


Abstract
It is now more than 15 years since the molecular structures of the major prostanoid receptors were elucidated. Since then, substantial progress has been achieved with respect to distribution and function, signal transduction mechanisms, and the design of agonists and antagonists (http://www.iuphar-db.org/DATABASE/FamilyIntroductionForward?familyId=58). This review systematically details these advances. More recent developments in prostanoid receptor research are included. The DP2 receptor, also termed CRTH2, has little structural resemblance to DP1 and other receptors described in the original prostanoid receptor classification. DP2 receptors are more closely related to chemoattractant receptors. Prostanoid receptors have also been found to heterodimerize with other prostanoid receptor subtypes and nonprostanoids. This may extend signal transduction pathways and create new ligand recognition sites: prostacyclin/thromboxane A2 heterodimeric receptors for 8-epi-prostaglandin E2, wild-type/alternative (alt4) heterodimers for the prostaglandin FP receptor for bimatoprost and the prostamides. It is anticipated that the 15 years of research progress described herein will lead to novel therapeutic entities.
I. Introduction
A. Receptor Classification (circa 1994)
1. Receptor Subtypes.
The major prostaglandins (PGs1), PGD2, PGE2, PGF2α, prostacyclin (PGI2), and thromboxane A2 (TxA2) preferentially interact with dedicated receptors designated DP, EP, FP, IP, and TP, respectively (Kennedy et al., 1982; Coleman et al., 1984). Although largely based on functional studies using a limited range of agonists, and an even more limited range of antagonists, the original classification of prostanoid receptors has entirely withstood the tests of time and scrutiny. Prostanoid receptor subtypes were also proposed. Four subtypes of EP receptor (EP1, EP2, EP3, and EP4) have been described (Coleman et al., 1994b). Two PGD2-sensitive receptors were suggested, but only DP1 was described in 1994, although diverse pharmacological evidence described a second PGD2-sensitve receptor (Jones, 1976a,b, 1978; Narumiya and Toda, 1985; Woodward et al., 1990, 1993b; Rangachari and Betti, 1993; Fernandes and Crankshaw, 1995).
2. Molecular Structure.
Prostanoid receptors are G protein-coupled receptors. Although the overall homology between those receptors cloned in the 1990s was not high, there were several conserved regions. The first prostanoid receptor to be structurally identified was TP. This was achieved by purifying the TP receptor protein using the high-affinity, radiolabeled ligand S-145 (Ushikubi et al., 1989). Based on a partial amino acid sequence, the cDNA for the TP receptor was obtained (Hirata et al., 1991). The other prostanoid receptors were cloned by homology based screening and by 1994, DP1, FP, IP, and all PGE2-sensitive receptors had been structurally identified (Coleman et al., 1994b; Hirata et al., 1994; Nakagawa et al., 1994; Regan et al., 1994b).
The deduced amino acid sequences for human DP1, EP1–4, FP, IP, and TP receptors, together with DP2, are compared in Fig. 1. Hydrophobicity analysis of the sequences indicated seven membrane-spanning segments, an extracellular N terminus, and an intracellular −COOH terminus typical of rhodopsin-type, G-protein-coupled receptors. Regions of significant homology occur in the seventh transmembrane domain and the second extracellular loop. The highly conserved arginine in the seventh transmembrane (TM) domain has been proposed as the interaction site for the carboxylate group, which is common to all natural prostanoids. Additional determinants of prostanoid receptor binding have been suggested as follows. Two conserved Cys residues in the first and second extracellular loops are believed to form a disulfide bridge critical for stabilization of the receptor conformation (Narumiya et al., 1999). The second extracellular loop connecting TM4 and TM5 contains an invariant Trp-Cys-Phe triplet (Pierce et al., 1995). There are one or more consensus sequences for N-glycosylation of arginine residues in the amino terminal region (Narumiya et al., 1999). N-glycosylation may also be important for ligand binding, at least for TP receptors (Chiang and Tai, 1998).

Deduced amino sequence of human prostanoid receptors, in alignment. Amino acid residues with 100% homology between all receptors are highlighted in green. Amino acid residues with complete homology, except for DP2, are highlighted in blue.
Studies on the molecular evolution of prostanoid receptors suggest a PGE2-sensitive entity as the ancestral receptor (Regan et al., 1994b; Boie et al., 1995; Toh et al., 1995; Foord et al., 1996). This may have occurred not only by gene duplication but also by chromosomal duplication (Duncan et al., 1995). These phylogenetic analyses suggested two major branches of prostanoid receptor evolution: one group Gs-coupled (DP1, EP2, EP4, and IP) and the other group Gi- (EP3) or Gq-coupled (EP1, FP, and TP), providing three clusters (Table 1). For most receptors, mRNA splicing variants have been identified.
Primary G protein coupling for prostanoid receptors
The DP2 receptor, although recognizing PGD2 as a primary natural ligand, has no significant homology with DP1 or other prostanoid receptors described in the original classification (Abe et al., 1999; Hirai et al., 2001; Hata et al., 2005). DP2 (CRTh2) is more closely related to chemoattractant receptors, such as C5α and N-formyl peptide receptors (Abe et al., 1999; Hirai et al., 2001). The arginine residue in the seventh membrane-spanning domain, which is conserved in all receptors of the original classification, is not present in DP2 (Fig. 1). Replacement of the corresponding serine by mutation to alanine had little effect on PGD2 binding (Hata et al., 2005). In contrast, replacement of lysine 209 in the fifth membrane-spanning domain by alanine greatly reduced PGD2 binding to DP2 receptors. Interaction of the carboxylate of PGD2 with Lys-209 would place PGD2 in an opposite orientation in the binding pocket to that proposed for other prostanoid receptors (Hata et al., 2005).
3. Second Messenger Signaling.
Early signal transduction studies and a paucity of receptor selective ligands provided little insight into the pharmacology of prostanoid-sensitive receptors. The cloning of each prostanoid receptor and their transfection into and expression in cultured cells led to rapid elucidation of their G protein coupling characteristics, at least with respect to Gq, Gs, and Gi. DP(1), EP2, EP4, and IP receptors were classified as Gs-coupled; EP1, FP, and TP receptors were designated as Gq-coupled; EP3 receptors seemed capable of coupling to both Gq and Gi (Coleman et al., 1994b). During the past decade and a half, the DP2 (CRTh2) receptor was discovered as a second Gi-coupled receptor. These findings are summarized in Table 1. The signaling cascades associated with prostanoid receptor stimulation have been further investigated to reveal diverse pathways.
4. Agonist and Antagonist Drugs.
The pharmacological characterization of prostanoid receptors was built on isolated tissue and cultured cell studies (Coleman et al., 1984). Despite an inherent reliance on pharmacological intuition and deduction and on low-throughput assays, highly selective ligands were obtained for certain receptors. 5-(6-Carboxyhexyl)-1-(3-cyclohexyl-3-hydroxypropyl)hydantoin (BW 245C) and 3-(3-[1,1′-biphenyl]-4-yl-3-hydroxypropyl)-2,5-dioxo-4-imidazolidineheptanoic acid, ethyl ester (BW A868C) (Giles et al., 1989) were discovered as a selective agonist and antagonist, respectively, for DP1 receptors, previously designated DP receptors. trans-2-(4-(1-Hydroxyhexyl)phenyl)-5-oxocyclopentaneheptanoic acid (AH 13205) (Nials et al., 1993) and butaprost (Gardiner, 1986) were selective EP2 agonists and (4-benzamidophenyl)-(Z)-7-[(1R,2R,3R)-3-hydroxy-2-[(2R)-2-hydroxy-3-phenoxypropoxy]-5-oxocyclopentyl]hept-5-enoate (GR 63799X) was a potent and selective EP3 agonist (Bunce et al., 1991). Fluprostenol (Dukes et al., 1974; Coleman et al., 1984) and 17-phenyl PGF2α (Woodward et al., 1995a) were known as selective FP agonists and, ultimately, formed the structural platform for launching antiglaucoma drugs. Above all, several potent and selective TP receptor antagonists were invented based on their potential utility for treating cardiovascular disease. Indeed, S-145 was sufficiently potent and selective to enable isolation of the TP receptor protein (Ushikubi et al., 1989; Hirata et al., 1991).
B. Summary of New Developments
1. Prostaglandin D2 (CRTh2) Receptors.
The eight prostanoid receptors described by the pharmacology-based classification were rapidly discovered by homology-based screening after structural identification of TP receptors. This made it unlikely that further similar receptors would emerge, and this has proved to be the case. More recently discovered prostanoid receptors are quite different. A second PGD2-sensitive receptor was long suggested by functional studies but, when discovered, was found to be structurally quite distinct (Hirai et al., 2001). The DP2 receptor, also and originally designated (CRTh2), is more closely related to chemo-attractant receptors. The DP2 receptor mediates effects that are opposed to those produced by DP1 receptor stimulation in some instances. Given the lack of structural identity between DP1 and DP2 (CRTh2) receptors, it is not surprising that ligand recognition can markedly diverge. For example, the cyclooxygenase (COX) inhibitor indomethacin actually stimulates DP2 (CRTh2) receptors (Hirai et al., 2002; Stubbs et al., 2002), albeit weakly.
2. Prostanoid Receptor Heterodimerization.
The discovery that G protein-coupled receptors may heterodimerize has explained certain pharmacological anomalies. Receptor heterodimerization offers the option of creating novel binding sites without evolution of a dedicated encoding gene. It also provides a means of closely regulating the activity of local hormones simultaneously released by intimately combining their receptors. IP/TP receptor heterodimerization provides an example of both phenomena: 1) a recognition site for 8-epi-PGE2 is created and 2) the pathological effects of TP are limited because cAMP levels are increased by TxA2 activating the associated heterodimeric protein (Wilson et al., 2004).
The pharmacology of PGF2α-ethanolamide (prostamide F2α), and its analog bimaprost, was elucidated by a longer and more traditional route. Numerous agonist studies suggested a pharmacological identity distinct from the classic prostanoid FP receptor; this was confirmed by the eventual discovery of selective antagonists (Woodward et al., 2008). All this led to the same place in that the bimatoprost recognition site was modeled by cotransfecting the wild-type FP receptor and an alternative mRNA splicing variant thereof (Liang et al., 2008).
3. Gene Deletion Studies.
Gene deletion studies with mice lacking each of the individual prostanoid receptors have enabled further elucidation of prostanoids in health and disease. Moreover, they have revealed important functions that had not been previously appreciated. The roles of prostanoids in inflammation and immune regulation provide a good example. It is now clearer that prostanoids exert both pro-inflammatory and anti-inflammatory effects and regulate gene expression in mesenchymal and epithelial cells at inflammatory sites (Narumiya, 2009). This self-regulatory role, with prostanoids exerting a dual role in a singular process, is often seen in immunology-based reactions. It had been held that prostanoids exerted very little role in immunity, but gene deletion studies revealed that prostanoids work at many levels in immune responses (Narumiya, 2003). A striking example of the contribution of gene deletion is the discovery of EP1 receptor involvement in suppressing impulsive behavior in response to stress (Furuyashiki and Narumiya, 2009). These findings will provide novel direction from discovering additional prostanoid-based therapeutics.
Gene deletion studies are an important step forward in elucidating the role of prostanoids in physiology and pathophysiology, especially when viewed in the context of small-molecule–based research. There are potential pitfalls in using small molecules to determine the pharmacological basis of experimental disease and thereby to discover new therapeutic approaches. False-negative results may occur by virtue of inadequate bioavailability at the target tissue: metabolic disposition and pharmacokinetics are traditionally a terminal step in the drug discovery process that occurs before clinical evaluation. Given the very high attrition rate in drug discovery, false-positive animal model data are clearly commonplace. Seemingly beneficial effects in animal models of human disease may be misleading for several reasons. Off-target pharmacology can never be fully assessed, even by screens aimed at 400 to 500 targets. Drug-induced toxicity is rarely overt in acute animal models but is likely to produce misleading positive effects, certainly in pain and inflammation models. Indications of toxicity could be obtained from such living animal studies but this rarely seems to be the case. Blood pressure and heart rate could readily be monitored by tail cuff instrumentation. Circulating leukocyte viability could be determined by in vitro studies on inflammatory cells that routinely determine cell viability. Studies employing genetically modified mice are more reliable in that there can be confidence that there is 1) no hidden off-target pharmacology, 2) no toxicity of unknown origin, and 3) no bioavailability issue. Thus, the use of genetically modified mice provides a solution to certain drug discovery complications; species differences and potential compensatory mechanisms remain a concern. Finally, the choice of disease models to complement gene deletion studies is also an important consideration. Many animal models, for example LPS-induced uveitis (Caspi, 2006), seem to be models in search of a human disease to mimic.
4. New Agonists and Antagonists.
The introduction of recombinant receptor technologies permitted high-throughput assays. This has resulted in selective and potent antagonists for all known prostanoid receptors, with the exception of EP2. Potent and selective agonists have now been discovered for all prostanoid receptors, with the possible exception of EP1. These pharmacological tools, with information provided by gene deletion studies, will result in therapies based on informed modulation of prostanoid-mediated events. Prostanoid-based therapeutics will realize its full potential.
Parallel to fitting ligands to known receptors, unexplained pharmacological characteristics associated with certain prostanoids have been successfully studied. The activities of certain “orphan” prostanoids, although not actually described as such, are now understood. The surprisingly high potency of the PGD2 metabolites 13,14-dihydro 15-keto PGD2 (Jones, 1976a,b; Jones and Wilson, 1978) and PGJ2 (Woodward et al., 1990) was explained by the discovery of the DP2 (CRTh2) receptor. Heterodimerization between IP and TP receptors provides a site for interaction with 8-epi-PGE2 (Wilson et al., 2004). Likewise, PG-ethanolamides, and their analog bimatoprost, have a predilection for cells cotransfected with wild type and an alternative mRNA splicing variant of the FP receptor (Liang et al., 2008).
5. Cell Signaling.
Elucidation of the cell signaling pathways associated with prostanoid receptors has provided an additional dimension to the body of information required to conceptualize therapeutic applications. In many instances, the repertoire of signaling pathways is now known to be quite expansive for some receptors. These will be described for each individual prostanoid receptor.
II. Receptor Types, Subtypes, and mRNA Splicing Variants
A. DP1 Receptors
1. Second Messenger Signaling.
DP1 receptors are Gs-coupled and stimulate cAMP formation (Gorman et al., 1977; Whittle et al., 1978; Schafer et al., 1979; Halushka et al., 1989; Goh and Nakajima, 1990; Hirata et al., 1994; Boie et al., 1995). Cells expressing DP1 receptors also elicit an increase in [Ca2+]i when stimulated by PGD2 or the selective DP1 agonist BW 245C, but this is cAMP-dependent (Boie et al., 1995). This increase in [Ca2+]i may result from activation of protein kinase A (PKA) and subsequent involvement of L-type Ca2+ channels and the ryanodine receptor (Zaccolo, 2009). This seems to be a minor pathway for transducing DP1 receptor signaling, cAMP/PKA activation being almost invariably involved. No evidence for the involvement of exchange proteins activated by cAMP (Epacs) seems to have emerged.
2. Distribution and Biological Functions.
Among the classic prostanoid receptors, DP1 is the least abundant in tissues and exhibits a relatively narrow distribution. Northern blot analyses detected expression only in the retina and small intestine (Boie et al., 1995). In mice, DP1 receptor expression is moderate in the ileum; weak in the stomach, lung, and uterus; and absent elsewhere (Hirata et al., 1994). Functional studies, however, have described DP1 receptors present in certain cells. In some cases, transcripts have provided supportive data. Human platelets express functional DP1 receptors, which inhibit aggregation (Whittle et al., 1983; Giles et al., 1989; Trist et al., 1989; Seiler et al., 1990). Effects of PGD2 on vascular smooth muscle vary according to species (Giles and Leff, 1988). In humans, effects were restricted to facial flushing and nasal congestion (Heavey et al., 1984), with no meaningful alteration in blood pressure. Similar effects were apparent for the DP1 agonist BW 245C (Giles and Leff, 1988). Thus, despite evidence for presynaptic DP receptors that would enhance norepinephrine release (Molderings et al., 1994), this does not seem to translate into gross cardiovascular events. In contrast, the skin flushing associated with nicotinic acid used for treating dyslipidemia is DP1 receptor-mediated (Cheng et al., 2006).
Compelling pharmacological evidence for DP1 receptors in the human myometrium has been advanced (Senior et al., 1992; Fernandes and Crankshaw, 1995). DP1 receptor stimulation would mediate tocolysis. It is noteworthy that Northern blotting did not identify a DP1 receptor transcript in the human uterus (Boie et al., 1995). There are additional examples of DP1 receptors being identified in specific cell types or localized regions in which Northern blot analyses of tissue would suggest otherwise.
The initial studies on DP1 receptor distribution showed undectable to very low levels in the brains of both mice and humans (Hirata et al., 1994; Boie et al., 1995). Because PGD2 has well documented activity in the CNS, these data were interpreted to mean that expression was limited to certain areas and/or specific cells (Narumiya et al., 1999). This has proven correct, when subject to careful examination, for not only the brain but also other tissues (Gerashchenko et al., 1998; Wright et al., 1998). A pharmacological rationale for the various central effects of PGD2 and its analogs is therefore tenable. These effects include sleep regulation (Urade and Hayaishi, 1999), neuroprotection (Liang et al., 2005b; Saleem et al., 2007b; Thura et al., 2009) allodynia (Minami et al., 1997), and hyperalgesia (Telleria-Diaz et al., 2008). DP1 receptor involvement in neurotransmission is not limited to the CNS, and a local effect on pruritus has been reported (Arai et al., 2004, 2007; Sugimoto et al., 2007). The pruritic activity associated with scratching, in an atopic dermatitis-like model in NC/Nga mice, was inhibited by PGD2 but not metabolites known to stimulate DP2 receptors (Arai et al., 2004). These effects on experimental pruritus were confirmed using a DP1 agonist, (4-((1R,2S,3R,5R)-5-chloro-2-((S)-3-cyclohexyl-3-hydroxyprop-1-ynyl)-3-hydroxycyclopentyl)butylthio) acetic acid monohydrate (TS-022), and the antagonist BW A868C to establish the pharmacological basis of the attenuated itch response (Arai et al., 2007). In addition, accelerated repair of the disrupted cutaneous barrier was reported for TS-022 (Arai et al., 2007), an effect ascribed not only to DP1 but also to EP3 and EP4 receptors in mice (Honma et al., 2005). A physiological role for PGD2 has also been contemplated, in that PGD2 released by mechanical scratching may be autoinhibitory, limiting the extent of the scratching response and preventing skin damage (Sugimoto et al., 2007; Takaoka et al., 2007). The NC/Nga model may, at least in part, be operational with respect to the antipruritic effects of DP1 agonists by virtue of a reduced level of endogenous PGD2 production (Takaoka et al., 2007). The decrease in PGD2 production in the late phase of dermatitis and scratching in NC/Nga mice, together with increased DP1 receptor expression assists in explaining the potent antipruritic activity of TS-022 (Sugimoto et al., 2007). Related to atopic dermatitis, DP1 receptor stimulation impedes TNF-α-induced migration of human Langerhans cells (LCs) and additional chemotactic events, which strongly decreases the recruitment of inflammatory cells in a model of murine atopic dermatitis (Angeli et al., 2004). The beneficial effects of TS-022 and BW 245C cannot be attributed solely to amelioration of pruritus by scratching behavior.
A study in mice also revealed that DP1 receptor stimulation inhibited airway inflammation and suppressed asthma by modulating dendritic cells and inducing regulatory T cells (Hammad et al., 2007). PGD2 also affects human dendritic cell differentiation and modulates the pattern of immunoregulatory cytokine production, favoring naive T cells toward a Th2 phenotype (Gosset et al., 2005). PGD2 may contribute to the control of allergic reactions and tumor formation (Gosset et al., 2005; Torres et al., 2008).
DP1 antagonists have also been the subject of anti-inflammatory studies. PGD2 is the major prostanoid produced by mast cells; this presents an attractive target for DP-receptor drug design. Despite the evidence for DP1 receptor-mediated pathological increases in blood flow and engorgement of blood vessels in the nasal mucosa, clinical trials on the DP1 antagonist laropiprant demonstrated no efficacy in patients with allergic rhinitis or asthma (Philip et al., 2009). For therapeutic modalities based on attenuating the activity of PGD2, consideration of DP2 (CRTh2)-mediated events is probably of greater importance. The significance of DP1 receptor activation in inflammation and immune responses is best appreciated when considered in the context of DP2 (CRTh2) receptors.
DP1 receptor expression is high in the retina (Boie et al., 1995), but it could be argued that this finding has not been followed up to its full extent. PGD2 induces heme oxygenase-1 expression in the retinal pigmented epithelium, an enzyme important for photoreceptor survival (Satarug et al., 2008). DP1 receptors exerted only a marginal influence on heme oxygenase-1 expression, the DP2 (CRTh2) receptor being important (Satarug et al., 2008). In addition to the retina, DP receptor expression occurs in the iris and ciliary body (Gerashchenko et al., 1998). Expression in the ciliary body entirely correlates with the ocular hypotensive activity of DP1 agonists and their mechanism of action (i.e., increased uveoscleral outflow) (Toris et al., 2006). DP1 receptor mRNA is also located in mucus-secreting goblet cells and the columnar epithelium of the rat gastrointestinal tract (Wright et al., 1998), but this does not transition into the eye, where goblet cell secretion is attributable to DP2 receptor pharmacology (Woodward et al., 1990, 1993b). Likewise, the DP1 receptor has been implicated in eosinophil trafficking (Schratl et al., 2007), but this was not observed in ocular studies (Woodward et al., 1990, 1993b). The most prominent effect of DP1 receptor stimulation in the eye is on intraocular pressure (IOP) (Goh et al. 1988; Nakajima et al., 1991; Woodward et al., 1993b). DP1 effects are summarized in Table 2 in the therapeutics section.
Potential therapeutic application of DP1 agonists
3. Gene Deletion Studies.
One line of DP (DP1)-deficient mice has been generated (Matsuoka et al., 2000). Using this line of mice, physiological roles of DP1 in allergy and immunity, sleep induction and other brain functions, and tumor progression and angiogenesis have been examined. Given that PGD2 is a major PG produced by mast cells and released in large amount after antigen challenge, Matsuoka et al. (2000) examined its role in allergic inflammation using ovalbumin (OVA)-induced asthma model. Sensitization and aerosol challenge of DP(−/−) mice with OVA-induced increases in the serum concentration of IgE similar to those observed in wild-type mice. However, the DP(−/−) animals developed substantially reduced asthmatic responses in this model; the concentrations of Th2 cytokines and the extents of lymphocyte accumulation and eosinophil infiltration in the lungs of the mutant animals after OVA challenge were greatly reduced compared with those apparent in the wild type. These observations indicate that PGD2 serves as a mediator of asthmatic responses. The authors found that DP1 is induced in airway epithelial cells after the challenge and suggested that PGD2 acts on epithelial cells to induce various allergy-associated genes to facilitate inflammation. On the other hand, Angeli et al. (2001), studying Schistosoma mansoni infection in the skin, found that parasite-derived PGD2 acts at DP1 receptors in LCs to induce their retention in the epidermis. They further showed that this retention can be mimicked by administration of a DP1 agonist, BW 245C, during local TNF-α treatment and that the retention of LCs in the skin by S. mansoni infection or by BW 245C was impaired in DP1-deficient mice. In DP1-deficient mice, LCs migrated to draining lymph nodes and triggered a Th2 response (Hervé et al., 2003). Such inhibitory modulation of dendritic cell (DC) function by DP stimulation is also found in DCs in the lung. Hammad et al. (2007) found that inhalation of BW 245C in mice during OVA challenge attenuated the asthmatic response and that this inhibitory effect of BW 245C was dependent on DP1 expression by lung DCs. They suggested that this DP1-dependent modulation of lung DCs operates in the actual process of allergic inflammation in the OVA-induced asthma response, because chimeric mice with DP(−/−) hematopoietic cells exhibited an enhanced airway response. Therefore, there is both enhancement and attenuation of PGD2-DP1 signaling-dependent pathways by different types of cells in allergic inflammation, and the final outcome shown in the study by Matsuoka et al. (2000) seems to represent the net effects of these pathways. Given the sleep-inducing effect of PGD2, Mizoguchi et al. (2001) examined localization of DP1 in the brain and examined whether DP1 is involved in PGD2-mediated sleep. They found that DP1 in the brain is mainly localized in arachnoid trabecular cells in the leptomeninges of the basal forebrain, and that infusion of PGD2 into the subarachnoid space of this region increased the extracellular adenosine level and induces an increase in the amount of nonrapid eye movement sleep in wild-type mice and not in DP1-deficient mice. Although this study unequivocally demonstrated the involvement of DP1 in PGD2-induced sleep, the baseline sleep-wake patterns were essentially identical between WT and DP(−/−) mice. These results suggest either that the DP1-mediated system may not be crucial for physiological sleep regulation or that some other system may effectively compensate for the loss of the DP system involved in the regulation of sleep. The same group of researchers (Qu et al., 2006) reported that administration of SeCl4, as in patients with insomnia, inhibits PGD2 production and induces insomnia in WT mice but not in DP(−/−) mice. Qu et al. (2006) further showed that administration of a specific DP1 antagonist, ONO-4127Na (pA2, 9.73 for human DP1) (Torisu et al. 2003a,b), markedly reduced the sleep period in rats (Qu et al., 2006). On the basis of these findings, they concluded that the PGD2-DP1 signaling is involved in regulation of physiological sleep. The importance of DP in the brain has also been studied from the viewpoint of neuroinflammation. Mohri et al. (2006) detected significantly higher PGD2 concentration in the brain of the genetic demyelination twitcher mouse and found that this was due to induction of hematopoietic type PGD synthase in the microglia of these mice. They further found that DP1 was induced on astroglia, and loss of DP1 impaired astrogliosis and demyelination and lessened clinical severity in this line of mice. The same group (Taniguchi et al., 2007) also examined the role of the PGD2-DP1 signaling in hypoxic ischemic encephalopathy in neonatal mice. They subjected 7-day old pups of WT mice or mice deficient in l-PGDS, HPGDS, both l-PGDS and HPGDS, DP1, or CRTh2 to left carotid artery ligation and hypoxia exposure and examined the infarct size. They found that the infarct size was significantly enhanced in mice deficient in both l-PGDS and HPGDS or mice deficient in DP1. Given the induction of DP1 in endothelial cells and more severe damage of endothelial cells in DP(−/−) mice, these authors suggested that the PGD2-DP1 signaling exerts a protective action against hypoxic ischemic injury by an action on endothelial cells. Finally, Murata et al. (2008) used DP(−/−) mice and reported that the PGD2-DP1 signaling is involved in suppression of tumor-associated angiogenesis and hyperpermeability. In this experiment, they implanted Lewis lung cancer cells onto the back of WT and DP(−/−) mice and found enhanced tumor growth in DP(−/−) mice. Furthermore, they found decreased apoptosis and increased angiogenesis and vascular leakage associated with tumors in DP1(−/−) mice. Given the expression of DP1 in endothelial cells and attenuation of angiogenesis and permeability in model systems, they suggested that PGD2-DP1 signaling exerts inhibitory actions in tumor-associated angiogenesis and plasma leakage.
4. Agonists and Antagonists.
As a general premise, position, type and configuration of oxygen substituents on the cyclopentane ring determines the receptor specificity of natural prostanoids, although no single prostanoid is truly specific. In addition, high agonist potency is generally dependent on a C1-carboxylate, trans geometry of the side-chains (8α,12β as conventionally drawn), and a 15S-15-hydroxy moiety. The recent work of Ungrin et al. (2001) on the human EP1 receptor illustrates these relationships well. There are, however, important deviations from this general picture, including the retention of high potency in C1-primary alcohol and 16-hydroxy prostanoids. In addition, quite small alterations to the ω terminus often results in large changes in potency. For example, the EP1 binding affinity of 17S,20-dimethyl-3,7-dithia PGE1 is 63-fold higher than its 17R epimer (Maruyama et al., 2002a). Modification of the ω chain has, therefore, been an attractive and successful strategy in searching for selective agonists, especially because the synthetic approach is usually ring construction → α-chain attachment → ω-chain attachment. Nonprostanoid agonists have also been described for DP2, EP2, EP3, and IP receptors.
Replacement of the ω-pentyl terminus with a cyclohexyl moiety has been a popular strategy in DP1 agonist development. For example, the hydantoin ring analog BW 245C (Town et al., 1983; Whittle et al., 1983) is commonly used as a standard agonist; it exhibits minimal DP2 agonism (Monneret et al., 2001; Yoshimura-Uchiyama et al., 2004). Restricting the conformational mobility of the α-chain in hydantoin-type analogs conserves DP1 agonist potency [e.g., BW-587C (Fig. 2); Barraclough et al., 1996], whereas the introduction of methyl, ethyl, and n-propyl groups at N10 progressively reduces efficacy (Leff and Giles, 1992); N10-benzyl substitution affords pure DP1 antagonism (BW A868C). Other potent DP1 agonists containing a 15-cyclohexyl group include RS-93520 (Fig. 2), a prostacyclin analog with completely inverted stereochemistry in the bicyclic ring (Alvarez et al., 1991; Crider et al., 1999) and SQ-27986 (Fig. 2), a PGH2 analog with correspondingly inverted stereochemistry (see Fig. 2; Seiler et al., 1990). Although the 9β-chloro-11α-hydroxy ring system in ZK-110841 (Fig. 1) (Thierauch et al., 1988; Ney and Schrör, 1991) and its 3-oxa analog ZK-118182 (Darius et al., 1994) affords high DP1 agonism, high binding affinity for EP1 receptors is also found (Wright et al., 1998; Ungrin et al., 2001; Sharif and Davis, 2002). ZK-110841, ZK-118182, and the 13,14-dihydro analog of the latter [9-chloro-15-cyclohexyl-11,15-dihydroxy-3-oxa-16,17,18,19,20-pentanor-5-prostenoic acid (AL-6556)] also seem to behave as partial agonists in human functional EP2 systems (EC50, ∼200–700 nM) (Sharif et al., 2000, 2004); compare with structure of ONO-AE1-259 in Fig. 4.
