cys6,d-Ala11,Cys14]Bn6–14 had >400 fold greater potency for activation of the BB2 receptor than the BB1 receptor, suggesting that the constrained conformation induced by cyclization resembled more closely the active conformation for the BB2 receptor than that for the BB1 receptor. It also suggested that the active conformations for BB2 and BB1 receptor are significantly different (Lin et al., 1996). The substitution of d-Ala in position 11 of bombesin for glycine would be expected to stabilize the folding in the above proposed model and therefore not lead to a decrease in affinity/potency (Lin et al., 1996). The finding that [d-Ala11]bombesin was equipotent to native bombesin for the BB2 receptor, but resulted in a marked decrease in affinity for the BB1 receptor, supports both the folded conformation model proposed for the GRP/Bn COOH terminus (Coy et al., 1988) and also suggests the active conformation of bombesin for these two receptors is very different (Lin et al., 1996).
To elucidate the molecular basis of BB2 receptor agonist selectivity and high-affinity and receptor activation both a chimeric receptor approach (Tseng et al., 1995a,b; Maughfling et al., 1997; Tokita et al., 2002) either alone or followed by site-directed mutagenesis (Tokita et al., 2002), a comparison of receptor selectivity for agonists combined with homology screening after computer alignment of bombesin receptor family members (Akeson et al., 1997; Nakagawa et al., 2005), and site-directed mutagenesis of specific residues (Benya et al., 1993, 1994d; Slice et al., 1994; Donohue et al., 1999; Lin et al., 2000; Schumann et al., 2003) have been used. A study (Maughfling et al., 1997) of chimeric BB2/BB1 receptors demonstrated receptor regions between the end of TM3 and TM6 were responsible for the high affinity and selectivity of neuromedin C (GRP18–27) for the BB2 receptor. A subsequent detailed study (Tokita et al., 2002) examined both GRP loss- and gain-of-affinity chimeric BB2/BB1 receptors followed by site-directed mutagenesis and demonstrated differences in the extracellular (EC) domain 3 (where the N terminus is EC1), indicating that EC3 was the specific critical region for determining GRP high affinity and selectivity (Fig. 4). Site-directed mutagenesis (Tokita et al., 2002) of each of the 20 amino acid differences between the BB2 and BB1 receptor in the EC3 demonstrated that two amino acid differences were the most important (i.e., the substitution of Phe185 in the BB2 receptor for Ile in the comparable position in the BB1 receptor and of Ala198 in the BB2 for Ile in the comparable position of the BB1 receptor) (Fig. 4). Additional point mutations in these positions (Tokita et al., 2002) demonstrated that an amino acid with an aromatic ring in position 185 of the BB2 receptor was the most important of these two changes, whereas the size of the backbone substitution in position 198 was the difference from the BB2 receptor at this position, but it was less important than the position 185 difference for determining high affinity for GRP. The mechanism (Tokita et al., 2002) of the effect of aromatic substitution in position was not studied in detail, but it was proposed it might be due to cation-π or π-receptor interaction.
Important amino acids for GRP selectivity/high affinity were also identified using a different approach of comparison of receptor selectivity for agonists combined with homology screening after computer alignment of bombesin receptor family members (Akeson et al., 1997; Nakagawa et al., 2005). This approach made use of the fact that the BB2, BB1, and frog BB4 receptors all have relatively high affinity for bombesin, whereas the BB3 receptor has a very low affinity. In the first study (Akeson et al., 1997) nine amino acids that were the same in BB1, BB2, and frog BB4 receptor but differed in the BB3 receptor were identified,. Site-directed mutagenesis (Akeson et al., 1997) demonstrated the occurrence of Arg288 in the BB2 receptor or comparable position of the other receptors with high affinity for bombesin, instead of a histidine in the comparable position of the BB3 receptor (i.e., R288H change), a glutamine in position 121 instead of arginine (Q121R), a proline in position 199 instead of a serine (P199S change), and an alanine in position 308 of instead of a serine (A308S change) as the critical differences accounting for high affinity for bombesin (Fig. 4). Of these four critical differences the Q121R and R288H change had the most profound effect on determining both the affinities of GRP and bombesin for the BB2 receptor (Akeson et al., 1997). Molecular modeling (Akeson et al., 1997) demonstrated that Q121,R288, and A308 all lie in a plane pointing inward toward the binding pocket. Furthermore, the critical Q121 lies in the same position in the BB2 receptor in TM3 as the highly conserved aspartate in biogenic amine receptors, which has been shown to be critical for their high-affinity interaction, suggesting that a similar interaction is critical for GRP high affinity. In a second study (Nakagawa et al., 2005) a modification of the above approach was used, in which amino acid differences from receptors with high affinity for GRP (BB2 receptor and frog BB4 receptor) were identified and compared with the BB3, which has low affinity for GRP. Fourteen amino acid differences (Nakagawa et al., 2005) were found and each was analyzed by site-directed mutagenesis with the results compared with the effects of the Q121R, P199S, R288H, and A308S point mutations described above (Akeson et al., 1997). This study (Nakagawa et al., 2005) demonstrated that the selectivity of GRP for the BB2 receptor was primarily determined by K101,Q121,A198,P199,S293,R288, and T297 of the BB2 receptor (Fig. 4). Molecular modeling of the BB2 receptor (Nakagawa et al., 2005) demonstrated that the backbone substitutions of 8 of the 14 amino acids identified using this approach were facing inward to the binding pocket and were within 6 Å including the Q121, A193, S293, and R288 which were especially important for GRP affinity. A phylogenetic analysis of the structures of the BB2 receptor from 21 species was performed and compared with that for other bombesin receptor family members and other GPCRs (Baldwin et al., 2007). This analysis (Baldwin et al., 2007) demonstrated the sequence GVSVFTLTALS (125–136 in murine BB2 receptor) in the cytoplasmic side of TM3 is unique to the bombesin receptor family and is retained by all members; the cysteines residues in positions C94, C114, C197, C277, and C317 in the murine BB2 are highly conserved in all BB2 receptors, and the important amino acids described for determining GRP affinity are generally well conserved in all BB2 receptors.
BB2 receptor mutations are reported to occur in human colon and gastric cancer and a number of these have identified and characterized (Carroll et al., 1999a, 2000b; Glover et al., 2003). In the human BB2 receptor P145Y, P198L, P200S, and V316E mutations (equivalent to positions 146, 199, 210, and 317 in murine BB2 receptor (Fig. 4) are found in colon and/or gastric cancers (Carroll et al., 1999a, 2000b; Glover et al., 2003), and each resulted in no ligand binding of the expressed BB2 receptor, demonstrating that these amino acids in the BB2 receptor are essential for either receptor expression and/or binding.
A number of studies have attempted to examine the important amino acids in BB2 receptor-mediating activation as well as in the stimulation of various receptor modulatory processes (internalization, down-regulation. and/or desensitization) (Benya et al., 1994a; Tseng et al., 1995a; Donohue et al., 1999; Schumann et al., 2003). Because the BB2 receptor as well as the BB1 and BB3 receptors have a conserved aspartate residue at position 98 (D98) just at the extracellular border of TM 2 and a arginine residue (R309) at the top of TM7 (Fig. 4), the effects of these on receptor binding and activation were explored using site-directed mutagenesis, binding studies, and an in situ reconstitution assay. The results (Donohue et al., 1999) demonstrated that these residues are not only important for high-affinity binding, but they are also critical for efficient coupling of the BB2 receptor to Gαq. The authors (Donohue et al., 1999) suggested that these results are consistent with the existence of a salt bridge interaction between these two polar and oppositely charged amino acids that maintains the proper BB2 receptor conformation necessary to interact with G proteins. The importance of the second and third intracellular domains (IC2 and IC3) of the BB2 receptor for affinity, activation, and internalization were examined by making BB2 receptor/m3 muscarinic cholinergic receptor chimeras (Tseng et al., 1995a). Replacement of the IC2 and/or IC3 domain alone or together in the BB2 receptor had minimal or no effect on receptor affinity or the occurrence of the high-affinity receptor binding state; however, replacement of IC3, but not IC2, dramatically decreased the ability of the BB2 receptor to internalize bombesin or to activate the receptor and stimulate phospholipase A2 or C (Tseng et al., 1995a). It was proposed from these results that agonist activation of a similar conformational state is required for BB2 receptor G protein-coupling and internalization but is not needed for generation of a high-affinity binding state (Acs et al., 2000). The BB2 receptor, as well as other bombesin receptors and many GPCRs, have a retained DRY sequence at the beginning of the second intracellular domain and a conserved alanine in the distal third intracellular domain (Benya et al., 1994a), which have been shown in a number of GPCRs to be important for G protein coupling and cell signaling (Benya et al., 1994a). Site-directed mutagenesis (Benya et al., 1994a, 1995a) was used to make a R139G and A263E mutant (Fig. 4) to explore the importance of these conserved residues for BB2 receptor affinity, cell signaling, and activation of receptor modulatory processes (internalization, down-regulation, and desensitization) (Benya et al., 1994a, 1995a). Both of these mutations decreased BB2 receptor affinity for bombesin by 9-fold, neither receptor could activate phospholipase C, and the R139G, but not the A263E mutant, was uncoupled from G-proteins. Both mutant receptors demonstrated impaired internalization, however the impairment was much greater with the R139G mutant. These results demonstrated that BB2 receptor internalization occurs by both phospholipase-dependent and phospholipase-independent mechanisms and that both are dependent on G protein coupling of the activated BB2 receptor. In contrast (Benya et al., 1995a), each of these mutant BB2 receptors demonstrated no bombesin-stimulated receptor down-regulation, whereas the wild-type receptor underwent a >75% decrease in receptor number when exposed to agonist. These results demonstrated that BB2 receptor internalization and down-regulation are at least partially mediated by different signaling mechanisms. In studies of the muscarinic cholinergic M3 receptor the central portion of IC2 is important for G protein coupling and internalization (Moro et al., 1993, 1994). Results of a systematic analysis of this region of the BB2 receptor (amino acids 142–148) (Fig. 4) have been reported (Schumann et al., 2003). In this study (Schumann et al., 2003) each amino acid was mutated to an alanine either alone or in combination. The mutations had minimal (<2-fold) to no effect on agonist receptor affinity; however, five mutants showed decreased efficacy for activation of phospholipase C (Schumann et al., 2003). Two mutations, the IM143,147AA and VM144,147AA, showed markedly decreased abilities to activate phospholipase C. The IM double mutant had defective internalization, whereas the R145A mutant had enhanced internalization (Schumann et al., 2003). Both double mutants and three single mutants also had decreased down-regulation. Maximal changes in phospholipase C were significantly correlated with maximal down-regulation, but not with internalization. Therefore, amino acids within the IC2 of the BB2 receptor are important for activation of phospholipase C and support the proposal that internalization and down-regulation have a different dependence on phospholipase C activation and are largely independent processes (Schumann et al., 2003). Kinetic analysis of the effect of the R145A mutation on BB2 receptor binding and internalization support the conclusion that the R145 in the native receptor is having a restraining effect on internalization and its mutation deceased receptor recycling without altering the endocytotic rate (Schumann et al., 2003).
