


Abstract
Orexin signaling is essential for normal regulation of arousal and behavioral state control and represents an attractive target for therapeutics combating insomnia. Alternatively termed hypocretins, these neuropeptides were named to reflect sequence similarity to incretins and their potential to promote feeding. Current nomenclature reflects these molecular and biochemical discovery approaches in which HCRT, HCRTR1, and HCRTR2 genes encode prepro-orexin, the orexin 1 receptor (OX1) and the orexin 2 receptor (OX2)—gene names designated by the Human Genome Organization and receptor names designated by the International Union of Basic and Clinical Pharmacology. Orexinergic neurons are most active during wakefulness and fall silent during inactive periods, a prolonged disruption in signaling most profoundly resulting in hypersomnia and narcolepsy. Hcrtr2 mutations underlie the etiology of canine narcolepsy, deficiencies in orexin-producing neurons are observed in the human disorder, and ablation of mouse orexin neurons or the Hcrt gene results in a narcolepsy-cataplexy phenotype. The development of orexin receptor antagonists and genetic models targeting components of the orexin pathway have elucidated the OX2 receptor-specific role in histamine-mediated arousal and the contribution of both receptors in brainstem pathways involved in vigilance state gating. Orexin receptor antagonists of varying specificity uncovered additional roles beyond sleep and feeding that include addiction, depression, anxiety, and potential influences on peripheral physiology. Combined genetic and pharmacological approaches indicate that orexin signaling may represent a confluence of sleep, feeding, and reward pathways. Selective orexin receptor antagonism takes advantage of these properties toward the development of novel insomnia therapeutics.
I. Introduction
Orexin/hypocretin signaling has a pre-eminent role in the regulation of arousal and vigilance state. Disruption of the genes encoding the orexin 2 receptor or orexin ligands themselves are associated with canine (Lin et al., 1999) and murine (Chemelli et al., 1999) narcolepsy, and orexin deficiency is associated with the human disorder (Nishino et al., 2000). These initial findings not only demonstrated that orexin governs the normal regulation of arousal but also set off a range of investigations aimed at defining the role of this signaling pathway in arousal and the regulation of sleep. Before the genetic link to mammalian narcolepsy was uncovered, these hypothalamic peptides had been discovered and named based on their similarity to incretins (“hypocretin”) (de Lecea et al., 1998) and their propensity to promote feeding (“orexin” after the Greek word orexis for appetite) (Sakurai et al., 1998). Although the designation of hypocretin may be appropriate from a molecular and perhaps a functional standpoint, the term orexin is much more pervasive in the biological, chemical, patent, and popular literature. The present work addresses the nomenclature of these neuropeptides and their cognate receptors as well as the small molecules targeting pathways associated with orexin signaling. Studies using these small-molecule antagonists in concert with genetic manipulation have also been invaluable toward dissecting the function of orexin signaling in arousal, vigilance state, and the mechanisms regulating sleep in general. In addition to its function in arousal, feeding, and energy homeostasis, remarkable progress has also been made toward understanding the role of orexin in addiction and psychiatric function, as well as peripheral influences on nociception, metabolism and cardiovascular physiology that may or may not be a secondary consequence of its central roles. From this work, the therapeutic potential of modulating orexin signaling for the selective treatment of insomnia and related sleep disorders has become evident, but so too have the possibilities for the treatment of disorders in which sleep/wake dysregulation occurs. This therapeutic potential contrasts with the current standard of care including GABAA receptor modulators, which have less selectivity for mechanisms controlling sleep/wake regulation.