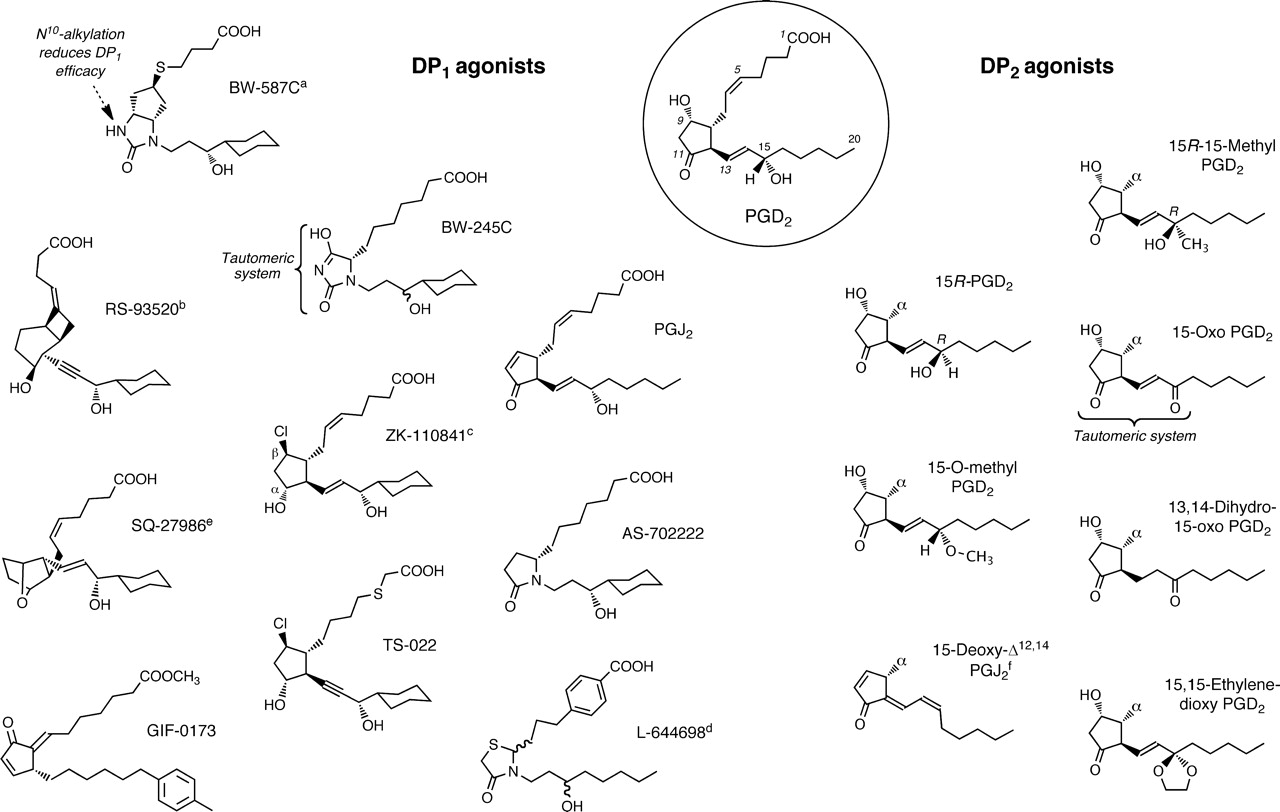
Structures of agonists for prostanoid DP1 and DP2 receptors. PGD2, the most active natural agonist, is shown in the circle. Promotion of DP2-selectivity mainly involves alterations to C15; α indicates natural 2-series side-chain. a, ring system related to 6β-PGI1. b, PGI2 analog with unnatural ring chirality. c, EP1 and EP2 agonism retained. d, some EP2 agonism remains. e, PGH2 analog with unnatural ring chirality. f, 14-cis isomer is major component.
Dehydration of the ring system of PGD2 to give PGJ2 results in retention of DP1 agonism (Fukushima et al., 1982; Mahmud et al., 1984; Wright et al., 1998) and approximately a 5-fold loss of DP2 agonism (Monneret et al., 2002). The related 4-oxo-thiazolidine L-644698 (Fig. 2) shows high DP1/DP2 selectivity but still retains some affinity for EP2 receptors (Ki, 270 nM; human recombinant) (Wright et al., 1998); indeed, complete elimination of EP2 agonism in the search for a selective DP1 agonist is not an easy task.
The first useful DP1 antagonist was the xanthone-carboxylic acid AH-6809 (Fig. 5) (Keery and Lumley, 1988). However, it has low affinity (pA2, 5.9–6.6), shows considerable EP1 antagonism and also nonspecificity in the low micromolar range. Subsequently, BW A868C (Fig. 3), which is related to the DP1 agonist BW 245C, emerged as a selective, competitive blocker of high affinity (human DP1: pA2, >9) (Giles et al., 1989; Lydford et al., 1996); it is still a first choice antagonist.

Structures of representative DP1 and DP2 (CRTH2) antagonists.
The last 10 years has seen the development of other chemical classes of DP1 antagonist (Fig. 3) (Medina and Liu, 2006), driven by renewed interest in the pathogenic role of PGD2 in allergic disorders (see Pettipher, 2008). Emerging from bicyclic prostanoid analogs showing TP antagonism (Narisada et al., 1988), a pinane ring system with a sulfonamido linkage to an aromatic group led to high affinity, as in S-5751 (Fig. 3) (Arimura et al., 2001). A quite different approach has involved the use of indole-3-acetic acid, present in nonsteroidal anti-inflammatory drugs such as indomethacin, as a pharmacophore. In ONO-AE3-237 (Fig. 3), the acetate unit is located on C4; its binding pKi for the human rc-DP1 receptor is 7.7 (Torisu et al., 2004). In the Merck series of DP1 antagonists, the positions of the carboxylic group and the substituents on the benzene ring of the indole template were optimized to yield MK-0524 (laropiprant) (Sturino et al., 2007). Laropiprant has very high affinity for the human rc-DP1 receptor (pKi, 10.5); it also has ∼300-fold lower affinity for the corresponding TP receptor, which requires consideration when using it to characterize prostanoid receptors.
5. Therapeutics.
DP1 receptor agonists are not currently used therapeutically. DP1 agonists are not used to treat glaucoma because of the initial ocular hypertensive spike and an unacceptable incidence of ocular surface hyperemia (Nakajima et al., 1991). Perhaps the most promising of the newly advanced medical hypotheses is their potential utility in treating pruritus and atopic dermatitis (Angeli et al., 2004; Arai et al., 2004, 2007; Sugimoto et al., 2007). DP1 receptor antagonists, notably BW A868C, have been available for a long time (Giles et al., 1989). The report that laropiprant has no efficacy in patients with allergic rhinitis and asthma (Philip et al., 2009) is discouraging. It is likely that combined DP1/DP2 therapies offer greater promise for treating allergic diseases (Mitsumori, 2004; Pettipher, 2008; Jones et al., 2009). One therapeutic concept that emerged with a positive clinical outcome was the utility of a DP1 antagonist to limit the cardiovascular side effects of niacin. The investigational product Cordaptive was not approved as a drug in the United States but seems to have fared better as Tredaptive in Europe (Jones et al., 2009). Thus, the potent vasodilator effects of DP1 receptor stimulation discovered so many years ago (Coleman et al., 1994b) have successfully transitioned into treatment for nicotinic acid-induced flushing in patients with dyslipidemia (Cheng et al., 2006; Paolini et al., 2009). Finally, DP1 receptors have been implicated in the development of thyroid eye disease, which affects approximately 40% of patients with Graves hyperthyroidism. DP1 receptors were found to be an important factor in promoting hyaluronan production, which would be a major contribution to exophthalmos (Guo et al., 2010). Thus, laropiprant would be of potential value in this debilitating ophthalmic condition. The potential therapeutic utilities of DP1 receptor agonists and antagonists are summarized in Tables 2 and 3, respectively.
Potential therapeutic application DP1 antagonists
B. Prostaglandin E2 Receptors
1. EP1 Receptors.
a. Second messenger signaling.
The EP1 receptor has long been linked to Ca2+ mobilization, with a negligible PI response (Funk et al., 1993; Watabe et al., 1993; Katoh et al., 1995). Ca2+ mobilization patterns appear variable according to cell type studied. It is still not entirely clear which G protein(s) may be involved. The involvement of the phospholipase C (PLC)/PKC pathway in EP1-mediated trophoblast and osteoblast stimulation implies that EP1 receptors may couple to Gq (Nicola et al., 2005; Tang et al., 2005). Compared with other prostanoid receptor studies, the signaling properties of EP1 receptors have received little attention. EP1 receptor mediated dephosphorylation of phosphatase and tensin homolog deleted on chromosome 10 and protein kinase B (Akt) (Zhou et al., 2008), and NO/cGMP pathway effects have been reported (Bachteeva et al., 2007).
The coexpression of more than one prostanoid receptor or isoform adds to the diversity of effects already inherent to prostanoid receptor stimulation. These include, but are not limited to, transactivation and cross-desensitization. EP1 receptor stimulation results in desensitization of TP receptors by PKC-mediated phosphorylation of C-terminal residues (Kelley-Hickie and Kinsella, 2004). An EP1 receptor-mediated transactivation of epidermal growth factor receptors, with resultant Akt activation, has also been reported (Han and Wu, 2005). The potential for transactivation obviates the need to distinguish such an event from signaling cascades directly emanating from activation of EP1 receptors per se.
b. Distribution and biological functions.
Contractile EP1 receptors do not have a widespread distribution in higher species and are more common in guinea pigs and murine species (Coleman et al., 1994b). In human tissues and cells, functional EP1 receptors have been demonstrated in the myometrium (Senior et al., 1991), pulmonary veins (Norel et al., 2004), mast cells (Wang and Lau, 2006) colonic longitudinal muscle (Smid and Svensson, 2009), and keratinocytes (Konger et al., 2009).
Northern blotting revealed EP1 transcription in the lung, stomach, and kidney of mice (Watabe et al., 1993). Functional studies in knockout mice and the employment of pharmacological tools have provided evidence for physiological participation for EP1 receptors in each of these organs. EP1 receptors produce airway constriction in mice, but this seems to be neuronally mediated rather than a direct smooth muscle effect (Tilley et al., 2003). EP1 receptors are also claimed to mediate PGE2-induced surfactant secretion from rat alveolar type II cells (Morsy et al., 2001). In the stomach, EP1 receptors seem to have detrimental and beneficial effects. Histamine-induced gastric injury, in the form of increased vascular permeability, is worsened by EP1 receptor activation (Hase et al., 2003). On the other hand, the EP1 receptor affords cytoprotection to the gastric mucosa against hemorrhagic lesions produced by indomethacin and HCl/C2H5OH injury (Kunikata et al., 2001; Takeuchi et al., 2001a,c). EP1 receptors are essential for HCO3− secretion in response to mucosal acidification in the stomach (Takeuchi et al., 2006). A dual role for EP1 receptors in esophagitis is clearer, whereby PGE2 has a protective effect at low doses and a deleterious effect at high doses (Yamato et al., 2005). In the kidney, PGE2 activates EP1 receptors to inhibit Na+ absorption by the renal collecting duct (Guan et al., 1998). EP1 receptors up-regulate transcription of the Na,K-ATPase β subunit in Madin-Darby canine kidney cells (Matlhagela and Taub, 2006) and attenuate up-regulation of epithelial sodium channel mRNA in inner medullary collecting duct cells by aldosterone (González et al., 2009). EP1 antagonists have been claimed as useful in the prevention of diabetic nephropathy (Makino et al., 2002) and hypertension-induced renal injury (Suganami et al., 2003). Severe renal impairment has been reported in glomerulonephritic EP1 knockout mice, underscoring an important role for EP1 in pathological renal conditions (Rahal et al., 2006). Although a few arguably beneficial effects may be ascribed to EP1 receptor activation, most effects are pathophysiological.
Prevention of EP1 receptor activation has been purported as an attractive proposition for many diseases but colon cancer, systemic hypertension and, above all, inflammation and associated pain have received the most attention. Cyclooxygenases and their products have long been considered to play a role in colon carcinogenesis. Animal models have provided a direct link between EP1 receptors and colon cancer. Aberrant crypt foci, induced by azoxymethane, were reduced by the EP1 antagonists 6-((2S,3S)-3-(4-chloro-2-methylphenysulfonylaminomethyl)-bicyclo(2.2.2)octan-2-yl)-5Z-hexenoic acid (ONO-8711) (Watanabe et al., 2000; Kawamori et al., 2001a) and ONO-8713 (Fig. 5) (Watanabe et al., 2000). A reduction in intestinal polyps was observed in the adenomatous polyposis coli gene knockout mouse model of tumorigenesis (Watanabe et al., 1999; Kitamura et al., 2003b).
A role for EP1 receptor in cardiovascular homeostasis is indicated by knockout mouse studies (Audoly et al., 1999; Stock et al., 2001). Physiological regulation by presynaptic EP1 receptors has been recently implicated in nitrergic neurovascular transmission (Jadhav et al., 2009). In models of hypertension, blockade of EP1 receptors or gene deletion seems to confer antihypertensive effects in diabetic mice (Rutkai et al., 2009) and spontaneously hypertensive rats (Guan et al., 2007). EP1 antagonist treatment also dramatically improved arteriolar lesions (Suganami et al., 2003).
The involvement of EP1 receptors in inflammation, inflammatory pain and hyperalgesia, and neuropathic pain has been a major research focus. The anti-inflammatory activity of EP1 antagonists has been reviewed extensively (Jones et al., 2009). EP1 receptors contribute to neuronal sensitization at peripheral sites (Omote et al., 2001) and at several levels in the CNS. EP1 receptors are localized in dorsal root ganglion neurons (Nakayama at el., 2004). Intrathecal PGE2 causes hyperalgesia in response to innocuous mechanical stimuli, an effect found to be EP1 receptor-mediated (Minami et al., 1994; Nakayama et al., 2004). Intrathecal administration of the EP1 antagonist ONO-8711 was shown to inhibit only the late phase of the mechanical hyperalgesic response associated with carrageenan-induced rat paw edema (Nakayama et al., 2002) and postoperative pain (Omote et al., 2002). These results correlate with a study on intra-articular Kaolin injection, in which spinal application of an EP1 agonist caused hyperexcitability 7 to 11 h after administration of the inflammatory stimulus (Bär et al., 2004). This time-dependent late phase response was not observed for EP2 and EP4 agonists (Bär et al., 2004). Set against these findings with respect to EP1 receptors in the spinal dorsal horn mediating hyperalgesia, microinjection of PGE2 into the ventromedial hypothalamus produced an EP1 receptor-mediated antinociceptive effect (Hosoi et al., 1999). Electrophysiological evidence has been provided for EP1-mediated hypoalgesia in response to noxious pinching of facial skin after lateral cerebroventricular administration of a receptor selective agonist and antagonists (Oka et al., 1997). These findings suggest that spinal processing of peripheral input may be subsequently relayed by EP1 receptors to higher centers, where the same (EP1) receptors attenuate transmission. A strong, centrally mediated override by EP1 receptors does not, however, seem to be the case because systemically administered EP1 antagonists are widely reported to be analgesic and antiallodynic (Hall et al., 2007a; Jones et al., 2009).
The EP1 receptor plays additional significant roles in the CNS. Of considerable interest is the role of EP1 receptors in controlling stress-induced impulse behavior. Thus, in mice lacking EP1 receptors, stress induces impulsive aggression, an exaggerated acoustic startle response, impaired cliff avoidance, and social dysfunction (Matsuoka et al., 2005). This behavioral phenotype was reproduced in wild-type mice by an EP1 antagonist and corrected by a dopaminergic antagonist (Matsuoka et al., 2005), establishing a link between EP1, DP1, and D2 receptor function (Kitaoka et al., 2007). EP1 receptor stimulation has also been shown to cause hyperthermia (Oka and Hori, 1994; Oka et al., 2003b).
Beyond studies on tissues and living animals, EP1 expression and functional analyses in individual cell types has produced interesting results. In human primary keratinocytes, EP1 receptors evoked intracellular Ca2+ mobilization and were shown to be expressed in the epidermis (Konger et al., 2005a). The growth of malignant keratinocytes (Thompson et al., 2001) and regulation of keratinocyte differentiation (Konger et al., 2009) seem to be EP1 receptor-dependent. EP1 receptor stimulation caused differentiation of uncommitted T cells to a Th1 phenotype, which are involved in cell-mediated immune reactions, such as dinitrofluorobenzene (DNFB) contact sensitivity (Nagamachi et al., 2007). Blockade of EP1 receptors has been shown to inhibit receptor activator of nuclear factor-κB ligand (RANKL)-induced osteoclastogenesis (Tsujisawa et al., 2005). Hypoxia induces increased EP1 receptor expression in osteoclasts (Lee et al., 2007a) by a signal transduction pathway involving HIF-1α (Genetos et al., 2009). A systematic study on EP1 receptor-mediated up-regulation of HIF-1α implicated Gi coupling with activation of a PI3K/Akt/mammalian target of rapamycin signaling pathway, and HIF-1α induction was associated with phosphorylation of the ribosomal protein 56 (Ji et al., 2010). Most noteworthy is perhaps the involvement of EP1 receptors in the proliferation of growth plate chondrocytes, the growth plate functioning to ossify long bones (Brochhausen et al., 2006).
c. Gene deletion studies.
EP1(−/−) mice appeared quite normal in all respects, including morphological characteristics (Ushikubi et al., 1998). Prostanoid EP1 receptors have long been heavily implicated in all aspects of nociception, from hyperalgesia to neuropathic pain. The advent of knockout mice has enabled follow-up of these observations, which were made with agonists and antagonists and the inherent complications of off-target pharmacology, drug-induced toxicity, and substandard experimental design. A role for EP1 receptors in pain was supported by studies on the stretching/writhing response to noxious chemical stimuli (Stock et al., 2001). Nevertheless, other EP1 gene deletion studies failed to confirm the substantive role of EP1 receptors in pain and inflammation suggested by agonist/antagonists studies in living animal models (Jones et al., 2009). According to gene deletion studies, IP receptors would be regarded as more important mediators of pain and inflammation (Murata et al., 1997; Ueno et al., 2000; Honda et al., 2006). Studies on EP1(−/−) mice actually indicated an increase in thermal nociception, EP1 receptors being suggested to exert centrally mediated control of thermal pain sensitivity (Popp et al., 2009).
Perhaps the most intriguing aspect of CNS function that emerged from gene deletion studies is the role of EP1 receptors in regulating stress responses (Furuyashiki and Narumiya, 2009). Stress, defined as a condition where body homeostasis is perturbed (Furuyashiki and Narumiya, 2009), elicits an adaptive response. This may take the form of febrile, neuroendocrine, and behavioral responses, and EP1 receptors participate in all of these. Although EP3 seems to be the dominant receptor in mediating fever (Ushikubi et al., 1998; Oka et al., 2003a; Furuyashiki and Narumiya, 2009), EP1 and EP3 receptors are equally important in PGE2-evoked adrenocorticotropic hormone and glucocorticoid release via the activation of corticotrophin-releasing hormone-containing neurons in the paraventricular nucleus of the hypothalamus (Matsuoka et al., 2003). The behavioral effects observed in EP1(−/−) mice are notable. Social withdrawal, impulse aggression, impaired cliff avoidance, and an enhanced acoustic startle response were apparent (Matsuoka et al., 2005). These behaviors were attributed to a lack of inhibition of impulsive activity, implying that EP1 receptors suppress impulsive activity under stress (Matsuoka et al., 2005). The EP1 receptor is also neurotoxic and has been postulated as the essential downstream effector of COX-2-induced neurocytotoxicity (Kawano et al., 2006). In EP1 receptor-deficient mice, it was found that COX-2-derived PGE2 does not mediate NMDA cytotoxicity (Kawano et al., 2006). EP1 receptor gene deletion also ameliorated brain injury produced by oxygen/glucose deprivation (Kawano et al., 2006) and ischemic damage produced by middle cerebral artery occlusion (Ahmad et al., 2006a; Kawano et al., 2006). In contemplating EP1 receptor participation in the middle cerebral artery occlusion model, it is important to take into consideration that cerebral blood flow is significantly increased in EP1(−/−) mice (Saleem et al., 2007a). EP1 receptors have also been implicated in innate immune responses in the CNS. PGE2, signaling via either EP1 or EP2 receptors, is essential for Toll-like 4 receptor-mediated depletion of intermediate progenitor cells from the hippocampal subgranular zone (Keene et al., 2009).
A role for EP1 receptors in carcinogenesis has been confirmed by gene deletion studies. In the azoxymethane colon cancer model, formation of aberrant crypt foci was reduced by approximately 40% in EP1(−/−) mice (Watanabe et al., 1999), and the number of tumors formed was essentially halved (Kawamori et al., 2005). In the methylcholanthrene-induced sarcoma model (MGC 101 mice), long-term growth was attenuated in EP1-deficient mice (Axelsson et al., 2005; Wang et al., 2005). The effects of cyclooxygenase inhibition on tumor progression were primarily on cell proliferation and apoptosis; angiogenesis was not an obvious primary determinant of onset and progression of tumor development (Axelsson et al., 2005).
Resting systolic blood pressure was reduced by approximately 10 mm Hg in EP1 receptor-deficient mice (Stock et al., 2001). These findings transitioned into cardiovascular hypertension, where EP1 receptor disruption spared the protein kinase N locus and reduced the acute vasopressor response and chronic hypertension produced by angiotensin II (Guan et al., 2007). The hypotension in EP1-deficient mice, notably male mice, was reported to elicit physiological compensation that manifested as increased pulse rate, increased renin mRNA levels in the kidney, and increased plasma renin activity (Stock et al., 2001). In experimental glomerulonephritis, there was reduced urine osmolality, and overall renal damage was more acute in EP1(−/−) mice (Rahal et al., 2006). PGE2, via EP1, modulates urine concentration not modulated in the renal collecting duct but within the hypothalamus to promote arginine vasopressin biosynthesis in response to water deprivation (Kennedy et al., 2007). In a model of bladder outlet obstruction, detrusor hyperactivity was negligible in EP1 receptor knockout mice (Schröder et al., 2004).
Two further findings from EP1(−/−) mice are noteworthy: 1) EP1 receptors seem to be critically involved in shifting the Th1/Th2 balance to Th1 dominance (Nagamachi et al., 2007); this was demonstrated in a therapeutic sense by reduced DNFB-induced contact sensitivity in EP1 receptor-deficient mice (Nagamachi et al., 2007). 2) Adaptive gastric cytoprotection is apparently mediated by EP1 receptors (Takeuchi et al., 2001a,c).
d. Agonists and antagonists.
17-Phenyl PGE2 (Fig. 4) has modest EP1/EP3 selectivity (Lawrence et al., 1992) and is a useful agonist in Schild antagonism protocols because of its high potency. ONO-DI-004 (Fig. 4) is a more selective EP1 agonist (Okada et al., 2000; Suzawa et al., 2000), resulting from development of 6-oxo PGE1 via its 17S,20-dimethyl (methyl ester) analog [OU-1308 (ornoprostil)] (Kobayashi et al., 1991). 6a-Carba analogs of prostacyclin, such as carbacyclin and iloprost, are unexpectedly potent EP1 agonists (Dong and Jones, 1982; Dong et al., 1986; Lawrence et al., 1992), iloprost showing partial agonism in some systems (Dong and Jones, 1982; Dong et al., 1986; Boie et al., 1997). Functional studies with rat and human recombinant EP1 receptors in either reporter gene (Durocher et al., 2000) or aequorin-based Ca2+ flux assays (Boie et al., 1997; Ungrin et al., 2001) have confirmed and expanded these structure-activity relationship data.

Structures of agonists for prostanoid EP receptor subtypes. PGE2, the most active natural agonist, is shown in the circle. 17-Phenyl PGE2 and sulprostone have modest EP1/EP3 and EP3/EP1 selectivities, respectively. CP-533536 and compound 9 are EP2 agonists with nonprostanoid structures. a, Belley et al. (2005). b, Shimazaki et al. (2000). c, Blovin et al. (2010).
The first EP1 receptor antagonist was 8-chloro-dibenzo(Z)[b,f][1,4]oxazepine-10(11H)-carboxylic acid, 2-acetylhydrazide (SC-19220), which is a dibenzoxazepine hydrazide (Sanner, 1969). It has low affinity (pA2 = 5.5), but proved useful in the early characterization of EP receptor pharmacology. Modification of SC-19220, notably removal of the acetyl group, led to the thioether SC-51222 (Fig. 5), which was much more potent than the corresponding sulfone (Hallinan et al., 1994). Within the Searle series, SC-51322 has become the agent of choice for EP1 receptor pharmacology studies (Fig. 5).

Structures of representative EP1, EP3, and EP4 receptor antagonists. Heterocycle substitution in DG-041 maintains high EP3 affinity (Hategan et al., 2009).a, Asada et al. (2010). ONO-AE3-240, T. Maruyama, personal communication.
An alternative early EP1 antagonist was AH-6809 (Fig. 5). Over a 0.1 to 10 μM concentration range, it is selective for EP1 receptors (pA2, 7.4) (Coleman et al., 1987; Eglen and Whiting, 1988; Lawrence et al., 1992) and does not block EP3 receptors (Lawrence et al., 1992; Racké et al., 1992; Qian et al., 1994). AH-6809 was later reported to antagonize the human EP2 receptor (Woodward et al., 1995b) and is now probably more useful for this purpose, given the diverse array of potent and selective EP1 antagonists currently available.
EP1 antagonists designed by Ono Pharmaceuticals show an interesting progression from the TP antagonist 11α-carba-12-(2′S-hydroxy-3′-phenylpropylamino)-9α,11α-isopropylideno-ω-octanor-prost-5Z-enoic acid (ONO-11120) (Katsura et al., 1983) to a related pinane analog (ONO-NT-012; Minami et al., 1995) showing EP1, FP, and TP antagonism (and EP3 agonism) to a bicyclo[2.2.2]octane analog (ONO-8711) showing EP1/EP3 antagonism and ultimately to the nonprostanoids ONO-8713 (Fig. 5), with high selectivity for the EP1 receptor. ONO-8711 and ONO-8713 possess Kd values for mouse recombinant EP1 receptors of 1.7 and 0.3 nM, respectively (Watanabe et al., 1999, 2000; Naganawa et al., 2006). Small modifications may dramatically increase EP3 antagonist affinity. This series has also been widely employed to elucidate the therapeutic utility of EP1 antagonists.
Pharmacophores possessing potent EP1 antagonism with good CNS penetration have emerged over the last 10 years. The series reported by Merck (Ruel et al., 1999) contains a tricyclic system similar to the Searle series. A large number of 1,2-diaryl-thiophene/-cyclopentene analogs have been prepared as highly potent EP1 antagonists, including 1-(5-{3-[2-(benzyloxy)-5-chlorophenyl]-2-thienyl}pyridin-3-yl)-2,2,2-trifluoroethane-1,1-diol (MF-266-1) (Ducharme et al., 2005; Clark et al., 2008) and GW-848687 (Giblin et al., 2007), shown in Fig. 5. Nonacidic analogs of GW-848687 (e.g., pyridylmethyl-amides) have been reported (Hall et al., 2007b,c).
e. Therapeutics.
Very few potential medical uses for EP1 agonists have been presented to date. Moreover, the prospect of major unwanted side effects would be anticipated. Probably the most promising clinical application is for EP1 agonist control of impulsive behavior in psychiatric patients (Matsuoka et al., 2005). EP1 antagonists are quite a different matter and several therapeutic applications have been put forward. Indeed, EP1 receptors have been described as the downstream effectors of COX-2-induced neurotoxicity (Kawano et al., 2006). These proposed therapeutic applications are listed in Table 4, with only a brief reference to antinociceptive and anti-inflammatory activities, because these have been extensively reviewed (Jones et al., 2009). Despite considerable effort devoted to the design, synthesis, and testing of EP1 antagonists, no convincing evidence of clinical efficacy seems to have emerged. Clinical success seems limited to acid-induced visceral pain hypersensitivity (Sarkar et al., 2003), which arguably portends little for other indications.
Potential therapeutic application of EP1 antagonists
2. EP2 Receptors.
a. Second messenger signaling.
EP2 receptors are Gs-coupled and mediate increases in cAMP (Regan et al., 1994b). A positive feedback loop whereby cAMP signaling enhances EP2 receptor expression has been suggested (Sagana et al., 2009). Changes in cAMP levels produce pleiotropic effects by activating cAMP-binding proteins. These include PKAs, Epacs, and cAMP response element-binding protein regulation of gene transcription. The nature of the cellular responses to cAMP is also dependent on compartmentalization; in fact, the EP2 receptor seems to be excluded from caveolin-rich membrane fractions (Ostrom et al., 2001). The potential repertoire of EP2-mediated responses via cAMP-Epacs-PKA-cAMP response element-binding protein has yet to be elucidated for many cells. The potential diversity may include ion channel function, [Ca2+] signaling, ion transport, exocytosis, cell adhesion, and gap junction function formation (Holz et al., 2006). In addition, cAMP can influence several transcription factors by both PKA dependent and independent mechanisms (Sands and Palmer, 2008). Beyond Gs, it is probably better to describe cell signaling on a “cell-to-cell” basis. Finally, EP2 receptors inhibit the formyl-Met-Leu-Phe–induced phospholipase D pathway activation of neutrophils (Burelout et al., 2004) and cause Th1 cell differentiation (Yao et al., 2009), which are dependent on PI3K rather than cAMP.
b. Distribution and biological functions.
EP2 receptors seem widely distributed, according to functional studies on isolated tissues, where they almost invariably produce relaxation (Coleman et al., 1994b). The earliest studies on mRNA expression suggested relatively low abundance and an uncertain distribution pattern (Regan et al., 1994b; Narumiya et al., 1999; Smock et al., 1999). In the past decade, a number of studies have indicated widespread distribution and important functions.
In the absence of potent and selective antagonists, gene deletion studies are of essential value in understanding the physiological and pathological roles of the EP2 receptor in living animals. In isolated tissues and cell culture, bioavailability is not an issue and therefore the EP2/EP1 antagonist AH-6809 (Fig. 5) may be used despite its low potency (Woodward et al., 1995b; Jones et al., 2009). In conjunction with selective agonists, the role of EP2 receptors is now better understood in many biological systems. EP2 receptors exert many inhibitory functions. In the general context of PG release and activity, the EP2 receptor could be considered to provide a major regulatory component in many instances.
PGE2 has long been known to be a bronchodilator with potential for treating asthma (Wasserman, 1981; Gardiner, 1986). The prospects improved with the discovery of selective EP2 agonists (Gardiner, 1986; Nials et al., 1993). PGE2-induced bronchodilation has been confirmed as EP2 receptor-mediated relaxation in isolated human bronchial preparations (Norel et al., 1999) and mouse airways (Sheller et al., 2000; Tilley et al., 2003; Hartney et al., 2006). EP4 receptors do not seem to mediate PGE2-induced relaxation of human bronchi (Norel et al., 1999). EP2 receptors also mediate substance P- and ATP-induced airway relaxation (Fortner et al., 2001). EP2 receptor activation on human airway smooth muscle cells may indirectly produce anti-inflammatory effects. IL-1β releases granulocyte macrophage–colony-stimulating factor from human airway smooth muscle cells, which is inhibited by EP2 receptor stimulation (Clarke et al., 2004). Thus, the survival of infiltrating leukocytes by granulocyte macrophage–colony-stimulating factor would be abrogated by the action of PGE2 at EP2 receptors.