Residues in the cytoplasmic carboxyl terminus of the receptor are important for various receptor modulatory processes such as internalization or desensitization in numerous GPCRs (Benya et al., 1993; Tseng et al., 1995b). Two different approaches have been used with the BB2 receptor to investigate the importance of this region. In one study (Benya et al., 1993) serial truncation mutants of the BB2 receptor COOH terminus were constructed as well as site-directed mutation of PKC consensus sites, a potential palmitoylation site and of Ser/Thr residues. None of these mutations altered receptor affinity or altered the ability of the expressed mutant to activate phospholipase C. Longer truncations (at residue 358 or more proximal) resulted in increasing impairment of internalization, whereas the mutation of the potential palmitoylation site had no effect. Mutation of the distal PKC consensus site moderately reduced internalization (approximately 50%), whereas mutation of all Ser/Thr residues in the COOH tail almost completely inhibited internalization (Benya et al., 1993). These results (Benya et al., 1993) show that BB2 receptor internalization is dependent on residues in the COOH terminus and suggest that it is partially PKC-dependent but completely dependent on the presence of at least some Ser or Thr residues in this region. A second approach used to examine the importance of the COOH terminus in BB2 receptor function was to make BB2 receptor/m3 muscarinic cholinergic receptor chimeras or BB2 receptor/CCKA receptor chimeras by substituting the COOH terminus of these receptors for that of the BB2 receptor (Tseng et al., 1995b). Each of the chimeric receptors demonstrated affinities similar to those of the wild-type BB2 receptor for bombesin and similar potencies for activation by bombesin. Ligand internalization as well as receptor recycling by the chimeric BB2 receptors generally assumed the characteristics of the donor receptor (Tseng et al., 1995b). This study (Tseng et al., 1995b) demonstrated that carboxyl-terminal structures determine both the internalization of the ligand-receptor complex and the subsequent recycling. The BB2 receptor undergoes rapid down-regulation and desensitization in addition to internalization with agonist stimulation (Benya et al., 1994b, 1994d, 1995a; Kroog et al., 1995a). A number of studies have explored the receptor structural elements involved in stimulation of these receptor modulatory processes as well as the signaling cascades involved. The latter will be discussed later in section IV.G. on BB2 cell signaling mechanisms. In a number of GPCRs a conserved NPXnY motif in the TM7 is important for mediating receptor internalization and/or resensitization (Slice et al., 1994). Mutation of T324 within this motif in the rat BB2 receptor did not affect receptor internalization or its resensitization (Slice et al., 1994), demonstrating that this motif is not universally involved in receptor internalization.
The importance of the COOH terminus of the BB2 receptor for mediating chronic desensitization or down-regulation was explored by using mutant BB2 receptors with increasing COOH-terminal truncations, a distal PKC consensus mutation, a deletion of all COOH-terminal Ser/Thr residues, or mutations that either prevent BB2 receptor-activated phospholipase C activation (R139G and A263E) or G protein-coupling (R139G) (Benya et al., 1995a). Receptor mutants that did not activate phospholipase C did not show down-regulation or desensitization and removal of the distal PKC consensus sequence markedly attenuated both processes (Benya et al., 1995a). These results led the authors to conclude that PKC activation was essential for chronic desensitization and down-regulation and that no evidence was provided for the involvement of second messenger-independent mechanisms driving these receptor modulatory processes.
2. BB2 Receptor Antagonist Binding.
Numerous structure-function studies of primarily peptide antagonists demonstrated that the COOH-terminal amino acid of GRP or bombesin was not required for high-affinity interaction with the BB2 receptor; however it was required to activate the receptor (Coy et al., 1988; Heimbrook et al., 1989; Wang et al., 1990a, 1992). A number of results from these studies and molecular modeling studies supported the model proposed by Coy et al. (1988) in which the COOH terminus of GRP existed in a folded conformation, stabilized by hydrogen bonding, with the rest of the amino acid chains arranged as an antiparallel β-pleated sheet, 1988). Computer-generated molecular modeling (Kull, et al., 1992) of the COOH terminus of various GRP/Bn pseudopeptides and correlation with whether they behaved antagonists or partial agonists for the BB2 receptor, supported the Coy model (Coy et al., 1988). In detailed studies of [desMet14]bombesin amides and alkylamides (Wang et al., 1990a), the resultant antagonist activity could also be explained by the proposed model (Coy et al., 1988) with the loss of the COOH-terminal carbonyl group disrupting hydrogen bonding and modifying the conformation from the active form. The effect of this disruption is similar to the introduction of pseudopeptide bonds, which were proposed to result in a conformation shift of the position 14 carboxamide groups in the receptor-bound peptide promoted by the increased rotational freedom and flexibility introduced (Coy et al., 1988; Wang et al., 1990a).
In contrast to agonists, only two studies have examined the BB2 receptor structural elements responsible for BB2 receptor high affinity or selectivity for antagonists(Maughfling et al., 1997; Tokita et al., 2001b) (Fig. 4). A chimeric approach using BB2/BB1 receptor combinations was used to examine the region of the BB2 receptor responsible for the 500-fold selectivity of [d-Phe6]Bn6–13 ethylamide for the human BB2 receptor over the human BB1 receptor (Maughfling et al., 1997). The region from the NH2 terminus to the end of TM2 and regions in the EC4 and TM7 were primarily responsible for this antagonist selectivity. Using BB2/BB1 receptor chimeras, site-directed mutagenesis, and molecular modeling, the molecular basis was examined for the >3000-fold and >5000-fold selectivity of the two class 3 BB2 receptor antagonists JMV641 and JMV594, which contains a pseudopeptide bond that mimics the transition state analog (Azay et al., 1996; Lamharzi et al., 1998). Both loss-of-affinity and gain-of-affinity chimera studies showed that only differences in EC4 contributed to the BB2 selectivity of these antagonists. Each of the 11 amino acid differences between BB2 and BB1 in EC4 was mutated one at a time. The important differences for determining the selectivity of each antagonist were the presence of Thr297 in BB2 instead of a proline in the comparable position in the BB1 receptor, the presence of Phe302 in BB2 instead of a Met in the BB1 receptor, and the presence of Ser305 instead of Thr in the BB1 receptor (Fig. 4). Receptor modeling showed that each of these three amino acids faced inward toward the binding pocket, and each was within 5 Å of the putative binding pocket (Tokita et al., 2001b). These results suggest that both receptor-ligand cation-π interactions and hydrogen bonding are important for the high selectivity of these antagonists.
G. BB2 Receptor Signaling, Activation, and Modulatory Processes (Internalization, Down-Regulation, and Desensitization)
The human BB2 receptor (Moody et al., 1986, 1996b; Corjay et al., 1991; Williams and Schonbrunn, 1994; Benya et al., 1995b), as well as the rat (Deschodt-Lanckman et al., 1976; Matozaki et al., 1991; Garcia et al., 1997; Tapia et al., 2006), mouse (Huang et al., 1990; Garcia et al., 1997), guinea pig (Jensen et al., 1978, 1988a,b; Jensen, 1994; Garcia et al., 1997), and canine BB2 receptors (Seensalu et al., 1997) are coupled to phospholipase C, resulting in breakdown of phosphoinositides, generation of diacylglycerol, stimulation of the mobilization of cellular calcium, and PKC activation (Klein et al., 1979; Rozengurt, 1988, 1998a; Jensen, 1994). BB2 receptor stimulation activates both phospholipase β1 and β3, and this is dependent on Gαq (MacKinnon et al., 2001). Activation of the BB2 receptor also results in activation of phospholipase D (Cook et al., 1991; Briscoe et al., 1994) and phospholipase A2 (Currie et al., 1992; Nishino et al., 1998) and is reported to stimulate increased cAMP in some tissues (Rozengurt and Sinnett-Smith, 1983; Bjøro et al., 1987; Millar and Rozengurt, 1988; Garcia et al., 1997). The increase in cAMP in Swiss 3T3 cells was reduced by PKC down-regulation and inhibition of cyclooxygenase, suggesting that these pathways were involved (Rozengurt et al., 1987). However, a systematic study demonstrated the activation of BB2 receptors in normal pancreas from three species (rat, mouse, and guinea pig) (Garcia et al., 1997) and the transfected human or mouse BB2 receptor did not stimulate an increase in cAMP (Benya et al., 1994b, 1995b). These results compared with those in a number of studies in the literature led the authors (Garcia et al., 1997) to propose that the BB2 receptor may be coupled differentially to different adenylate cyclases in different tissues in the same species. Downstream diacylglycerol leads to the activation of both classic and novel PKCs, which catalyze the phosphorylation of a number of membrane-bound and cytosolic proteins. Furthermore, specific protein kinase cascades are triggered, including the Raf/MEK/ERK kinase cascade, activation of protein kinase D, and rapamycin-sensitive p70s6k, which result in increased expression of immediate early response genes (i.e., c-myc, c-jun, and c-fos), leading to the regulation of the cell cycle and cell proliferation (Rozengurt, 1998a).
BB2 receptor stimulation also results in the activation of tyrosine kinases and tyrosine phosphorylation of a number of proteins including p125 focal adhesion kinase and PYK2, paxillin, ERK kinase, and P130CAS (Rozengurt, 1998a,b). Paxillin and P130CAS function as important adaptors with paxillin promoting protein-protein interactions and P130CAS interacting with Src and c-Crk and with numerous proteins that have SH2 and SH3 binding domains (Turner, 1994; Harte et al., 1996). BB2 receptor stimulation of P125FAK tyrosine phosphorylation occurs largely independent of PKC activation (Sinnett-Smith et al., 1993) but is dependent on the small GTP-binding protein Rho and the integrity of the actin cytoskeleton and focal adhesion plaques (Rozengurt, 1998a). In addition to BB2 receptor activation stimulating the formation of focal adhesion plaques via a Rho-dependent mechanism, it also stimulates actin proliferation, resulting in membrane ruffling via rac proteins (Nobes et al., 1995). In contrast with P125FAK and paxillin tyrosine phosphorylation due to BB2 receptor stimulation, stimulation of ERK activation and tyrosine phosphorylation is not dependent on Rho or the other factors listed above (Seufferlein et al., 1996a). GRP-induced activation of ERK is dependent on PKC (Rozengurt, 1998b) and transactivation of the EGF receptor (MacKinnon et al., 2001; Lui et al., 2003; Thomas et al., 2005) which may be mediated by Gi proteins (MacKinnon et al., 2001). Recent studies provide evidence that BB2 receptor stimulation of tyrosine phosphorylation of P125FAK, paxillin, and P130CAS occurs via an interaction with Gα12/13 and Rho (Rozengurt, 1998a). BB2 receptor stimulation also leads to coupling to Gα12 to elicit c-Jun N-terminal kinase activation (MacKinnon et al., 2001; Chan and Wong, 2005).
BB2 receptor stimulation results in a rapid activation of Src kinase family members (Rodríguez-Fernández and Rozengurt, 1996; Vincent et al., 1999; Pace et al., 2006), which is not dependent on either PKC or mobilization of calcium, nor is it dependent on Rho or the integrity of the cytoskeleton (Rodriguez-Fernandez and Rozengurt, 1996). Blockade of Src family kinases decreases BB2 receptor-stimulated transactivation of the EGFR as well as MAP kinase stimulation (Vincent et al., 1999). The EGFR transactivation by BB2 receptor activation in head and neck squamous cancers is dependent on Src-mediated cleavage and release of transforming growth factor-α and amphiregulin and is essential for invasion and growth of these cancers (Vincent et al., 1999).