II. Discovery and Nomenclature of Orexin Signaling Components
A. Orexin-A and -B Are Products of the Hypocretin Gene
Even before the genetic association with narcolepsy was discovered, orexin neuropeptides were simultaneously described by two different groups using distinct molecular and biochemical approaches. From mRNA enriched from rat hypothalamus, de Lecea et al. (1998) identified, cloned, and sequenced a 569-nucleotide transcript encoding a 130-amino acid prepro-peptide based on sequence similarity to secretin, a gut hormone involved in osmoregulation. Because of its CNS expression restricted to large cell bodies of the dorsal lateral hypothalamic area and its sequence similarity to the incretin family of peptide hormones, the gene was termed hypocretin (Hcrt). Two peptide products predicted from proteolytic cleavage sites were confirmed by immunohistochemistry to be present in cell bodies and efferent fibers, and one of the synthetically generated peptides was able to elicit depolarizing currents in primary cultures of hypothalamic neurons (de Lecea et al., 1998). In an effort to “deorphanize” a panel of G-protein-coupled receptors, a second group (Sakurai et al., 1998) identified one such receptor capable of mediating Ca2+ transients in response to crude rat brain extracts. Purification and sequencing of the biological activity capable of activating this receptor revealed a sequence encoding a precursor peptide processed into two related peptides. Because the mRNA was found to be expressed in the lateral hypothalamus (LH), an area implicated in feeding regulation (Bernardis et al., 1993; Bernardis and Bellinger, 1996), and because intraventricular administration of both peptides dose-dependently induced food intake, the peptides were designated orexin-A and -B (OX-A1 and OX-B) after the Greek word orexis, for appetite. These deorphanized receptors were identified as orexin 1 and orexin 2 (OX1 and OX2) receptors (Sakurai et al., 1998). These findings ignited a large body of work aimed at deciphering the role of these signaling components in appetite control (Edwards et al., 1999; Sweet et al., 1999), energy homeostasis (Beck and Richy, 1999), and metabolism (Lubkin and Stricker-Krongrad, 1998; Takahashi et al., 1999) such that the term “orexin” seemed appropriate, yet the nomenclature debate had already begun (Nisoli et al., 1998). The subsequent discoveries of the genetic link to narcolepsy and the predominant role of orexin in arousal (Mieda et al., 2004), however, raised the possibility that orexin-induced feeding observed in preclinical models may be secondary to heightened wakefulness (Ida et al., 1999), further fueling the debate.
B. Nomenclature Recommendations
A parsimonious resolution to the nomenclature debate is to designate hypocretin the gene and mRNA name (human abbreviation: HCRT; rodent: Hcrt) and the precursor peptide and processed peptides after orexin (orexin A, Ox-A, orexin B, Ox-B) (Table 1). Likewise, the same is true for the two known G-protein-coupled receptors for these peptides; the HCRTR1 and HCRTR2 genes (Hcrtr1 and Hcrtr2 in rodents) encode the protein products OX1 and OX2 receptors, respectively. Although this nomenclature may be confusing to those new to the field, it does recognize the identification of the hypocretin mRNA and gene by molecular biology approaches (de Lecea et al., 1998) and the biochemical discovery of orexin peptides and their now deorphanized G-protein-coupled receptors (Sakurai et al., 1998). These distinct genetic and protein product designations are also now a matter of practical necessity. HCRT, HCRTR1, and HCRTR2 are now the accepted gene symbols in all genetic databases, including GenBank and HUGO. As for orexin-related protein products, the formal nomenclature from the International Union of Basic and Clinical Pharmacology Nomenclature Committee designates “OX-A” and “OX-B” as pharmacological ligands for “OX1” and “OX2” receptors, respectively (http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=51). The biological literature primarily uses the “orexin” designation, with references as well to “hypocretin” and/or the accepted gene name. Where the term “orexin” has been used much more exclusively, however, is in both the chemistry and patent literature, which has expanded substantially in the past 15 years as the therapeutic potential of modulating the orexin system has become increasingly evident (Fig. 1). Although early patents were filed for orexin peptide ligand derivatives and increasingly thereafter for molecular probes (e.g., microarray and quantitative polymerase chain reaction probes and primers), compound patents became more prominent with a growing interest in the development of small-molecule therapeutics. As seen in Fig. 1, the number of “orexin”-only patent filings has far exceeded that of “hypocretin”-only patents since 1999, the year the genetic link with narcolepsy was made. At the peak of patent filings in 2009, only 6 of a total of 965 patents filed mentioned “hypocretin” only, whereas 866 were exclusively for “orexin” and 93 for both. These figures demonstrate that the orexin and orexin receptor protein designations are the most widely accepted pharmacological designations.
Nomenclature of orexin signaling components
The IUPHAR (International Union of Basic and Clinical Pharmacology) ID was retrieved from http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=51. The HGNC gene name is that approved by the Human Genome Nomenclature Committee. Chromosomal location is based on fluorescent in situ hybridization mapping (human) and ISCN (International System for Cytogenetic Nomenclature) lengths (mouse and rat) from UCSC Genome Bioinformatics (http://genome.ucsc.edu). ChEMBL is from the MedChem literature data on drug-like molecules and their targets.