The tocolytic effect of EP2 agonists has also received attention. Suppression of spontaneous uterine activity has been reported with selective EP2 receptor agonists: 19(R)-OH PGE2 in rabbit (Spilman et al., 1977; Woodward et al., 1993a) and butaprost in human preparations (Senior et al., 1991; Duckworth et al., 2002). 19(R)-OH PGE2 increased uterine motility in a second-trimester pregnant monkey (Spilman et al., 1977), but this effect did not transition into the human pregnant myometrium, where EP2 receptor stimulation produced tocolysis (Duckworth et al., 2002). Temporal and regional changes in EP2 receptor expression have also been implicated in pregnancy maintenance and labor-associated events. EP2 receptors decline toward term gestation (Brodt-Eppley and Myatt, 1999; Leonhardt et al., 2003), although they remain unaltered (Brodt-Eppley and Myatt, 1999; Astle et al., 2005: Sooranna et al., 2005) or increase during parturition (Grigsby et al., 2006). EP2 mRNA and nuclear EP receptors were most abundant in lower compared with upper segment tissues (Astle et al., 2005: Grigsby et al., 2006). This corresponds with a caudal decline in contractile responsiveness to PGE2, suggesting that EP2 receptors (Wikland et al., 1984) facilitate uterine distension for delivery of the fetus during labor. Rabbit oviductal motility was also suppressed by 19(R)-OH PGE2 (Spilman et al., 1977). Should this effect occur in humans, with a resultant slowdown in Fallopian tube movement, it could disrupt the timing for implantation. It could even result in an ectopic pregnancy, an unwanted event made more likely by EP2 receptor signaling as a contributory factor for fertilization and implantation (Lim and Dey, 1997; Hizaki et al., 1999; Tamba et al., 2008).
EP2 receptors have long been known to relax vascular smooth muscle and produce a vasodepressor response (Armstrong et al., 1976; Audoly et al., 1999). The renin-angiotensin system also regulates systemic blood pressure, and EP2 receptors are involved. In addition to controlling electrolyte and water homeostasis, the kidney also regulates blood pressure, and EP2 and EP4 receptors participate in PGE2-induced renin release (Schweda et al., 2004). At the juxtaglomerular cell level, EP2 and EP4 receptors were found to produce renin exocytosis (Friis et al., 2005). By measuring afferent arteriolar diameter, it was concluded that EP2 receptors partially mediate PGE2-induced vasodilatation (Imig et al., 2002). It has been claimed that EP2 receptors do not regulate overall renal hemodynamics, according to studies involving direct injection into mouse renal arteries and ultrasonic flowmetry to measure blood flow (Audoly et al., 2001). Intramedullary PGE2 infusion resulted in EP2-mediated renal Na+ excretion (Chen et al., 2008). In mice lacking EP2 receptors, salt-sensitive hypertension occurs (Kennedy et al., 1999). These results suggest that EP2 receptors produce natriuresis and promote normotension in a high-salt dietary regimen. Protection of renal cystic epithelial cells from apoptosis has implicated the EP2 receptor in polycystic kidney disease (Elberg et al., 2007).
PGE2 has been proposed to influence a number of CNS functions by activating EP2 receptors. These range from activity-dependent synaptic plasticity against oxidative stress and acute excitotoxicity to a role in the development of COX-2-induced neurotoxicity. Evidence for EP2 and EP3 receptor involvement in long-term potentiation and long-term depression has been advanced; α-amino-3 hydroxy-5-methyl-4-isoxazole-propionic acid receptor trafficking in the postsynaptic membrane was implicated as an underlying mechanism (Andreasson, 2010). The behavioral phenotype in EP2 receptor knockout mice was associated with a deficit in hippocampal long-term depression (Savonenko et al., 2009). EP2 receptors have also been implicated in PGE2 pain transmission (Ahmadi et al., 2002; Harvey et al., 2004; Reinold et al., 2005), the mechanism involving blockade of inhibitory glycine receptors. Calcitonin-gene related peptide (CGRP) release from trigeminal neurons may be evoked by EP2, DP1, and IP receptors (Jenkins et al., 2001).
Because both EP2 and EP4 receptors are Gs-coupled G-protein-coupled receptors, it is not surprising that they share certain neurological functions. These include modification of tetrodotoxin-resistant Na+ currents in neonatal rat nodose ganglia (Matsumoto et al., 2005) and protection against oxidative stress and amyloid β-peptide neurotoxicity (Echeverria et al., 2005). Nevertheless, there are several instances in which EP2 receptors are the singular cognate receptor mediating PGE2-mediated events.
EP2 receptor activation protects neurons against NMDA-receptor-mediated cytotoxicity, according to in vitro (Akaike et al., 1994; McCullough et al., 2004; Liu et al., 2005; Ahmad et al., 2006b) and living animal studies (Ahmad et al., 2006). EP2 receptors have actually been claimed to exacerbate NMDA-mediated cytotoxicity in the very same cells, cultured rat cortical neurons (Takadera and Ohyashiki, 2006), in which EP2-mediated neuroprotection was originally described (Akaike et al., 1994). In hippocampal neurons, caspase-dependent apoptosis seems to be produced by PGE2 acting through EP2 receptors (Takadera et al., 2004). Nevertheless, the majority of studies on neuronal cytotoxicity suggest that EP2 receptors are neuroprotective. In neuronal cells, EP2 signaling affords significant neuroprotection by a cAMP-PKA mechanism, whereas microglial EP2 receptor activation may lead to secondary neurotoxicity by up-regulating proinflammatory genes (Andreasson, 2010). Likewise, EP2 receptors may contribute to either neuroprotection or neurotoxicity by inducing brain-derived neurotrophic factor release from microglial cells and astrocytes (Hutchinson et al., 2009). In astrocytes, it was reported that EP2 receptor stimulation elicits Ca2+ release from intracellular stores (Di Cesare et al., 2006).
A proinflammatory neurotoxic function for EP2 receptors has emerged during the past few years, notably with respect to innate immunity. LPS has been used to model innate immunity, because binding to CD14 and toll-like receptor 4 up-regulates pro-inflammatory genes, including COX-2 (Andreasson, 2010). EP2 receptors are highly inducible in the cerebral cortex and hippocampus (Zhang and Rivest, 1999). In hippocampal slice preparations, EP2 receptors exacerbate LPS-induced neurotoxicity (Wu et al., 2007). EP2, and EP1, receptors are very important in Toll-like receptor 4-induced depletion of intermediate progenitor cells destined for maturation in the hippocampal subgranular zone (Keene et al., 2009). In terms of hippocampal neurotransmission, postsynaptically synthesized PGE2 modulates transmission via presynaptic EP2 receptors (Sang et al., 2005). Activated microglial EP2 receptors seem to play a central role in the generation of reactive oxygen species and secondary neurotoxicity (Andreasson, 2010). LPS-induced cerebral oxidative damage was selectively abolished in EP2 gene deletion studies (Montine et al., 2002). Additional, mostly deleterious, EP2 effects on microglia include α-synuclein neurotoxicity (Jin et al., 2007), regulation of amyloid precursor protein (Pooler et al., 2004; Liang et al., 2005a), and inhibition of ATP-induced microglia migration (Nagano et al., 2008).
Neuroprotection has also attracted attention in vision research because sight is entirely dependent on optimal ocular and central neurotransmission. Experiments have been largely designed to assess vision-sparing potential in glaucoma (Woodward, 2000) and retinal disease (Andrade da Costa et al., 2009; Mori et al., 2009; Osborne et al., 2009). The underlying EP2-regulated signaling mechanisms involved in ocular neuroprotection have received little attention but are probably likely similar to those reported for CNS neurons. The link between sight-threatening ocular diseases and immune regulation is tenuous at best, and no attention has been paid to innate immunity.
Generally EP2 receptors tend to play anti-inflammatory roles, which is in contrast to their participations in innate immunity in the CNS. EP2 receptors inhibit T-cell proliferation according to MLR studies (Nataraj et al., 2001), regulate antigen-presenting cell function (Nataraj et al., 2001), inhibit TNFα release from bone marrow-derived dendritic cells (Vassiliou et al., 2003), inhibit major histocompatibility complex class II expression in dendritic cells (Harizi et al., 2003), suppress IFNα release by natural killer cells (Walker and Rotondo, 2004), are particularly effective in inhibiting Th1 and Th2 polarized antigen-specific T-cell responses (Okano et al., 2006), and augment the signaling and function associated with the anti-inflammatory cytokine IL-10 (Cheon et al., 2006). Several immune regulatory functions assigned to EP2 receptors are shared with EP4. These include Th1 differentiation (Yao et al., 2009), Th2 polarization (Kubo et al., 2004), and Th17 differentiation (Boniface et al., 2009; Napolitani et al., 2009; Narumiya, 2009; Yao et al., 2009). It should also be noted that numerous immunomodulatory effects of PGE2 are reported but not pharmacologically defined. Studies in living animals seem somewhat consistent with findings on immune competent cells: EP2(−/−)mice treated with an EP4 antagonist exhibited a therapeutic effect in the collagen-induced arthritis model (Narumiya, 2009), which is consistent with in vitro T-cell studies.
In polymorphonuclear leukocytes and monocytes/macrophages, EP2 receptors largely exert down-regulatory functions. EP2 receptors inhibit neutrophil chemotaxis, superoxide generation, LTB4 release, and aggregation (Nials et al., 1993; Wheeldon and Vardey, 1993; Talpain et al., 1995; Yamane et al., 2000). LPS-stimulated murine peritoneal neutrophils also secrete cytokines, and EP2 receptor activation augments IL-6 and granulocyte cell-stimulating factor release (Yamane et al., 2000; Sugimoto et al., 2005) but suppresses TNFα production to some extent (Yamane et al., 2000). Efferocytosis, ingestion of apoptotic cells by phagocytes, triggers the release of cytokines, NO, and PGE2. Although this process may recover tissue homeostasis after injurious stimuli, it renders the lung, in particular, susceptible to secondary infection, because alveolar macrophages are impaired by apoptotic cells. PGE2, acting via EP2 receptors, mediates efferocytosis-induced inhibition of pulmonary macrophage antibacterial function (Medeiros et al., 2009). Such an effect may be mitigated by EP2-mediated monocyte/macrophage survival against free radical damage by peroxynitrite, a superoxide-nitric oxide coupling product that is an extremely reactive free radical (Tommasini et al., 2008). More pertinent to asthma and other type 1 allergies, EP2 receptors inhibit lung mast cell degranulation (Kay et al., 2006) and eosinophil trafficking (Sturm et al., 2008).
PGE2 has been implicated in osteoclast, chondrocyte, and synoviocyte function. EP2 and EP4 receptors often mediate similar effects, but responses to EP4 stimulation are typically more prominent. Small animal cell lines are used extensively, but results are contradictory (Graham et al., 2009). EP2 effects include osteoblast differentiation, osteoclast-induced bone resorption, and bone anabolic activity in living animal studies (Graham et al., 2009). It is also pertinent to note that PGE2 strongly inhibits human osteoclast formation (Take et al., 2005). In human and rat chondrocytes, EP2 receptors have quite opposite effects on proteoglycan accumulation: suppression (Li et al., 2009b) and enhancement (Miyamoto et al., 2003), respectively. EP2 receptors have been identified in synovial fibroblasts obtained from rheumatoid arthritis tissue specimens (Kojima et al., 2009).
Studies on EP2 receptor function in fibroblasts usually involves fetal or patient-derived lung fibroblasts. EP2 receptors have been shown to produce diverse effects that may limit fibrosis and scar formation. It is noteworthy that EP2 receptors inhibit the transition of human lung fibroblasts to myofibroblasts, which are the hallmark of pulmonary fibrotic disease (Kolodsick et al., 2003). Furthermore, PGE2 inhibits fibroblast proliferation and collagen expression in patient-derived lung fibroblasts (Huang et al., 2007b) and migration (White et al., 2005) via EP2 receptor activation. It is pertinent that bleomycin, which produces fibrogenesis, produces a loss of EP2 expression in pulmonary fibroblasts with a resultant loss of the inhibitory effect of PGE2 on collagen biosynthesis and proliferation (Moore et al., 2005). Finally, it is of interest that EP2 receptors protect human lung fibroblasts from cigarette smoke-induced apoptosis (Sugiura et al., 2007). Tissue destruction associated with periodontitis and healing may also involve EP2 receptors expressed by fibroblasts (Noguchi et al., 2002; Weinberg et al., 2009). In view of the importance of fibroblasts in cutaneous wound healing and scar formation, dermal fibroblasts have also received attention. Maintenance of the migratory phenotype may be important for remodeling and regenerative repair, an effect achievable by EP2 receptor activation (Parekh et al., 2007). Reduced EP2 expression causes increased collagen synthesis in keloid fibroblasts (Hayashi et al., 2006).
The role of EP2 receptors in skin tumor development has been the subject of several investigations. EP2 receptors are claimed to induce COX-2 (Ansari et al., 2007) and to participate in tumor formation (Sung et al., 2006; Chun et al., 2009). Loss of EP2 receptors from keratinocytes has been suggested as a mechanism for neoplastic progression resulting from a more invasive phenotype, although EP2 receptors protect against UV-induced carcinogenesis (Konger et al., 2002; Brouxhon et al., 2007). The immune regulatory effects associated with EP2 receptors, which may be beneficial in relieving cutaneous hypersensitivity, can result in diminished antitumor immune responses by virtue of reduced cytotoxic T-cell responses and inhibition of dendritic cell differentiation (Yang et al., 2003).
It seems to be generally accepted that COX inhibitors are of value in treating colorectal cancer (Clevers, 2006). A significant role for PGE2 in producing a signaling cascade involving phosphorylation of glycogen synthase kinase 3, β-catenin translocation to the nucleus, and resultant Tcf/Lef transcription and COX-2 up-regulation in the development of chronic inflammation and colon cancer has been elucidated (Fujino et al., 2002; Regan, 2003) and subsequently confirmed (Castellone et al., 2005). Because EP2 receptors are claimed to have a central role in colon cancer cells (Sonoshita et al., 2001; Seno et al., 2002), the role of EP2 receptors in other carcinomas has been the subject of intense activity.
The EP2-dependent angiogenesis associated with mouse intestinal tumors (Seno et al., 2002) has been further studied in pulmonary endothelial cells, aortic rings, and the cornea (Kamiyama et al., 2006). It was concluded that EP2 receptors are a major factor in endothelial cell motility (Kamiyama et al., 2006). EP2 receptors, in addition to EP4 receptors, produce angiogenesis in prostate cancer (Jain et al., 2008). EP2 and EP4 have also been implicated in VEGF expression and increased invasiveness of ovarian carcinoma cells by stimulating tumor-associated matrix metalloproteases (Spinella et al., 2004). EP2 receptors may play a role in breast cancer by inducing aromatase (Brueggemeier et al., 2001; Subbaramaiah et al., 2008) and VEGF induction and hyperplasia in mammary tumor cells (Chang et al., 2005a,b). EP2 receptor deficiency decreased the growth, angiogenesis, and metastasis of mammary tumors in mice; increased EP2 receptor expression by TGFβ increased mammary epithelial cell invasion, growth, and resistance to TGFβ-mediated cytostasis (Tian and Schiemann, 2010). Proliferation of squamous cell carcinoma has been ascribed to EP2 receptors (Donnini et al., 2007; Yu et al., 2008a, 2009). PPARγ ligands were shown to inhibit lung carcinoma cell proliferation by suppressing EP2 receptor expression (Han and Roman, 2004). In complete contrast, EP2 and EP4 receptors inhibit human gastric carcinoma cell lines (Okuyama et al., 2002).
c. Gene deletion studies.
No less than three different lines of EP2(−/−) mice have been generated (Hizaki et al., 1999; Kennedy et al., 1999; Tilley et al., 1999). EP2 receptor-deficient mice exhibited reduced fertility; they became pregnant and delivered at term, but the litter sizes were reduced (Hizaki et al., 1999; Kennedy et al., 1999; Tilley et al., 1999). This seems to be due to failure of the released ovum to be fertilized by virtue of incomplete expansion of the cumulus (Hizaki et al., 1999; Tilley et al., 1999); cumulus cells are needed for oocyte maturation-associated reduction in TNFα-stimulated gene 6 (TSG-6) expression, which seems to be the EP2 receptor-mediated effect (Ochsner et al., 2003).
EP2 receptor deletion also affects blood pressure. Resting blood pressure for mice on a normal diet is reduced but may be restored by providing excess salt in the diet (Tilley et al., 1999). Neither plasma renin activity nor renin mRNA were elevated in EP2(−/−) animals (Tilley et al., 1999). The vasodilator responses to PGE2, per se, seems to be EP2 receptor-mediated, and PGE2 produces considerable hypertension in EP2(−/−) mice (Kennedy et al., 1999). In one study, a high-salt diet resulted in a rapid and sustained increase in blood pressure (Kennedy et al., 1999). The degree of salt-sensitive hypertension recorded in the two studies was different and, in contrast to Tilley et al. (1999), Kennedy et al. (1999) reported an elevation in resting blood pressure. These findings are not readily explained; they may reflect different genetic backgrounds of the mice used in each study. There is also incongruence with respect to renal hemodynamics. Deletion of EP2 receptors had little effect on PGE2-induced renal vasodilation and did not alter resting renal blood flow (Audoly et al., 2001). Quite different and unexpected results were obtained by monitoring afferent renal arteriolar caliber. The vasodilator response to PGE2, as measured by arteriolar diameter, in wild-type mice was not merely abolished by EP2 receptor deletion but was converted to a vasoconstrictor response (Imig et al., 2002). The precise reason for this apparent discrepancy is not clear. It may reflect different technologies: ultrasonic flowmeter (Audoly et al., 2001) versus measurement of arteriolar diameter by transillumination videomicroscopy (Imig et al., 2002). EP2 receptors have been shown also to be involved in natriuresis, according to gene deletion studies. Thus, a high-salt diet increases PGE2 biosynthesis in the renal medulla, which promotes EP2 receptor-mediated sodium excretion (Chen et al., 2008).
The CNS has been a major focus of EP2 receptor deficiency studies, where the neuroprotective properties of EP2 receptors have been confirmed and extended. Thus, gene deletion resulted in a greater infarct volume in the cerebral artery occlusion-reperfusion model of transient forebrain ischemia (McCullough et al., 2004). EP2 receptor deletion reduced oxidative damage and amyloid burden in a model of Alzheimer's disease, however (Liang et al., 2005a). In EP2-deficient mice, there is altered long-term synaptic plasticity in the hippocampus and impaired spatial learning (Yang et al., 2009). In a separate study using the same test (i.e., the Morris water maze to test hippocampal dependent spatial memory), EP2(−/−) mice demonstrated no deficits in spatial memory (Savonenko et al., 2009). In studies of EP2 receptor involvement behavior and cognition, there were cognitive deficits in tests for fear and social memory (Savonenko et al., 2009). EP2(−/−) mice also exhibited impaired prepulse inhibition and heightened anxiety. The complex behavioral phenotype observed was attributed to a long-term depression deficit in the hippocampus (Savonenko et al., 2009). It is noteworthy that mice lacking EP2 receptors completely lacked spinal hyperalgesia in response to PGE2 (Reinold et al., 2005). This was identified by electrophysiology as diminished synaptic inhibition of excitatory dorsal horn neurons.
Neuronal cytotoxicity is intimately associated with innate immunity. LPS, which stimulates innate immunity by binding to CD14 and activating toll-like receptor 4, produces cerebral oxidative damage that was almost abolished in EP2 receptor-deficient mice (Montine et al., 2002). Innate immunity-mediated neurodegeneration produced by LPS was subsequently determined to be critical to EP2 receptors expressed by microglia (Shie et al., 2005). COX-2 and inducible nitric-oxide synthase are also implicated in LPS-induced neurodegeneration. The roles of EP2 receptors in acquired immunity are arguably worthy of further study, notably because excessive concentrations of PGE2 have been used in some key human cell studies. An important role for the EP2 receptor in inhibition of dendritic cell differentiation and function has been claimed (Yang et al., 2003). In the MC26 tumor model, there was improved dendritic cell function and number in tumor-bearing EP2(−/−) mice (Yang et al., 2003). This reflects EP2 receptor-mediated tumor immunosuppression and inhibition of the host reaction to tumor progression. In a different setting, 7,12-dimethylbenz(a)anthracene/12-O-tetradecanoylphorbol-13-acetate–induced skin tumors, EP2(−/−) mice showed suppressed skin tumor development associated with decreased proliferation, angiogenesis, inflammation, and cell survival (Sung et al., 2005). The link between COX-2 and EP2 receptors is also manifest in a study on mammary hyperplasia, which was reduced in EP2(−/−) mice (Chang et al., 2005a).
Bone deposition and resorption has been a popular avenue of study using EP2-deficient mice. In cocultures of spleen and calvarial osteoblasts, the response to PGE2, or even PTH, was greatly reduced when both of these cell types were obtained from EP2(−/−) mice; the data in total suggest that EP2 receptors activate osteoblastic cells to stimulate osteoclast formation (Li et al., 2000). Osteoclast formation from cells of an osteoblastic lineage requires up-regulation of RANKL and macrophage colony-stimulating factor. PGE2-induced increases in RANKL expression were reduced in cells derived from both EP2(−/−) and EP4(−/−) mice (Li et al., 2002); this was the major effect observed. A biphasic effect of PGE2 on osteoclast formation was apparent, the secondary stimulatory effect being EP2 receptor-mediated (Ono et al., 2005). Finally, in EP2(−/−) mice, bone exhibited weak biomechanical properties compared with that obtained from wild-type control mice (Akhter et al., 2001).
d. Agonists and antagonists.
Misoprostol (Collins et al., 1985), in which the 15-hydroxyl group of PGE1 is displaced to C16, shows modest selectivity for EP2 and EP3 receptors, whereas the related butaprost (TR4979) is EP2-selective (Gardiner, 1986). However, tissue-dependent hydrolysis of its C1 ester is required to obtain full bioactivity (Ki: butaprost, 3500 nM; butaprost-free acid, 91 nM for human rc-EP2 receptor; Abramovitz et al., 2000). There has been some confusion over the configuration at C16 of butaprost used in the original reports (see http://www.caymanchem.com/app/template/Product.vm/catalog/10006045 for resolution of this issue). Commercially available butaprost-FA (Fig. 4) is the more active (racemic) 16S epimer. The “2-series” analog of butaprost-FA (CAY-10399) also shows high EP2/EP4 selectivity and is devoid of the IP agonism present in butaprost-FA (Tani et al., 2002a,b). Retaining the 16S configuration and replacing the 9-ketone with a β-chloro group generated highly potent and selective EP2 agonists (Tani et al., 2002a,b). One of these, ONO-AE1-259 (Fig. 4), has proved valuable in characterizing inhibitory EP2 systems (Cao et al., 2002; Clarke et al., 2004) and has proved useful when both EP4 and IP receptor systems are also present (Jones and Chan, 2005).
Acyclic derivatives of PGE1 with a N8-methylsulfone structure exhibited weak, but selective, EP2 agonism (Jones et al., 1977). CP-533536 (Fig. 4) is a related nonprostanoid with subnanomolar EC50 for the rat rc-EP2 receptor (Li et al., 2003; Cameron et al., 2009). EP2 full agonism was also found for another nonprostanoid (Fig. 4, compound 9; from Belley et al., 2005) during screening for EP3 antagonism.
There are no potent and selective EP2 receptor antagonists available. AH-6809 (Fig. 5) remains the most useful compound in this class (Woodward et al., 1995b), despite significant activity at EP1 receptors (Coleman et al., 1994b). Presumably, there has been inadequate rationale and commercial incentive to design potent and selective EP2 antagonists. Potential uses, such as for innate immunity and secondary pulmonary infection, would likely be effectively treated by COX-2 inhibitors.
e. Therapeutics.
The therapeutic potential of EP2 agonists has been under consideration for more than 2 decades; so far, however, no clinically useful drugs have emerged. Indications for EP2 agonist therapy involving relief of smooth muscle spasm have been known for at least 2 decades. These include tocolysis, for the prevention of preterm labor and dysmenorrhoea (Senior et al., 1993; Duckworth et al., 2002) and bronchodilatation for asthma treatment (Gardiner, 1986; Nials et al., 1993), which would be analogous to β2-adrenoceptor therapy for relieving bronchospasm. An additional dimension to EP2 agonist therapy is the inhibitory effect on eosinophil infiltration (Sturm et al., 2008) and lung mast cell degranulation (Kay et al., 2006). AH 13205 was not a clinical success and caused airway irritation in human volunteers (Nials et al., 1993). Although highly selective, AH 13205 is not a potent EP2 agonist (Regan et al., 1994b). Perhaps asthma therapy should be revisited using the more potent compounds that have been discovered during the past few years.
Bone anabolic therapy with both EP2 and other prostanoid agonists has been studied for several years (Hartke and Lundy, 2001; Graham et al., 2009). EP2 agonists, even within the prostanoid class of drugs, are not the only options for treating osteoporosis and enhancement of fracture healing. Likewise, several prostanoid receptors mediate ocular hypotension. EP2 agonists are, however, particularly efficacious at lowering intraocular pressure, and even butaprost can restore laser-induced ocular hypertension to an ocular normotensive state (Nilsson et al., 2006). A novel series of EP2 agonists of a nonprostanoid structure have been reported to be extraordinarily potent, efficacious, and long-acting (Coleman and Middlemiss, 2009). The potential therapeutic utility of EP2 agonists is summarized in Table 5.
Potential therapeutic application of EP2 agonists
3. EP3 Receptors.
a. Second messenger signaling.
Pharmacological characterization of EP3 receptors revealed smooth muscle contractibility (Coleman et al., 1994b) and pertussis toxin sensitivity (Sonnenburg et al., 1990), which predicted a more promiscuous G protein-coupling repertoire. This has been proven to be the case to some extent. The major signaling pathway for EP3 receptors is Gi-induced adenylate cyclase inhibition (Narumiya et al., 1999). Numerous alternatively spliced EP3 variants have been identified (Sugimoto et al., 1993; Breyer et al., 1994; Regan et al., 1994a; Takeuchi et al., 1994; Kotani et al., 1995; Schmid et al., 1995; Kotelevets et al., 2007). EP3 mRNA splicing variants are reported to subserve diverse receptor functions. The EP3 receptor carboxyl terminus is essential for G protein activation (Irie et al., 1994) and alternative splicing variants thereof determine agonist and constitutive Gi activity (Hasegawa et al., 1996; Negishi et al., 1996; Hizaki et al., 1997; Jin et al., 1997). Moreover, these alternative splicing variants confer a wide repertoire of signaling pathways. In addition to decreasing cAMP levels, PI turnover, and increased intracellular Ca2+ have been reported for EP3 isoforms (Namba et al., 1993; An et al., 1994; Takeuchi et al., 1994; Schmid et al., 1995; Yamaoka et al., 2009). EP3 isoforms also activate Rho via pertussis toxin-sensitive G protein(s) (Katoh et al., 1996; Hasegawa et al., 1997), via G12 and possibly G13 (Hasegawa et al., 1997; Macias-Perez et al., 2008). EP3 receptor stimulation produces neurite retraction via small GTPase Rho (Katoh et al., 1996). A role for Rho kinase in EP3 receptor-induced smooth muscle contraction was indicated by agreement between the potencies of Rho kinase inhibitors for suppression of sulprostone-induced contraction in guinea pig aorta and their reported potencies on the isolated enzyme system (Shum et al., 2003). However, in the case of “silent” EP3 contraction/synergism in the rat femoral artery preparation, Rho kinase inhibition suppressed both the priming agent response and the enhanced response to sulprostone, implicating Rho kinase as a common late-stage transduction process (Hung et al., 2006). Synergism between agonists on smooth-muscle EP3 agonist-dependent Gs activity also occurs (Negishi et al., 1996), which seems to depend on interaction between the arginine residue in the seventh transmembrane-spanning segment and the carboxylate anion of the ligand (Negishi et al., 1995), as reported for the EP3D isoform. The differential function of EP3 mRNA splicing variants even extends to receptor internalization (Bilson et al., 2004) and membrane targeting (Hasegawa et al., 2000). No consistent nomenclature for alternative splicing variants of EP3 or other prostanoid receptors has been agreed upon.
b. Distribution and biological functions.
In situ hybridization, Northern blotting, and functional studies have revealed a wide distribution for EP3 receptors. They seem to play important roles in the CNS, cardiovascular system, reproductive system, kidney, and urinary bladder. In common with most other prostanoid receptors, EP3 receptors have been implicated in cancer and inflammation/immune regulation.
EP3 receptors are widely expressed in the CNS and specifically localized to neurons (Sugimoto et al., 1994b). EP3 mRNA was most abundant in neurons in the sensory ganglia (Sugimoto et al., 1994b). This would be consistent with studies on acute herpetic pain, where PGE2 content is increased in the dorsal root ganglia and analgesia was produced by an EP3 antagonist and in EP3(−/−) mice (Takasaki et al., 2005). Gene deletion studies have also revealed a role for EP3 receptors in PGE2-induced hyperalgesia (Minami et al., 2001), LPS enhanced acetic acid-induced writhing (Ueno et al., 2001), and allodynia produced by intrathecal administration of HIV-1 glycoprotein gp120 (Minami et al., 2003). A mechanistic study on sensory ganglia demonstrated that EP3 receptors attenuate desensitization of B2 receptors, thereby restoring the response to bradykinin (Kozaki et al., 2007). EP3 receptors located in higher centers have been implicated in mediating hyperalgesia (Oka et al., 1994; Hosoi et al., 1997; Oliva et al., 2006).
Aspirin has been used to reduce fever for over a century, now a detailed insight into the pharmacology is available. EP3 receptors play a major role in hyperpyrexia. An impaired febrile response in EP3(−/−) mice was reported (Ushikubi et al., 1998). EP3 receptors in the median preoptic nucleus have been demonstrated as critical for sickness-induced fever (Lazarus et al., 2007; Furuyashiki and Narumiya, 2009). Studies on EP receptor-specific ligands also implicate EP3 receptors in producing fever but, somewhat surprisingly, also implicate EP1 receptors (Oka et al., 2003b). Subsequent studies have focused on EP3 receptors. EP3 receptors have been proposed to mediate brown adipose tissue thermogenesis, although supportive pharmacological evidence is lacking (Yoshida et al., 2003). The preoptic area expresses EP3 receptors that provide direct pyrogenic input to two hyperpyrexia generating sympathoexcitatory brain regions, the dorsomedial hypothalamus (Nakamura et al., 2005), and the rostral raphe pallidus nucleus (Nakamura et al., 2009). It has been suggested that EP3 receptors cause a decrease in preoptic GABAA expression as a mechanism for PGE2-induced fever (Tsuchiya et al., 2008).
The abundance (Sugimoto et al., 1992) and widespread distribution of EP3 receptors in the brain (Sugimoto et al., 1994b) point to multiple CNS functions and even opportunities for novel therapies. This opportunity does not seem to have been pursued to its fullest extent. Nevertheless, some progress has been made. EP3 receptor mRNA was found to be associated with monoaminergic neurons in the brainstem, and EP3 receptors were postulated as performing a modulatory function (Narumiya et al., 1999). In mouse cerebral cortex slices, EP3 and histamine H3 receptors located presynaptically inhibited norepinephrine in a partially exclusive manner (Schlicker and Marr, 1997), but beyond this EP3, modulation of monoamine neurotransmitter release does not seem to have been diligently pursued. One study points to PGE2-activated sympathetic nerve activity in the brain stem, with resultant tachycardia and hypertension (Ariumi et al., 2002). Behavioral suppression produced by IL-1β on naloxone-induced withdrawal jumping in morphine-dependent mice (Nakagawa et al., 1995) and Δ8-tetrahydrocannabinol on lever-pressing behavior (Yamaguchi et al., 2004) both seem to involve PGE2 acting through EP3 receptors. Finally, reduced brain injury has been reported after cerebral ischemia in mice lacking EP3 receptors (Saleem et al., 2009b) and ischemic excitotoxicity was reduced by the EP3 antagonist ONO-AE3-240 (Fig. 5) (Ikeda-Matsuo et al., 2010).