Acute and chronic BB2 receptor stimulation results in an activation of a number of receptor modulatory processes (internalization, down-regulation, or desensitization) (Lee et al., 1980; Pandol et al., 1982; Millar and Rozengurt, 1990; Walsh et al., 1993; Benya et al., 1994b; Briscoe et al., 1994; Kroog et al., 1995a), and a number of studies have investigated the cell signaling processes involved. In cells containing human (Benya et al., 1995b), mouse (Zachary and Rozengurt, 1987; Brown et al., 1988; Benya et al., 1993, 1994d; Wang et al., 1993; Tsuda et al., 1997a; Acs et al., 2000), or rat BB2 receptors (Zhu et al., 1991), with agonist exposure the receptor-ligand complex is rapidly internalized (t0.5 5 min) with 80 to 85%, 70 to 90%, and 50%, respectively, of the bound ligand internalized. In epithelial cells transfected with the murine BB2 receptor, agonist ligand and receptor were internalized by 5 min into early endosomes, after 10 min both were in perinuclear vesicles, and after 60 min the BB2 receptor had recycled back to the surface (Grady et al., 1995). In this study (Grady et al., 1995) and in others (Benya et al., 1994b, 1995a) there was a rapid down-regulation of cell surface receptors, and the recovery was decreased by acidotropic agents but not by inhibitors of new protein synthesis. The internalization of the BB2 receptor is partially dependent on phospholipase C activation (Benya et al., 1994a; Williams et al., 1998; Schumann et al., 2003) and requires clathrin-coated pits because it is inhibited by hyperosmolar sucrose as well as phenylarsine oxide (Grady et al., 1995). Acute desensitization of the BB2 receptor occurs within seconds to minutes of agonist exposure (Walsh et al., 1993; Briscoe et al., 1994) and is reported to occur with stimulated phospholipase D activity as well as with stimulation of phosphoinositide breakdown (Briscoe et al., 1994; Williams et al., 1998) and for stimulation of changes in cytosolic calcium, with the latter shown to be homologous in nature (Walsh et al., 1993). In some tissues acute desensitization and down-regulation of the BB2 receptor are caused by hormones/neurotransmitters activating phospholipase C such as carbachol and cholecystokinin (Younes et al., 1989; Vinayek et al., 1990). Chronic BB2 receptor desensitization occurs after prolonged incubation with agonist (1–2 h) and is homologous in nature (Lee et al., 1980; Benya et al., 1995a). The receptor structure-function studies reviewed above provide strong support for the conclusion that down-regulation and chronic desensitization are coupled processes being affected by similar receptor structural alterations and cellular signaling cascades and have a mechanism distinct from that causing internalization (Benya et al., 1994d, 1995a; Tsuda et al., 1997a; Schumann et al., 2003). The results of these studies provided no evidence for second messenger-independent processes in mediation of down-regulation or desensitization, whereas internalization is equally stimulated by second messenger-dependent and -independent processes and the presence of the COOH-terminal serines and threonines was essential for mediating these effects. In HIT-T15 cells BB2 receptor-mediated desensitization was closely coupled to down-regulation (Swope and Schonbrunn, 1990).
Studies in the β-adrenergic receptor and a number of GPCRs demonstrate that receptor phosphorylation, primarily by G protein-coupled receptor kinases (GRKs) and subsequent binding of arrestins are critical for receptor internalization and deactivation during acute desensitization (Krupnick and Benovic, 1998; Ferguson, 2001; Premont and Gainetdinov, 2007). Studies demonstrate that BB2 receptor activation results in rapid phosphorylation of the receptor (Kroog et al., 1995b, 1999; Williams et al., 1996; Ally et al., 2003) as does stimulation of the BB2 receptor-containing cells by the phorbol ester, 12-O-tetradecanoyl-phorbol-13-acetate (Kroog et al., 1995b; Williams et al., 1996; Ally et al., 2003). However, agonist and 12-O-tetradecanoyl-phorbol-13-acetate-induced BB2 receptor phosphorylation occur at different receptor sites (Williams et al., 1998). GRKs are serine-threonine kinases that preferentially phosphorylate agonist occupied, active conformation GPCRs and lead to uncoupling from G protein and endocytosis (Szekeres et al., 1998; Premont and Gainetdinov, 2007). Bn/GRP stimulates BB2 receptor phosphorylation at serine/threonine residues in the COOH terminus but does not stimulate tyrosine phosphorylation in the BB2 receptor (Williams et al., 1996; Ally et al., 2003). With BB2 receptor activation arrestin translocation occurs to the plasma membrane (Ally et al., 2003) and requires an intact DRY sequence in the second intracellular domain of the BB2 receptor (Ally et al., 2003). BB2 receptor internalization has been proposed to play a key role in acute BB2 receptor desensitization (Swope and Schonbrunn, 1990) because the kinetics of each is identical. Furthermore, the kinetics of BB2 receptor phosphorylation correlate closely with both internalization and acute desensitization (Kroog et al., 1995b; Williams et al., 1996, 1998). Phosphorylation of the BB2 receptor after GRP/Bn stimulation is reported in one study (Williams et al., 1996) but not another (Kroog et al., 1995b) to be mediated by both a PKC-dependent and a PKC-independent process (probably a GRK family member).
Studies demonstrate that radiolabeled GRP/Bn is rapidly degraded by the BB2 receptor (Swope and Schonbrunn, 1987; Zachary and Rozengurt, 1987; Brown et al., 1988; Zhu et al., 1991; Wang et al., 1993; Williams et al., 1998). This degradation is best inhibited by the general inhibitor bacitracin or the thermolysin-like metalloproteinase inhibitor, phosphoramidon, and to a less degree by leupeptin and bestatin > chymostatin > amastatin (Wang et al., 1993). The lysosomal proteinase inhibitor, choroquine, also inhibits degradation (Swope and Schonbrunn, 1987; Williams et al., 1998).
Activation of the BB2 receptor results in growth of both normal and neoplastic tissues (Moody et al., 2003a; Jensen and Moody, 2006). The cell signaling cascades involved have been studied extensively in both Swiss 3T3 cells and in numerous tumors cells. In 3T3 cells and a number of tumor cells (prostate, head and neck squamous cell cancer, and non-small cell lung cancer cells) activation of the BB2 receptor results in stimulation of phosphorylation of Akt (Liu et al., 2007) and ERK phosphorylation (Sakamoto et al., 1988; Rozengurt, 1998b; Koh et al., 1999; Vincent et al., 1999; Lui et al., 2003; Thomas et al., 2005), which has been shown in some cells to be dependent on the transactivation of the EGF receptor, which in turn depends on Src and changes in cytosolic calcium in some cases. Mitogenesis in 3T3 cells is dependent on BB2 receptor-stimulated changes in cytosolic calcium, activation of PKC, PKD, and ERK, and release of arachidonic acid (Rozengurt, 1998b). BB2 receptor stimulation of ERK phosphorylation is dependent on Ras but not Rap1 in prostate tumor cells (Sakamoto et al., 1988). The transactivation of the EGF receptor by BB2 receptor activation is dependent on PKC and PKD activation in some cells (Seufferlein et al., 1996b; Rozengurt, 1998b; Sinnett-Smith et al., 2004, 2007). EGF receptor transactivation upon BB2 receptor stimulation as well as by a number of other GPCRs occurs via metallo-proteinase-dependent cleavage and release of EGF-related peptides that then activate the receptor (Sakamoto et al., 1988; Vincent et al., 1999; Lui et al., 2003). The inhibition of either EGF receptor transactivation or ERK activation inhibited BB2 receptor-stimulated DNA synthesis in these tumor cells (Sakamoto et al., 1988). BB2 receptor activation stimulates the invasion and cell migration of tumor cells (Vincent et al., 1999; Thomas et al., 2005; Zheng et al., 2006). This stimulation occurs via Gα13, leading to activation of RhoA and Rho-associated coiled-coil forming protein kinase (Zheng et al., 2006). BB2 receptor activation promotes progression from the G1 to the S phase of the cell cycle by increasing the expression of cyclin D1 and E through the early growth response protein Egr-1, down-regulating the cyclin-dependent kinase inhibitor p27kip1 and hyperphosphorylating the retinoblastoma protein (Mann et al., 1997; Rozengurt, 1998b; Xiao et al., 2005).
H. BB2 Receptor Function in Various Tissues and in Vivo
A major difficulty in assessing the effects of BB2 receptor activation in vivo and in a number of tissues in vitro is the fact that they frequently possess both classes of bombesin receptors, and bombesin, the agonist frequently used, has high affinity for both receptor subtypes. Recently a number of developments have contributed to solving this problem. Selective receptor antagonists for the BB2 receptor are described, studies on BB2 receptor knockout animals are being increasing performed, more selective BB2 receptor agonists such as GRP are being used, and with the cloning of the mammalian bombesin receptors, it has become clear that some widely studied tissues such as Swiss 3T3 cells and pancreatic acinar cells only possess BB2 receptors.
Many effects of GRP are observed both in vivo and in vitro, but it remains unclear in many cases which are pharmacological or which are physiological. Studies support a role for the BB2 receptor in numerous gastrointestinal functions, including regulation of gastric acid secretion via both stimulation of gastrin release from antral G cells and somatostatin release from D cells and stimulation of acid secretion (Schubert et al., 1991; Hildebrand et al., 2001; Schubert, 2002); regulation of gastrointestinal motility, especially gastric emptying, small intestinal transit, and gallbladder emptying (Degen et al., 2001; Yeğen, 2003); stimulation of pancreatic secretion (Niebergall-Roth and Singer, 2001; Nathan and Liddle, 2002), insulin release (Persson et al., 2002), and colonic ion transport (Traynor and O'Grady, 1996); and stimulation of the secretion of a variety of hormones (gastrin, somatostatin, CCK, pancreatic polypeptide, enteroglucagon, pancreatic glucagon, and gastric inhibitory polypeptide) (Modlin et al., 1981; Ghatei et al., 1982; Pettersson and Ahren, 1987; Bunnett, 1994). BB2 receptor activation has number of immunologic effects including functioning as a chemoattractant in peritoneal macrophages, monocytes, and lymphocytes (Ruff et al., 1985; Del Rio and De la Fuente, 1994), stimulating lymphocyte proliferation (Del Rio et al., 1994), and stimulating natural killer and antibody-dependent cellular cytotoxicity in leukocytes (De la Fuente et al., 1993). BB2 receptors are reported to be important for fetal lung development including lung branching, cell proliferation, and differentiation (Subramaniam et al., 2003) as well as in a number of lung diseases, which will be discussed in section IV.I. BB2 receptors are widely expressed in the CNS and in the spinal cord, and numerous central effects have been described with their activation including effects on satiety, regulation of circadian rhythm, thermoregulation, grooming behaviors, modulation of stress, fear, and anxiety response, memory, and gastrointestinal function such as acid secretion (Martinez and Tache, 2000; Yeğen, 2003; Moody and Merali, 2004; Karatsoreos et al., 2006; Roesler et al., 2006a,b; Kallingal and Mintz, 2007; Presti-Torres et al., 2007). The satiety effect of BB2 receptors has been extensively studied (Gibbs et al., 1979; Gibbs and Smith, 1988; Flynn, 1997; Fekete et al., 2007; Ladenheim and Knipp, 2007). A recent study (Ladenheim and Knipp, 2007) showed that the satiety effect of peripherally administered NMB, but not that of GRP, is inhibited by capsaicin pretreatment, suggesting that the neural pathways involved in BB2 receptor-mediated satiety are either capsaicin-insensitive neurons or involve direct activation of BB2 receptors in the CNS (Ladenheim and Knipp, 2007). The importance of BB2 receptors in mediating the satiety effects of GRP was demonstrated by the ability of a specific BB2 receptor antagonist administered in the hindbrain of rats to inhibit the satiety effects of peripherally administered GRP (Ladenheim et al., 1996a).