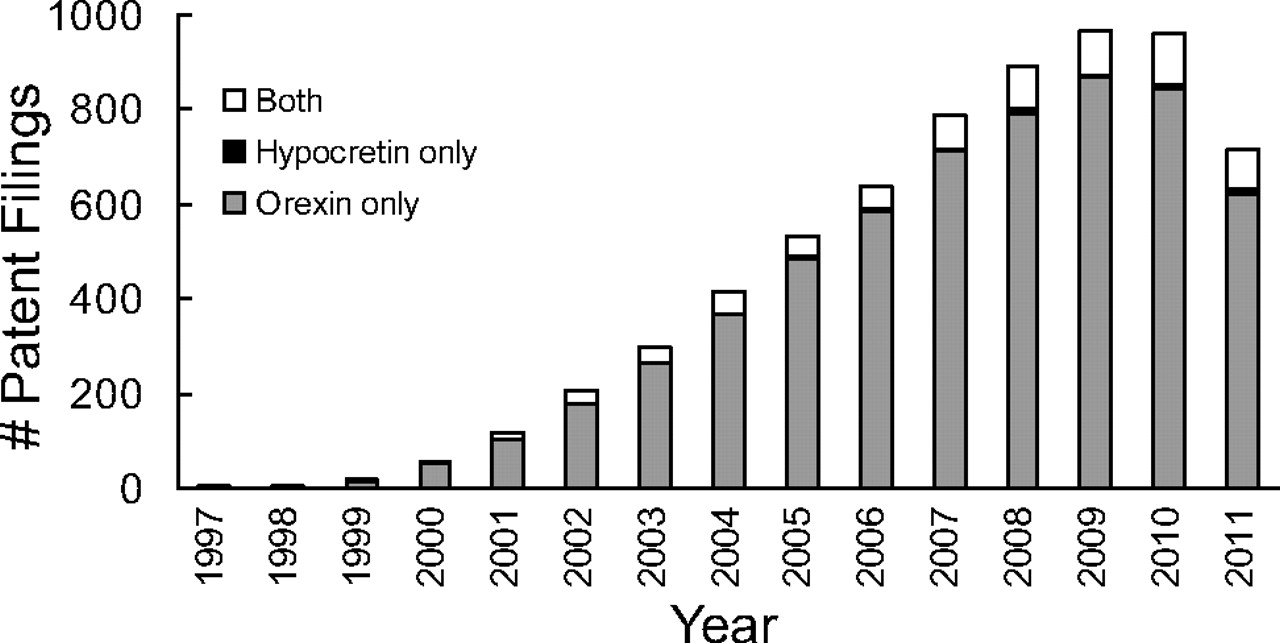
“Orexin” is used preferentially in the patent literature over “hypocretin.” The number of patent filings containing, in the full text, the words “orexin” (gray), “hypocretin” (black), or both (open) per year are shown. Results do not include patents in which the word “orexinergic” appears without either of the above.
C. Orexin-A and -B Structures Are Highly Conserved
Both OX-A and OX-B neuropeptides are derived from the same prepro-orexin precursor encoded by the HCRT gene. The structure and organization of the hypocretin gene has been largely conserved through evolution. In all vertebrates examined, the gene is composed of two exons with the intron splice falling within the early portion of the open reading frame encoding the secretory signal sequence (Fig. 2). Hcrt genes from multiple organisms, including teleost fish, avian, and mammalian species, are located within chromosomal loci having considerable synteny and, along with neighboring genes, may have experienced considerable pressure for functional conservation through evolution (Wong et al., 2011).

OX-A and OX-B are encoded by the HCRT gene. Structures of the human gene [from UCSB genome browser (http://genome.ucsc.edu); intronic sequence is shown at 1/10th scale of exon sequence], mRNA, and protein gene products shown. IC50 values for radioligand binding by OX-A and OX-B are depicted with the exception of the affinity of OX-B for OX1 receptors (420 nM, not shown), which is ∼10-fold less than for OX2 receptor (36 nM) (Sakurai et al., 1998).
The organization of prepro-orexin precursor peptide and the sequence of mature OX-A and OX-B ligands that are derived from it are highly conserved. The translated 131-amino acid human prepro-orexin peptide consists of nearly contiguous sequences encoding the secretory signal sequence, 33-amino acid OX-A, and 28-amino acid OX-B (Sakurai et al., 1999), and this organization, along with cleavage site sequences, are exactly conserved among all vertebrate organisms examined, including frog, chicken, and fish. This includes consensus “Gly-basic-basic” cleavage and C-terminal Gly-Lys-Arg amidation motifs, separating the OX-A and OX-B sequences, and Gly-Arg-Arg sequences, marking the termination of OX-B (Wong et al., 2011). Among mammals, the sequence of the mature OX-A ligand is entirely conserved among all species examined and contains two disulfide bridges, one formed by cysteines 6 and 12 and another between cysteines 7 and 14. These four residues are also 100% conserved from humans to amphibians (Wong et al., 2011). Mature OX-A is further post-translationally modified with an N-terminal pyroglutamic acid (Sakurai et al., 1998). Mammalian OX-B sequences, on the other hand, are very well conserved but have two points of differentiation: a serine reside at the second amino acid position in rodents, canines, and bovines is replaced by a proline in the human sequence, and a serine in the 18th position is replaced by an asparagine in rodents (Wong et al., 2011). Sequence diversity outside the secretory signal, OX-A, and OX-B sequences substantiates the functional importance of these defined regions.