The major foci of cardiovascular EP3 receptor research have been myocardial injury and platelets. The cardiovascular system, however, may provide a large-scale operational model of EP3 receptor influence on monoaminergic neuronal function in the CNS. This would be provided by the pithed rat model, where presynaptic EP3 receptors inhibit the release of catecholamines and the resultant vasopressor response (Malinowska et al., 1994). Platelet aggregation studies have largely concentrated on TP and IP receptors, but EP3 receptors seem to have a more subtle role in platelet aggregation. PGE2 exerts a dual action on platelets, inhibition at high doses, and potentiation of the effect of proaggregatory agents (Armstrong, 1996). This potentiating effect was ascribed to EP3 receptors (Matthews and Jones, 1993), which has been amply confirmed (Fabre et al., 2001; Ma et al., 2001; Gross et al., 2007; Singh et al., 2009). EP3 agonists have been shown to reduce infarct size and reduce myocardial injury (Zacharowski et al., 1999; Hohlfeld et al., 2000), with supportive evidence from cardiospecific EP3 receptor overexpression (Martin et al., 2005). In the kidney, an EP3 vasoconstrictor effect has been observed for the intralobular arteries (van Rodijnen et al., 2007).
In 2008, a series of articles implicated EP3 receptors in bladder micturition. First, a report on increased bladder capacity in EP3(−/−) mice was reported (McCafferty et al., 2008). Infusion of the EP3 agonist GR 63799X into the bladder of wild-type mice reduced bladder capacity, implicating EP3 receptors as a contributing factor to overactive bladder (McCafferty et al., 2008). At the same time, two studies on peripheral and central neuronal control of bladder function were published. The EP3 antagonist DG-041 (Fig. 5) selectively inhibited responses of mechanosensitive afferent nerves to urinary bladder distension, inhibited the visceromotor response to bladder distension, and reduced the frequency of rhythmic bladder motility (Su et al., 2008a). A second study demonstrated that intrathecal or intracerebroventricular administration of potent EP3 antagonists reduced the frequency of bladder contractions, albeit not their amplitude (Su et al., 2008b). The visceromotor reflex response was more effectively inhibited by intrathecal dosing, suggesting that bladder nociception primarily involves spinal EP3 receptors (Su et al., 2008b). The potential utility of EP3 antagonists for treating detrusor hyperactivity and pain associated with bladder disorders may be offset by a study showing that EP3 receptors cause hypercontractility in obstructed urethra (Ankem et al., 2005).
EP3 receptor mRNA has been found in parietal and chief cells of the gastric fundic epithelium (Narumiya et al., 1999). This is consistent with EP3-mediated inhibition of gastric acid secretion (Bunce et al., 1991; Perkins et al., 1991; Savage et al., 1993; Yokotani et al., 1996; Kato et al., 2005; Dey et al., 2006) in rats. This EP3 inhibitory mechanism seems widely held to be true for humans, although actual functional evidence cannot be found. EP3 receptors, however, have been shown to be expressed throughout the human gastric epithelium (Takafuji et al., 2002). It was proposed that EP3 receptors may control gastrointestinal smooth muscle contraction because expression was found in longitudinal smooth muscle and in neurons of the myenteric ganglia (Narumiya et al., 1999). EP3 receptors increase slow-wave peristaltic frequency in mice (Forrest et al., 2009). The presence of EP3 receptors in the duodenum seems essential for HCO3− secretion to counteract acid-induced mucosal damage (Takeuchi et al., 1999). Species differences may occur in the gastrointestinal functions of prostanoid receptors (Dey et al., 2006).
In cancer, EP3 receptors have been found to exert both facilitatory and inhibitory effects. In human colon cancer specimens, EP3 receptor mRNA was reduced by 28% compared with the normal colon mucosa (Shoji et al., 2004). Studies in the human colon cancer cell line HCA-7 showed that a selective agonist decreased viable cell numbers by 30%, providing supportive evidence for EP3 receptor down-regulation playing a permissive role in colon carcinogenesis (Shoji et al., 2004). In a detailed study on individual mRNA splicing variants of the EP3 receptor, overexpression of each individual isoform decreased tumorigenic potential in cell lines and stably transfected human embryonic kidney 293 or HCT 116 cells exhibited decreased tumor growth in vivo (Macias-Perez et al., 2008). Analysis of second-messenger signaling revealed a G12-RhoA pathway for all three variants (Macias-Perez et al., 2008). EP3 receptors decreased aromatase activity in human adipose stromal cell lines, pointing to an inhibitory role in breast cancer (Richards and Brueggemeier, 2003). A role for EP3 receptors in tumor development is supported by other studies. In a model of tumor-stromal angiogenesis, tumor growth and angiogenesis were inhibited in EP3(−/−) mice and by a selective antagonist (Amano et al., 2003). A role for EP3 receptors in up-regulating VEGF (Amano et al., 2003) has been confirmed with respect VEGF and VEGF receptor expression (Taniguchi et al., 2008; Amano et al., 2009). In addition, a contributory role for EP3 receptors has been suggested in oral squamous cancer cell growth (Hoshikawa et al., 2009).
Studies on edema formation have yielded tissue-specific results. In edema formation in rat and mouse skin, EP3 receptor stimulation potently inhibited the response to zymosan-activated serum and platelet-activating factor (Ahluwalia and Perretti, 1994). PGE2-induced mouse paw edema was EP3 receptor-mediated (Claudino et al., 2006). In the rat adjuvant arthritis model, synoviocytes expressed the EP3B isoform, which mediated FL-6 release (Kurihara et al., 2001). EP3 antagonists are also reported to be active in standard models of pain and inflammation (Jones et al., 2009).
Inflammatory cell studies seem largely restricted to T cells and mast cells. Prolactin stimulates Ig and cytokine release from T cells and PGE2 enhances prolactin transcription via EP3 and EP4 receptors (Gerlo et al., 2004). EP3 receptors also regulate expression and release of matrix metalloproteinase-9 in early T cells (Zeng et al., 1996). Both of these studies relied on cultured cell lines. The influence of EP3 receptors on mast-cell function seem of greater pathophysiological significance. The first studies employed mouse bone marrow derived mast cells, which expressed all four EP receptor subtypes (Nguyen et al., 2002). However, only EP3 receptors were involved in potentiation of antigen-induced degranulation and IL-6 production (Nguyen et al., 2002). The importance of EP3 receptors in allergic airway inflammation was demonstrated by 1) suppression of eosinophil infiltration and antigen-induced mediator release by the EP3 agonist ONO-AE-248 (Fig. 4) and 2) development of a more prominent eosinophil and mononuclear cell infiltrate and increased cytokine release in EP3(−/−) mice (Kunikata et al., 2005). In dermal mast cells, EP3 receptor-induced degranulation occurred only in older mice, suggesting reprogramming with age (Nguyen et al., 2005). Studies in human mast cells provide at least partial confirmation in that PGE2 potentiates IgE-mediated histamine release via EP3 receptors but also EP1 receptors (Wang and Lau, 2006). Studies on the recruitment of mast cell progenitors showed that EP3 receptors are also chemoattractant for mast cells (Weller et al., 2007).
Beyond EP3 receptor-mediated down-regulation of cutaneous mast cell function and a potential role in type 1 allergies such as urticaria, EP3 receptors have also been implicated in keratinocytes and indirectly in atopic dermatitis. This is based on increased neutrophin 4 expression in keratinocytes, a factor that may participate in the hyperinnervation that occurs in atopic dermatitis (Kanda et al., 2005). Although the choice of pharmacological agents chosen for these studies was not ideal for unambiguously distinguishing EP3 from EP1 involvement, the use of antisense oligonucleotides provided strong evidence for responses exclusively mediated by EP3 receptors (Kanda et al., 2005). In addition, EP3 receptors were shown to inhibit the growth of human primary keratinocytes (Konger et al., 2005b). Three EP3 mRNA splicing variants were expressed in the intact human epidermis and keratinocytes. Predominant expression in the proliferative zone of the intact epidermis further supported involvement in regulating proliferation (Konger et al., 2005b).
Very diverse activities have been attributed to EP3 receptors in the lung. EP3 agonists constrict the human pulmonary artery (Qian et al., 1994). PGE2-induced sensory nerve activation, as assessed by depolarization of vagus nerves, was found to be EP3 receptor-mediated (Maher et al., 2009). Apnea in human neonates has been related to increased PGE2 release and EP3-mediated modulation of respiratory neurons in the brainstem (Hofstetter et al., 2007). EP3 receptors may not only participate in infection (sepsis)-induced apnea (Hofstetter et al., 2007) but also may contribute to mortality caused by infection with Streptococcus pneumoniae, according to EP3 gene deletion studies (Aronoff et al., 2009). The EP3 agonist GR 63799X induced S-phase arrest and inhibited fibroblast growth (Sanchez and Moreno, 2006), which is possibly relevant to fibrotic disease of the lungs and fibrosis in general.
Prostaglandins are very important in uterine function and reproduction. EP3 receptors contract the myometrium (Senior et al., 1991). Two EP3 receptor isoforms were found to be expressed in the human uterus, which signaled via Gi and MAP kinase (Kotani et al., 2000). EP3 receptors have also been implicated in cervical ripening and misoprostol is used clinically for labor induction and cervical ripening (Sanchez-Ramos et al., 1997).
c. Gene deletion studies.
Two independent lines of EP3(−/−) mice have been generated (Fleming et al., 1998; Ushikubi et al., 1998). The progeny of EP3(−/−) mice were normal in all respects. Mice were born at the anticipated Mendelian frequency, survived normally, and were observed to be normal. Histological examination of tissues from EP3 receptor-deficient mice revealed no pathological changes (Fleming et al., 1998). Fertility and reproduction were normal in all respects (Fleming et al., 1998; Ushikubi et al., 1998). Nonsteroidal anti-inflammatory drugs have been known as antipyretics, and the generation of prostanoid receptor knockout mice has enabled further insight into the pharmacology of febrile responses. These investigations were guided by the original proposal that PGE1 acted as a central mediator of fever (Milton and Wendlandt, 1970). Mice lacking EP3 receptors failed to mount a febrile response to PGE2 or to IL-1β or LPS (Ushikubi et al., 1998). In contrast, PGE2 induced fever in EP1(−/−), EP2(−/−), and EP4(−/−) mice (Ushikubi et al., 1998). After these studies on body temperature using a rectal probe (Ushikubi et al., 1998), febrile responses and thermoregulation in EP1(−/−) and EP3(−/−) mice were studied by telemetric measurement of core temperature (Oka et al., 2003). In this more systematic study, the EP1 receptor was implicated in the febrile response. The secondary phase of the hyperthermia to LPS was blunted in EP1(−/−) mice, whereas only the initial hyperthermic phase was affected in EP3(−/−) mice (Oka et al., 2003a). Agonist studies also implicated both EP1 and EP3 involvement in hyperthermia (Oka et al., 2003b).
EP3 receptors have also been implicated in urine production, osmolality, and bladder function. EP3 receptors are not essential for regulation of urinary osmolality, but in animals treated with indomethacin, urinary osmolality was increased in EP3(+/+) but not EP(−/−) mice (Fleming et al., 1998). Enhanced bladder capacity and PGE2-induced bladder hyperactivity were reduced in mice lacking EP3 recepors (McCafferty et al., 2008). Thus, EP3 receptors may contribute to overactive bladder disorders. EP3 receptor deletion studies have shown that the EP3 receptor is essential for maintaining duodenal HCO3− secretion and mucosal integrity (Takeuchi et al., 1999).
The majority of studies have implicated EP3 receptors in inflammation, pain, and associated events. Mice lacking EP3 receptors develop more pronounced allergic airway inflammation than wild-type or other knockout mice (Kunikata et al., 2005). Allergic inflammation was suppressed by an EP3 agonist (Kunikata et al., 2005). In complete contrast, EP3 receptor deletion has equally revealed proinflammatory roles. Systematic study of all EP knockout mice identified only the EP3 receptor as responsible for PGE2-induced mast cell activation and associated proinflammatory signaling pathways (Nguyen et al., 2002). Arachidonic acid-induced cutaneous microvascular exudation and edema was attenuated in EP3(−/−) but not EP1(−/−), EP2(−/−), or EP4(−/−) mice (Goulet et al., 2004), Exudate formation is also partially mediated by EP3 receptors in the carrageenin-induced pleurisy model (Yuhki et al., 2004). Additional studies on the lung have found that EP3 receptor deletion protects against severe S. pneumoniae infection (Aronoff et al., 2009) and virtually abolishes PGE2-induced depolarization of isolated vagus nerves (Maher et al., 2009). EP3 receptors have also been implicated in angiogenesis. Thus, in full-thickness skin wounds, EP3 receptor deletion delayed wound closure, re-epithelialization, and angiogenesis (Kamoshita et al., 2006). Accordingly, CD31 and VEGF expression were reduced (Kamoshita et al., 2006). COX-2 and EP3 receptors have also been found to be involved in acute herpetic pain: in contrast to EP3(−/−) mice, the allodynia and hyperalgesia were not altered in EP1(−/−), IP(−/−), or TP(−/−) mice (Takasaki et al., 2005). Tumor progression was also attenuated in EP3-deficient mice (Shoji et al., 2005).
As a possible connection between inflammatory signaling and obesity, effects observed in EP3 receptor-deficient mice are of interest. The body weight illustration and histogram data are quite striking, the EP3-deficient mice being clearly obese compared with wild-type littermate control mice (Sanchez-Alavez et al., 2007). Adult EP3(−/−) mice were feeding more frequently during the day and developed an obese phenotype on a normal fat diet. Increased locomotor activity did not affect the obesity. Obesity was accompanied by elevated leptin and insulin levels (Sanchez-Alavez et al., 2007).
The presence of functional EP3 receptors that potentiate platelet aggregation and inhibit adenylate cyclase has been recognized for some time (Matthews and Jones, 1993). Consistent with these findings, mice lacking EP3 receptors show an increased bleeding tendency and decreased susceptibility to thromboembolism (Ma et al., 2001). Finally, few studies on prostanoid receptor overexpression have been reported. In transgenic mice with cardiospecific EP3 receptor overexpression, increased calcineurin and NFAT activity with cardiac hypertrophy were found (Meyer-Kirchrath et al., 2009).
d. Agonists and antagonists.
Phenoxy substitution (particularly p-halo-phenoxy) at C16 on the PGE template modestly promotes EP1, EP3, and FP agonism but markedly accentuates TP agonism. 16-p-Chlorophenoxy-ω-tetranor PGE2 (ICI-80205) is a good example of these trends (Jones et al., 1982; Lawrence et al., 1992). 11-Deoxy-16-phenoxy PGE1 (MB-28767) shows reasonably good EP3-versus-EP1 selectivity but is still a moderately potent TP agonist (Banerjee et al., 1985; Lawrence and Jones, 1992; Lawrence et al., 1992; Boie et al., 1997). Sulprostone, in which the C1 carboxylate is converted to the acidic methylsulfonamide (Schillinger et al., 1979), has modest EP3-versus-EP1 selectivity and minimal TP agonism (Coleman et al., 1987; Coleman and Sheldrick, 1989). The combination of sulprostone and 17-phenyl PGE2 (Lawrence et al., 1992) has often been used to discriminate EP1 and EP3 receptors but is clearly not ideal. In a comprehensive structure-activity relationship study using guinea pig vas deferens and binding to mouse recombinant EP receptor subtypes, Shimazaki et al. (2000) showed that replacement of the C1-carboxylate in 13,14-didehydro-16-phenoxy PGE1 by a primary alcohol group (Fig. 4, compound 7b) results in only modest loss of agonist potency coupled with high EP3 selectivity; the C1-methyl ketone analog showed a similar profile.
Other routes to EP3-selective agonism have been explored. Deriving from misoprostol, SC-46275 (Fig. 4) is a highly potent and selective EP3 agonist (Savage et al., 1993) used in a limited number of investigations (Jones et al., 1998); the orientation of the 16-hydroxyl is opposite that of ONO-AE-259. Again, de-esterification may be required for full bioactivity. Finally, methylation of both 11- and 15-hydroxyls in PGE2 (ONO-AE-248; Fig. 4) imparts high EP3 selectivity (Okada et al., 2000; Suzawa et al., 2000), but potency is only modest (Jones et al., 2008).
The development of EP3 antagonists has recently been reviewed (Jones et al., 2009) and is therefore summarized only briefly. The acryloylsulfonamides L-798106 and L-826266 emerged from a combinatorial approach (Fig. 5) (Gallant et al., 2002; Belley et al., 2005). They are both highly lipophilic with slow onsets on certain isolated smooth muscle preparations (Jones et al., 2008). The related EP3 antagonist DG-041 (Heptinstall et al., 2008; Singh et al., 2009) is even more lipophilic (log P = 7.67); affinity is maintained when the acryloyl unit is replaced by a heterocycle (Fig. 5, Hategan et al., 2009). More water-soluble antagonists are present within the DeCode Genetics series (O'Connell et al., 2009); one of these showed faster block than L-798106/L-826266 (Jones et al., 2011). An EP3 antagonist of a different class is represented by compound 49 (Fig. 5) (Asada et al., 2010); it showed good in vivo activity against PGE2-induced contraction of pregnant rat uterus.
e. Therapeutics.
EP3 receptor pharmacology has successfully resulted in small-molecule therapeutics in current use, both of which contain misoprostol (Collins et al., 1985). Misoprostol is used for cervical ripening and labor induction (Sanchez-Ramos et al., 1997; Woodward and Chen, 2004). Misoprostol is effective gastric ulcer therapy (Rachmilewitz et al., 1986) and is combined with a nonsteroidal anti-inflammatory agent to ameliorate gastric irritation and ulceration and is commercially available as Arthrotec (G. D. Searle, Peapack, NJ) (Woodward and Chen, 2004). Misoprostol is a potent EP3 agonist but does not really possess adequate receptor selectively for modern therapeutic application. Given the complexity of PG-mediated effects (e.g., EP3 receptors and EP2/EP4 receptors having opposing effects on cAMP), a highly selective agent may be preferable for new uses. A decade of recent EP3 research has witnessed diverse findings. EP3 agonists protect against ischemic myocardial injury and reduced infarct size (Hohlfeld et al., 1997, 2000; Zacharowski et al., 1999), culminating in a study on transgenic mice with cardiospecific EP3 overexpression (Martin et al., 2005). Reduction in tumor development may be achieved with EP3 agonists (Amano et al., 2003; Macias-Perez et al., 2008), and down-regulation of EP3 receptors may enhance colon carcinogenesis in later stages (Shoji et al., 2004). Suppression of allergic inflammation by EP3 receptor activation has also been demonstrated (Kunikata et al., 2005). Finally, EP3 agonists may offer treatment for opiate withdrawal syndrome (Nakagawa et al., 1995). The potential of the EP3 agonist-based therapeutic options (Table 6) must be viewed from the standpoint that EP3 receptor stimulation may result in many pathophysiological events, as indicated by the potential uses of EP3 antagonists.
Potential therapeutic application of EP3 agonists
Potential therapeutic uses of EP3 receptor antagonists, identified and confirmed in animal models with compounds, are summarized in Table 7. These include pain and inflammation (Jones et al., 2009), type 1 allergy (Nguyen et al., 2002), lung infection (Aronoff et al., 2009), cough (Maher et al., 2009), overactive bladder (McCafferty et al., 2008; Su et al., 2008), hyperpyrexia (Ushikubi et al., 1998), and cancer (Amano et al., 2003). Most recently, EP3 antagonists have emerged as a new target for antiplatelet agents in atherothrombotic disease, without prolonged bleeding (Heptinstall et al., 2008; Singh et al., 2009).
Potential therapeutic application of EP3 antagonists
4. EP4 Receptors.
a. Second messenger signaling.
EP4 receptors are widely distributed (Narumiya et al., 1999). The pharmacologically defined EP4 receptor was originally designated EP2 (Honda et al., 1993; Bastien et al., 1994) until the authentic EP2 receptor was cloned (Regan et al., 1994b). Studies on second-messenger signaling demonstrated functional coupling to cAMP via Gs (Narumiya et al., 1999). Certain EP4 receptor-mediated effects seem to exclusively employ the cAMP-PKA pathway (Southall and Vasko, 2001; Gray et al., 2004; Ziemann et al., 2006; Boniface et al., 2009). Direct comparison of EP4 and EP2 receptor signaling demonstrated that the functional coupling to cAMP seems less efficient for EP4 compared with the EP2 subtype (Fujino et al., 2002, 2005). The prospect of a second EP4 signaling pathway was realized when a PI3K signaling pathway was discovered coupled to the pertussis toxin-sensitive G protein Gi (Fujino et al., 2002, 2003, 2005; Fujino and Regan, 2005). This also provides a mechanism for limiting the cAMP response to EP4 stimulation (Fujino and Regan, 2006). It should be noted that several cAMP-independent signaling cascades have been reported for EP4 receptor activation (Fiebich et al., 2001; Pozzi et al., 2004; Mendez and LaPointe, 2005; Frias et al., 2007; George et al., 2007; Rao et al., 2007). Evidence for the EP4 signaling cascade PI3K-ERK-early growth response-1 (Fujino et al., 2003) seems operative in cell growth (Pozzi et al., 2004; Mendez and LaPointe, 2005; Frias et al., 2007; Rao et al., 2007). Serum-deprived apoptosis in Jurkat cells was reduced by EP4 receptors through PI3K and cAMP pathways may be operative, as in EP4-mediated inhibition of apoptosis (Leone et al., 2007).
Epac signaling has received little attention from the standpoint of prostanoid receptor signal transduction. A recent study has indicated both PKA and Epac 1 signaling in rheumatoid synovial fibroblasts (Kojima et al., 2009) for both EP2 and EP4 receptors. Both EP receptor subtypes activate the small GTPase Rap 1 (Kojima et al., 2009). Rap was also involved in EP4-mediated brain natriuretic peptide expression (Qian et al., 2006). EP4 receptors have also been shown to be involved in COX-2 mRNA induction and stabilization and stimulation of translation (Faour et al., 2001; Martineau et al., 2004), p38 MAP kinase playing a key role. EP4 receptor activation of p38 MAP kinase has been found in certain cells: astrocytes, podocytes (Fiebich et al., 2001; Martineau et al., 2004), RAW 264.7 cells (Chen et al., 2006), Caco-2 cells (Leone et al., 2007), and colonic myofibroblasts (Hoang et al., 2007). Such an EP4 effect on p38 MAP kinase is not always observed, for example in tracheobronchial epithelial cells (Gray et al., 2004) and neonatal ventricular myocytes (Qian et al., 2006). In cardiac myocytes, EP4 receptor-mediated hypertrophy (Mendez and LaPointe, 2005) and BNP promoter regulation (Qian et al., 2006) both involve p42/44 MAP kinase. The potential complexities of EP4 activating signaling cascades is illustrated by the concomitant activation of PKC in some instances (Fiebich et al., 2001; Chen et al., 2006).
b. Distribution and biological functions.
EP4 receptors were originally pharmacologically characterized predominantly from studies on smooth muscle (Coleman et al., 1994a). Thereafter, EP4 receptor-mediated effects on smooth muscle tone have received little attention. EP4 receptors mediate vasorelaxation of pulmonary arterial veins but produce no effect on pulmonary arteries (Foudi et al., 2008). EP4 receptors, however, do not play an entirely passive role in the human pulmonary artery: COX-2 induction by bradykinin in human pulmonary arterial smooth muscle cells involves cAMP response element activation by EP4 and EP2 agonists (Bradbury et al., 2003). In the rat aorta, EP4 receptor-mediated vasorelaxation was endothelium-dependent and involved endothelial nitric-oxide synthase and cGMP (Hristovska et al., 2007). The most therapeutically significant study demonstrated EP4 receptor-mediated vasodilation of the human middle cerebral and meningeal arteries and obviated the potential clinical utility of EP4 antagonists for treating migraine (Davis et al., 2004; Maubach et al., 2009). EP4 overexpression in atherosclerotic plaque results in an unstable phenotype prone to inflammation and instability (Cipollone et al., 2005). Endothelial cell migration and angiogenesis in vivo are also produced by EP4 receptor activation (Rao et al., 2007; Jain et al., 2008).
An interesting aspect of EP4 cardiovascular biology is its potential involvement in cardiac hypertrophy. EP4 receptor activation produces cardiomyocyte hypertrophy, as measured by increased protein synthesis (Mendez and LaPointe, 2005; Miyatake et al., 2007; He et al., 2010), cell size and surface area (Frias et al., 2007; Miyatake et al., 2007; He et al., 2010), re-expression of fetal genes, and activation of hypertrophic marker genes such as BNP (Qian et al., 2006; Miyatake et al., 2007; He et al., 2010). This was confirmed in living animals, where cardiospecific EP4 receptor deletion resulted in decreased hypertrophy and fibrosis after experimental myocardial infarction (Qian et al., 2008). The transcription factor STAT3 was activated and correlated with hypertrophy and fibrosis (Frias et al., 2007; Qian et al., 2008). Cardiac function, paradoxically, was worsened in hearts lacking EP4 receptor expression (Qian et al., 2008), which actually correlates with EP4 protection from ischemia-reperfusion injury (Xiao et al., 2004).
The ductus arteriosus is a shunt in the fetus between the pulmonary artery and the aorta. Closure of the ductus arteriosus in the newborn is vital to prevent pulmonary hypertension-induced lung edema and congestive heart failure. The EP4 receptor is critical for remodeling of the ductus arteriosus at birth (Nguyen et al., 1997). Likewise, EP4 agonists reopen the ductus arteriosus in neonates dependent on placental oxygenation (Momma et al., 2005). Analogous to some extent, the uterine cervix also plays a crucial role in pregnancy; closed during gestation, soft and dilated during labor. PGE2 has been used for inducing cervical ripening for many years (Woodward and Chen, 2004), and the changes that occur resemble those that are observed in physiological ripening. EP4 receptor expression has been shown to be maximum at parturition in rats (Chien and Macgregor, 2003) and has been implicated in LPS-induced cervical ripening (Fukuda et al., 2007). Glycosaminoglycan biosynthesis is an important part of cervical ripening, and EP4 receptor stimulation produces this effect in human cervical fibroblasts via a PKA-independent pathway (Schmitz et al., 2001).
Results on EP4 receptor involvement in mucus secretion are mixed. EP4 and EP1 receptors evoke mucin exocytosis from central mucous cells (Ohnishi et al., 2001), whereas an EP4 agonist inhibited LPS-induced mucus secretion from airway epithelial cells (Hattori et al., 2008). PGE2 was claimed to protect guinea pig gastric mucosal cells from ethanol-induced apoptosis (Hoshino et al., 2003): set against this finding, the EP4 antagonist (S)-4-(1-(5-chloro-2-(4-fluorophenyoxy)benzamido)ethyl)benzoic acid (CJ-42794) did not damage the rat gastric mucosa or worsen the response to aspirin or stress (Takeuchi et al., 2007). Cytoprotective duodenal HCO3− secretion is also EP4 receptor-mediated in rats (Aoi et al., 2004) and humans (Larsen et al., 2005).
There is an extensive volume of literature on PGs and bone formation, and EP4 agonists occupy a prominent place. EP4 receptors mediate not only bone formation but also bone resorption, as indicated in EP4(−/−) mice (Miyaura et al., 2000). Nevertheless, a litany of reports shows EP4-mediated bone formation, augmentation of bone morphogenetic protein-induced bone mass, and beneficial effects on fracture healing. These findings suggest that, physiologically, EP4 receptors favor resorption, but exogenously administered EP4 agonists have an anabolic effect on bone formation. It may be speculated that exogenous EP4 agonists locate receptors not under PGE2 regulation under normal physiological circumstances. The potential therapeutics associated with EP4 and EP2 agonists for treating bone loss and accelerating bone repair have been reviewed (Li et al., 2007; Graham et al., 2009). It should also be noted that findings in vitro may not transition into in vivo studies; thus, although EP4 receptors are essential for anabolic responses to PGE2, in osteoblasts, they are not essential for bone remodeling in living animals (Gao et al., 2009). In quite marked contrast, the EP4 antagonist N-(((2-(4-(2-ethyl-4,6-dimethyl-1H-imidazo(4,5-c)pyridin-1-yl)phenyl)ethyl)amino)carbonyl)-4-methylbenzenesulfonamide (CJ-023,423) reduced bone destruction in the rat adjuvant-induced arthritis model (Okumura et al., 2008). In mouse collagen-induced arthritis, the EP4 antagonist ONO-AE3-208 (Fig. 5) did not alter the arthritis in wild-type mice (Honda et al., 2006).These results indicate that EP4 receptor participation in bone remodeling is disease-model specific.
Endochondral bone formation involves chondrocytes as well as coordinated bone formation and mineralized matrix resorption by osteoblasts and osteoclasts. In the growth plate, chondrocytes undergo a maturation process. Resting chondrocytes transition into proliferating chondrocytes, which express type II collagen mRNA and synthesize proteoglycan. Chondrocytes then mature into hypertrophic cells characterized by a 5- to 10-fold increase in cell volume. Terminal differentiation is associated with expression of genes associated with calcification of bone matrix, such as osteocalcin. Terminally differentiated chondrocytes undergo apoptosis, and the calcified cartridge left behind provides a template for primary bone formation (Cheung et al., 2003). An understanding of PG involvement in each phase of chondrocyte maturation is of significance for osteoarthritis and rheumatoid arthritis. EP4 receptor stimulation alone did not up-regulate type II collagen expression or increase proteoglycan in rat growth plate chondrocytes; concomitant EP2 receptor activation was required (Miyamoto et al., 2003). Other murine chondrocyte studies are not entirely in agreement (Clark et al., 2005; Brochhausen et al., 2006), these studies using more physiological concentrations. The concentrations of PGE2 in studies on human chondrocytes are also sufficiently high to be in the nonselective range.
Fibroblast-like synoviocytes are important in the pathogenesis of rheumatoid arthritis, where they proliferate and secrete enzymes involved in joint degradation and cytokine/chemokine production. In human synovial fibroblasts, IL-1α (Yoshida et al., 2001) and IL-1β (Faour et al., 2001) effects are mediated by PGE2 release. EP2 and EP4 receptors are consistently expressed in human synovial fibroblasts (Yoshida et al., 2001; Mathieu et al., 2008; Kojima et al., 2009). IL-1β-induced up-regulation of COX-2 was ascribed to interaction of released PGE2 with EP4 receptors (Faour et al., 2001). IL-1β also increases PGE2 production in human tendon fibroblasts, which mediates down-regulation of type 1 collagen via EP4 receptors, which in turn may disturb tendon homeostasis (Thampatty et al., 2007).