BB2 receptor knockout mice have been described and limited study results are available (Wada et al., 1997; Hampton et al., 1998). In the initial study performed with these mice (Hampton et al., 1998), no developmental abnormalities were seen; however, bombesin failed to suppress glucose intake, whereas it caused a dose-dependent decrease in normal mice (Hampton et al., 1998). In a second study (Wada et al., 1997) the intracerebroventricular administration of GRP failed to cause hypothermia in the BB2 receptor knockout mice as observed in the wild-type mice. Furthermore, the BB2 receptor knockout mice demonstrated abnormal behaviors and altered spontaneous activity during darkness (Wada et al., 1997). In a more detailed study of feeding behavior in these mice, neither GRP, NMB, nor bombesin altered satiety in the knockout mice; however, the satiety response to cholecystokinin was present and in fact enhanced (Ladenheim et al., 2002). In a long-term study (Ladenheim et al., 2002) BB2 receptor knockout mice ate more food than normal mice because of a defect in terminating meals and had greater weight gain, supporting the conclusion that the BB2 receptor has important roles in satiety. These mice were used to study the effects of BB2 receptor activation on islet function (Persson et al., 2000, 2002). BB2 receptor knockout mice had impaired glucose tolerance, a defect in early insulin release (Persson et al., 2000), and their plasma glucagon-like peptide-1 response to gastric glucose administration was significantly reduced, suggesting that the BB2 receptor had an important role in normal glucagon-like peptide-1 release and insulin and glucose responses after a glucose meal. In a second study (Persson et al., 2002) GRP was found to potentiate glucose-stimulated insulin release in wild-type but not BB2 receptor knockout mice. This study (Persson et al., 2002) demonstrated that BB2 receptor activation contributes to insulin secretion induced by activation of autonomic nerves and that the deletion of the BB2 receptor is compensated for by increased cholinergic sensitivity (Persson et al., 2002). These results are consistent with earlier studies, which demonstrated that GRP potentiated glucose-induced insulin release by both a ganglionic and direct effect but did not alter glucagon or pancreatic somatostatin release (Hermansen and Ahren, 1990; Gregersen and Ahren, 1996; Karlsson et al., 1998). BB2 receptor knockout mice were used to study possible behavioral effects of GRP (Shumyatsky et al., 2002). In one study the BB2 receptor was found in wild-type but not knockout mice to be highly expressed in the lateral nucleus of the amygdala, which is important in mediating fear responses. BB2 receptor knockout mice showed more persistent long-term fear responses (Shumyatsky et al., 2002), supporting other study results, which suggest that the BB2 receptor has an important role in memory and fear responses (Roesler et al., 2006a). Other behavior changes seen in BB2 receptor knockout mice include increased social investigatory behavior (Yamada et al., 2000b), preference for conspecific odors (Yamada et al., 2000b), and altered social preferences in females (Yamada et al., 2001). BB2 receptor knockout mice have also been used to investigate the role of this receptor in specific diseases, which will be discussed in the next section.
BB2 receptor activation has important growth effects on normal and neoplastic tissues (Moody et al., 1992; Jensen and Moody, 2006). BB2 receptor activation stimulates growth of normal endometrial stomal cells (Endo et al., 1991), bronchial epithelial cells (Willey et al., 1984; Siegfried et al., 1993), melanocytes (Terashi et al., 1998), chondrocytes (Hill and McDonald, 1992), and normal enterocyte growth and turnover after small bowel resection (Chu et al., 1995; Sukhotnik et al., 2007) as well as normal development of the intestinal villus (Carroll et al., 2002) and normal fetal lung development (Emanuel et al., 1999; Shan et al., 2004). The effects of BB2 receptor activation on neoplastic growth have been extensively studied (Moody et al., 1992; Jensen et al., 2001; Patel et al., 2006). This widespread interest occurred after human small cell lung cancers were shown to possess high-affinity BB2 receptors (Moody et al., 1985), and bombesin was shown to have an autocrine growth effect on these cells (Cuttitta et al., 1985). Subsequent studies demonstrated such an autocrine growth effect, for which the tumor cells not only possessed BB2 receptors but also secreted bombesin-like peptides, resulting in a growth stimulatory effect (Moody et al., 2003a; Jensen and Moody, 2006; Patel et al., 2006) in a large number of cells from various types of cancer including neuroblastomas (Kim et al., 2002), squamous head and neck tumors (Lango et al., 2002; Lui et al., 2003), pancreatic cancer (Wang et al., 1996; Murphy et al., 2001), colon cancer (Chave et al., 2000), prostate cancer (Plonowski et al., 2000), human glioblastomas (Sharif et al., 1997), and non-small cell lung cancer (Siegfried et al., 1999). Furthermore, many human cancers or the blood vessels in the cancers either overexpress or ectopically express BB2 receptors, and the stimulation or inhibition of these receptors is reported to affect growth/differentiation (Jensen et al., 2001; Moody et al., 2003a; Heuser et al., 2005; Jensen and Moody, 2006; Patel et al., 2006; Fleischmann et al., 2007). The potential clinical importance of ectopic expression and overexpression will be discussed further in the next section. The role of the ectopic expression or overexpression in various cancers may be different with different tumors. Whereas many of the studies referred to in the following paragraph emphasize the growth stimulatory effects of BB2 receptor on tumor cells, other studies, especially in colon cancer, support the conclusion that the ectopic expressing of the BB2 receptor has a morphogenic effect rather than a mitogenic effect (Jensen et al., 2001). Whereas in normal colonic mucosal epithelial cells, the BB2 receptor is not found (Preston et al., 1995; Ferris et al., 1997; Carroll et al., 1999b), in 40 to 100% of colon cancers (Carroll et al., 1999b) the BB2 receptor is aberrantly expressed. BB2 receptor activation on some colon cancer cells is reported to result in proliferation (Radulovic et al., 1991b; Frucht et al., 1992; Narayan et al., 1992). However, in detailed studies, although 62% of the tumors expressed both GRP and the BB2 receptor, their coexpression was equally frequent in early- or late-stage cancers and was rarely detected in metastases (Carroll et al., 1999b). However, GRP/BB2 receptor expression was seen in all well differentiated tumors, whereas poorly differentiated tumors never coexpressed GRP/BB2 receptors (Carroll et al., 1999b). Furthermore, no difference in survival occurred in patients with cancers expressing or not expressing the GRP/BB2 receptor (Carroll et al., 1999b). In a study (Carroll et al., 2000a) of BB2 receptor knockout mice with colon tumors induced by azoxymethane, larger tumors were better differentiated in wild-type mice than in BB2 receptor knockout mice. From these studies and others it was proposed that BB2 receptor activation in these cells is functioning primarily as a morphogenic or differentiating factor (Carroll et al., 1999b; Jensen et al., 2001). More recent studies show that this morphogenic effect is mediated by activation of p125FAK, which inhibits invasion/metastases by enhancing cell attachment (Glover et al., 2004), most likely by up-regulating the expression of intracellular adhesion protein-1 (Taglia et al., 2007). Subsequent studies showed that BB2 receptor mutations occurred frequently in poorly differentiated colon tumor, resulting in the formation of inactive receptors, and the generation of these mutations correlated inversely with the differentiation of the tumor, suggesting that their production represents a new mechanism allowing for the differentiation of tumors (Carroll et al., 2000b; Glover et al., 2003).
A recent study (Ruginis et al., 2006) used a proteomic approach to identify proteins selectively up-regulated in human colorectal cancer cells subsequent to BB2 receptor activation. This study took advantage of the fact that human colorectal cancer cells such as Caco-2 and HT-29 only secreted GRP and expressed BB2 receptors when they are preconfluent and not when they are postconfluent (Glover et al., 2005). Total cellular proteins were isolated from preconfluent, GRP and BB2 receptor-expressing cells in the presence and absence of the specific BB2 receptor antagonist [d-Phe6]bombesin6–13 methyl ester and from postconfluent cells not expressing GRP or BB2 receptors. By using two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (Ruginis et al., 2006), at least six separate proteins were up-regulated subsequent to BB2 signaling when this receptor is aberrantly expressed in human colorectal cancer: gephyrin, heat shock protein 70, heterochromatin associated protein 1, intracellular adhesion protein-1–1, and ThiL/acetyl-CoA acetyltransferase. These findings hold promise for further definition of the mechanism whereby aberrantly expressed BB2 receptors in human colorectal cancer promote tumor cell differentiation and improve patient outcome.
I. BB2 Receptor in Diseases
BB2 receptor activation has been proposed to be important in the mediation of a number of human disorders including disorders of lung development, various pulmonary diseases, CNS disorders, and the growth/differentiation of human cancers. The tumor differentiation effects of BB2 receptor activation were discussed in the previous section, and the growth effects and effects of BB2 receptor overexpression will be considered here. BB2 receptors are ectopically expressed or overexpressed in a large number of tumors including 85 to 100% of small cell lung cancers, 74 to 78% of non-small cell lung cancer cells, 38 to 72% of breast cancer, 75% of pancreatic cancer cell lines and 10% of pancreatic cancers, 62 to 100% of prostate cancers, 100% of head/neck squamous cell cancers, and 72 to 85% of neuroblastomas/glioblastomas (Jensen and Moody, 2006; Patel et al., 2006). Bn-like peptides are critical to the growth of some, but not all, small cell lung cancers (Cuttitta et al., 1985) and as discussed above have been shown to have an autocrine growth effect on a large number of tumors as well as stimulate growth in a large number of these tumors (Jensen and Moody, 2006; Patel et al., 2006). In some tumors such as prostate cancer, BB2 receptor overexpression correlates with neoplastic transformation (Markwalder and Reubi, 1999). Bn peptides also function as proangiogenic factors in various tumors (Levine et al., 2003; Kanashiro et al., 2005; Martínez, 2006). The production of GRP-related peptides and/or overexpression of BB2 receptors by tumors are playing a potential role in a number of aspects of the treatment and management of these tumors. These aspects include functioning as targets for antitumor treatment, as prognostic factors, as targets to image the tumors, and as targets to deliver cytotoxic treatment selectively to the tumor (Smith et al., 2003; Schally and Nagy, 2004; Jensen and Moody, 2006). Attempts to inhibit the autocrine growth effect of GRP-like peptides on tumor growth are reported in human and/or animal studies using monoclonal antibodies to GRP, antisense constructs, BB2 receptor antagonists, or other inhibitors (Zhou et al., 2004). Infusion of the monoclonal antibody 2A11 directed against the biologically active COOH terminus of GRP is safe in humans (Chaudhry et al., 1999), and 2A11 was given to 13 patients with small cell lung cancer (Kelley et al., 1997). One patient had a complete remission and four patients had radiologically stable disease, and further evaluation was recommended (Kelley et al., 1997). In a recent study (Schwartsmann et al., 2006) the BB2 receptor antagonist, RC-3095 was administered to 25 patients with different advanced malignancies. No side effects occurred, and there were no tumor responses; however, maximal doses could not be reached by the methods used despite dose escalation (Schwartsmann et al., 2006). EGF receptor transactivation upon BB2 receptor stimulation may also be a target for therapeutic intervention. Experimental studies demonstrate that activation of the BB2 receptor rescues tumor cells from the growth-inhibiting effect of the EGF receptor inhibitor, gefitinib, by stimulating the release of amphiregulin and activation of the Akt pathway (Liu et al., 2007). When a BB2 receptor antagonist is combined with an EGF receptor inhibitor (erlotinib), there is marked enhanced antitumor activity (Zhang et al., 2007b), suggesting that such an approach may be useful in some cancers such as head/neck tumors or lung tumors. In a number of studies plasma levels of GRP precursors such as pro-GRP or assessment of GRP expression in tumors provides prognostic information (Hamid et al., 1990; Okusaka et al., 1997; Sunaga et al., 1999; Shibayama et al., 2001; Yonemori et al., 2005).
Recent clinical and laboratory studies with somatostatin receptors demonstrate that many endocrine tumors overexpress or ectopically express these receptors and that radiolabeled analogs of somatostatin can be used to localize these tumors as well as for somatostatin receptor-mediated cytotoxicity (Breeman et al., 2007; Van Essen et al., 2007). Somatostatin analogs coupled to 111In are now widely used to image neuroendocrine tumors, and numerous studies have demonstrated that they have greater sensitivity than conventional imaging modalities (computed tomography, magnetic resonance imaging, angiography, or ultrasound) and that their routine use changes patient management in 20 to 47% of cases (Gibril et al., 1996; Gibril and Jensen, 2004; Breeman et al., 2007). Recent studies with somatostatin analogs coupled to 111In, 90Yt, and 177Lu show promising results for somatostatin receptor-mediated cytotoxicity in patients with advanced neuroendocrine tumors, and they have entered phase 3 studies (Breeman et al., 2007; Forrer et al., 2007; Van Essen et al., 2007). Unfortunately, many common tumors (colon, pancreas, head/neck, prostate, and lung) may not overexpress somatostatin receptors; however they frequently overexpress Bn receptors, especially the BB2 receptor (Jensen et al., 2001; Reubi et al., 2002; Moody et al., 2003a; Heuser et al., 2005; Jensen and Moody, 2006; Patel et al., 2006; Fleischmann et al., 2007). This observation has led to considerable interest in the possibility of developing radiolabeled analogs of Bn that could be use for localization of the tumors containing Bn receptors or the development of radiolabeled Bn analogs or Bn analogs coupled to cytotoxic agents that could be used to treat tumors overexpressing Bn receptors through bombesin receptor-mediated cytotoxicity (Breeman et al., 2002; de Jong et al., 2003; Cornelio et al., 2007; de Visser et al., 2007a). Numerous radiolabeled (111In, 68Ga, 177Lu, 64Cu, 86Yt, 18F, and 99mTc) GRP analogs with enhanced stability that bind with high affinity to BB2 receptors have been reported, as well as their ability to image various human tumors in vivo using gamma detectors or positron emission tomography (Breeman et al., 2002; Nock et al., 2003; Smith et al., 2003, 2005; Johnson et al., 2006; Lantry et al., 2006; Zhang et al., 2006, 2007a; de Visser et al., 2007a; Dimitrakopoulou-Strauss et al., 2007; Garrison et al., 2007; Parry et al., 2007; Prasanphanich et al., 2007; Waser et al., 2007). In some preliminary studies in humans, tumors were imaged in the majority of patients, and in some cases, tumors that were not seen with other commonly used imaging modalities were detected using radiolabeled Bn analogs (De Vincentis et al., 2004; Scopinaro et al., 2004, 2005; Dimitrakopoulou-Strauss et al., 2007). At present no study has established the value of imaging using radiolabeled Bn analogs.
A number of Bn analogs coupled to radiolabeled compounds (e.g., 177Lu) (Smith et al., 2005; Johnson et al., 2006; Lantry et al., 2006; Zhang et al., 2007a) and to cytotoxic agents (camptothecin, a topoisomerase inhibitor, doxorubicin analogs, and paclitaxel) (Schally and Nagy, 1999; Breeman et al., 2002; Moody et al., 2004, 2006b; Schally and Nagy, 2004; Engel et al., 2005; Buchholz et al., 2006; Nanni et al., 2006; Panigone and Nunn, 2006; Safavy et al., 2006; Engel et al., 2007) have been described. These analogs retain their high affinity for Bn receptors and are internalized by Bn receptor-bearing tissues, for the possibility of delivering Bn receptor-mediated tumoral cytotoxicity. Many of these compounds have been shown to cause tumor cytotoxicity in animal studies, and one study has provided evidence that it is due to specific interaction with the BB2 receptors overexpressed on the tumor (Moody et al., 2006b). At present it is unclear whether this approach will be effective in vivo in human tumors whether alone or in combination with other antitumor treatments. A recent study using a chemically identical active and inactive cytotoxic GRP analog (i.e., camptothecin-L2-[d-Tyr6,β-Ala11,Phe13,Nle14]bombesin6–14 (where L2 = [N-(N-methyl-amino ethyl)glycine carbamate]) or its d-Phe13 inactive form, demonstrated that specific tumor receptor interaction is important in mediating the tumor cytotoxicity of these compounds (Moody et al., 2006b). Various studies have demonstrated that such an approach can inhibit the growth of pancreatic lung, prostate, and gastric cancers (Schally and Nagy, 1999, 2004; Breeman et al., 2002; Moody et al., 2004). At present the usefulness of GRP or the BB2 receptor in management of human tumors in each of the areas discussed here has not been established (Jensen and Moody, 2006).
BB2 receptor activation, GRP secretion, or abnormalities of either have been proposed to be important in a number of other diseases. In a recent study (Sun and Chen, 2007) evidence that activation of the BB2-receptor in the dorsal spinal cord is important for mediating pruritus was presented. GRPR knockout mice showed significantly decreased scratching behavior in response to pruritogenic stimuli, whereas other responses were normal. Furthermore, administration of a BB2 receptor antagonist into the spinal fluid inhibited scratching behavior in three different models of itching (Sun and Chen, 2007). The authors (Sun and Chen, 2007) point out that the BB2 receptor may represent the first molecule identified that is dedicated to mediating the itch response in the spinal cord and may provide an important therapeutic target for the treatment of chronic pruritic conditions. Abnormalities of GRP, BB2 receptors, and other bombesin-like peptides and/or their receptors are proposed to be important in normal lung development and mediation of the lung injury in premature infants with bronchopulmonary dysplasia (Li et al., 1994; Sunday et al., 1998; Emanuel et al., 1999; Cullen et al., 2000; Ashour et al., 2006; Ganter and Pittet, 2006; Subramaniam et al., 2007). In one recent study (Ashour et al., 2006) GRP given to newborn mice induced features of human BPD including interstitial pulmonary fibrosis and alveolarization. In a hyperoxic baboon model of BPD (Subramaniam et al., 2007) the early overproduction of Bn-like peptides correlated with the development of BPD-like histological features and the blockage of GRP partially reversed these effects, leading the authors to suggest that such an approach could have important implications for preventing BPD in premature infants. GRP has been shown to be protective to the small intestine in various injury models (Assimakopoulos et al., 2004, 2005a,b; Kinoshita et al., 2005; Kimura et al., 2006b), enhance gut barrier function, prevent the atrophy of enteric ganglia caused by FK506 in small bowel (Assimakopoulos et al., 2005a; Higuchi et al., 2006; Kimura et al., 2006a,b), and in a recent study (Fujimura et al., 2007) to prevent the atrophy of Peyer's patches and dysfunction of M cells in rabbits receiving long-term parenteral nutrition. These studies suggest that GRP agonists may have a potential therapeutic role in diseases causing this type of injury. Numerous studies in rodents provide evidence that GRP/BB2 receptor activation is important for memory as well as for a number of social behaviors (learning, grooming, and stereotypy) (Roesler et al., 2006a,b). These results were supported by a recent study (Presti-Torres et al., 2007) in which the administration of BB2-receptor antagonists in neonatal rats resulted in marked impairment of memory, and social interaction. These changes have led one group (Roesler et al., 2006a) to propose that the BB2 receptor should be consider a therapeutic target in a subset of human CNS diseases, especially those involving memory, learning, and fear. Specifically, in the CNS it has been proposed that alterations in either the GRP and/or BB2 receptor may be important in schizophrenia, Parkinson's disease, anxiety disorders, anorexia, bulimia, and mood disorders (Merali et al., 1999, 2006; Frank et al., 2001; Yeğen, 2003; Moody and Merali, 2004; Roesler et al., 2006a).
V. BB3 Receptor
A. Early Studies of the BB3 Receptor
Before the identification of the BB3 receptor when it was cloned in 1992 from guinea pig uterus (Gorbulev et al., 1992), no pharmacological or functional studies suggested its existence.
B. Cloned BB3 Receptor and Receptor Structure
The human BB3 receptor is a 399-amino acid protein (Fathi et al., 1993b), and it shows 95% amino acid identities with the rhesus BB3 receptor (Sano et al., 2004), 80% amino acid identity with the rat BB3 receptor that shows 92% with the mouse BB3 receptor, and 77% with the sheep BB3 receptor (Liu et al., 2002) (Table 2). The human BB3 receptor has 51% amino acid identities with the human BB2 receptor and 47% with the human BB1 receptor (Fathi et al., 1993b). The human BB3 receptor has a predicted molecular mass of 44.4 kDa (Fathi et al., 1993b), and there are two potential N-linked glycosylation sites at Asn10 and Asn18 and a consensus site for potential PKC phosphorylation in the third cytoplasmic loop and carboxyl terminus (Fathi et al., 1993b; Whitley et al., 1999). A putative palmitoylation site existed at C347 and C348 (Fathi et al., 1993b; Whitley et al., 1999). Hydropathy plots yielded results consistent with a seven-transmembrane structure typical for a G protein-coupled receptor (Fathi et al., 1993b). The BB3 receptor has been cloned from rat (Liu et al., 2002), mouse (Ohki-Hamazaki et al., 1997a), sheep (Whitley et al., 1999), and guinea pig (Gorbulev et al., 1992). In the chicken a receptor was cloned that has similarities to both the mammalian BB3 receptor and the frog BB4 receptor and has been termed the chBRS-3.5 receptor (Iwabuchi et al., 2003). No cross-linking studies have been performed on the mature BB3 receptor so the extent of glycosylation or type is not known at present.
C. BB3 Receptor Genomic Organization
The human BB3 receptor gene is localized at human chromosome Xq25 and in the mouse on chromosome XA7.1–7.2 (Fathi et al., 1993b; Gorbulev et al., 1994; Weber et al., 1998). The human BB3 receptor gene (Fathi et al., 1993b; Gorbulev et al., 1994; Weber et al., 1998) contained two introns and three exons similar to the sheep (Whitley et al., 1999), rhesus (Sano et al., 2004), mouse (Ohki-Hamazaki et al., 1997a), and rat BB3 receptor genes (Liu et al., 2002). In the mouse the BB3 receptor gene spanned more than 5 kb with exon 1 of the BB3 gene separated from exon 2 by 1.6 kb and this in turn separated from exon 3 by 1.6 kb (Weber et al., 1998). In human, sheep, monkey, rat, mouse, and guinea pig the exon/intron splice sites occurred at Arg145 in the second intracellular loop and at Ile263 in the third intracellular domain (Gorbulev et al., 1994; Weber et al., 1998; Sano et al., 2004).
D. BB3 Receptor Expression
Expression levels of the BB3 receptor mRNA have been reported in the rat (Fathi et al., 1993b; Liu et al., 2002; Jennings et al., 2003), sheep (Whitley et al., 1999), mouse (Ohki-Hamazaki et al., 1997a), monkey (Sano et al., 2004), and guinea pig (Gorbulev et al., 1992). In the monkey in which it was studied in detail, BB3 mRNA is found in the greatest amount in the CNS and in the testis (Sano et al., 2004). This high expression in the testis is not seen in the sheep (Weber et al., 2003) or mouse (Ohki-Hamazaki et al., 1997a) but is similar to that in the rat (Fathi et al., 1993b) in which it was localized to the secondary spermatocytes and was not present in the Sertoli cells or different maturation stages of the spermatogonia (Fathi et al., 1993b). Detectable levels were also found in the monkey pancreas, thyroid, and ovary in peripheral tissues, and it was either undetectable or found in very low amounts in other tissues showing a very different distribution from that for the BB1 receptor or BB2 receptor (Fathi et al., 1993b; Sano et al., 2004).
In the CNS BB3 receptor mRNA was expressed in a restricted distribution (Ohki-Hamazaki et al., 1997a; Liu et al., 2002; Jennings et al., 2003; Sano et al., 2004). In the rat and mouse (Ohki-Hamazaki et al., 1997a; Liu et al., 2002) the BB3 receptor was present in the highest amounts in the hypothalamic area, notably the paraventricular, arcuate, striohypothalamic, dorsal hypothalamic, and dorsomedial hypothalamic nuclei, medial and lateral preoptic areas, and lateral/posterior hypothalamic areas. In the rat expression was also detected in the medial habenula nucleus in one study (Liu et al., 2002) and a second study (Jennings et al., 2003) in the nucleus accumbens and the thalamus. In the monkey brain (Sano et al., 2004) polymerase chain reaction quantitation showed that the BB3 receptor mRNA was present in the highest amounts in the hypothalamus followed by the pituitary gland, amygdala, hippocampus, and caudate nucleus.
Specific BB3 receptor antibodies have been used to localize the receptor in the tunica muscularis of the rat gastrointestinal tract (Porcher et al., 2005) and the rat CNS (Jennings et al., 2003). In the gastrointestinal tract tunica muscularis BB3 receptor IR was observe in all regions studied (i.e., antrum, duodenum, ileum and colon) in nerves and non-neuronal cells but not in muscle cells (Porcher et al., 2005). It was detected in both myenteric and submucosal ganglia as well as in nerve fibers interconnecting myenteric ganglia (Porcher et al., 2005). BB3 receptor IR was observed in the cell bodies and processes of the c-kit interstitial cells of Cajal, leading the authors to propose that the BB3 receptor was probably involved in the regulation of gastrointestinal motility through the enteric nervous system and possibly in the pacemaker function of the gastrointestinal smooth muscle (Porcher et al., 2005). In the CNS, particularly strong BB3 receptor IR was observed in the cerebral cortex, hippocampal formation, hypothalamus, and thalamus (Jennings et al., 2003).