Studies on leukocyte EP4 receptor function have been largely restricted to mononuclear cells. In mouse peritoneal neutrophils, EP4 and EP2 receptors suppress TNFα production, and EP2 receptors augment IL-6 production (Yamane et al., 2000). In human eosinophils, EP4 mRNA was found to be significantly higher than EP2 (Mita et al., 2002). In human blood monocytes, EP2 and EP4 receptors up-regulated C-C chemokine receptor 7 mRNA, which is essential for migration to secondary lymphoid tissues (Côté et al., 2009). In a further study on human macrophages, only EP4 receptors were expressed and therefore considered to mediate PGE2 inhibition of macrophage inhibitory proteins 1α and 1β, IL-8, monocyte chemotactic protein-1, and IL-10 release (Takayama et al., 2002).
PGE2 was also found to be a key factor for increased C-C chemokine receptor 7 mRNA expression in monocyte-derived dendritic cells (Scandella et al., 2002), although the relative importance of EP4 and EP2 receptors is uncertain because of indeterminate pharmacological definition. PGE2 is regarded as essential for the development of a migratory phenotype of human dendritic cells, and this is ascribed to EP4 and EP2 receptor mediation (Luft et al., 2002; Harizi et al., 2003; Legler et al., 2006; McIlroy et al., 2006). Polarization into Th-2 helper cells seems to involve both EP4 and EP2 receptors (Kubo et al., 2004; McIlroy et al., 2006; Krause et al., 2007). The choice and concentrations of the pharmacological “tools” in these studies was not always ideal, and further investigation would be worthwhile. EP4 receptors were shown to initiate cutaneous immune responses by promoting Langerhans cell maturation and migration, in a comprehensive study employing EP4(−/−) mice and selective EP4 agonist and antagonist compounds (Kabashima et al., 2003b).
T-cell functions are also regulated by PGE2 and EP4 receptor activation. PGE2 was shown to suppress Th1 and Th2 T-helper cell activity. Both EP4 and EP2 receptors acted additively to suppress Th1 cell proliferation and IFN-γ release. In Th2 T-helper cells, independent EP2 and EP4 receptor activation virtually abolished proliferation, but combined EP2/EP4 agonism was most effective in inhibiting IL-4 production (Okano et al., 2006). Some studies have implicated EP4 and EP2 receptors in development of the Th17 phenotype from naive T cells. Th17 T cells are distinct from Th1 and Th2 subsets. Human Th17 T-helper cell differentiation is controlled by the retinoic acid receptor-related orphan receptor-γt, and PGE2 seems to synergize with IL-1β and IL-23 to up-regulate retinoic acid receptor-related orphan receptor-γt and down-regulate T cell-specific T box transcription factor, IFN-γ and the anti-inflammatory cytokine IL-10 (Boniface et al., 2009; Napolitani et al., 2009). No role for EP1 or EP3 receptors was apparent. A clear role for EP2 receptors was established, but the choice of EP4 agonists and the high concentrations used cannot rule out EP2/EP4 mutual inhibitory activity, EP2/EP4 synergism, or other nonspecific activity. In an in vivo transplantation model, a combination of EP2, EP3, and EP4 agonists was needed to match the immunosuppressive effect of PGE2 (Fujimoto et al., 2005). A singular EP4 receptor-mediated event was found in HIV-1-infected T cells with respect to pro-viral DNA activation (Dumais et al., 1998). Based on Pt ger 4 (EP4) behaving as a strongly expressed, delayed early gene that inhibits B cell proliferation (Murn et al., 2008), EP4 receptors could represent a novel target for treatment of B cell malignancies.
Numerous reports have linked EP4 receptor activation to cancer. EP4 receptors promote cancer in many dimensions: cell proliferation (Cherukuri et al., 2007; Zheng et al., 2009), cell survival (George et al., 2007), invasiveness (Spinella et al., 2004; Pan et al., 2008), angiogenesis (Jain et al., 2008), migration (Kim et al., 2010), and tumor metastasis (Ma et al., 2006; Yang et al., 2006). PGE2 produces proliferation of colon cancer cell lines H-29 (Chell et al., 2006) and HCA7 (Cherukuri et al., 2007). This is consistent with the idea that EP4 is the most abundant transcript in both H-29 and HCA7 cells, which also biosynthesize PGE2 via COX-2 (Doherty et al., 2009). EP4 receptors stably transfected into HT-29 cells promoted anchorage-independent growth and increased resistance to apoptosis and the formation of fluid-filled cysts (Hawcroft et al., 2007). Transfection of EP4 receptors into a human adenoma cell line (RG/C2) also introduces anchorage-independent growth (Chell et al., 2006). PGE2 also caused EP4-mediated cell growth in untransfected RG/C2 adenoma cells (Chell et al., 2006). COX-2 (Gustafsson et al., 2007; Yuan et al., 2008; Doherty et al., 2009) and EP4 receptor expression are increased in colorectal cancer (Chell et al., 2006), although the latter result was not confirmed (Gustafsson et al., 2007). Animal models provide evidence for a major role for EP4 receptors in the development of colon cancer (Mutoh et al., 2002; Kitamura et al., 2003b; Yang et al., 2006). EP4 receptors have been implicated in many forms of cancer: these include lung (Han et al., 2007; Zheng et al., 2009), upper urinary tract (Miyata et al., 2005), stomach (Okuyama et al., 2002), prostate (Jain et al., 2008), breast (Timoshenko et al., 2003; Ma et al., 2006; Pan et al., 2008; Robertson et al., 2008; Subbaramaiah et al., 2008), cervix (Sales et al., 2001; Muller et al., 2006; Oh et al., 2009), ovary (Spinella et al., 2004), and nonmelanoma skin cancer (Lee et al., 2005).
Prostanoid EP4 receptors have been extensively implicated in mediating hyperalgesia and allodynia (Jones et al., 2009). All EP subtypes are expressed in sensory neurons, but EP4 may be regarded as the most important because it causes sensitization (Southall and Vasko, 2001) and is exclusively expressed in a subset of primary sensory dorsal root ganglia, which increases in subchronic inflammation (Lin et al., 2006). EP4 receptors protect against NMDA-induced acute excitotoxicity (Ahmad et al., 2005). A dichotomy of effects was found with respect to EP4 receptors in Alzheimer's disease. PGE2 was found to stimulate amyloid-β peptide production via EP4 receptor internalization (Hoshino et al., 2009). However, cell death produced by β-amyloid was attenuated by EP4 and EP2 receptor stimulation (Echeverria et al., 2005).
c. Gene deletion studies.
Two lines of systemic-null EP4-deficient mice were generated independently (Nguyen et al., 1997; Segi et al., 1998). Most EP4-deficient mice on a C57BL/6 background die within 3 days after birth because of marked pulmonary congestion and heart failure due to a patent ductus arteriosus. It is well known that administration of indomethacin to maternal mice during late pregnancy induces closure of the ductus in wild-type mice, which led to the proposal that endogenous PGs maintains the ductus open. However, indomethacin treatment did not induce closure of the ductus in EP4-deficient fetuses, indicating that a PG-independent dilatory mechanism comes into play in the absence of EP4. These findings led the authors to suggest that opening of this vessel during the embryonic period is mediated by the PGE2-EP4 signaling and that in its absence, the compensatory dilatory mechanism is mobilized, and this mechanism continues to maintain the vessel open after the birth, resulting in a paradoxical patent ductus arteriosus in the EP4-deficient mice. However, in contrast to this general idea on the patent ductus arteriosus phenotype of EP4(−/−) mice, Trivedi et al. (2006) suggested an alternative possibility that the PGE2-EP4 signaling functions close, not open, the ductus. Their suggestion was based on their findings that COX-2 is induced in the ductus around term; that the loss of COX-2 or long-term treatment with COX inhibitors leads to its opening, not closure; and that the COX-2 expression in the ductus around term was attenuated in EP4-deficient mice.
Survival of EP4-deficient mice can be improved using the mixed genetic background C57BL/6 and 129s/v. Therefore, colonies of EP4(−/−) mice are maintained by intercrossing surviving EP4(−/−) mice, and progenies of these colonies are used with progenies of the littermate wild-type mice as a control. Using these mice, functions of EP4 in several physiological processes have been examined, one being that in bone metabolism. Osteoclasts develop from precursor cells of the macrophage lineage in the bone microenvironment. Factors such as PTH, vitamin D, IL-1, and IL-6 act on osteoblasts to induce the synthesis of RANKL, which in turn stimulates the formation of mature osteoclasts from hematopoietic precursors through cell-cell interaction. These factors induce COX-2 expression in osteoblasts, and their induction of osteoclast differentiation is inhibited, at least in part, by aspirin-like drugs; such inhibition is reversed by the addition of PGE2, implicating PGE2 in this process (Tai et al., 1997). Sakuma et al. (2000) and Miyaura et al. (2000) examined the identity of the EP subtype responsible for mediating this action of PGE2. Sakuma et al. (2000) found that PGE2-induced osteoclast formation was impaired in cocultures of osteoblasts from EP4-deficient mice and osteoclast precursors from the spleen of wild-type mice. IL-1β, TNF-α, and basic fibroblast growth factor also failed to induce osteoclast formation in these cultures. Miyaura et al. (2000) added PGE2 to cultures of parietal bone from mice deficient in each of the EP subtypes, as well as from wild-type mice, and examined bone resorption by measuring the release of Ca2+ into the medium. They found that the induction of bone resorption by PGE2 was greatly impaired, whereas bone resorption in response to dibutyryl cAMP was unaffected, in bone from EP4-deficient mice. These studies unequivocally established a role for EP4 receptors in the induction of osteoclast differentiation factor and in PGE2-dependent bone resorption. On the other hand, Li et al. (2000) showed that the osteoclastogenic response to PGE2, PTH, or 1,25-dihydroxyvitamin D in vitro was impaired in cultures of cells derived from EP2-deficient mice. These findings probably reflect redundant roles of the two relaxant PGE receptor subtypes. In addition to bone resorption, systemic administration of PGE2 has long been known to induce bone formation in vivo. To examine the identity of the EP receptor in this process, Yoshida et al. (2002) infused PGE2 into the periosteal region of the femur of wild-type mice or mice-deficient in each EP subtype with a miniosmotic pump and found that, after 6 weeks, PGE2 induced extensive callus formation on the femur at the site of infusion in wild-type as well as EP1-, EP2-, and EP3-deficient mice but not in EP4-deficient mice. Infusion of an EP4-selective agonist, but not those specific for other EP subtypes, consistently induced bone formation in wild-type mice with increased density of both osteoblasts and osteoclasts. These findings suggest that EP4 is responsible for both bone resorption and bone formation induced by PGE2 and that activation of EP4 in situ integrates these two actions for bone remodeling.
Given the marked induction of COX-2 and high production of PGE2 in the heart during myocardial infarction and preferential expression of EP4 among EPs in this organ, Xiao et al. (2004) subjected EP4-deficient mice to an ischemia-reperfusion model by ligation of the left anterior descending coronary artery for 1 h, followed by 24-h reperfusion. They found that EP4-deficient mice developed larger infarct sizes than wild-type mice and that, conversely, administration of ONO-4819CD (Xiao et al., 2004), an EP4 agonist, administered 1 h before or 50 min after occlusion reduced the infarct size in wild-type mice. Based on these findings, the authors suggested that EP4 exerts cardioprotective actions under ischemia-reperfusion conditions in the heart. Another study using EP4-deficient mice in cardiovascular diseases concerned its role in atherosclerosis. Because macrophages that accumulate in early lesions of atherosclerosis are capable of producing a large amount of PGE2, and cells in atherosclerotic plaques express EP2 and EP4 among EP subtypes, Babaev et al. (2008) generated chimera mice in which fetal liver cells from either EP2(−/−) or EP4(−/−) mice were transplanted into lethally irradiated LDLR(−/−) mice. They then fed these mice with western chow for 8 weeks to develop atherosclerosis. They found that atherosclerotic lesions were significantly smaller in mice transplanted with cells of EP4-deficient mice [EP4(−/−) → LDLR(−/−)] than either WT → LDLR(−/−) or EP2(−/−) → LDLR(−/−) mice with significantly more apoptotic cells in the lesions. Further analysis revealed that macrophages from EP4(−/−) chimeras are more sensitive to apoptotic stimuli, which may derive from attenuated PI3K and nuclear factor-κB signaling in this line. These findings have led the authors to suggest that the macrophage EP4 signaling may be a target for suppressing development of atherosclerosis.
Studies on EP4-deficient mice also revealed a variety of actions of this EP subtype in immune inflammation. One action is on dendritic cells. Kabashima et al. (2003b) found that migration of Langerhans cells in the skin to draining lymph nodes on hapten application is impaired in EP4(−/−) mice, and this impairment can be mimicked by treatment of animals with an EP4-selective antagonist. Consequently, contact hypersensitivity to the hapten was suppressed in EP4-deficient mice. Further analysis revealed that the PGE2-EP4 signaling facilitates mobilization, migration, and maturation of Langerhans cells after initial antigen application. Honda et al. (2006) found that EP4 also plays a role in collagen-induced arthritis. In this model, the PGI2-IP signaling and the PGE2 signaling through EP2 and EP4 additively mediate joint inflammation through regulation of expression of arthritis-related genes, including those for IL-6, vascular endothelial growth factor-A, and RANKL, in synovial fibroblasts. More recently, Yao et al. (2009) used T cells from mice deficient in EP2 or EP4 and found that EP2 and EP4 receptors redundantly facilitate IL12-mediated Th1 differentiation and IL-23-mediated Th17 expansion. EP4 also functions in production of IL-23 from activated dendritic cells. Development of immune inflammation in experimental allergic encephalomyelitis is consistently and significantly suppressed by treatment of mice with EP4-selective antagonist or in EP4-deficient mice. On the contrary to these proinflammatory actions in immune inflammation, EP4 can exert anti-inflammatory action. Kabashima et al. (2002) found that EP4-deficient mice developed severe colitis in response to treatment with 3% dextran sodium sulfate, a dose that can be tolerated in wild-type animals. Again, this phenotype was mimicked by administration of an EP4-selective antagonist to wild-type mice. EP4 deficiency was shown to result in impairment of mucosal barrier function that was associated with epithelial loss, crypt damage, and accumulation of neutrophils and CD4+ T cells in the colon. DNA microarray analysis revealed increased expression of genes associated with immune responses and reduced expression of genes associated with mucosal repair and remodeling in the colon of EP4-deficient mice. Given the elevated level of PGE2 and high expression of COX-2 in the brains of patients with Alzheimer's disease, Hoshino et al. (2007) examined the role of PGE2 in processing β-amyloid precursor protein in cultured cells and found that PGE2 stimulated production of amyloid-β peptide by activating γ-secretase and that this action occurs via EP2 and EP4. They then cross-mated an Alzheimer's disease model of APP-23 transgenic mice with either EP2(−/−) mice and EP4(−/−) mice and found that the levels of Aβ peptides were significantly lower in the presence of either deletion. Besides these actions in complex inflammatory diseases, EP4 is also implicated in a simple form of acute inflammation. Kabashima et al. (2007) subjected the ear of mice deficient in each of the EP subtypes to ultraviolet B irradiation and examined the extent of skin inflammation. They found that ear swelling caused by UV irradiation was significantly suppressed with reduced inflammatory cell infiltration and blood flow in EP2(−/−) and EP4(−/−) mice compared with wild-type mice and that the effect of EP4 deficiency was mimicked by administration of an EP4 antagonist to wild-type mice. They further found that blockade of EP2 and EP4 is additive, suggesting that they function redundantly.
Epidemiological as well as experimental studies have implicated COX isoforms and PGs in familiar adenomatous polyposis and development of colon cancer. To gain an insight into the receptor involved in this process, Watanabe et al., (1999) and Mutoh et al. (2002) examined azoxymethane-induced formation of aberrant cryptic foci in mice deficient in each prostanoid receptor. They found that the formation of such foci was suppressed in both EP1(−/−) and EP4(−/−) mice but not in those deficient in other receptor types or subtypes. In both instances, the number of foci was reduced to 50 to 60% of that apparent in wild-type mice.
Although systemically null EP4-deficient mice have contributed to our understanding of physiological functions of this receptor, their mixed genetic background has often limited their use. To conquer this weakness, Schneider et al. (2004) generated conditional EP4flox/flox mice in which deletion of the EP4 gene can be achieved by expression of Cre recombinase. Using endothelial cells derived from these mice and rendered EP4-null by transfection with adenovirus harboring Cre, Rao et al. (2007) found that EP4 is required for PGE2-mediated migration, in vitro formation of capillary-like structure, cAMP production, and ERK activation. Combining these in vitro findings with in vivo findings that EP4 agonists can induce angiogenesis in sponge implanted into mice, they argued proangiogenic potential for PGE2-EP4 signaling. Gao et al. (2009) used this EP4flox/flox mouse line and generated mice in which one allele of EP4 was globally deleted and the other was targeted in osteoblasts. They found that this line of KO mice developed normal bone and exhibited no change in bone volumes or bone formation, whereas osteoblasts of these mice lost their responsiveness to PGE2 in vitro. They argued that either EP4 signaling may not be required for physiological regulation of bone development and maintenance or the loss of EP4 may be compensated for by other mechanisms. Their findings are not compatible with the previous report by Li et al. (2005), who found osteopenia and impaired fracture healing in aged globally KO EP4-deficient mice and that by Akhter et al. (2001), who reported that EP4-deficient mice have small distal femur and vertebral bone volume and exhibit reduced structural and apparent material strength in the femoral shaft and vertebral body. Another study focusing on EP4 in bone using conditional knockout mice examined the action of EP4 in periplastic osteolysis and the identity of the cell type mediating this action. Tsutsumi et al. (2009) implanted polyethylene beads to the periosteal surface of calvaria in EP1(−/−) mice, EP2(−/−) mice, and mice with conditionally deleted EP4 in FSP1+ fibroblasts and examined osteolysis. They also prepared fibroblasts and osteoblasts from these mice, stimulated them with titanium beads or PGE2, and assessed production of RANKL. They found that polyethylene-bead–induced osteolysis is impaired only in conditional EP4 KO mice. It is noteworthy that this conditional knockout exhibited reduced RANKL production only in fibroblasts and not in osteoblasts. On the basis of these findings, the authors suggested that osteolysis associated with total joint replacement is induced by RANKL produced by fibroblasts at the tissue-implant interface. Another example of the use of EP4flox/flox mice is generation of mice with loss of EP4 selectively in cardiomyocytes. Qian et al. (2008) generated this line of mice by crossing EP4flox/flox with mice carrying Cre recombinase driven by α-myosin heavy chain promoter. This line of cardiac-specific EP4 KO mice does not show any abnormality but exhibits less hypertrophy and less fibrosis with attenuated STS3 activation in a model of myocardial infarction induced by left anterior descending coronary artery ligation. They do, however, show reduced ejection function. A later study by the same group (Harding et al., 2010) revealed that male mice of this line of conditional EP4 knockouts spontaneously develop dilated cardiomyopathy at 23 to 33 weeks of age. Therefore, the cardiospecific EP4-null mice exhibit a cardiac phenotype different from that of systemic EP4-null mice.
d. Agonists and antagonists.
Selective EP4 agonists typically contain a 16-phenyl group, the importance of which to vasodilator activity was recognized early (Johnson et al., 1980). Within the ONO series, a 3,7-dithia substitution pattern in the α-chain was found to dramatically favor EP4/EP3 selectivity (Maruyama et al., 2002a). Addition of large groups (e.g., m-phenyl) to the 16-phenyl ring resulted in retention of EP4 binding affinity but reduction of functional potency, perhaps because of loss of efficacy (intrinsic activity) (Maruyama et al., 2002b). ONO-AE1-329, with a m-methoxymethyl substituent, emerged as a highly selective EP4 full agonist (Cao et al., 2002). Prostanoids with a 8-aza-9-oxo functionality were originally identified as potent ligands for the rat kidney prostaglandin E receptor (Smith et al., 1977). In the Merck series of selective EP4 agonists, this ring system is combined with a 16-phenyl group, and an acidic 5-tetrazole ring replaces the carboxylate, thereby preventing β-oxidation (Billot et al., 2003; Young et al., 2004); L-902688 (Fig. 4) seems to be the preferred molecule. CP-734432 (Fig. 4), the active metabolite of 5-(3-(2-(3-hydroxy-4-(3-(trifluoromethyl)phenyl)butyl)-5-oxopyrrolidin-1-yl)propyl)thiophene-2-carboxylate (PF-04475270), has an EC50 value of 1 nM in a human rc-EP4 assay (Prasanna et al., 2009). However, 8-aza-9-oxo prostanoids, with a 15,16-didehydro-15-methyl structure, switch from EP4 to EP2 agonist selectivity (Brugger et al., 2008). Finally, compound 12 (Fig. 4) in Blouin et al. (2010), which moves away from the classic prostanoid template, is an EP4 full agonist.
The first EP4 antagonist reported was 7-(5-(((1,1-biphenyl)-4-yl)methoxy)-2-(4-morpholinyl)-3-oxocyclopentyl)-4-heptanoic acid (AH 23848) (Coleman et al., 1994a), and it played a pivotal role in the early pharmacological definition of the EP4 receptor. AH 23848 was the prototype and has been overtaken by more selective and much more potent compounds, some of which contain a diaryl-acylsulfonamide as a key component of the scaffold: L-161982 (Fig. 5) bears structural resemblance to GW 627368 and BGC-20-1531 (Fig. 5) (Jones et al., 2009). Other EP4 antagonists are carboxylic acids, such as MK-2894 (Blouin et al., 2010) (Fig. 5). Finally, ER-819762 (Fig. 5) is an EP4 antagonist containing a novel spiro ring system (Chen et al., 2010).
e. Therapeutics.
The most prominent utility for EP4 antagonists is for treating inflammatory diseases, related hyperalgesia, and allodynia (Jones et al., 2009). In common with most prostanoid receptors, EP4 receptors have been implicated in carcinogenesis, and EP4 antagonists are shown to be effective in animal models (Table 8). It is noteworthy that Yanni et al., (2009) have presented data showing that EP4 receptor antagonists may be useful for treating neovascular eye disease.
Potential therapeutic application of EP4 antagonists (inflammation/pain is reviewed in Jones et al., 2009)
The potential uses of EP4 agonist are many and diverse (Table 9). There is enormous diversity: hearing loss (Hori et al., 2009), nephritis (Nagamatsu et al., 2006) glaucoma (Woodward et al., 2009), and myocardial infarction (Xiao et al., 2004). This wide array of EP4 agonist and antagonist activities portends a potential catalog of unwanted side effects, especially associated with oral systemic administration. Beneficial and deleterious side effects may occur in the same target tissue in response to EP4 receptor agonist administration. Although EP4 receptors are cardioprotective in ischemia-reperfusion injury (Xiao et al., 2004), they are likely to exacerbate hypertrophy that occurs as an adaptive response to cardiovascular disease (Mendez and LaPointe, 2005; Frias et al., 2007). EP4 receptors stimulate the BNP promoter and could result in an antifibrotic action in the heart (Qian et al., 2006), which may be compensatory. It seems difficult to assess the pros and cons of treating a patient with heart failure with an EP4 agonist. Added to all this, EP4 agonists are likely to lower blood pressure in humans, because they are well known as potent vasodilators and may also contribute to atherosclerotic plaque destabilization (Cipollone et al., 2005).
Potential therapeutic application of EP4 agonists
Potential cardiovascular and other safety risks are likely to be avoided where local drug delivery is feasible. An EP4 agonist may be useful in treating colitis (Nitta et al., 2002; Jiang et al., 2007), and a compound could perhaps be designed so that systemic absorption is limited. Set against this, EP4 receptors are implicated in colon carcinogenesis (Mutoh et al., 2002; Chell et al., 2006; Doherty et al., 2009), a highly undesirable side effect. In glaucoma therapy, where local topical drug administration is routinely employed, the highly efficacious ocular hypotensive effects produced by EP4 agonists are accompanied by ocular surface hyperemia and corneal neovascularization (Aguirre et al., 2009; Prasanna et al., 2009; Woodward et al., 2009). Although some redness of the eyes is not harmful, the patients do not necessarily want it. Perhaps the most therapeutically and commercially successful utility may be for the recently reported utility of EP4 agonists for sensorineural hearing loss (Hori et al., 2009). This would involve local therapy, probably an implant.
C. FP Receptors
1. Second Messenger Signaling.
The prostanoid FP receptor is predominantly Gq-coupled, with activation of the classic pathway. Thus, after PLCβ activation, there is PI turnover, with resultant diacylglycerol-mediated PKC activation and a Ca2+ transient signal in response to inositol trisphosphate formation (Nakao et al., 1993; Abramovitz et al., 1994; Ito et al., 1994; Sugimoto et al., 1994a; Woodward and Lawrence, 1994; Carrasco et al., 1996). Downstream of Gq, other protein kinases are activated and receptors transregulated. Elevated [Ca2+]i results in calmodulin-mediated myosin light-chain kinase activation (Ansari et al., 2003). PKC activates the Raf/MEK/MAP kinase signaling pathway (Chen et al., 1998; Bos et al., 2004; Husain et al., 2005; Xu et al., 2008). In addition to activating MAP kinases, a PKC/Ca2+-calcineurin-nuclear factor of activated T cells pathway has been implicated in PGF2α-mediated cell growth (Horsley and Pavlath, 2003; Sales et al., 2009). FP receptors may also activate MAP kinases via PLC-mediated phosphorylation of the epidermal growth factor (EGF) receptor (Sales et al., 2004).
The repertoire of FP receptor G protein coupling extends beyond Gq. It also includes activation of Rho via G12/G13 (Pierce et al., 1999). The FP receptor is also reported to couple to Gi (Melien et al., 1998; Hébert et al., 2005), providing an alternative route to the Raf/MEK/MAP kinase pathway (Bos et al., 2004). The early response gene Cyr61 is also up-regulated by FP receptor stimulation (Liang et al., 2003) by a pathway sequentially involving Ras/Raf signaling and Tcf transcription independent of MEK/ERK (Xu et al., 2009).
2. Distribution and Biological Functions.
FP receptors have a wide distribution and subserve many important functions. The importance of these functions is reflected by the fact that, among the prostanoid receptors, the FP receptor has been the most successful therapeutic target. It has prominent functions in reproduction. The FP receptor is highly expressed in ovarian tissue, with mRNA expressed exclusively in the corpus luteum (Sugimoto et al., 1994a). Although the FP receptor is central to corporal luteal regression and regulating the estrous cycle in farm animals (Coleman et al., 1994b), this does not seem to be the case in mice, because mice lacking FP receptors exhibited an unchanged estrous cycle and were fertile (Sugimoto et al., 1997). FP receptors are also present in the human corpus luteum (Narko et al., 1997; Väänänen et al., 1998), but their function is uncertain and certainly not dramatic. In mice lacking the FP receptor, parturition was abolished (Sugimoto et al., 1997). This has stimulated interest in the use of the FP antagonists for preventing preterm labor.
Functional expression of FP receptor in the myometrium mediating contraction has long been known (Senior et al., 1992; Carrasco et al., 1996). FP receptors are also expressed in the human endometrium, where PGF2α is biosynthesized in endometrial epithelial cells and causes cell proliferation (Asselin et al., 1997; Milne and Jabbour, 2003). FP receptor expression has been reported in human deciduae, and this has been suggested to contribute to parturition (Makino et al., 2007). By far the most prominent implication of FP receptors in disease is in uterine cancer, specifically endometrial adenocarcinomas. The role of the FP receptors in the progression of endometrial adenocarcinoma includes potentiation of angiogenesis by EGF receptor transactivation and induction of VEGF mRNA expression (Sales et al., 2005), alteration of adhesion, morphology, and migration (Sales et al., 2008, 2009).
In the CNS, PGF2α given intracisternally alleviates kainic acid-induced seizures potentiated by COX-2 inhibitors, suggesting that PGF2α behaves as an endogenous anticonvulsant (Kim et al., 2008). In contrast, FP receptors are claimed to significantly contribute to brain damage associated with focal brain ischemia (Saleem et al., 2009a). The colocalization of the FP receptor and PGF synthase 1 in the spinal cord suggest a role in pain, notably because of intense FP immunostaining in spinal laminae I and II of the dorsal horn (Suzuki-Yamamoto et al., 2009). Indeed, FP receptors mediate αB-methylene ATP-evoked allodynia (Kunori et al., 2009). These data are supported by the finding that spinal intrathecal administration of PGF2α produces allodynia by activating FP receptors (Muratani et al., 2003).
Renal expression of FP receptors is high (Sugimoto et al., 1994a; Saito et al., 2003). It is most abundant in the distal convoluted tubule and aquaporin-2-positive cortical collecting ducts (Saito et al., 2003; Hébert et al., 2005). This distribution is consistent with known FP-mediated effects on water and solute transport in these segments of the kidney. Renal FP receptors seem to signal via a pertussin toxin-sensitive mechanism (Hébert et al., 2005).
The FP receptor is widely distributed in the human eye (Schlötzer-Schrehardt et al., 2002). Despite high FP receptor expression in the corneal epithelium, ciliary epithelium, and iridial stroma and smooth muscle (Schlötzer-Schrehardt et al., 2002), FP agonists are functionally important only in those cells involved in aqueous humor outflow. Thus, ciliary smooth muscle and trabecular meshwork cells are targeted by FP-based therapeutics, without a litany of unwanted side effects. The success of FP agonists, in the form of ester prodrugs, as first-line antiglaucoma agents originated from the vision and determination of Bito (2001). The mechanism by which FP agonists lower intraocular pressure is now entirely understood. FP agonists increase aqueous humor outflow from the anterior segment of the eye, predominantly through the uveoscleral pathway (Stjernschantz et al., 2001; Toris et al., 2005). Effects on trabecular outflow have also been noted in human subjects (Ziai et al., 1993; Toris et al., 2007) and anterior segment preparations (Bahler et al., 2008). Uveoscleral outflow occurs via a widening of the interstitial spacing between ciliary muscle fiber bundles and a controlled remodeling that creates organized drainage channels (Richter et al., 2003). Consistent with a controlled remodeling process, tissue inhibitor of metalloproteinases regulation (Anthony et al., 2002), altered matrix metallopeptidase transcription, and translation products occur (Ocklind, 1998; Sagara et al., 1999; Gaton et al., 2001; Weinreb and Lindsey, 2002). Morphological changes are not restricted to the anterior portion of the ciliary body; they also occur in the trabecular meshwork (Richter et al., 2003). It is not surprising, therefore, that tissue inhibitor of metalloproteinases and matrix metallopeptidase regulation occurs in trabecular meshwork cells (Oh et al., 2006). Gene regulation associated with FP receptor stimulation, however, is not identical in ciliary muscle and trabecular meshwork with respect to genes that would influence aqueous humor dynamics (Zhao et al., 2003). More specifically related to tissue remodeling, Cyr61, connective tissue growth factor, epidermal growth factor receptor-1, and HIF-1α are up-regulated by FP receptor stimulation in ciliary muscle cells (Liang et al., 2003; Hutchinson et al., 2010). The full repertoire of genes that regulate ciliary body remodeling and increased uveoscleral outflow remains to be elucidated.