With assessment of BB3 mRNA (Fathi et al., 1993b) and/or binding studies (Reubi et al., 2002), BB3 receptors have been shown to exist on a number of different human tumors (Fathi et al., 1993b; Reubi et al., 2002), including small cell and non-small cell lung cancers (Fathi et al., 1993b; Toi-Scott et al., 1996; Ryan et al., 1998b; Reubi et al., 2002), carcinoids (lung) (Fathi et al., 1993b; Reubi et al., 2002), renal cell cancers (Reubi et al., 2002), Ewing sarcomas (Reubi et al., 2002), pancreatic cancer (Schulz et al., 2006), pituitary tumors (Schulz et al., 2006), ovarian cancer (Sun et al., 2000b), and prostate cancer (Sun et al., 2000a; Schulz et al., 2006). BB3 receptors have also been shown to exist on normal bronchial epithelial cells (DeMichele et al., 1994; Tan et al., 2006), human islets (Fleischmann et al., 2000), and rat kidney cells (Dumesny et al., 2004).
E. BB3 Receptor Pharmacology
1. BB3 Receptor Agonists.
In the original studies describing the ability of GRP, neuromedin C, or NMB to interact with the expressed cloned guinea pig BB3 receptor (Gorbulev et al., 1992) or the ability of GRP and NMB to activate the cloned human BB3 receptor expressed in Xenopus oocytes (Fathi et al., 1993b), it was clear that this receptor had low affinity for these peptides (Table 2). Similar results were later reported (Liu et al., 2002) with the rat BB3 receptor. A later study (Wu et al., 1996) demonstrated that human BB3 receptors expressed in BALB 3T3 cells had low affinity for all bombesin-related peptides tested (i.e., ranatensin, litorin, NMB, GRP, bombesin, and alytesin), but at concentrations >1 μM, each could activate the BB3 receptor and stimulate changes in cytosolic calcium. In 1997 Mantey et al. (1997) performed a detailed study of the ability of all naturally occurring bombesin-related peptides and a number of novel synthetic analogs of bombesin to interact with the human BB3 receptor. Because no cell lines with wild-type BB3 receptors existed, to check that the correct pharmacology and cell signaling were being obtained, in this study (Mantey et al., 1997) human BB3 receptors were expressed in BALB 3T3 cells, which have been shown with transfected BB1 (Benya et al., 1992) and BB2 receptors (Benya et al., 1994b) to have characteristics similar to those of the wild-type receptors and overexpressing BB3 receptors in human non-small cell lung cancer cells, NCI-H1299. In this study (Mantey et al., 1997) none of the 15 naturally occurring bombesin-related peptides had a affinity of >1 μM for the human BB3 receptor. Furthermore, none of the 26 synthetic bombesin analogs that functioned as BB1 or BB2 receptor agonists or antagonists had a high affinity for the BB3 receptor, including [d-Phe6]Bn6–13 propylamide (Ki 2 μM), which had been reported in another study (Wu et al., 1996) assessing changes in cellular calcium to have a relatively high affinity of 84 nM for the human BB3 receptor. In this study (Mantey et al., 1997) one novel bombesin analog, [d-Phe6,β-Ala11,Phe13,Nle14]-bombesin6–14 was discovered, which had high affinity (Ki 4 nM) and potency for activating the BB3 receptor, and its Tyr6 analog retained high affinity and could be radiolabeled to study the pharmacology and ligand receptor interaction in detail. With this radioligand it was demonstrated (Mantey et al., 1997) that binding to the BB3 receptor fit a single site-binding model; it was rapid and temperature-dependent, with slow dissociation, supporting ligand internalization; and the binding affinities of all agonists and antagonists for the BB3 receptor could be determined for the first time and compared with those for the BB1 and BB2 receptors. These results demonstrated that the BB3 receptor has a unique pharmacology and does not interact with high affinity with any known naturally occurring bombesin peptide, supporting the conclusion that the natural ligand is either an undiscovered member of the bombesin family with significant structural differences or an unrelated peptide (Mantey et al., 1997). In a subsequent study (Ryan et al., 1996) two human lung cancer cell lines, NCI-N417 and NCI-H720, were found to possess sufficient wild type BB3 receptors to allow assessment of the pharmacology of the native BB3 receptor using the 125I-[d-Tyr6,β-Ala11,Phe13,Nle14]bombesin6–14 ligand described earlier. Pharmacology for all agonists and antagonists of the native BB3 receptor was found to be similar to that reported previously with the BB3 receptor transfected cell lines (Mantey et al., 1997) with only the agonist, [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 demonstrating high affinity (Ki 7.4 nM).
Subsequent studies demonstrated that the synthetic bombesin analog [d-Phe6,β-Ala11,Phe13,Nle14]-bombesin6–14, in addition to having high affinity for the human BB3 receptor, also has high affinity for the human BB1 receptor, the human BB2 receptor, the BB1 receptor, BB2 receptors from all species studied, and the fBB4 receptor (Mantey et al., 1997; Pradhan et al., 1998; Katsuno et al., 1999; Reubi et al., 2002; Iwabuchi et al., 2003) (Table 2). When the rat BB3 receptor was cloned (Liu et al., 2002) a surprising finding was that [d-Phe6,β-Ala11,Phe13,Nle14]-bombesin6–14 had a low potency for this receptor (EC50 2 μM). In the chicken (Iwabuchi et al., 2003) a receptor that had high affinity for [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14, moderate affinity for bombesin, and low affinity for GRP and NMB and showed structural similarity to both mammalian BB3 receptor and the amphibian BB4 receptor was found and thus was called chBRS-3.5. A subsequent study demonstrated that the monkey BB3 receptor (Sano et al., 2004) had a high potency for [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 (EC50 5.6 nM) similar to that of the human receptor. The molecular basis for the difference in affinity of [d-Phe6,β-Ala11,Phe13, Nle14]bombesin6–14 between human/monkey and rat BB3 receptors has been studied (Liu et al., 2002) and will be discussed in section. V.F.
Because of the lack of selectivity of the high-affinity agonist, [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 for the human BB3 receptor, there have been a number of groups who have attempted to develop more selective BB3 receptor ligands. Each of the different groups used [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 as the starting point to identify BB3 receptor selective agonists. In one study (Mantey et al., 2001) rational peptide design was used by substituting conformationally restricted amino acids into the prototype peptide, [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 or its d-Tyr6 analog. A number of BB3 receptor-selective agonists were identified with two peptides with either an (R)- or (S)-amino-3-phenylpropionic acid substitution for β-Ala11 in the prototype ligand having the highest selectivity (i.e., 17- to 19-fold) (Mantey et al., 2001). Molecular modeling demonstrated that these two selective BB3 receptor ligands had a unique conformation of the position of the 11β-amino acids, which probably accounted for their selectivity (Mantey et al., 2001). In a second study (Mantey et al., 2004) two strategies were used to attempt to develop a more selective BB3 receptor ligand: substitutions on the phenyl ring of Apa11 and the substitution of additional conformationally restricted amino acids into position 11 of [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 or its d-Tyr6 analog. One analog, [d-Tyr6,Apa-4Cl11, Phe13,Nle14]bombesin6–14 retained high affinity for the BB3 receptor and was 227-fold more selective for the BB3 receptor than for the human BB2 receptor and 800-fold more selective than the human BB1 receptor (Mantey et al., 2004). With [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 or its d-Tyr6 analog as the prototype, three studies (Weber et al., 2002, 2003; Boyle et al., 2005) reported shortened analogs with selectivity for the BB3 receptor as assessed by calcium or fluorometric imaging plate reader calcium assays. A recent study has assessed the selectivity of four of the most selective of these shortened [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 analogs by binding assays and by assessment of phospholipase C potencies (Mantey et al., 2006). Three analogs, which were reported to be selective in calcium assays for the BB3 receptor [H-d-Phe,Gln, d-Trp,NH(CH2)2C6H5 and H-d-Phe,Gln, d-Trp,Phe-NH2, compounds 68 and 54 in Weber et al. (2002), and 3-phenylpropionyl-Ala,d-Trp,NH(CH2)2C6H5, compound 17d in Weber et al. (2003)] were found (Mantey et al., 2006) in binding studies and studies of potency for activation of phospholipase C to have affinities >5 μM for all three human bombesin receptor subtypes and therefore not to be useful. The novel compound Ac-Phe,Trp,Ala,His(tBzl), Nip,Gly,Arg-NH2 [compound 34 in Boyle et al. (2005)] had 14- and 20-fold higher affinities for the BB3 receptor than for the BB1 receptor BB2 receptor, respectively (Mantey et al., 2006); however, its selectivity for the BB3 receptor was less than that of [d-Tyr6,Apa-4Cl11,Phe13,Nle14]-bombesin6–14 (i.e., >100 fold selectivity) (Mantey et al., 2006) (Table 2).
2. BB3 Receptor Antagonists.
No specific or potent antagonists of the BB3 receptor exist. In four studies (Ryan et al., 1996, 1998a, 1999; Mantey et al., 1997) none of the members of the different classes of potent BB2 or BB1 receptor antagonists had an affinity of <3 μM for the human BB3 receptor. In one study (Ryan et al., 1996) the d-amino acid-substituted somatostatin analog, d-Nal,Cys,Tyr,d-Trp,Lys,Val,Cys,Nal-NH2, had an affinity of 1 μM for the human BB3 receptor and was 30-fold more potent at inhibiting activation of the BB3 receptor than any other compound (Table 2). Unfortunately, this compound also functions as a BB1 receptor antagonist and as a somatostatin and μ-opioid receptor agonist (Orbuch et al., 1993; Ryan et al., 1999).
F. BB3 Receptor Structural Basis of Receptor Binding/Activation
1. BB3 Receptor Agonist Binding/Activation.
At present, because the natural ligand of the BB3 receptor is unknown, there is minimal information available on the importance of amino acid residues in BB3 receptor activation or on determining high-affinity interactions. For the only ligand known with high affinity for the BB3 receptor, [d-Phe6,β-Ala11,Phe13,Nle14] bombesin6–14 (Ryan et al., 1996, 1998a; Mantey et al., 1997), limited structure-function studies have suggested that it is unlikely that the deletion of the first five amino acids in [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14, the insertion of the d-Phe6, or the presence of either Phe13 or Nle14 moieties is determining the high affinity for the BB3 receptor compared with Bn, because other bombesin analogs with these substitutions do not have high affinity (Ryan et al., 1996; Mantey et al., 1997). These results suggest that the position 11 substitution (i.e., β-Ala11 or Apa-4Cl11) in bombesin analogs is the key substitution for determining high-affinity interaction with the BB3 receptor. At present the basis for the high affinity with these substitutions is not known.
One study (Liu et al., 2002) investigated the molecular basis for the high affinity of [d-Phe6,β-Ala11,Phe13, Nle14]bombesin6–14 for the human BB3 receptor but low affinity for the rat BB3 receptor. By using a chimeric receptor approach in which the individual extracellular loops of the rat BB3 receptor were replaced with the corresponding human sequences, the important residues were localized to the fourth extracellular domain (first = N-terminus) (Liu et al., 2002). Within this region, with the use of site-directed mutagenesis (Liu et al., 2002), the mutation of Y298E299S330 (rat) to S298Q299T300 (human) or of D306V307H308 (rat) to A306M307H308 (human) partially mimics the effect of switching the entire fourth extracellular domain. These results indicate that variations in the fourth extracellular domains of the rat and human BB3 receptor are responsible for the differences in affinity for [d-Phe6,β-Ala11,Phe13,Nle14]bombesin6–14 (Liu et al., 2002).
Whereas there is no information on the molecular basis of the selectivity of the various [d-Phe6,β-Ala11, Phe13,Nle14]bombesin6–14 analogs for the BB3 receptor, a number of studies have assessed the molecular basis for the low affinity of the human BB3 receptor for the naturally occurring high-affinity BB1 receptor and BB2 receptor agonists (GRP, bombesin, and NMB). These studies have used an alignment of the receptor structures of the various bombesin receptors and identified key amino acid differences between the BB3 receptor, which has low affinity for GRP, bombesin, or NMB, and the BB2 receptor, BB1 receptor, or fBB4 receptors, which have high affinities for these ligands (Akeson et al., 1997; Sainz et al., 1998; Nakagawa et al., 2005). The results of these studies are summarized earlier in sections III.F.1 and IV.F.1. No studies have been performed to investigate the structural basis for BB3 receptor activation.
2. BB3 Receptor Antagonist Binding.
No potent selective antagonists exist for the BB3 receptor.
G. BB3 Receptor Signaling, Activation, and Modulatory Processes (Internalization, Down-Regulation, and Desensitization)
The human BB3 receptor (Fathi et al., 1993b; Ryan et al., 1996, 1998a; Wu et al., 1996), as well as the monkey (Sano et al., 2004) and rat BB3 receptors (Liu et al., 2002) is coupled to phospholipase C, resulting in breakdown of phosphoinositides, mobilization of cellular calcium, and presumed activation of protein kinase C.
BB3 receptor activation results in the stimulation of phospholipase D (Ryan et al., 1996) but does not activate adenylate cyclase (Ryan et al., 1996, 1998a). BB3 receptor stimulation also results in activation of tyrosine kinases (Ryan et al., 1998a; Weber et al., 2001), stimulating tyrosine phosphorylation of p125FAK by a mechanism that is not dependent on either limb of the phospholipase C cascade (i.e., activation of PKC or mobilization of cellular calcium) (Ryan et al., 1998a). Activation of BB3 receptor also stimulates MAP kinase activation, resulting in rapid tyrosine phosphorylation of both 42- and 44-kDa forms, which is inhibited by the MEK-1 inhibitor PD98059 (Weber et al., 2001). In BB3 receptor-transfected NCI-1299 lung cancer cells, activation of the BB3 receptor by [d-Phe6,β-Ala11,Phe13, Nle14]bombesin6–14 resulted in stimulation of Elk-1 in a MEK-1-dependent manner as well as a 47-fold increase in c-fos mRNA (Weber et al., 2001). These results demonstrated that BB3 receptor activation causes increased nuclear proto-oncogene expression and upstream events including activation of MAP kinase and Elk-1 activation (Weber et al., 2001). There have been no studies of BB3 receptor modulatory processes (internalization, down-regulation, or desensitization).
H. BB3 Receptor Function in Various Tissues and in Vivo
At present the function of the BB3 receptor in normal physiology and pathological conditions is largely unknown because the natural ligand is still not known. An important insight into possible BB3 receptor function was provided by studies of BB3 receptor knockout mice. In the initial study (Ohki-Hamazaki et al., 1997b) mice lacking the BB3 receptor developed mild obesity, associated with hypertension and impairment of glucose metabolism. These changes were associated with reduced metabolic rate, increased feeding behavior, a 5-fold increase in serum leptin levels, and hyperphagia (Ohki-Hamazaki et al., 1997b) and the results suggested that the BB3 receptor might play an important role in the mechanisms responsible for energy balance and control of body weight. A number of studies have been performed subsequently on BB3 receptor knockout mice to attempt to establish the mechanism of these effects. BB3 receptor knockout mice were shown to have altered taste preference (Yamada et al., 1999), which was proposed to be due to the lack of BB3 receptor expression in the medial and central nuclei of the amygdala and the hypothalamic nuclei, which are known to be involved in taste perception (Yamada et al., 1999) and to possibly be a contributory factor to the obesity. BB3 receptors are present on pancreatic islets (Fleischmann et al., 2000), and BB3 receptor knockout mice have a 2.3-fold increase in plasma insulin levels (Matsumoto et al., 2003) (Table 2). One study (Matsumoto et al., 2003) concluded that the BB3 receptor contributes to regulation of plasma insulin concentration/secretion and that dysregulation in this contribution in these mice contributes to obesity (Matsumoto et al., 2003). In a second study (Nakamichi et al., 2004) it was concluded that the impaired glucose metabolism in BB3 receptor knockout mice is mainly due to impaired glucose transporter 4 translocation in adipocytes.
I. BB3 Receptor in Diseases
At present there are no diseases in which activation or alterations of the BB3 receptor have been shown to be involved. BB3 receptor activation has been proposed to be important in the mediation of a number of human disorders including disorders of lung development, various pulmonary diseases, CNS disorders, and the growth/differentiation of human cancers. The tumor differentiation effects of BB3 receptor activation were discussed in the previous section; the growth effects and effects of BB3 receptor overexpression will be considered here. In human cancer cells or cancers BB3 receptors are not only ectopically expressed in a large number of tumors, as reviewed earlier (Fathi et al., 1993b, 1996; Toi-Scott et al., 1996; Sun et al., 2000b; Reubi et al., 2002; Schulz et al., 2006), but their activation alters lung cancer behavior by increasing MAP kinase activation and nuclear oncogene expression (Weber et al., 2001) and increasing adhesion of lung cancer tumor cells, which was proposed to contribute to increased tumor invasion and metastases by these tumors (Hou et al., 2006). In BB3 knockout mice (Maekawa et al., 2004) the hyperphagic response to melanin-concentrating hormone (MCH) is impaired, but this impairment is not seen in BB2 receptor knockout mice. Furthermore, the levels of the MCH receptor and prepro-MCH mRNAs in the hypothalamus of BB3 receptor knockout mice were higher than those of controls, suggesting that up-regulation of the MCH receptor and MCH occurs in the knockout mice, which triggers hyperphagia and probably upsets the mechanism by which leptins decrease MCH receptors and feeding (Maekawa et al., 2004). Studies of BB3 receptor knockout mice suggest that this receptor is important in various behavioral effects, including the neural mechanisms that regulate social isolation (Yamada et al., 2000a), and are important in modulating emotion including forms of anxiety (Yamada et al., 2002a).
BB3 receptors as well as BB1 receptor and BB2 receptor are expressed in developing primate and murine fetal lungs (Emanuel et al., 1999; Shan et al., 2004). Studies (Tan et al., 2006, 2007) demonstrate that BB3 receptors are expressed in the airway in response to ozone injury and that wound repair and proliferation of bronchial epithelial cells is accelerated by BB3 receptor activation, suggesting that it may mediate wound repair. The mechanism of lung ozone injury mediation of the up-regulation of BB3 receptors has been studied by examining proteins interacting with the BB3 receptor gene promoter region (Tan et al., 2007). Activator protein-2α and peroxisome proliferator-activated receptor-α increased the ozone-inducible DNA binding on the BB3 receptor gene promoter, suggesting that they are specifically involved in the BB3 receptor up-regulation (Tan et al., 2007). BB3 receptors are expressed on small cell and non-small cell lung cancers (Fathi et al., 1993b; Toi-Scott et al., 1996; Ryan et al., 1998b; Reubi et al., 2002) as well as lung carcinoids (Fathi et al., 1993b; Reubi et al., 2002). In the small cell lung cancer cell line, NCI-N417, which is known to possess functional BB3 receptors (Ryan et al., 1998b), [d-Phe6,β-Ala11,Phe13,Nle14]-bombesin6–14 stimulated tumor cell adhesion, probably by stimulation of focal adhesion formation (Hou et al., 2006). It was proposed (Hou et al., 2006) that BB3 receptor activation in these cells may be important for their invasion and development of metastases.
Although the function of BB3 receptors in the gastrointestinal tract is largely unknown, specific BB3 receptor antibodies localized the receptor in the tunica muscularis of the rat gastrointestinal tract (Porcher et al., 2005). BB3 receptors were detected in both myenteric and submucosal ganglia as well as in nerve fibers interconnecting myenteric ganglia (Porcher et al., 2005). BB3 receptor IR was observed in cell bodies and processes of the c-kit interstitial cells of Cajal, leading the authors to propose that the BB3 receptor was probably involved in the regulation of gastrointestinal motility.
One study screened 104 Japanese obese men for defects in the BB3 receptor gene, but no mutations or polymorphisms were found (Hotta et al., 2000), suggesting BB3 receptor gene mutations are unlikely to be a major cause of obesity in humans.
The above studies and those reviewed in the previous section suggest that BB3 receptor activation could be involved in human disorders of energy metabolism, including obesity, glucose homeostasis, blood pressure control, lung injury, tumor growth, and possibility motility disorders. However, all of these possibilities remain unproven at present.
VI. Therapeutic Implications of Bombesin Receptors
This topic was partially covered under the sections dealing with disease for each of the three receptor classes, but a few important summary points will be made here. The principal therapeutic interests are in the BB2 receptors, to a lesser extent in the BB3 receptor, and least in the BB1 receptor. In the case of the BB2 receptor the recent study (Sun and Chen, 2007) providing evidence that activation of the BB2 receptor in the spinal cord may be an important pathway in mediating pruritic signals has profound clinical implications. Chronic itching is a very common problem (Yosipovitch et al., 2007): in a population survey of 18,770 adults in Norway (Dalgard et al., 2007a,b) itching was the most common skin problem occurring in 7%, and it is associated with poor general health. Often existing therapies provide limited relief, and there are no general-purpose antipruritic drugs (Yosipovitch et al., 2007); therefore, identification of the BB2 receptor as a possible central target has significant therapeutic implications for this disorder. The tumoral growth effects and frequent overexpression or ectopic expression of all of the Bn receptors have important clinical implications, particularly for the BB2 receptor, which is the most frequently overexpressed, and has been the most extensively investigated for its growth effects on different human tumors (Jensen and Moody, 2006; Lantry et al., 2006; Patel et al., 2006; Schulz et al., 2006; Cornelio et al., 2007; Engel et al., 2007). Studies demonstrating that GRP and NMB can have autocrine growth activity, that in some tumors BB2 receptor activation results in stimulation of the EGF receptor, that continued stimulation through the BB2 receptor can counter the inhibitory effects of EGFR blockade on tumor growth, and that the combination of a BB2 receptor blockade and EGF receptor inhibition can have profound inhibitory effects on tumor growth all have important therapeutic implications (Santiskulvong et al., 2001, 2004; Madarame et al., 2003; Santiskulvong and Rozengurt, 2003; Xiao et al., 2003; Stangelberger et al., 2005; Thomas et al., 2005; Jensen and Moody, 2006; Patel et al., 2006; de Visser et al., 2007b; Liu et al., 2007; Zhang et al., 2007b). As discussed in detail in the sections III.I., IV.I., and V.I., the overexpression of BB2 receptors in particular by many common tumors (breast, colon, head and neck squamous cancers, various CNS tumors, lung, prostate, ovary, and renal) has important therapeutic implications. This is particularly true for the Bn family of receptors, because they are one of the classes of G protein-coupled receptors most frequently present on these tumors. Furthermore, in many cases existing therapies are inadequate with these tumors as they frequently stop responding to current first-line treatments, and therefore new approaches are needed. There are potential therapeutic implications not only for development of labeled Bn analogs for enhanced tumor imaging and staging (Breeman et al., 2002; Nock et al., 2003; Smith et al., 2003, 2005; Johnson et al., 2006; Lantry et al., 2006; Zhang et al., 2006; de Visser et al., 2007a; Dimitrakopoulou-Strauss et al., 2007; Garrison et al., 2007; Parry et al., 2007; Prasanphanich et al., 2007; Waser et al., 2007; Zhang et al., 2007a), but also for use for bombesin receptor-mediated cytotoxicity, either with radiolabeled compounds, as is being widely evaluated with somatostatin analogs in phase 3 studies (Breeman et al., 2007; Forrer et al., 2007; Van Essen et al., 2007) or for use of Bn analogs coupled to other cytotoxic agents such as doxorubicin analogs, paclitaxel, or camptothecin (Schally and Nagy, 1999, 2004; Breeman et al., 2002; Moody et al., 2004, 2006b; Engel et al., 2005, 2007; Buchholz et al., 2006; Nanni et al., 2006; Panigone and Nunn, 2006; Safavy et al., 2006). The participation of BB3 receptors in energy balance and in glucose homeostasis as manifested by BB3 receptor knockout animals developing obesity and diabetes (Ohki-Hamazaki et al., 1997a) has potential important clinical implications. At present there has been increased understanding of the mechanisms of these effects (section V.H.) (Yamada et al., 1999; Matsumoto et al., 2003; Nakamichi et al., 2004), but possible progress in extending this understanding to a clinical application is limited by the lack of identification of the natural ligand for this receptor.