The side effects associated with FP receptor antiglaucoma therapy are also well understood. The major side effect of latanoprost is iridial hyperpigmentation. This results from a benign eumelanogenic effect, with larger and more mature melanosomes present in iridial melanocytes (Stjernschantz, 2001). Mechanistic investigation demonstrated that latanoprost does not directly stimulate melanocytes but rather activates FP receptors on neighboring fibroblasts (Smith-Thomas et al., 2004). This is quite different from dermal melanocytes, where FP receptor activation directly produces dendricity and increases tyrosinase activity (Scott et al., 2005). FP receptor-induced ocular surface hyperemia involves NO-mediated, endothelium-dependent vasodilation (Chen et al., 1995). Latanoprost also produces hypertrichosis of the eyelashes (Johnstone and Albert, 2002).
FP receptor stimulation has long been known to stimulate fibroblast proliferation. FP receptor activation in normal rat kidney fibroblasts adds a new dimension. Here FP receptor stimulation caused membrane depolarization of transformed normal rat kidney cells (Almirza et al., 2008). A positive feedback loop involving FP receptors and COX-2 up-regulation was proposed (Almirza et al., 2008). A most interesting recent report shows that FP receptors facilitate bleomycin-induced pulmonary fibrosis (Oga et al., 2009). FP, via a Rho kinase signaling pathway, produces fibrosis independent of TGFβ, which, until now, has been considered the dominant profibrotic mediator.
Rat ventricular cardiomyocytes undergo hypertrophic growth in response to FP receptor stimulation (Adams et al., 1996; Lai et al., 1996; Pönicke et al., 2000). PGF2α has also been claimed to mediate inflammatory tachycardia (Takayama et al., 2005). Studies on the contractility of the intact heart have revealed that FP receptors produce a negative inotropic effect, which could contribute to cardiac dysfunction (Jovanović et al., 2006). PGF2α seems to have pathophysiological consequences for the cardiovascular system, because it also elevates blood pressure and promotes atherosclerosis (Yu et al., 2009b). PGF2α also increases skeletal muscle cell growth (Horsley and Pavlath, 2003).
Most prostanoid receptors have been implicated in cancer, and FP is no exception. PGF2α stimulates the motility and invasiveness of colorectal tumor cells, with potency equal to that of PGE2 (Qualtrough et al., 2007). PGF2α-mediated COX-2 up-regulation has been suggested to potentiate tumorigenesis with respect to endometrial adenocarcinoma (Jabbour et al., 2005). Numerous early studies on PGF2α have shown a mitogenic effect on fibroblast cell lines, and this was often suggested as contributory to tumor growth (De Asua et al., 1975).
Studies on murine osteoclast development suggest that PGF2α would inhibit bone resorption (Kamon et al., 2008). FP receptor expression has been detected in primary human osteoblasts (Sarrazin et al., 2001), and in UMR-106 cells, functional FP receptors were identified (Yamaguchi et al., 1988). Finally, FP receptor activation potently inhibits adipose cell differentiation according to studies on adipocyte precursors obtained from murine inguinal fat pads (Serrero and Lepak, 1997).
3. Gene Deletion Studies.
Sugimoto et al. (1997) generated a line of FP-deficient mice by replacing the second exon of the FP gene with β-galactosidase- and neomycin-resistance genes, and this line has been used in all studies below. PGF2α is known as a luteolytic substance in animals. Initial analysis of this line of mice revealed that FP(−/−) female mice did not show any abnormality in the luteal cycle but exhibited parturition failure (Sugimoto et al., 1997). The failure was apparently due to the lack of labor, and these mice did not exhibit prepartum decline in the plasma progesterone level. Ovariectomy 24 h before the expected term decreased the progesterone level and induced normal parturition in FP(−/−) dams. Given the high expression of FP in the corpora lutea that produces progesterone and the luteolytic action of PGF2α, these findings suggest that the PGF2α-FP pathway triggers parturition by inducing luteolysis in the ovary.
Several PGF analogs are used as antiglaucoma drugs. FP(−/−) mice were used to examine the identity of the receptor mediating IOP-lowering activity of these PG analogs. These mice were resistant not only to FP agonists such as latanoprost and travoprost but also to bimatoprost and unoprostone, PG analogs suggested not to bind to FP with high affinity, although these compounds lowered IOP to a comparable extent in wild-type C57BL/6 mice (Crowston et al., 2004, 2005; Ota et al., 2005). These results indicate that FP mediates IOP-lowering action of all of these PG analogs at least in mice. Whereas FP mediates potent reduction of IOP in response to exogenously applied FP agonists, FP(−/−) mice showed no abnormality in diurnal variation of IOP compared with wild-type mice, indicating that the PGF2α-FP pathway is not involved in physiological regulation of IOP (Crowston et al., 2007). Furthermore, administration of ONO-AE1-259 (Fig. 4), an EP2 agonist, and ONO-AE1-329 (Fig. 4), an EP4 agonist, induced reduction of IOP equally well in wild-type and FP(−/−) mice, indicating the presence of a PG-dependent pathway different from that of FP in lowering IOP (Saeki et al., 2009).
Systemic inflammation induces many adaptive symptoms, one being tachycardia. Takayama et al. (2005) used mice deficient in individual prostanoid receptors and examined their involvement in this inflammatory tachycardia. They first found that PGF2α and a TP agonist, I-BOP, could elevate the heart rate of mice through FP and TP, respectively, and that this action was exerted locally in the atria. They then showed that tachycardia induced by systemic administration of lipopolysaccharide was abrogated partially in either TP(−/−) or FP(−/−) mice and completely in mice deficient in both FP and TP receptors. Inflammatory tachycardia was believed to result from increased sympathetic discharge, and the results by Takayama et al. (2005) have changed this traditional view. The function of PGF2α in the cardiovascular system was further elucidated by the use of FP(−/−) mice (Yu et al., 2009b). Systolic blood pressure in FP(−/−) mice was significantly lower than that of wild-type mice, with significantly lower plasma concentrations of renin and angiotensin-1 in FP(−/−) mice. To explore the basis of this phenotype, they examined FP receptor localization in the kidney and found that FP is expressed in the preglomerular artery (Yu et al., 2009b), in addition to previously identified localization in distal tubules and collecting ducts (Saito et al., 2003); stimulation of FP increased renin mRNA expression in isolated JG cells, though it did not stimulate renin release. In addition, FP is expressed in the medial smooth muscle layer of resistant arterioles, and infusion of PGF2α elevated blood pressure in wild-type but not in FP(−/−) mice. Yu et al. (2009b) further cross-mated FP(−/−) with mice deficient in the LDL receptor, and reported that atherosclerosis was significantly attenuated on the FP(−/−) background. Although macrophage infiltration and inflammatory cytokine expression in atherosclerotic plaques were diminished in FP(−/−) mice, it remained unclear how FP is involved in atherogenesis. Saleem et al. (2009a) subjected FP(−/−) mice to transient brain ischemia-reperfusion by reversible ligation of the middle cerebral artery. They found that the loss of FP does not alter physiological parameters such as cerebral blood flow, PaO2, PaCO2, mean arterial blood pressure, and body temperature before, during, or after brain ischemia. Nonetheless, FP(−/−) mice exhibited significantly less neurological deficit and smaller infarct volume than wild-type mice. To examine whether this difference is due to excitoneurotoxicity associated with transient ischemia-reperfusion, they then injected NMDA into the striatum unilaterally and found that FP(−/−) mice exhibited significantly smaller infarct volumes than wild-type mice again under these conditions, indicating that FP somehow regulates the excitation of glutamatergic neurons after ischemic injury. Consistent with these findings, they showed that administration of latanoprost widened the infarction and worsened the deficit. Finally, Oga et al. (2009) subjected mice deficient in individual prostanoid receptor to bleomycin-induced pulmonary fibrosis and found significant suppression of fibrosis in FP(−/−) mice. They found that pulmonary inflammation comparable with that in wild-type mice was induced by bleomycin, but subsequent fibrosis was suppressed in FP(−/−) mice. Microarray analysis revealed significant attenuation of the induction of genes associated with fibrosis, such as various isoforms of collagen and other matrix proteins in FP(−/−) mouse lung, and FP stimulation in cultured lung fibroblasts facilitated cell proliferation and collagen production in vitro. It is noteworthy that the loss of FP receptors did not affect expression and activation of TGF-β, a critical cytokine in fibrosis, and PGF2α and TGF-β contributed additively to fibrosis. On the basis of these results, the authors suggested that PGF2α functions as a mediator of fibrosis independent of TGF-β, and FP may be a drug target for idiopathic pulmonary fibrosis in humans.
4. Agonists and Antagonists.
Early studies established the high FP selectivity of the (racemic) 16-m-trifluoromethyl-phenoxy analog of PGF2α (ICI-81008, fluprostenol; Dukes et al., 1974), particularly its minimal TP agonism (Welburn and Jones, 1978). Fluprostenol continues to be the FP agonist of choice and is commercially available as the more active (+)-enantiomer. The 16-m-chlorophenoxy analog of PGF2α (cloprostenol) is the most potent FP agonist reported but retains considerable EP3 agonism; change to a 3-hydroxy-tetrahydofuran ring with shift of the 5,6-cis bond to the 4 position as in AL-12180 (Fig. 6) improved the selectivity ratio (Sharif et al., 2006). 13,14-Dihydro-17-phenyl PGF2α (latanoprost-free acid) also has high FP selectivity, showing 22-fold less EP1 agonism than 17-phenyl PGF2α (Ungrin et al., 2001). Introduction of a 2-indanyl group into the ω-chain has resulted in claims for FP antagonism (Griffin et al., 1999; Sharif et al., 2000; AL-3138, AL-8810), although partial agonism seems to be a better description (Woodward et al., 2007). Structures are given in Fig. 6. Woodward et al. (2000) reported that although the C1-alcohol and C1-methyl ether analogs of PGF2α were very weak agonists at FP-receptors of cat and human, they showed high potency on the cat lung parenchyma preparation, an action that could not be ascribed to other known prostanoid receptors. Subsequent studies have suggested the existence of a new receptor type, the prostamide F receptor, which recognizes carboxyl, alcohol, ethanolamide, and alkylamide moieties at C1, PGF2α-ethanolamide (Fig. 6) being a potential natural ligand (Woodward et al., 2008). In cat iris sphincter digests, 17-phenyl PGF2α-ethylamide (bimatoprost) activated one set of cells and 17-phenyl PGF2α and PGF2α, another (Spada et al., 2005).

Structures of agonists for the prostanoid FP receptor and the corresponding prostamide F receptor. PGF2α potently activates both receptors, whereas its C1-ethanolamide activates only the prostamide F receptor. a, partial agonist.
C-1 amine and amide derivatives of PGF2α show weak FP antagonism in some systems, but their utility is severely limited (see Jones et al., 2009). 15-Indanyl-ω-pentanor PGF analogs, such as AL-3138 and AL-8810 (Fig. 7), represent improvements (Griffin et al., 1999; Sharif et al., 2000). However, AL-8810 has modest affinity for FP receptors (IC50, 8.7 μM in human ciliary muscle cells; Sharif et al., 2006) and blocks TP receptors in mouse uterus (Hutchinson et al., 2003), pig ciliary artery (Vysniauskiene et al., 2006), and human recombinant receptor assays (Krauss and Woodward, unpublished). Partial block of epidermal growth factor-induced contraction of guinea pig trachea by AL-8810 (Schaafsma et al., 2005) may be explained by TP antagonism rather than the proposed FP antagonism. Moreover, both PGF analogs often exhibit FP partial agonism (Griffin et al., 1999; Hutchinson et al., 2003), and AL-8810 was even a full agonist in the cat iris preparation, an action not blocked by a prostamide F antagonist (Woodward et al., 2007). More potent, selective prostamide F antagonists, such as AGN 211334 (Fig. 7) have more recently been reported (Wan et al., 2007; Woodward et al., 2008).

Structures of FP antagonists and the prostamide antagonist (AGN 211334).
Peptides of the THG series (Chemtob and Peri, 2006; Peri et al., 2009) block PGF2α-induced responses, but this may not necessarily involve competition for the FP receptor. The octapeptide THG-113.31 [Ile-Leu-Gly-His-(d-Cit)-Asp-Tyr-Lys] insurmountably blocked PGF2α-induced contraction of pig retinal blood vessels, while having minimal effect on contraction to 17-phenyl PGE2, U-46619, phenylephrine, Angiotensin II and endothelin-1. However, the affinity of TGH-131.31 for the human recombinant FP receptor was poor (∼13% at 10 μM). Moreover, TGH-131.31 (10 μM) weakly antagonized contraction of sheep myometrium induced by PGF2α and had no effect on PGF2α-induced contraction of human pregnant myometrium but inhibited spontaneous and oxytocin-induced contractions at much lower concentrations (Friel et al., 2005). At 10 to 50 μM, TGH-113.31 enhanced BKCa channel opening in human uterine myocytes, which was reversed by iberiotoxin (Doheny et al., 2007). Simpler peptides (e.g., TGH-113.824; Fig. 7) also block PGF2α-induced contraction (Peri et al., 2009).
5. Therapeutics.
By far the greatest therapeutic success of prostanoid pharmacology is the use of FP receptor agonist prodrugs for the treatment of glaucoma. Latanoprost was the first in its therapeutic class (Bito, 2001; Stjernschantz, 2001). This was followed by the isopropyl ester of fluprostenol, named travoprost (Hellberg et al., 2001) and tafluprost (Takagi et al., 2004; Cirillo et al., 2007). Comparisons between these drugs are widely available and are not reviewed here.
An alternative therapeutic use for FP agonists emerged from studies on glaucoma: hair growth. Latanoprost and related compounds cause eyelash growth (Johnstone and Albert, 2002). Studies in the stump-tailed macaque model of androgen-induced scalp alopecia showed latanoprost to be effective (Uno et al., 2002). This opportunity does not seem to have been commercially realized. The reasons for this are obscure. For this use, the domain of pharmacocosmetics (Woodward et al., 2008) is accessed. In the domain of cosmetics, unlike treating pain, inflammation, cancer, etc., the options are not restricted to drug intervention and surgery. Cosmetic aspirations can be instantly gratified by colorants, surgery, or implantation/replacement. This may have discouraged development of latanoprost for male pattern baldness. However, acceptance of cosmetic intervention and instant results is widely accepted by women but not by men. A gradual improvement in natural scalp hair is likely to be more socially acceptable to men because it would avoid derision from friends and colleagues. Regrowth of natural scalp hair would take a long time, because anaphase extends over years. In addition, like any other disease, early intervention is essential. These may have been major discouraging factors. On the other hand, the psychological impact of male pattern baldness cannot be underestimated, and balding men are typically highly motivated and inclined toward subtle and natural intervention. FP receptor expression has been detected in the human dermal papillae (Colombe et al., 2008), which supports a favorable outcome for human clinical investigation. Fat reduction/antiobesity is a further potential pharmacocosmetic/medical use of FP agonists, but this is still at the adipocyte precursor stage essentially (Serrero and Lepak, 1997).
With the exception of osteoporosis, the effects of FP receptor activation are pathophysiological, and antagonist therapy would be the requirement. The major focus has been the prevention of preterm labor. These antagonists include the peptide THG-113.31 and derivates (Olson, 2005; Chemtob and Peri, 2006; Jones et al., 2009). The non-PG structure FP antagonist AS-604872 (Fig. 7) (Cirillo et al, 2007) seems to have a better commercial prospect as an orally bioavailable small molecule. It showed no agonism in a human FP receptor-inositol phosphate assay (IC50, 47 nM for PGF2α). In vivo, AS-604872 (1–30 mg/kg i.v.) inhibited PGF2α-induced uterine contraction in the nonpregnant rat: inhibition of oxytocin-induced contractions was slight (Chollet et al., 2007). These apart, no serious attention seems to have been given to FP antagonist design. This may change in light of reports implicating FP receptors in cardiovascular disease (Yu et al., 2009b) and fibrosis (Oga et al., 2009).
D. IP Receptors
1. Second Messenger Signaling.
The IP receptor has long been known to be both Gs- and Gq-coupled, resulting in increased cAMP formation, PI turnover, and Ca2+ signaling (Coleman et al., 1994; Namba et al., 1994; Narumiya et al., 1999). IP receptor activation of the cAMP-PKA pathway seems to be exclusively responsible for some prostacyclin-mediated events (Nasrallah et al., 2001; Ritchie et al., 2004; Kamio et al., 2007; Muja et al., 2007). Solution structure studies indicate that the first and third intracellular loops of the IP receptor are in contact with C-terminal residues of Gαs to initiate cAMP signaling (Zhang et al., 2006a; Zhang et al., 2006b). N-glycosylation is involved in both adenylyl cyclase and inositol phosphate formation (Zhang et al., 2001).
IP receptors are not unique in activating dual or multiple signaling but seem to be among the best studied. Gi and Gq coupling to IP receptors has been reported to be dependent on Gs coupling as a prerequisite, the key event being phosphorylation of Ser357 of the IP receptor by PKA (Lawler et al., 2001). This mechanism is consistent with investigations invoking both cAMP and cAMP-independent mechanisms in many instances. These include MaxiK channel activation (Yamaki et al., 2001) and STAT3 phosphorylation (Lo et al., 2008), in addition to straightforward analysis of dose-dependent effects on cAMP formation (Accomazzo et al., 2002). PKA-mediated switching from Gs to Gq and/or Gi coupling, however, does not seem to be a universal phenomenon (Chow et al., 2003). G-protein coupling seems to be cell-specific. No evidence for Gi coupling could be found in NG108-15 and SK-N-SH cells and in Chinese hamster ovary or human embryonic kidney 293 cells expressing recombinant IP receptors (Chow et al., 2003). In marked contrast, Gi-coupled IP receptors were found exclusively in human erythroleukemia HEL cells with respect to STAT1 and STAT3 phosphorylation (Lo et al., 2006) and rat medullary thick ascending limb cells (Hébert et al., 1998). Finally, but beyond the intended scope of this review, there are reports claiming that prostacyclin interacts with peroxisome proliferator-activated receptors to exert a portion of its activities (Lim and Dey, 2002; Ali et al., 2006).
2. Distribution and Biological Functions.
The role of prostacyclin in cardiovascular homeostasis is well publicized. Thus, COX-2 inhibitors may present a cardiovascular hazard by depressing prostacyclin production by vascular endothelial cells without a concomitant reduction in platelet COX-1-derived TxA2. This results in a potentially deleterious effect on thrombosis and blood pressure and accelerated atherogenesis (Cheng et al., 2002; Dogné et al., 2005; Wang et al., 2005). Of particular relevance is that COX-2 expression and PGI2 production in human coronary arterial endothelial cells may be increased by proinflammatory stimuli (Tan et al., 2007). These findings imply that local PGI2 produced in the coronary vasculature may directly counteract the vasoconstriction and platelet aggregation production by TxA2 during episodic vascular insult. The cardioprotective action of prostacyclin has been proposed to extend to preventing atherosclerosis, intimal hyperplasia, and restenosis (Fetalvero et al., 2007).
Vascular smooth muscle cells are not terminally differentiated and can proliferate and further differentiate. IP receptors inhibit vascular smooth muscle cell proliferation (Lin et al., 2008) by inhibiting G1-to-S-phase progression (Fetalvero et al., 2007) and inducing apoptosis (Li et al., 2004). Prostacyclin also induces genes characteristic of a contractile phenotype and inhibits vascular smooth muscle cell migration (Blindt et al., 2002; Fetalvero et al., 2007). Vascular endothelial cells are, in addition to vascular smooth muscle cells, an important source of prostacyclin, which is induced by laminar shear stress (Di Francesco et al., 2009). Beyond prostacyclin effects on vascular smooth muscle and endothelial cells, the inhibitory effects on platelet aggregation and its vasodilator properties should not be understated. IP receptor stimulation increases retinal, choroidal (Mori et al., 2007), and coronary blood flow (Gwóźdź et al., 2007). In ApoE-deficient mice, IP receptors conferred protection against the initiation and progression of atherogenesis by limiting platelet activation and leukocyte-vascular endothelial cell interaction (Kobayashi et al., 2004). Mice lacking IP receptors showed augmented cardiac hypertrophy in response to pressure overload (Hara et al., 2005) and increased myocardial infarct size after ischemia-reperfusion injury (Xiao et al., 2001), suggesting a cytoprotective effect on cardiomyocytes independent of platelet and neutrophil inhibition. IP (−/−) mice also develop salt-sensitive hypertension and cardiac fibrosis, in addition to cardiac hypertrophy (Francois et al., 2005).
Continuing the subject of systemic hypertension, decreased susceptibility to renovascular hypertension was reported in mice lacking IP receptors (Fujino et al., 2004). Prostacyclin, therefore, indirectly produces hypertension in the two-kidney, one-clip hypertension model: the mechanism seems to involve increased renin mRNA expression and production according to studies on cicaprost in juxtaglomerular cells (Fujino et al., 2004). In addition to juxtaglomerular cells, IP message is also present in the renal cortex, outer and inner medulla, and inner medullary collecting duct, according to reverse transcription-polymerase chain reaction (Nasrallah et al., 2001). Localization studies using in situ hybridization detected IP receptor mRNA in the tubules of the inner and outer medulla, the vasculature, and in the arteries, glomeruli, and tubules of the cortex (Nasrallah et al., 2001). Renal IP receptor distribution in murine species does not necessarily correlate with that in humans (Nasrallah and Hébert, 2005). No obvious pathological renal condition is apparent in IP(−/−) mice. Prostacyclin, however, may also be involved in renal hypertrophy, fibrosis, and apoptosis (Nasrallah and Hébert, 2005).
The prostanoid IP receptor plays a prominent role in edema formation, hyperalgesia, and pain (Bley et al., 1998; Hata and Breyer, 2004). Nociceptive responses in the acetic acid writhing model are virtually abolished in mice lacking IP receptors (Murata et al., 1997). In models of rheumatoid arthritis, such as carrageenin-induced paw edema and collagen antibody-induced arthritis (Murata et al., 1997; Ueno et al., 2000; Honda et al., 2006; Pulichino et al., 2006), a pronounced reduction is observed in IP (−/−) mice. Likewise, pleurisy induced by carrageenin (Yuhki et al., 2004) and zymosan (Yuhki et al., 2008) is attenuated in mice lacking IP receptors. Results obtained with IP antagonists in rat models of pain and inflammation (Bley et al., 2006; Pulichino et al., 2006) are consistent with results obtained in the IP(−/−) mice studies. The involvement of IP receptors in pain and hyperalgesia are supported by IP expression studies (Bley et al., 1998; Doi et al., 2002) and functional studies on neurons conducting at C velocity (Smith et al., 1998). Studies on dorsal root ganglia support an IP receptor role in sensitization of sensory neurons (Smith et al., 1998; Rowlands et al., 2001; Nakae et al., 2005).
IP receptor expression in the thymus and spleen was reported more than a decade ago (Narumiya et al., 1999). Since then, some insight into IP receptor involvement in lymphocyte function has emerged. IP receptors tend to suppress Th2-mediated responses (Hata and Breyer, 2004). Thus, IP receptor stimulation inhibits Th2 cytokine (IL-4, IL-10, IL-13) and Th1 cytokine (IFNγ) production (Zhou et al., 2007). Plasmacytoid dendritic cells, which are believed critical for controlling adaptive immunity, also express functional IP receptors, which suppressed Toll-like receptor-mediated TNFα and INFγ production and enhanced IL-10 production (Hung et al., 2009). IP signaling has been claimed to prevent recruitment of Th2 cells into airways in an asthma model (Jaffar et al., 2007), but the use of highly unstable PGI2 confounds interpretation. Nevertheless, because allergic airway and cutaneous inflammation involving Th2 cells in the same species is augmented in IP receptor-deficient mice (Takahashi et al., 2002), it seems correct that IP receptors inhibit Th2 cell function. Further support is provided by a study that implicates prostacyclin in the anti-inflammatory activity of 1-methylnicotinamide in experimental contact hypersensitivity (Bryniarski et al., 2008). Finally, in the macrophage cell line Raw 264.7, IP activation via LPS-induced prostacyclin production generates VEGF (Park et al., 2007). Because this pathway involved Akt signaling, it is relevant to angiogenesis and cancer. IP receptors up-regulate angiogenic genes in the human endometrium (Smith et al., 2006).
IP receptors have been functionally characterized in the human myometrium (Senior et al., 1992). Prostacyclin synthase and IP receptor expression are increased during the menstrual phase (Battersby et al., 2004), indicating a possible role in normal and/or dysfunctional menstruation. Perhaps a better case could be made for PGI2 participation in pregnancy and labor by up-regulating contractile proteins and connexin 43 that could prime the myometrium for parturition (Fetalvero et al., 2008; Taggart et al., 2008). IP receptors have also been implicated in preimplantation embryo development (Huang et al., 2007a) and may contribute to embryo transport by decreasing the amplitude of oviductal contractility (Arbab et al., 2002).
Prostacyclin receptor deletion has been shown to aggravate hippocampal neuronal loss caused by ischemia (Wei et al., 2008) and cortical cell loss after traumatic brain injury (Lundblad et al., 2008). These experiments indirectly suggest that IP receptors could promote neuronal survival. The ulcerogenic response to ischemia-reperfusion was increased in severity in IP(−/−) mice, suggesting that IP receptors play a crucial role in gastric mucosal defense in vascular injury (Kotani et al., 2006). IP involvement in adaptive cytoprotection produced by mild irritants seems controversial (Boku et al., 2001; Takeuchi et al., 2001b).
3. Gene Deletion Studies.
Two lines of IP-deficient mice were generated independently (Murata et al., 1997; Cheng et al., 2002). A study on IP(−/−) mice (Kotani et al., 2006) showed that, although these animals develop and age normally, they manifest an increased thrombotic tendency in the presence of endothelial damage. These findings confirmed the long-held view of PGI2 as an endogenous antithrombotic agent and suggest that this antithrombotic system is activated in response to vascular injury. Indeed, Cheng et al. (2002) examined the interplay between IP and TP signaling in response to vascular injury by subjecting IP(−/−) mice and TP(−/−) mice to vascular injury by a balloon catheter. They found that IP deficiency increased, whereas TP deficiency decreased, injury-induced vascular proliferation and platelet activation. They further showed that the augmented response apparent in IP(−/−) mice was abolished by ablation of TP. Such augmented intimal hyperplasia was also seen in IP(−/−) mice subjected to common carotid artery ligation and transplant arteriosclerosis (Rudic et al., 2005). Furthermore, Xiao et al. (2001) subjected IP-deficient mice to cardiac ischemia-reperfusion injury and found that IP(−/−) mice exhibited a significantly larger size of myocardial infarct than wild-type mice, whereas TP(−/−) showed no difference, suggesting that receptor PGI2-IP signaling exerts a protective action during these conditions. As for the role of IP in chronic vascular disease, Kobayashi et al. (2004) examined progression of atherosclerosis in ApoE(−/−)/IP(−/−) double-knockout mice and found that atherosclerosis was accelerated in these mutant animals, despite the fact that they manifested similar plasma cholesterol and triglyceride concentrations. The lumen of the innominate artery was almost completely occluded in 45-week-old ApoE(−/−)/IP(−/−) mice. Mice deficient in IP do not show abnormalities of blood pressure under basal conditions. There are two reports suggesting the involvement of IP in development of hypertension under some conditions. Fujino et al. (2004) examined the contribution of prostanoids to the development of this condition by subjecting mice deficient in either IP or each of the four EP subtypes to a two-kidney, one-clip model of renovascular hypertension. They found that hypertension in this model was ameliorated in IP-deficient mice but not in any of the EP-deficient animals. Consistent with these observations, plasma renin activity, the abundance of renin mRNA in the kidney, and the plasma concentration of aldosterone were all substantially reduced in the IP knockout animals compared with those in the wild type. Given that IP is expressed in the afferent glomerular arterioles, that expression of the renin gene is expanded to these arterioles in response to reduced perfusion of the kidney, and that PGI2 induces renin release from cultured juxtaglomerular cells in vitro, these researchers suggested that PGI2-IP signaling directly stimulates renin release. Alternatively, such signaling may regulate the perfusion pressure of the juxtaglomerular apparatus locally and induce renin release indirectly. Francois et al. (2005) found that IP-deficient mice fed on a high-salt (6% NaCl) diet developed a significantly larger increase in blood pressure than wild-type mice without alteration in renin and aldosterone levels, and concomitantly developed cardiac hypertrophy and fibrosis. Their finding on salt-induced hypertension in IP(−/−) is consistent with that of Watanabe et al. (2005), who found that with a high-salt diet, systolic blood pressure in female IP(−/−) mice gradually increased, whereas that in the IP(+/+), TP(−/−), or TP(+/+) mice remained unchanged. On the other hand, Hara et al. (2005) examined cardiac hypertrophy and fibrosis in response to pressure overload. They subjected mice deficient in each prostanoid receptor to banding of the transverse aorta and found augmented hypertrophy and fibrosis only in IP(−/−) mice. Thus, a protective role of IP signaling in cardiac hypertrophy and fibrosis is found in mice on two different backgrounds and different models.
There are several studies using IP(−/−) mice to determine the role of IP in pain sensation and inflammatory swelling. Murata et al. (1997) found that in IP-deficient mice, the acetic acid writhing response was reduced to a level similar to that observed in wild-type mice treated with the COX inhibitor indomethacin. The capsaicin receptor TRPV1 is a nociceptor for heat and pH. Moriyama et al. (2005) found that thermal hyperalgesia in response to PGI2 is absent not only in IP(−/−) mice but also in TRV1(−/−) mice. They further found that capsaicin-activated current in dorsal root ganglia neurons was augmented by PGI2 as well as an IP agonist, and this augmentation was absent in neurons from IP(−/−) mice. It is noteworthy that the TRV1 augmentation is seen at a rather high concentration of PGI2, 1000 nM. As for inflammatory swelling, Murata et al. (1997) subjected IP-deficient mice to the carrageenin-induced paw swelling model and found that inflammatory swelling was reduced by ∼50% in these animals, an effect similar in magnitude to that achieved by treatment of wild-type mice with nonsteroidal anti-inflammatory drugs. Yuhki et al. (2004) showed that IP, as well as EP2 and EP3 receptors, mediate exudate formation in carrageenin-induced mouse pleurisy, suggesting that PGE2 and PGI2 elicit inflammatory responses in a context-dependent manner (i.e., one dependent on the type of stimulus and site in the body) and that their contribution may also change during the course of inflammation. The inflammatory action of IP is not limited to inflammatory swelling exerted through regulation of the peripheral circulation in acute inflammation. Honda et al. (2006) examined the role of prostanoids by back-crossing mice deficient in each prostanoid receptor on a DBA background and subjecting them to collagen-induced arthritis. They found that, whereas the incidence of arthritis was unaffected, the extent and progression of this condition were markedly suppressed in IP-deficient mice as well as in EP2-deficient mice treated with an EP4 antagonist. These findings thus indicated that the PGI2-IP signaling and PGE2 signaling at EP2 and EP4 receptors mediate joint inflammation in this model. Further analysis revealed that both PGI2 and PGE2 pathways regulate expression of arthritis-related genes, including those for IL-6, vascular endothelial growth factor-A, and RANKL, in synovial fibroblasts and thereby contribute to arthritic inflammation, bone destruction, and pannus formation. On the other hand, Pulichino et al. (2006) evoked arthritis by administering collagen antibodies to their IP(−/−) mice on a C57BL/6 background and found that arthritis was almost completely suppressed in this line of mice. They further used a newly developed IP antagonist [N-[4-(imidazolidin-2-ylideneamino)-benzyl]-4-methoxy-benzamide], administered it into heterozygous IP(+/−) mice before the antibody injection or after the onset of arthritis, and examined its prophylactic or therapeutic effects. They found that although the prophylactic administration of the IP antagonist exerted suppression of arthritis in IP(+/−) mice comparable with that in IP(−/−) mice, it showed no effects when administered after the disease onset. These results suggest that the PGI2-IP signaling functions as part of the process where self-reactive antibodies trigger the disease. However, whether this is the same mechanism as that identified by (Honda et al., 2006) remains unknown. In addition, starting with the finding that COX-2-deficient mice show exaggerated fibrotic response in bleomycin-induced pulmonary fibrosis, Lovgren et al. (2006) subjected mice deficient in either EP2, EP4, or IP to this model. They found that only IP-deficient mice exhibited significantly enhanced fibrosis and suggested that COX-2-derived PGI2 exerts a protective action against fibrosis in this model.