Numerous other actions of each of the three Bn receptors have potential importance for therapeutic interventions, but at present either the understanding of their participation in normal and pathological conditions is insufficient to specifically target these receptors or the drugs to do this are not available. In the case of the BB1 receptor such areas include involvement in thyroid function and alterations in thyroid disease (Ortiga-Carvalho et al., 2003; Pazos-Moura et al., 2003; Oliveira et al., 2006), behavior effects in mediating aspects of fear, anxiety, and stress responses (Ohki-Hamazaki et al., 1999; Merali et al., 2002, 2006; Yamada et al., 2003; Bédard et al., 2007); and satiety effects (Merali et al., 1999; Ladenheim and Knipp, 2007). For the BB2 receptor, such areas include its role in motility with mediation of the descending peristaltic reflex (Grider, 2004) its role in lung injury and development of lung diseases, particularly neonatal lung disease and bronchopulmonary dysplasia, in which Bn-like peptides and the BB2 receptor were shown to play an important role in various animal models (Li et al., 1994; Sunday et al., 1998; Emanuel et al., 1999; Cullen et al., 2000; Ashour et al., 2006; Ganter and Pittet, 2006; Subramaniam et al., 2007), its role in sepsis and in small intestinal mucosal protection and prevention of injury (Assimakopoulos et al., 2004, 2005a, b; Dal-Pizzol et al., 2006; Higuchi et al., 2006; Kimura et al., 2006a,b), its role in satiety effects (Merali et al., 1999; Ladenheim and Knipp, 2007), and its CNS effects on memory, learning, various behaviors, and response to stress (Merali et al., 1999, 2006; Yeğen, 2003; Moody and Merali, 2004; Roesler et al., 2004, 2006a,b; dos Santos Dantas et al., 2006; Luft et al., 2006Presti-Torres et al., 2007). For the BB3 receptor such areas include its possible role in lung development and responses to lung injury (Shan et al., 2004; Hou et al., 2006; Tan et al., 2006, 2007) and its possible role in regulation of aspects of gastrointestinal motility (Porcher et al., 2005).
VII. Unresolved Nomenclature Issues
The principal unresolved issue is that the natural ligand of the BB3 receptor remains unknown, and therefore its pharmacology and roles in normal physiology or pathological processes is unknown. Another unresolved issue is whether a receptor equivalent to the frog BB4 exists in human and mammals. Two studies have sought additional members of the bombesin receptor family, and none were found in mammals (Fathi et al., 1993b; Sano et al., 2004). With human and mouse genome sequences now known, it is high unlikely that any other mammalian BB receptors beside BB1,BB2, and BB3 will be found. An additional key issue unresolved at present is whether the COOH-terminal extended or precursor form or fragments of GRP or NMB have physiological or pathological effects that are not mediated by the three classes of mammalian receptors described in the current nomenclature. A number of recent studies (Dumesny et al., 2004, 2006; Patel et al., 2004, 2007a,b) provide evidence that nonamidated precursor forms of GRP can stimulate proliferation of different tumors/tissues. COOH-terminal precursor forms of GRP are reported to stimulate the proliferation and migration of the human colorectal carcinoma cell line DLD-1 (Patel et al., 2007a,b) through a BB2 receptor-independent mechanism and the growth of the prostate cancer cell line DU145 (Patel et al., 2007b). Furthermore, pro-GRP immunoreactivity is reported in 90% of resected colorectal carcinomas and all endometrial, prostate, and colon cancer cell lines tested, without any amidated forms being present (Dumesny et al., 2006). Recombinant pro-GRP stimulated proliferation of the colon cancer cell line DLD-1, activating MAP kinase, but did not stimulate phospholipase C activity nor bind to known bombesin receptors, suggesting it was stimulating the tumor growth through a novel receptor (Dumesny et al., 2006). At present no receptor has been isolated that mediates these actions, but they are not inhibited by BB2 receptor antagonists, raising the possibility they could be mediated by a novel receptor. A final key problem area that is unresolved at present is the roles of the three described mammalian bombesin receptors in normal physiology and pathological conditions, which are still largely unknown. This lack of knowledge is due in large part to lack of specific antagonists for all subclasses of bombesin receptors, especially high-affinity, selective nonpeptide receptor antagonists.
Acknowledgments
We thank Terry Moody, National Cancer Institute, for carefully proofreading the article and for making thoughtful suggestions.
Footnotes
-
↵1 Abbreviations: Bn, bombesin; GRP, gastrin-releasing peptide; NMB, neuromedin B; IR, immunoreactivity; CNS, central nervous system; GI, gastrointestinal; TSH, thyroid-stimulating hormone; GRP-R, GRP-preferring receptor (BB2); NMB-R, NMB-preferring receptor (BB1); BRS-3, bombesin receptor(s) subtype 3 (BB3); h, human; fBB4, frog bombesin receptor subtype 4; PK, protein kinase; kb, kilobase(s); BIM231127, [d-Nal-Cys-Tyr-d-Trp-Orn-Val-Cys-Nal-NH2]; PD 165929, 2-[3-(2,6-diisopropyl-phenyl)-ureido]3-(1H-indol-3-yl)-2-methyl-N-(1-pyridin-2-yl-cyclohexylmethyl)-proprionate; PD 168368, 3-(1H-indol-3-yl)-2-methyl-2-[3(4-nitro-phenyl)-ureido]-N-(1-pyridin-2-yl-cyclohexylmethyl)-propionamide; PD 176252, 3-(1-H-indol-3-yl)-N-[1-(5-methoxy-pyridin-2-yl)-cyclohexylmethyl]-2-methyl-2-[3-(4-nitro-phenyl)-ureido]-propionamide; TM, transmembrane region; p125FAK, p125 focal adhesion kinase; MAP, mitogen-activated protein; 5-HT, 5-hydroxytryptamine (serotonin); CCK, cholecystokinin; bp, base pairs; SP, substance P; GPCR, G protein-coupled receptor; Ψ bonds, pseudopeptide bonds; BW2258U89, (3-PhPr)-His,Trp,Ala,Val,d-Ala,His,d-Pro-Ψ(CH2NH)-Phe-NH2; RC-3950-II, [d-Phe6,Ψ13–14,Tac14]Bn6–14 (tac = thiazolidine-4-carboxylic acid); RC-3095, [d-Tpi6,Ψ13–14]bombesin6–14; JMV641, H-d-Phe, Gln,Trp,Ala,Val,Gly,His-NH-*CH[CH2-CH(CH3)2]-**CHOH-(CH2)3-CH3, where * is (S) and ** is 92% of S isomer; JMV594, d-Phe6, statine13]Bn6–14], where statine is 4-amino-3-hydroxy-6-methylhepatanonoic acid; EC, extracellular domain; IC, intracellular domain; MEK, mitogen-activated protein kinase kinase; ERK, extracellular regulated kinase; SH, Src homology; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; GRK, G protein-coupled receptor kinase; BPD, bronchopulmonary dysplasia; FK506, tacrolimus; PD98059, 2′-amino-3′-methoxyflavone; MCH, melanin-concentrating hormone.
-
This research was partially supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and National Institute on Deafness and Other Communication Disorders, National Institutes of Health; National Cancer Institute Grant CA-094346 (R.V.B.); and a Veterans Affairs merit review award (R.V.B.).
-
This article is available online at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.107.07108.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
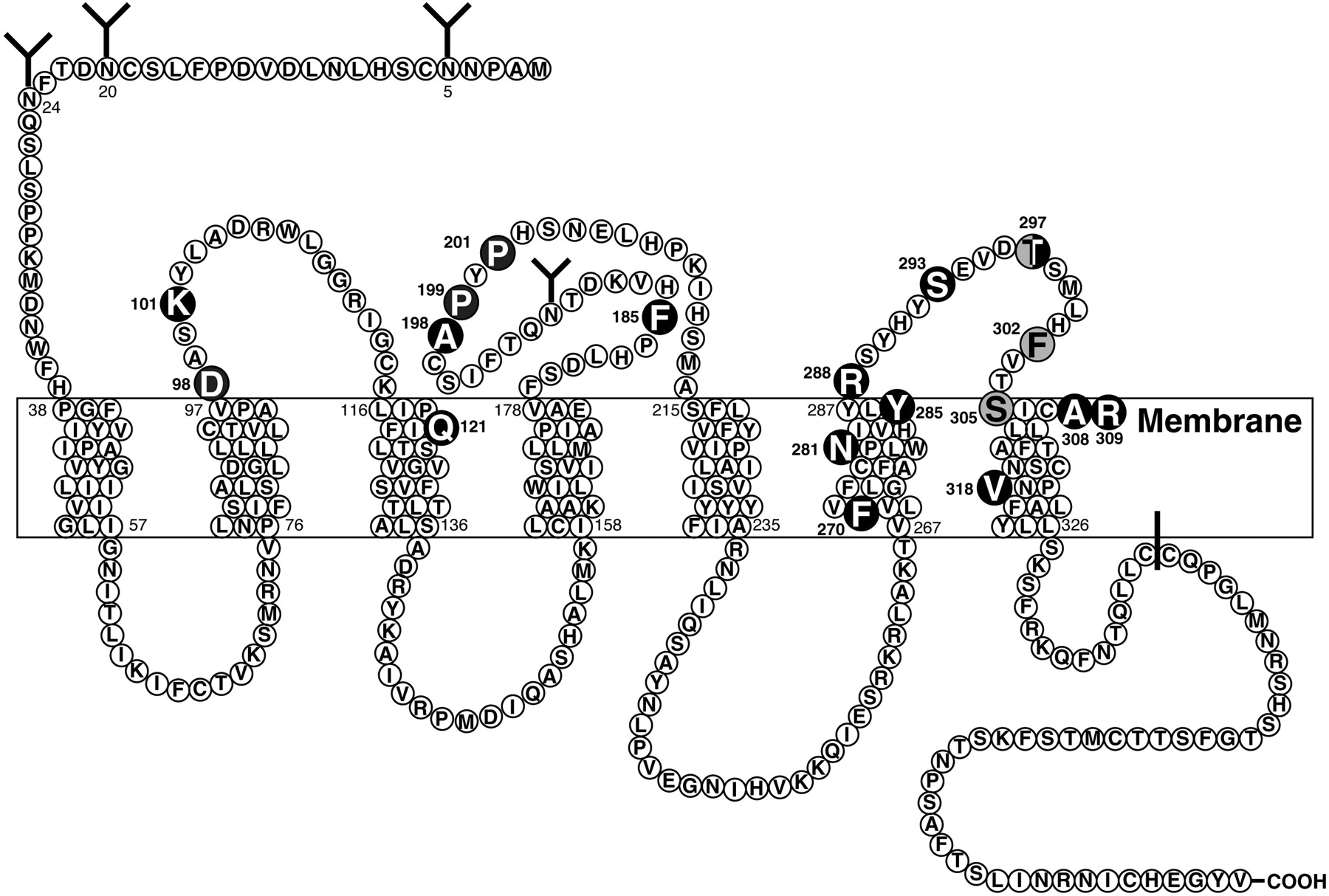{kind=link}