PGI2 has been shown to exert a protective effect on the gastric mucosa in response to injurious stimuli. IP-deficient mice have also been used to examine this phenomenon. Boku et al. (2001) and Arai et al. (2003) examined the role of IP in releasing CGRP in the stomach in response to a mild stimulant, 1 M NaCl, or capsaicin, which exerts protection to ethanol. They found that release of CGRP to both stimuli is impaired in IP(−/−) mice, and coadministration of capsaicin and beraprost enhanced the release, suggesting that endogenous PGI2-IP signaling functions to release CGRP for gastric protection. Furthermore, Terashima et al. (2009) used IP(−/−) mice and showed that iloprost can decrease histamine-stimulated acid secretion in the stomach in an IP-dependent manner. They further found that this IP-mediated decrease is dependent on the somatostatin SST2 receptor. On the basis of these findings, they suggested that PGI2 receptors may mediate somatostatin release in the gastric mucosa, which in turn suppresses acid secretion. Given expression of IP in various types of immune cells, including macrophages and T cells, Takahashi et al. (2002) subjected IP-deficient mice to the OVA-induced asthma model and examined its role. They found that IP(−/−) mice exhibited substantially higher plasma concentrations of antigen-specific and total IgE, indicating that PGI2-IP signaling is somehow involved in sensitization to IgE production. Finally, IP(−/−) mice were used to distinguish some actions of PGI analogs on IP and PPARβ. Ali et al. (2006) used lung fibroblasts from IP(−/−) mice and those from PPARβ(−/−) mice, examined inhibitory effects of treprostinil on their proliferation, and found that this activity was lost in PPARβ(−/−) cells but not in IP(−/−) cells.
4. Agonists and Antagonists.
The upper row in Fig. 8 shows stabilization of the vinyl ether of PGI2 by carbon replacement (iloprost), electron withdrawal with fluorine (AFP-07), and conjugation with or within aromatic moieties (beraprost, taprostene). In each case, orientation of the α-carboxyl terminus away from the ω-chain, which is crucial to potent IP agonism, is maintained; a more detailed treatment may be found in Wise and Jones (1996). A 16-methyl/18.19-triple bond structure has often been favored for the ω terminus. Iloprost and AFP-07 also show potent EP1 agonism and to a lesser extent EP3 agonism, whereas cicaprost is more selective (Dong et al., 1986; Lawrence et al., 1992; Abramovitz et al., 2000; McCormick et al., 2010) and is the preferred choice for an IP standard agonist. Taprostene is a IP partial agonist (Jones and Chan, 2001, 2005).
Structures of agonists for the prostanoid IP receptor. PGI2 (prostacyclin), the most active natural agonist, is shown in the circle. The diphenyl-heteroatomic unit critical to IP agonism in nonprostacyclin mimetics of prostacyclin is shown in blue. a, partial agonist. b, prodrug for MRE-269.
EP-157 (Fig. 8) is a PGH2 analog exhibiting both TP antagonism and IP agonism (Armstrong et al., 1986); the diphenylmethyl-heteroatomic group is critical to the latter activity (Jones et al., 1993). The related compounds in Fig. 4 also possess a similar or 1,2-diphenylethyl group and have been referred to as “nonprostanoid prostacyclin mimetics” (Meanwell et al., 1994). BMY-45778 (Fig. 8) is the most potent agent within this subclass (Jones et al., 1997; Seiler et al., 1997). Caution is necessary in using these agonists to characterize IP receptors because of their high lipophilicity, partial agonism (octimibate; Merritt et al., 1991a) and ability to inhibit Gq/PLC-driven effects (Chow et al., 2003). ONO-1301 (Kondo et al., 1995 also inhibits TxA2 synthase because of the presence of a 3-pyridyl group). MRE-269 is obtained by hydrolysis of the methylsulfonamide moiety in the prodrug NS-304 (Kuwano et al., 2007, 2008).
Chemical library screening was the starting point for two classes of selective IP antagonist (Bley et al., 2006): RO-1138452 is a 2-(phenylamino)-imidazoline and RO-3244019 is an N-substituted phenylalanine (Fig. 9). In addition to its high affinity for human native (platelet) and recombinant IP receptors (pKi, 9.3 and 8.7, respectively), RO-1138452 also binds to platelet-activating factor (pKi, 7.9) and imidazoline (pKi, 8.3) receptors (Bley et al., 2006). RO-1138452 exhibited surmountable antagonism in functional studies on blood vessel preparations from human, rabbit, and guinea pig, with pA2 values in the range 8.1 to 8.4 (Jones et al., 2006). Higher concentrations of RO-1138452 slightly suppressed the cicaprost maximum response, probably because of the EP3 agonist action of cicaprost. In contrast, insurmountable antagonism was found for RO-1138452 in a chemokine release assay involving human airway epithelial cells (Ayer et al., 2008). RO-1138452 inhibition of IP agonist-induced cAMP response element-dependent transcription was not reversed after a 20-h “washout” period. It was proposed that allosterism or a state of antagonist hemiequilibrium and may underlie this profile (Ayer et al., 2008).
Structures of IP receptor antagonists.
5. Therapeutics.
Prostacyclin infusion has been in medical use for some time for treating pulmonary hypertension (Wise and Jones, 1996; Olschewski et al., 2004; Gryglewski 2008; Mubarak, 2010). The stable prostacyclin analog iloprost provides several practical advantages for intravenous therapy and may be given by aerosol (Olschewski et al., 2004; Mubarak, 2010). Treprostinil was developed for subcutaneous administration, and beraprost is an orally active prostacyclin mimetic (Olschewski et al., 2004; Mubarak, 2010). Prostacyclin and its analogs are also useful for treating advanced critical limb ischemia that may occur in Buerger's disease, Raynaud's syndrome, and atherosclerosis (Zardi et al., 2005; Gryglewski, 2008). New IP agonists continue to be developed, the latest being NS-304 (Fig. 8). This is both long-acting and has better selectivity than beraprost or iloprost and represents better future therapy for pulmonary arterial hypertension (Kuwano et al., 2008). Beyond critical care, IP agonists have not enjoyed wide use as medical therapy. The potential for profound decreases in blood pressure and for IP receptor-induced pain and inflammation provide a significant barrier to widespread use. Other limited and certain local therapeutic applications may be possible.
There is also an IP receptor therapeutic focus on designing antagonists. The impetus for this is to provide analgesic/anti-inflammatory agents, with no effect on bleeding time (Bley et al., 2006; Pulichino et al., 2006; Brescia et al., 2007; Zhao et al., 2008; Jones et al., 2009). A potential use for IP antagonists for treating overactive bladder disorders has been advanced. Thus, RO-3244019 decreased bladder contraction frequency and increased the micturition threshold in isovolumetric bladder contraction and refill models, respectively (Cefalu et al., 2007), and improved intercontractile interval and voiding volumes as assessed by cystometry (Khera et al., 2007).
E. TP Receptors
1. Second Messenger Signaling.
TP receptors have been shown to couple to Gq, thereby initiating the PLCβ → inositol trisphosphate/diacylglycerol → [Ca2+]i/PKC signaling cascade (Hirata et al., 1991; Dorn and Becker, 1993; Kinsella et al., 1997; Offermanns, 2006; Nakahata 2008). Subsequent studies have strongly implicated G12/13-Rho as a major signaling pathway for TP receptors (Huang et al., 2004; Honma et al., 2006; Mir and Le Breton, 2008; Nakahata, 2008; Song et al., 2009; Zhang et al., 2009; Saito et al., 2010). More than any other prostanoid receptor, TP receptors have been subjected to extensive second messenger pathway analyses. Thus, at the molecular level, there is detailed understanding of TP receptor-G protein interactions obtained by using G protein fusion constructs and guanosine 5′-O-(3-thio) triphosphate binding studies (Hildebrandt, 2006; Zhang et al., 2006, 2009). The G protein coupling repertoire for TP receptors is beyond extensive; it seems all-encompassing. Other members of the Gq family are TP receptor-linked: G11 (Kinsella et al., 1997), G15, and G16 (Offermanns and Simon, 1995), activating the Gq-mediated signaling cascade. TP receptors may couple to Gs (Hirata et al., 1996; Walsh et al., 1998; Mir and Le Breton, 2008) and Gi (Ushikubi et al., 1994; Song et al., 2009). Finally, TP receptors are reported to couple to Gh (Vezza et al., 1999).
TP receptor signaling may result in transactivation, for example of ERK 1/2 (Nakahata, 2008) and EGF receptors (Uchiyama et al., 2009). Likewise, TP receptors possess a PKA/protein kinase G phosphorylation site and four PKC phosphorylation sites, providing mechanisms for modulation/desensitization (Huang et al., 2004). The type of G protein coupling may also alter TPα conformation, which may influence ligand binding and activation of the receptor (Zhang et al., 2006).
Human TP receptors exist in two isoforms, TPα and TPβ (Hirata et al., 1991; Raychowdhury et al., 1994). TPβ is an alternative mRNA splicing variant with an extended carboxyl terminus. The differences in intracellular termini may influence desensitization, internalization, and G protein coupling (Parent et al., 1999; Reid and Kinsella, 2007; Nakahata, 2008). Thus, the TPβ isoform may couple to Gi (Hirata et al., 1996). However, only the TPα isoform is translated in platelets (Habib et al., 1999). It is noteworthy that because TP receptors play a prominent role in mediating the activity of isoprostanes, heterodimerization of TPα and TPβ have been reported to enhance isoprostane signaling (Wilson et al., 2007).
2. Distribution and Biological Functions.
The platelet has always been a major focus of TP receptor research. The TP receptor was originally purified from human platelets (Ushikubi et al., 1989). TP receptor activation produces shape change and platelet aggregation, providing a positive feedback event for causing thrombus formation and thrombosis (Offermanns, 2006; Nakahata, 2008). TP receptor-deficient mice exhibit prolonged bleeding times and are unable to form stable thrombi (Thomas et al., 2001). It has been reported that TxA2 promotes soluble CD40 ligand release from platelets (Enomoto et al., 2010) The platelet TxA2-vascular endothelial PGI2 balance is of central importance to the rational for low-dose aspirin therapy and is the basis for COX-2 inhibitor cardiovascular side effects, underscoring the pathophysiological importance of TP receptors.
Beyond platelets, TP receptors are widely expressed in the cardiovascular system and represent a liability for cardiovascular disease at all levels. In endothelial cells, TxA2 accelerates the expression of adhesion proteins (Ishizuka et al., 1998) and impairs insulin signaling (Song et al., 2009). Increased TxA2 production may contribute to the development of endothelial dysfunction, with resultant vasoconstriction (Gendron and Thorin, 2007; Francois et al., 2008; Denniss and Rush, 2009; Félétou et al., 2009; Graham and Rush, 2009). TxA2 has also been implicated in atherogenesis (Dogné et al., 2005), and mice that are TP receptor-deficient develop fewer atherosclerotic lesions (Kobayashi et al., 2004). The presence of TP receptors on peripheral blood monocytes (Allan and Halushka, 1994) would contribute to the formation of atherosclerotic plaque. Cardiovascular TP receptor expression even extends to cardiomyocytes, and the TxA2 mimetic U-46619 (Fig. 10) causes arrhythmia (Wacker et al., 2009). TP receptors are also located on afferent sympathetic nerve endings in the heart and may participate in the sympathoexcitatory reflex that occurs during myocardial ischemia (Fu et al., 2008).
Structures of agonists for the prostanoid TP receptor. TxA2 and PGH2, the most active natural agonists, are shown in the circle. Only structural alterations relative to PGH2 and TxA2 are illustrated in the figure. The vertical bracket indicates the presence of 2-series α- and ω-chains. a, partial agonist.
Indirect evidence for functional TP receptor expression on peripheral sensory neurons is provided by the pruritogenic activity of U-46619 (Andoh et al., 2007). Introduction of U-46619 into the fourth ventricle rapidly produces emesis (Kan et al., 2008). In relation to the CNS, TP receptors are reported to be expressed on oligodendrocytes (Blackman et al., 1998; Mir and Le Breton, 2008) and human astrocytoma cells (Honma et al., 2006).
In addition to blood vessels, TP receptors are expressed in several smooth muscle types (Coleman et al., 1994b; Nakahata, 2008). Among these, the lung has always featured prominently. The presence of TP receptors on human bronchial smooth muscles was pharmacologically defined in 1989 (Coleman and Sheldrick, 1989), to provide early impetus to TP receptor studies in the bronchopulmonary system. TP receptors played a substantial role in mediating leukotriene-mediated bronchoconstriction (Piper and Samhoun, 1981; Weichman et al., 1982). TxA2 is a potent constrictor of bronchial smooth muscle and has been implicated in both asthma and chronic obstructive pulmonary disease (Rolin et al., 2006). TP receptors are also uterotonic (Senior et al., 1991) and seem to be particularly important in parturition, because PGF2α responsiveness, but not that of TxA2, is lost in the human myometrium during labor (Fischer et al., 2008). Finally, on the smooth muscle cell theme, TP receptors seem capable of differentiating human stem cells to smooth muscle-like cells (Kim et al., 2009).
In common with all prostanoid receptors, TP receptors have been implicated in cancer. U-46619 has been reported to stimulate endothelial cell migration in vivo and angiogenesis and lung metastasis in vivo (Nie et al., 2000). TP receptors also cause proliferation and growth of human lung cancer cells (Li and Tai, 2009; Wei et al., 2010). TP receptors are elevated in prostate cancer and stimulate motility of human prostate cancer cell lines (Nie et al., 2008). It is noteworthy that the TPβ isoform, but not TPα, promoted cell proliferation and migration and invasion by bladder cancer cells; moreover, TPβ receptor expression was increased in tissue obtained from bladder cancer patients (Moussa et al., 2008).
Among the additional functions attributed to TP receptors is their role in immune regulation. Naive T cells obtained from mice expressing TP receptors suppress interaction between dendritic cells and inhibit dendritic cell-dependent T cell proliferation (Kabashima et al., 2003a). In TP-deficient mice, immune responses to foreign antigenic stimuli were enhanced, a phenomenon reproduced in wild-type mice by TP receptor blockade during the sensitization period (Kabashima et al., 2003a). In contrast, splenocytes obtained from IP-deficient mice exhibited a reduced proliferative response to phytohemagglutinin or anti-CD3 antibody (Thomas et al., 2003). Although survival of transplanted hearts from wild-type mice into TP-deficient mice was not prolonged, there was reduced pathological severity in the allografts (Thomas et al., 2003). A role for TxA2 in inflammatory bowel disease is also a possibility (Rampton and Collins, 1993). The vasoconstrictor activity of TP receptors may also be a factor in renal disease (Michel et al., 2008; Araujo and Welch, 2009). TxA2 has also been advanced as a key regulator during Trypanosoma cruzi infection and may contribute to mortality and parasitism (Ashton et al., 2007). Finally, thromboxane synthase and TP receptors are expressed in the murine retina and vasoconstriction in the streptozotocin diabetes model was attenuated by TP receptor blockade (Wright et al., 2009a,b).
3. Gene Deletion Studies.
Two lines of TP(−/−) mice were generated independently (Thomas et al., 1998; Kabashima et al., 2003a). Both lines of mice show no abnormalities of blood pressure under the basal conditions but do show increased bleeding tendencies and are resistant to cardiovascular shock induced by intravenous infusion of arachidonic acid or the TP agonist U-46619. Given the antagonistic actions of TxA2 and PGI2 on platelets and blood vessels, TP signaling in cardiovascular homeostasis was studied using these lines of mice by comparing their phenotypes with those of IP(−/−) mice. Cheng et al. (2002) subjected IP(−/−) mice and TP(−/−) mice to vascular injury by a balloon catheter and found that IP deficiency increased injury-induced vascular proliferation and platelet activation, whereas TP deficiency decreased it. They further showed that the augmented response apparent in IP(−/−) mice was abolished by ablation of TP. These results showed that, once endothelial integrity is disrupted, IP functions protectively, whereas TP aggravates the remodeling. The same group also examined the involvement of TP signaling in vascular remodeling using the external carotid artery ligation model that retains endothelial integrity (Rudic et al., 2005). In this model, treatment of animals with nimesulide augmented the neointimal hyperplastic response of the artery, which was reduced by the loss of TP receptors, suggesting again that TxA2-TP signaling facilitates vascular remodeling after injury or stress. During the above ligation model, production of not only TxA2 but 8,12-iso-isoprotane F2α was increased. Isoprostanes (iPs) are free radical-catalyzed products of arachidonic acid that reflect lipid peroxidation in vivo. Among them, iPF2α-III and iPE2-II can activate platelets and increase vascular tone. The pressor response in vivo and platelet aggregation in vitro induced by these substances were abolished in TP(−/−) mice, suggesting that actions of these iPs are mediated by TP (Audoly et al., 2000). To test the involvement in chronic vascular diseases, Kobayashi et al. (2004) generated the ApoE(−/−)/TP(−/−) double-knockout mice, and examined the effects of TP deficiency on the progression of atherosclerosis. In contrast to acceleration of atherosclerosis in the ApoE(−/−)/IP(−/−) double-knockout mice, atherosclerosis was delayed in the ApoE(−/−)/TP(−/−) mice, despite the fact that they manifested similar plasma cholesterol and triglyceride concentrations. A recent bone marrow transfer experiment (Zhuge et al., 2006) indicated that the effects of TP deficiency attenuating atherogenesis cannot be attributed simply to bone marrow-derived cells such as macrophages. Finally, Francois et al. (2005) found that IP(−/−) mice developed salt-sensitive hypertension, which led to cardiac hypertrophy and severe cardiac fibrosis, and coincidental deletion of TP did not suppress hypertension but ameliorated the hypertrophy and abolished the fibrosis, suggesting that TxA2-TP signaling is responsible for hypertension-induced cardiac complications. These findings have verified that the presumed antagonistic roles of the PGI2-IP signaling and TxA2-TP signaling exists in not only subacute vascular remodeling but also chronic vascular lesions such as atherosclerosis and cardiac fibrosis and may explain the increased incidence of cardiovascular events that has been observed in clinical trials with COX-2 inhibitors. Thus, the loss of TP usually suppresses the progression of pathological vascular conditions. However, a paradoxical example was reported in kidney lesions induced by NG-nitro-l-arginine methyl ester. Francois et al. (2008) subjected TP(−/−) mice to the NG-nitro-l-arginine methyl ester-induced hypertension model and found that the extent of hypertension and resultant cardiac hypertrophy were significantly suppressed in TP(−/−) mice compared with wild-type mice but that the kidney lesion, including glomerulosclerosis, tubule vacuolization, and chronic intestinal inflammation, was worsened with the loss of TP, suggesting that TxA2-TP signaling exerts protective actions against kidney injury, at least in this model. In addition, as described in the FP section, the inflammatory tachycardia induced by either cytokines or LPS was suppressed in mice deficient in TP or FP and was almost completely abolished in mice deficient in both TP and FP. Because both PGF2α and I-BOP, a TP-selective agonist, induced potent positive chronotropic effects on direct application to the nodal area, these results suggest that both TxA2 and PGF2α are produced in response to inflammatory stimuli in situ in the heart and act on TP and FP receptors expressed on pacemaker cells to induce tachycardia. In addition, TxA2-TP signaling also regulates the microcirculation under pathological conditions. During endotoxemia, TNF-α induces dysfunction of the hepatic microcirculation. Katagiri et al. (2008) found that this TNF-α-induced dysfunction, examined as leukocyte adhesion to microvessel endothelial cells, increased the number of nonperfused sinusoids, which was significantly lessened in TP(−/−) mice, indicating that TNF-α mobilizes TxA2-TP signaling and impairs the microcirculation.
In addition to the cardiovascular actions of TP described above, studies on TP(−/−) mice revealed thus-far-unappreciated actions of TP in immunity and inflammation. Kabashima et al. (2003a) noticed lymphadenopathy and splenomegaly in aged TP(−/−) mice and found that immune response to foreign antigens was enhanced in TP(−/−) mice. They further found that stimulation of TP receptors enhanced chemokinesis of T lymphocytes and down-regulated dendritic cell-dependent T-cell proliferation in vitro. These findings led them to suggest that TxA2-TP signaling in T cells regulates the interaction between T cells and dendritic cells and thereby the extent of the immune response. On the other hand, Thomas et al. (2003) found that T cells deficient in TP receptor exhibited less proliferation in response not only to phytohemagglutinin or anti-CD3 antibody but also in the mixed lymphocyte response (MLR). Reduced proliferation of TP(−/−) lymphocytes in MLR was mimicked by the MLR of wild-type T cells treated with a TP antagonist, 7-(3-((2-((phenylamino)carbonyl)hydrazino)methyl)-7-oxabicyclo(2.2.1)hept-2-yl)-5-heptenoic acid (SQ-29548) or a thromboxane synthase inhibitor, carboxyhexyl imidazole. These findings are opposite those of Kabashima et al. (2003a), who found that T-cell proliferation in MLR was suppressed by treatment with a TP agonist, I-BOP. Thomas et al. (2003) reported that their TP(−/−) mice exhibited prolonged cardiac allograft survival compared with wild-type mice. We do not know why such a discrepancy arose using different lines of TP(−/−) mice. In addition to these functions of TP in the peripheral immune system, TxA2-TP signaling also seems to regulate the immune system centrally. Ushikubi et al. (1993) found that TP mRNA was markedly expressed in the thymus, particularly in CD4−/CD8− and CD4+/CD8+ immature thymocytes, and that the addition of a TP agonist, stable TxA2 analog, induced apoptosis of CD4+/CD8+ cells. Rocha et al. (2005) found that administration of LPS into mice markedly increased production of TxA2 and PGE2 in the thymus, and caused apoptotic deletion of CD4+/CD8+ cells there. They then demonstrated that thymocyte apoptosis in response to LPS was significantly attenuated in TP(−/−) mice, suggesting that thymocyte apoptosis mediated by TxA2-TP signaling functions physiologically.
In addition to these actions in the immune system, TxA2-TP signaling exerts its action in a different system to combat against infection. Ashton et al. (2007) found that a parasite, T. cruzi, is itself capable of producing TxA2 and that TP(−/−) mice exhibited higher mortality and more severe cardiac pathology and parasitism than wild-type mice after infection. Bone marrow transfer experiments showed that TP receptor in somatic cells, and not bone marrow-derived cells, is important for protection against parasites; how it functions remains to be determined.
4. Agonists and Antagonists.
Thromboxane A2 and its precursor endoperoxide PGH2 both activate TP receptors. Both are very chemically unstable and therefore of limited practical use. For this reason, stable analogs were developed. The methanoepoxy analog 9–11-dideoxy-11α,9a-epoxymethano-prostaglandin F2a (U-46619; Fig. 10) has been widely used. More potent compounds, such as I-BOP, are also available for study but may have an inconveniently slow onset and offset in isolated tissue preparations.
Both PGH2 and TxA2 activate TP receptors, the latter showing higher potency (Needleman et al., 1976; Svensson and Hamberg, 1976; Salzman et al., 1980; Armstrong et al., 1985; Vezza et al., 2002), although their chemical lability and the enzymatic conversion of PGH2 to PGD2, PGE2, PGF2α, and PGI2 complicate these measurements (Hornberger and Patscheke, 1989). Stabilization of the TxA2 ring structure by difluoro substitution at C10 yields a potent TP agonist (Morinelli et al., 1989). However, the majority of stable TP agonists derive from substitution of one or both ring oxygens in PGH2 or TxA2 with carbon (Fig. 10). U-46619 (the most commonly used standard agonist), 9,11-azo PGH2 and stable TxA2 analogs behave as full agonists on isolated vascular and respiratory smooth muscle and human platelet preparations (Tymkewycz et al., 1991), as does the cyclic carbonate AGN-191976 (Fig. 10) (Krauss et al., 1996). U-44069, 9,11-etheno PGH2, pinane TxA2, and carbocyclic TxA2 often show partial agonism on smooth muscle preparations; that is, they show a reduced maximum response and block the action of U-46619 without affecting the action of other contractile agonists (e.g., phenylephrine or histamine) (Jones et al., 1982; Krauss et al., 1996). They may also show partial agonism on human platelets, but results from this assay system must be interpreted cautiously. Thus, there are three distinct, concentration-dependent, functionally related components of “platelet activation,” namely shape change, primary (reversible) aggregation, and secondary aggregation as a result of release of mediators, including PGH2/TxA2 from de novo synthesis. The generated PGH2 could spontaneously decay to PGE2 and/or PGD2, with corresponding activation of EP3 receptors enhancing platelet activation (Oelz et al., 1977; Matthews and Jones, 1993) and DP1 receptors inhibiting platelet activation (Keery and Lumley, 1988) through changes in cAMP levels (see Gresele et al., 1988). True TP partial agonists could elicit either submaximal shape change or complete shape change and reversible aggregation, as is the case for 9,11-etheno PGH2 (Jones and Wilson, 1980). However, functional antagonism may also modulate the response as in the case of carbocyclic TxA2, which weakly raises the cyclic AMP level (possibly through activation of sensitive DP1 and/or IP systems) sufficient to suppress the maximum aggregation response (Armstrong et al., 1985). Finally, COX/thromboxane synthase involved in the release reaction may be inhibited by PGH/TxA analogs (Wlodawer et al., 1971). Taking these issues into account, Krauss et al. (1996) identified the C1-alcohol derivative AGN-192903 (Fig. 10) as an agent that behaved as a potent full agonist on rat aorta but only caused shape change in human platelets at high concentration. The authors discuss their findings in relation to the identification of two TP receptor subtypes (Takahara et al., 1990; Furci et al., 1991), one of which elicits smooth muscle contraction and platelet shape change, whereas the other elicits platelet aggregation (see also section on identification of TPα and TPβ subtypes).
Analogs with a 7-oxabicyclo[2.2.1]heptane ring system (Fig. 10, bottom right) were prepared as TP agonists on the basis of a favorably directed ring oxygen (Sprague et al., 1985). Of the eight possible α-oxy isomers, the “natural” 8α,12β,15S analog (SQ-26655; Fig. 10) is a potent TP agonist. Introduction of a 16-p-halophenoxy moiety into the ω terminus, as in EP-171 (Fig. 10) (Wilson et al., 1988) and I-BOP (Sessa et al., 1990), markedly enhances TP agonism. Near-maximal shape change of the human platelet elicited by EP-171 at 0.1 nM is only slowly reversed by a high concentration of TP antagonist, reflecting the slow dissociation of this agonist from the TP receptor (Jones et al., 1989). [125I]BOP is a useful radioligand for the TP receptor (Morinelli et al., 1989). The 8α,12α,15R analog retains TP agonism, whereas the 8α,12α,15S analog shows a switch to TP antagonism (Sprague et al., 1985) and was the starting point for potent and selective TP antagonists such as SQ-29548 and BMS-180291 (ifetroban; Fig. 11). The 8β,12α,15S analog shows broad inhibitory activity on human platelets, which was subsequently attributed to DP1 agonism (see Fig. 11).

Structures of representative antagonists for the prostanoid TP receptor.
TP antagonist design has been ongoing for more than 2 decades. Many potent and selective TP antagonists of diverse structure have been synthesized (Jones et al., 2009). Partial agonism is prevalent among carba-ring analogs of PGH2 or TxA2 (see Wilson and Jones, 1985), and further modification of the ω-chain often leads to pure antagonism. A prime example is I-SAP (Fig. 11), which contains a pinane ring akin to the dioxabicyclo [3.1.1]heptane ring of TxA2 and trans orientation of the side chains (Naka et al., 1992). In other prostanoid-like antagonists, a cis orientation of the side chains affords high affinity as in GR-32191 (Lumley et al., 1989) and BMS-180291(Ogletree et al., 1993) (Fig. 11). A benzene-sulfonamido moiety present in I-SAP features in simpler TP antagonist molecules, such as BM-13505 (daltroban; Yanagisawa et al., 1987), Z-335 (Tanaka et al., 1998) and S-18886 (terutroban; Fig. 11) (Cimetière et al., 1998); the distance from the C1-carboxylate group seems to be critical. 14,15-Epoxy-eicosatrienoic acid has been claimed to be an endogenous TP antagonist; it shows modest affinity for human DP1, FP, and TP receptors (Ki = 6.1, 5.3, and 3.2 μM) with lower affinity for other prostanoid receptors (≥13 μM) (Behm et al., 2009).
For many of these compounds, surmountable antagonism on isolated smooth muscle preparations points toward reversible-competitive mechanism, and conventional Schild analysis confirms this classification. However, a slow approach to steady-state block by high-affinity antagonists can confound pA2 estimation. Under the Cheng-Prusoff inhibition-curve protocol, 6-(2-(2-chlorophenyl-4-hydroxyphenyl)-1,3-dioxan-5-yl)hexenoic acid (ICI-192605) (Brewster et al., 1988) had not reached steady-state block on guinea pig aorta after 90-min incubation (pA2 = 10.25) (Jones et al., 2008). With higher affinity TP antagonists in human and rat platelet assays, surmountability is seen for the shape-change response, whereas insurmountability occurs for aggregation. This profile is still compatible with a reversible-competitive mechanism, because slow dissociation of the high-affinity antagonist from the TP receptor retards agonist (U-46619) occupancy in the early stage of the aggregation response, thereby favoring the disaggregation process and insurmountability; in contrast, the nonfading shape-change response affords a truer measure of dose ratios at steady state (Armstrong et al., 1985; Jones et al., 1989; Lumley et al., 1989; Tymkewycz et al., 1991; Ogletree et al., 1993).
There has been much debate about the existence of different TP receptor subtypes in platelet and vascular smooth systems (Mais et al., 1985, 1988; Swayne et al., 1988; Morinelli et al., 1989; Masuda et al., 1991; Tymkewycz et al., 1991; Folger et al., 1992). It is certainly clear that species heterology exists; for example, higher affinities are often found for human and rat platelet TP receptors compared with rabbit platelet TP receptors (Tymkewycz et al., 1991). However, the situation may be more complex. There is evidence for two saturable binding sites for TP agonists on human platelets using several radioligands (Armstrong et al., 1983; Pollock et al., 1984; Ahn et al., 1988; Hedberg et al., 1988). The high-affinity site was associated with the platelet shape change (and increase in cytosolic Ca2+), whereas the lower-affinity site was associated with aggregation (and activation of PLC) (Dorn, 1989; Takahara et al., 1990). 3H-labeled GR-32191 (vapiprost) was bound reversibly to the “shape change site” and irreversibly to the “aggregation site” (Takahara et al., 1990). Given the structure of GR-32191 (Fig. 11), it is unlikely that covalent bonding is involved. Exposure of human platelets to GR-32191 for 30 min resulted in approximately 50% loss of binding sites for either [3H]GR-32191 or [3H]SQ-29548, whereas neither SQ-29548 nor BM-13177 (sulotroban) affected Bmax. It was speculated that GR-32191 binds to internalized TP receptors (Armstrong et al., 1983). 4[2-(4-azido-benzenesulfonylamino)-ethyl[phenoxyacetic acid), a light-activated, covalent-binding TP antagonist, also discriminated these platelet sites by blocking aggregation but not shape change induced by U-46619 (Zehender et al., 1988). Two TP receptor isoforms (TPα and TPβ) have been identified using a human umbilical vein endothelial cDNA library (Raychowdhury et al., 1994), and mRNAs for the α and β isoforms have been detected in human platelets (Hirata et al., 1996). However, these isoforms, which arise by alternative gene splicing and differ only in their cytoplasmic tails, do not show the ligand discrimination typical of the high- and low-affinity binding sites. Finally, only the TPα isoform was found in human platelets (Habib et al., 1999).
TP antagonism associated with IP agonism, TX synthase inhibition and LT receptor antagonism in the same molecule are all known. The bicyclo[2.2.2]octene PGH2 analog EP-157 (Figs. 8 and 11) activates IP receptors in both platelet and vascular systems (Armstrong et al., 1986, 1989; Jones et al., 1993). The presence of a diarylhetero(cyclic) moiety in the ω-chain is crucial (Jones et al., 1993). Similar properties were found for octimibate, which lacks a prostanoid ring system (Merritt et al., 1991a,b) and is a member of a series of nonprostanoid prostacyclin mimetics (Meanwell et al., 1994; Seiler et al., 1997). Some of these agents also inhibit (nonprostanoid) Gq-PLC-driven responses (Chow et al., 2001).
Combined TP antagonism/thromboxane synthase inhibition usually requires the presence of a (N)-imidazole (dazoxiben; Randall et al., 1981) or a pyridin-3-yl group (isbogrel; Imura et al., 1990; ridogrel, Hoet et al., 1990; Fig. 11) to combine with the heme site of the synthase (see Hsu et al., 1999). Existing TP antagonists have also been modified to include similar reactive moieties: ZD-1542 (Brownlie et al., 1993), a relative of ICI-192605, contains a pyridin-3-yl group and GR-83783 (Campbell et al., 1991a), a relative of GR-32191, has a 4-(pyridin-3-yl)-phenyl moiety. In addition, sulotroban/daltroban moieties have been combined with ridogrel/isbogrel moieties (Campbell et al., 1991b; CGS-22652 (Bhagwat et al., 1993; Soyka et al., 1994) and the whole or part of the ICI-192605 nucleus has been tethered to either a dazoxiben or an isbogrel nucleus (Ackerley et al., 1995). More recently, compounds with dual antagonist properties have been designed. For example, (2-(N-(4-(4-chlorobenzenesulfonylamino)butyl)-N-(3-(4-isopropylthiazol-2-yl)methoxy)benzyl)sulfamoyl)benzoic acid (KP-496) (Mizutani et al., 2008; Ishimura et al., 2009) and YM-158 (Fig. 11) (Arakida et al., 1998) are dual TP/cysteinyl leukotriene antagonists.
5. Therapeutics.
The original purpose for designing TP antagonists was as cardiovascular therapy but low-dose aspirin has proven a more economically viable proposition (Jones et al., 2009). Nevertheless, even recently the TP antagonist terutroban was shown to exhibit superior antithrombotic activity compared with aspirin in humans (Bal Dit Sollier et al., 2009). Another early indication for TP antagonists was asthma, which met with some clinical success, albeit limited (Rolin et al., 2006; Jones et al., 2009). Other early indications do not seem to have met with any clinical success. These include cancer, glomerulonephritis, allergic rhinitis, inflammatory bowel disease, septic shock, and diabetes (Jones et al., 2009). The unique role of the TPβ receptor isoform in bladder cancer and the delayed onset and prolonged survival afforded by TP antagonist treatment in mice transfected with bladder cancer cells (Moussa et al., 2008) holds promise for therapeutic utility in at least one form of cancer.
There are other potential uses for TP antagonists that have emerged more recently. The TP antagonist seratrodast seems to possess antitussive properties (Xiang et al., 2002; Ishiura et al., 2003). Ramatroban has been reported to attenuate cough in subjects with cough variant asthma (Kitamura et al., 2003a). The potential use of TP antagonists for treating preterm labor is not new but deserves new impetus based on recent findings on the human myometrium that PGF2α responsiveness, but not TxA2 responsiveness, of the uterus is lost during labor (Fischer et al., 2008). The most recent potential uses for TP antagonists are listed in Table 10.
Recently identified potential therapeutic application of TP antagonists
F. DP2 Receptors (CRTR2)
1. Second Messenger Signaling.
Three independent but convergent research pathways led to the discovery of the DP2 (CRTH2) receptor subtype. A new surface marker on human Th2 cells in vivo (Nagata et al., 1999) was termed the “chemoattractant receptor-homologous molecule expressed on Th2 cells” (CRTH2). This proved to be identical to the orphan G protein-coupled receptor GPR44 (Marchese et al., 1999), which had sequence homology similar to that of typical chemoattractant receptors. Nevertheless, the naturally occurring ligand for the CRTH2 receptor was subsequently found to be PGD2 (Hirai et al., 2001; Monneret et al., 2001), and the term DP2 was introduced. There are now three descriptors: CRTH2, DP2, GPR44, with license for upper/lower case and subscripted/nonsubscripted variations. It has been suggested that the widespread distribution of this receptor, which extends far beyond the immune system, makes placement in the prostanoid receptor classification as DP2 more appropriate (Jones et al., 2009). The DP2 designation is used herein, given the topic of this review.
DP2 receptors are Gi-coupled, but signal transduction pathways have been subject to only limited investigation. DP2 receptor activation may result in pertussis toxin-sensitive decreases in cAMP levels (Sawyer et al., 2002; Gallant et al., 2007) and Ca2+ mobilization (Hirai et al., 2001; Sawyer et al., 2002). PI3K signaling has also been implicated in mediating DP2 effects (Hata et al., 2003; Xue et al., 2007). DP2 receptor trafficking has been studied and, interestingly, PGD2 induced DP2 but not DP1 receptor internalization (Gallant et al., 2007), which was decreased by inhibition of PKA and PKC. DP2 receptor internalization may be regulated by PKC, GRK2, GRK3, GRK6 and arrestin-3 (Gallant et al., 2007) and the determinants located in the carboxyl terminus have been studied (Roy et al., 2010).
2. Distribution and Biological Functions.
By combining functional data and transcription profiles together, it is clear that DP2 receptor expression is widespread. Northern blotting revealed high expression in the human stomach, small intestine, heart, and thymus; intermediate expression in the colon, spinal cord, and blood; and lower expression in the brain, skeletal muscle, and spleen (Sawyer et al., 2002). Functional studies demonstrate DP2 receptors present in smooth muscle, the cardiovascular system, the gastrointestinal tract, and the eye (Jones et al., 2009). The principal research focus during the past decade, after the cloning of DP2, however, has been inflammation.
PGD2 has long been known to cause eosinophil infiltration but with a pharmacological profile inconsistent with DP1 receptor mediation (Woodward et al., 1990, 1993b). The subsequent cloning of the receptor (Marchese et al., 1999; Nagata et al., 1999) and discovery of DP2 receptor as a Th2 cell chemoattractant (Hirai et al., 2001) provided important new dimensions. The role of DP1 and DP2 receptors in immunology and inflammation has been the subject of more than one review (Herlong and Scott, 2006; Kostenis and Ulven, 2006; Pettipher, 2008) and this topic is therefore discussed only briefly.
The discovery of PGD2 as an activator of Th2 lymphocytes via DP2 (CRTH2) receptors implicated them in immune regulation. The polarization of lymphocytes to the Th-2 phenotype is typically implicated in the development of allergic responses such as asthma and atopic dermatitis. DP2 receptor expression in CD4+ T cells is low in healthy humans but enhanced in atopic subjects and correlates with the severity of the disease (Kostenis and Ulven, 2006). In addition to recruitment of Th2 cells, DP2 receptor activation results in the production of cytokines such as IL-4, IL-5, IL-9, and IL-13 (Honda et al., 2003; Gallant et al., 2005; Xue et al., 2005; Kostenis and Ulven, 2006 Herlong and Scott, 2006). Recruitment and activation of eosinophils is a central feature of allergic responses, and they also express DP2 receptors. Activation of DP2 receptors leads to chemotaxis and degranulation of eosinophils (Gervais et al., 2001; Hirai et al., 2001; Monneret et al., 2001; Sugimoto et al., 2003; Böhm et al., 2004). DP2 receptor stimulation produces eosinophil infiltrates in living animals, and these are located at sites typically associated with allergy, such as the conjunctiva (Woodward et al., 1990, 1993b), cornea (Fujishima et al., 2005), lung (Almishri et al., 2005; Shiraishi et al., 2005; Spik et al., 2005), and skin (Spik et al., 2005). An important role for DP2 receptors in chronic cutaneous inflammation was revealed in DP2-deficient mice (Satoh et al., 2006). Prostaglandin D2 also activates basophils via DP2 receptors (Hirai et al., 2001; Cossette et al., 2007). A final comment on the roles of DP1 and DP2 in regulating allergy/inflammation (Kostenis and Ulven, 2006; Pettipher, 2008): the employment of a DP2 antagonist may switch PGD2 from a pro- to an anti-inflammatory mediator in many instances.
Although widely distributed, notably in the gastrointestinal tract (Sawyer et al., 2002), the focus has been almost exclusively on leukocytes. Studies in the eye demonstrate that even within the context of allergic inflammation, DP2 receptor stimulation produces more than leukocyte activation. Thus, DP2 receptors are also associated with goblet cell depletion and increased microvascular permeability in the conjunctiva (Woodward et al., 1990, 1993b). In human retinal pigmented epithelial cells, DP2 receptors induce heme oxygenase-1 expression (Satarug et al., 2008): these cells are essential for photoreceptor survival. DP2 has been suggested as a therapeutic target for delaying the onset of age-related macular degeneration and cerebral malaria (Satarug et al., 2008). Both DP receptor subtypes have been implicated in astrogliosis and demyelination and involved in the neuroinflammatory effects of PGD2 (Mohri et al., 2006).
3. Gene Deletion Studies.
DP2 knockout mice were generated independently by two groups (Chevalier et al., 2005; Satoh et al., 2006), and apparently different phenotypes were reported for these two lines. Chevalier et al. (2005) subjected their DP2-deficient mice to the OVA-induced allergic asthma model and found markedly increased eosinophil recruitment into the bronchoalveolar lavage (BAL) fluid of KO mice compared with WT mice. This was opposite to the known function of DP2 (CRTH2), in that it mediates the chemotactic action of PGD2 on eosinophils. Indeed, DP2 receptor stimulation has been shown to increase the degree of inflammation in mice (Spik et al., 2005). To examine this discrepancy, they found that IL-5 production by activated T cells from DP2-deficient mice in vitro was increased compared with that observed with wild-type cells. They suggested that DP2 indeed functions to facilitate allergy in situ at the site of inflammation, but that this receptor also regulates IL-5 production in the early phase of allergy development. On the other hand, Satoh et al. (2006) injected anti-dinitrophenyl-specific IgE into the skin of the ear lobe of their DP2-deficient mice, challenged them with DNFB, and found that this type of IgE-induced dermatitis was significantly suppressed in DP2-deficient mice. They further showed that acute as well as chronic contact hypersensitivity was partially suppressed in DP2-deficient mice. In the Discussion section of their article, Satoh et al. (2006) commented that their DP2-deficient mice did not exhibit enhanced eosinophilia when subjected to the asthma model and that splenocytes from their KO mice did not show exaggerated IL-5 production on activation. The DP2-deficient mice that Satoh et al. (2006) generated consistently showed less inflammatory response in allergic dermatitis induced by cutaneous application of Japanese cedar pollen (Oiwa et al., 2008) or allergic rhinitis induced by repeated intranasal sensitization with Cry j 1, Japanese cedar pollen antigen (Nomiya et al., 2008). In the latter study, suppression of the increase in serum Cry j 1-specific IgE, a significant reduction of IL-4, and a slight reduction of IL-5 produced by draining lymph node cells in DP2-deficient mice were reported. Furthermore, Shiraishi et al. (2008) examined the role of DP2 in the poly I:C-induced enhancement of allergic inflammation in the OVA asthma model and found that although DP2-deficient mice showed a comparable inflammatory eosinophil infiltration in BAL to WT mice after OVA challenge alone, they showed complete loss of the poly I:C-induced enhancement. No difference was found in the number of neutrophils and lymphocytes or the amounts of IL-5 and IL-13 in the BAL between WT and DP2-deficient mice. In addition to these studies focusing on the role of DP2 in eosinophil recruitment in allergic reactions, Tajima et al. (2008) found enhanced production of PGE2 and PGD2 during macrophage activation with LPS and examined the role of DP2 in LPS-induced migration of peritoneal macrophages. They reported that LPS-mediated migration was significantly suppressed in macrophages from DP2-deficient mice.
4. Agonists and Antagonists.
Potent and selective agonists for the DP2 receptor have been discovered or devised. Although no therapeutic utility for DP2 agonists has been established, they have provided invaluable pharmacological tools. Indeed, 13,14-dihydro,15-keto PGD2 was the first DP2 agonist to be discovered (Jones, 1976a,b), and over the past 3 decades, numerous unique natural ligands have been discovered (Pettipher, 2008). Synthetic selective DP2 agonists include 15R-methyl PGD2 (Monneret et al., 2003) and (9-((4-chlorophenyl)thio)-6-fluoro-2,3-dihydro-1H-pyrrolo(1,2-a)indol-1-yl)acetic acid (L-888,607) (Gervais et al., 2005). It is also interesting that indomethacin is a DP2 agonist (Hirai et al., 2002).
Attaining selective DP2 agonism has mainly involved alterations to the 15-hydroxyl group. The 15S stereochemistry is atypically not crucial to DP2 agonism, with 15R- and 15R-15-methyl PGD2 showing high DP2/DP1 selectivity (Jones, 1976,a,b, 1978; Monneret et al., 2001, 2003; Kim et al., 2005; Cossette et al., 2007). Moreover, 15-oxo structures (usually considered to be bioinactive products of 15-hydroxy PG dehydrogenase) retain high DP2 agonism. The biologically active form of 15-oxo PGD2 is not clear, however, because of its ready interconversion between 11,5-dioxo-13E-ene (as drawn in Fig. 2) and 11,15-dioxo-12-ene tautomers, of which there are two E and Z geometric isomers (Jones and Wilson, 1978); the corresponding conjugated enols (e.g., 11-oxo-12,14-diene-15-hydroxy) may even be active given the high potency of Δ12-PGJ2 (Monneret et al., 2002). Saturation of the 13,14-double bond obviates this chemical lability (Jones, 1976a,b, 1978; Rangachari et al., 1995) as does conversion of the 15-oxo group to an ethylene ketal (Jones, 1978). Loss of the 15-oxygen function also results in retention of DP2 agonism, as in 15-deoxy-Δ12,14-PGJ2 (Monneret et al., 2002). The variance allowed at C15 is not unlimited, however; 15S-15-methyl PGD2 shows only weak DP2 agonism (Monneret et al., 2003). Finally, replacement of the 11-oxo group in PGD2 by methylene essentially abolishes DP1 agonism and results in weak DP2 antagonism (Cossette et al., 2007).
DP2 antagonists have been a major focus for drug discovery in recent years. This has been reviewed (Jones et al., 2009) and therefore is not discussed in depth herein. Several distinct structural classes have been used as design templates. Indomethacin being a DP2 agonist (Hirai et al., 2002; Stubbs et al., 2002), indole acetic acids have been designed as antagonists. Likewise, the nonsteroidal anti-inflammatory drug fenclofenac was also used as a starting point for DP2 antagonist design (Jones et al., 2009). Additional scaffolds include tetrahydroquinolines (Jones et al., 2009) and thiazoleacetic acids (Grimstrup et al., 2010). Ramatroban (Bay U 3405) has played a central role in DP2 antagonist design, and elements of this core structure are incorporated into many structures (Jones et al., 2009), including recently described indole-based DP2 antagonists (Stearns et al., 2009). Ramatroban is also a TP antagonist and is marketed for the treatment of allergic rhinitis (Sugimoto et al., 2003). Development of ramatroban was first directed toward TP antagonism (McKenniff et al., 1988). Later studies revealed low-affinity DP2 antagonism (pA2, 7.44) (Sugimoto et al., 2003; Mathiesen et al., 2006). Modification of ramatroban led to TM-30089 (CAY-10471; Fig. 3), which has much higher DP2/TP-selectivity (Ulven and Kostenis, 2005). In assays of guanosine 5′-O-(3-[35S]thio)triphosphate binding/inositol phosphate accumulation and PGD2-induced eosinophil shape change, ramatroban and the related TM-30642 were surmountable competitive antagonists, whereas TM-30643 and TM-30089 (Ulven and Kostenis, 2005) show insurmountability. Indomethacin has also provided a template for DP2 blockers, based to some extent on its weak agonism for the DP2 receptor (Hirai et al., 2002; Stubbs et al., 2002). Many of these analogs are designed around an inverted indole template (Birkinshaw et al., 2006). This latter profile may be due to slow dissociation from the DP2 receptor (Mathiesen et al., 2006). The embodiment of both DP1 and DP2 receptor antagonism in a single molecule seems to offer a potentially more effective therapeutic approach (Pettipher, 2008). This approach for antiallergic drug design is in progress, with at least one positive outcome in the form of AMG 009 (Liu et al., 2009).
5. Therapeutics.
A variety of DP2 antagonists have been claimed active in models of allergic rhinitis, asthma, and atopic dermatitis (Table 11). Lung inflammation resulting from smoke inhalation has recently reported to be reduced by a DP2 antagonist (Stebbins et al., 2010).
Therapeutic applications of DP2 antagonists
G. Receptor Heterodimerization
It is now established that G protein-coupled receptors exist as dimers (Prinster et al., 2005; Lohse, 2010) that are precoupled to heterotrimeric proteins in living cell membranes (Nobles et al., 2005). IP receptor precoupling to Gs has been shown in the cell membrane (Nobles et al., 2005). Heterodimerization allows the receptor to extend its repertoire of G protein coupling and ligand binding. IP/TP heterodimerization provides an example of both phenomena. In terms of second-messenger signaling, PGI2-like characteristics are conferred on TxA2 mimetics, which occurs as a robust cAMP response (Wilson et al., 2004). IP/TP heterodimerization creates a new binding site, which recognizes isoprostane E2 (Wilson et al., 2004). Heterodimerization of a prostanoid receptor with a member outside of the prostanoid family has been observed in the form of EP1/β2-adrenoceptor dimerization (McGraw et al., 2006).
Heterodimerization of alternative mRNA splicing variants may also occur. Isoprostane responses were enhanced when TPα and TPβ were coexpressed (Wilson et al., 2007). Coexpression of the wild-type FP receptor and an alternative mRNA splicing variant resulted in ligand recognition that was quite different from wild-type FP receptors (Liang et al., 2008). Unlike FP receptors, wild-type/alternative FP/FP heterodimers were able to respond to the prostamide F2α mimetic bimatoprost, and the antagonist AGN 211335 (Liang et al., 2008) blocked responses to bimatoprost but not PGF2α. This pharmacology was entirely consistent with that observed for the prostamide receptor in ocular cells and isolated tissue preparations (Liang et al., 2003; Woodward et al., 2003, 2007; Matias et al., 2004; Spada et al., 2005; Wan et al., 2007; Stamer et al., 2010). Therapeutic uses for the prostamide F2α mimetic bimatoprost have extended beyond glaucoma to include trichomegaly (Tauchi et al., 2010). No potential medical uses for prostamide antagonists (Woodward et al., 2009) have been reported.
Acknowledgments
We thank L. Rubin for considerable assistance in preparing and formatting the manuscript and J. W. Wang for compilation and formation of Fig. 1.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Woodward, Jones, and Narumiya.
Footnotes
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.003517.
-
↵1 Abbreviations:
- AH 13205
- trans-2-(4-(1-hydroxyhexyl)phenyl)-5-oxocyclopentaneheptanoic acid
- AH 23848
- 7-(5-(((1,1-biphenyl)-4-yl)methoxy)-2-(4-morpholinyl)-3-oxocyclopentyl)-4-heptanoic acid
- Akt
- protein kinase B
- AL-6556
- 9-chloro-15-cyclohexyl-11,15-dihydroxy-3-oxa-16,17,18,19,20-pentanor-5-prostenoic acid
- ApoE
- apolipoprotein E
- BAL
- bronchoalveolar lavage
- Bay U 3405
- ramatroban
- BM-13177
- sulotroban
- BM-13505
- daltroban
- BMS-180291
- ifetroban
- BNP
- brain natriuretic protein
- BW 245C
- 5-(6-carboxyhexyl)-1-(3-cyclohexyl-3-hydroxypropyl)hydantoin
- BW A868C
- 3-(3-[1,1′-biphenyl]-4-yl-3-hydroxypropyl)-2,5-dioxo-4-imidazolidineheptanoic acid, ethyl ester
- CAY-10399
- “2-series” analog of butaprost-FA
- CGRP
- calcitonin gene-related peptide
- CJ-023,423
- N-(((2-(4-(2-ethyl-4,6-dimethyl-1H-imidazo(4,5-c)pyridin-1-yl)phenyl)ethyl)amino)carbonyl)-4-methylbenzenesulfonamide
- CJ-42794
- (S)-4-(1-(5-chloro-2-(4-fluorophenyoxy)benzamido)ethyl)benzoic acid
- CNS
- central nervous system
- COX
- cyclooxygenase
- CRTH2
- chemoattractant receptor-homologous molecule expressed on Th2 cells
- Cyr 61
- cysteine-rich angiogenic protein 61
- DC
- dendritic cell
- DNFB
- dinitrofluorobenzene
- EGF
- epidermal growth factor
- Epacs
- exchange proteins activated by cAMP
- ERK
- extracellular signal regulated protein kinase
- GR 63799X
- (4-benzamidophenyl)-(Z)-7-[(1R,2R,3R)-3-hydroxy-2-[(2R)-2-hydroxy-3-phenoxypropoxy]-5-oxocyclopentyl]hept-5-enoate
- GR-32191
- vapiprost
- GRK
- G protein-coupled receptor kinase
- HIF
- hypoxia-inducible factor
- HPGDS
- hematopoietic prostaglandin D synthase
- I-BOP
- [1S-[1α,2α(Z),3β(1E,3S*),4α]]-7-[3-[3-hydroxy-4-(4-iodophenoxy)-1-butenyl]-7-oxabi-cyclo[2.2.1]hept-2-yl]5-heptenoic acid
- ICI-192605
- 6-(2-(2-chlorophenyl-4-hydroxyphenyl)-1,3-dioxan-5-yl)hexenoic acid
- ICI-80205
- 16-p-chlorophenoxy-ω-tetranor PGE2
- ICI-81008
- fluprostenol
- IFN
- interferon
- IL
- interleukin
- IOP
- intraocular pressure
- iP
- isoprostane
- KO
- knockout
- KP-496
- (2-(N-(4-(4-chlorobenzenesulfonylamino)butyl)-N-(3-(4-isopropylthiazol-2-yl)methoxy)benzyl)sulfamoyl)benzoic acid
- L-888,607
- (9-((4-chlorophenyl)thio)-6-fluoro-2,3-dihydro-1H-pyrrolo(1,2-a)indol-1-yl)acetic acid
- LC
- Langerhans cell
- LPS
- lipopolysaccharide
- MAP
- mitogen-activated protein
- MB-28767
- 11-deoxy-16-phenoxy PGE1
- MEK
- mitogen-activated protein kinase kinase
- MF-266-1
- 1-(5-{3-[2-(benzyloxy)-5-chlorophenyl]-2-thienyl}pyridin-3-yl)-2,2,2-trifluoroethane-1,1-diol
- MK-0524
- laropiprant
- MLR
- mixed lymphocyte reaction
- NMDA
- N-methyl-d-aspartate
- ONO-11120
- 11α-carba-12-(2′S-hydroxy-3′-phenylpropylamino)-9α,11α-isopropylideno-ω-octanor-prost-5Z-enoic acid
- ONO-8711
- 6-((2S,3S)-3-(4-chloro-2-methylphenysulfonylaminomethyl)-bicyclo(2.2.2)octan-2-yl)-5Z-hexenoic acid
- OVA
- ovalbumin
- PF-04475270
- 5-(3-(2-(3-hydroxy-4-(3-(trifluoromethyl)phenyl)butyl)-5-oxopyrrolidin-1-yl)propyl)thiophene-2-carboxylate
- PG
- prostaglandin
- PGDS
- prostaglandin D synthase
- PGI2
- prostacyclin
- PI
- phosphatidylinositol
- PI3K
- phosphoinositide-3-kinase
- PKA
- protein kinase A
- PKC
- protein kinase C
- PLC
- phospholipase C
- PPAR
- peroxisome proliferator-activated receptor
- PTH
- parathyroid hormone
- RANKL
- receptor activator of nuclear factor-κB ligand
- rc
- recombinant
- Rho
- rhodopsin
- S-145
- 5,7-(3-phenylsulfonylamino(2.2.1)bicyclohept-2-yl)heptenoic acid
- S-18886
- terutroban
- SC-19220
- 8-chloro-dibenzo(Z)[b,f][1,4]oxazepine-10(11H)-carboxylic acid, 2-acetylhydrazide
- SQ-29548
- 7-(3-((2-((phenylamino)carbonyl)hydrazino)methyl)-7-oxabicyclo(2.2.1)hept-2-yl)-5-heptenoic acid
- STAT
- signal transducer and activator of transcription
- TGF
- transforming growth factor
- Th
- T helper
- THG-113.31
- Ile-Leu-Gly-His-(d-Cit)-Asp-Tyr-Lys
- TM
- transmembrane domain
- TNF
- tumor necrosis factor
- TR4979
- butaprost
- TS-022
- (4-((1R,2S,3R,5R)-5-chloro-2-((S)-3-cyclohexyl-3-hydroxyprop-1-ynyl)-3-hydroxycyclopentyl)butylthio) acetic acid monohydrate
- TxA2
- thromboxane A2
- U-46619
- 9–11-dideoxy-11α,9a-epoxymethano-prostaglandin F2a
- VEGF
- vascular endothelial growth factor
- WT
- wild type.
- © 2011 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
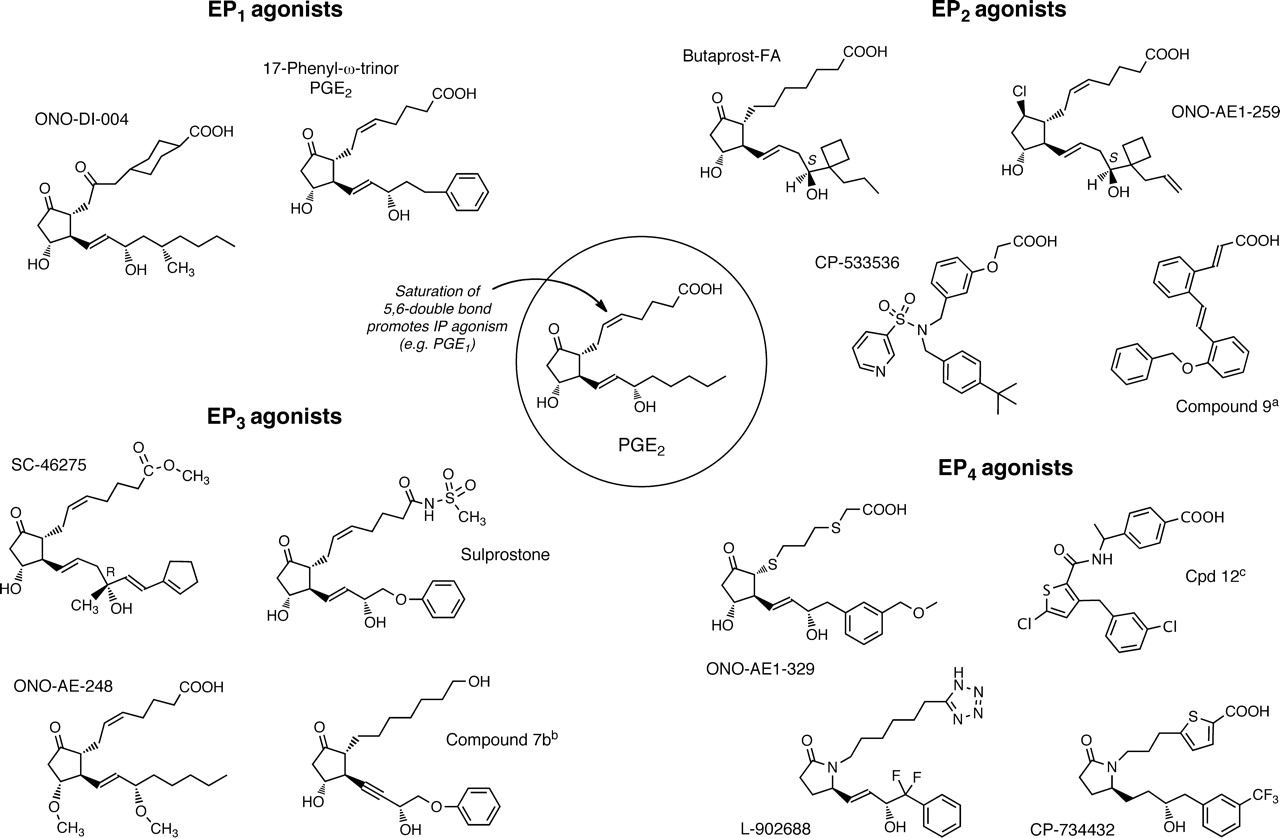{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}