OX-A and OX-B also share sequence similarity with one another, which is likely to underlie their ability to serve as ligands for both OX1 and OX2 receptors, albeit with differing affinities. In this regard, it is worth noting that mammalian OX-A and OX-B sequences are identical in the C-terminal portion of the mature peptides, including the nine-amino acid sequence Gly-Asn-His-Ala-Ala-Gly-Ile-Leu-Thr. They also share Arg-Leu and Leu-Leu sequences spaced two amino acids from one another and three amino acids N-terminal of the nine conserved positions mentioned above, suggesting that these residues may exist at one surface of an α-helical secondary structure (Sakurai et al., 1998; Wong et al., 2011). Because both peptides have measurable affinities for each of the OX1 and OX2 receptors, these observations indicate that these residues are essential for orexin receptor interaction. Human OX-A has nearly equal activity on both orexin receptors, with ligand binding affinities (IC50) of 20 and 38 nM for OX1 and OX2 receptors, respectively, and EC50 values of 30 and 34 nM in [Ca2+]i mobilization assays of cells transfected to express human OX1 and OX2, respectively (Fig. 2) (Sakurai et al., 1998). OX-B, however, has markedly less activity toward OX1 receptors with an IC50 for radioligand binding of 420 nM and an EC50 for [Ca2+]i of 2500 nM. It is more selective for OX2 receptors, exhibiting an IC50 of 36 nM and EC50 of 60 nM (Sakurai et al., 1998). This selectivity of OX-B for OX2 has been used to interpret the relative roles of OX2 and OX1 receptors in biological functions, because differential responses to microinjection of these peptides into brain regions indicates OX1 receptor function, whereas similar responses to both peptides may suggest OX2 receptor function. Definitive receptor selective function, however, is demonstrated only with genetic and/or highly selective orexin receptor antagonist reagents.
D. Orexin Receptor Structure Is Evolutionarily Conserved
1. Orexin 1 Receptor.
Orexin 1 and 2 receptors are found throughout mammalian species, and the core regions of these proteins are highly conserved. The greatest diversity occurs between rat and human OX1 receptor sequences, but even these proteins are 91% identical and 93% homologous (Table 2). As seen in the predicted structure of the OX1 receptor based on a β2-adrenergic receptor homology model (Fig. 3A), most of the divergent residues occur in the large cytoplasmic loop between transmembrane spanning helices five and six. Fewer residues within membrane spanning loops and the ligand-binding pocket diverge in rat, dog, and human sequences, and these changes are largely homologous. The exception is an Arg-to-His change at residue 205 within the second extracellular loop between transmembrane helices four and five of the predicted canine sequence, which may have the potential to affect ligand binding and antagonist activity. Differences in canine OX1 receptor sequences relative to human and rodent sequences may be of interest given the possible difference in phenotypes displayed by disruptions in orexin signaling in these species. Truncation of the dog OX2 receptor results in a narcoleptic phenotype accompanied by cataplexy (Lin et al., 1999), whereas deletion of the Hcrt gene or both orexin receptors in mice (Chemelli et al., 1999; Willie et al., 2003; Scammell et al., 2009) and orexin neuron loss in human narcoleptics is associated with this phenotype (Thannickal et al., 2003; Nishino et al., 2010). The Arg-to-His change in the dog OX1 receptor, however, does not seem to affect ligand activation or the activity of two different dual orexin receptor antagonists, [2(R,5R)-5-{[(5-fluoropyridin-2-yl)oxy]methyl}-2-methylpiperidin-1-yl][5-methyl-2-(pyrimidin-2-yl)phenyl]methanone (MK-6096) and DORA-22, toward the dog OX1 receptor (Winrow et al., 2012), indicating that any differences in the biological function of this receptor in canines may not be due to differences in receptor activity but might be explained by differential or regional expression changes.
Mammalian OX1 receptor protein homology
The indicated annotated protein sequences were compared with the human OX1 receptor sequence using a BLASTP algorithm with default comparative parameters (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Identity and similarity values are the percentage of identical or homologous amino acids shared with the core region of the human OX1 receptor.
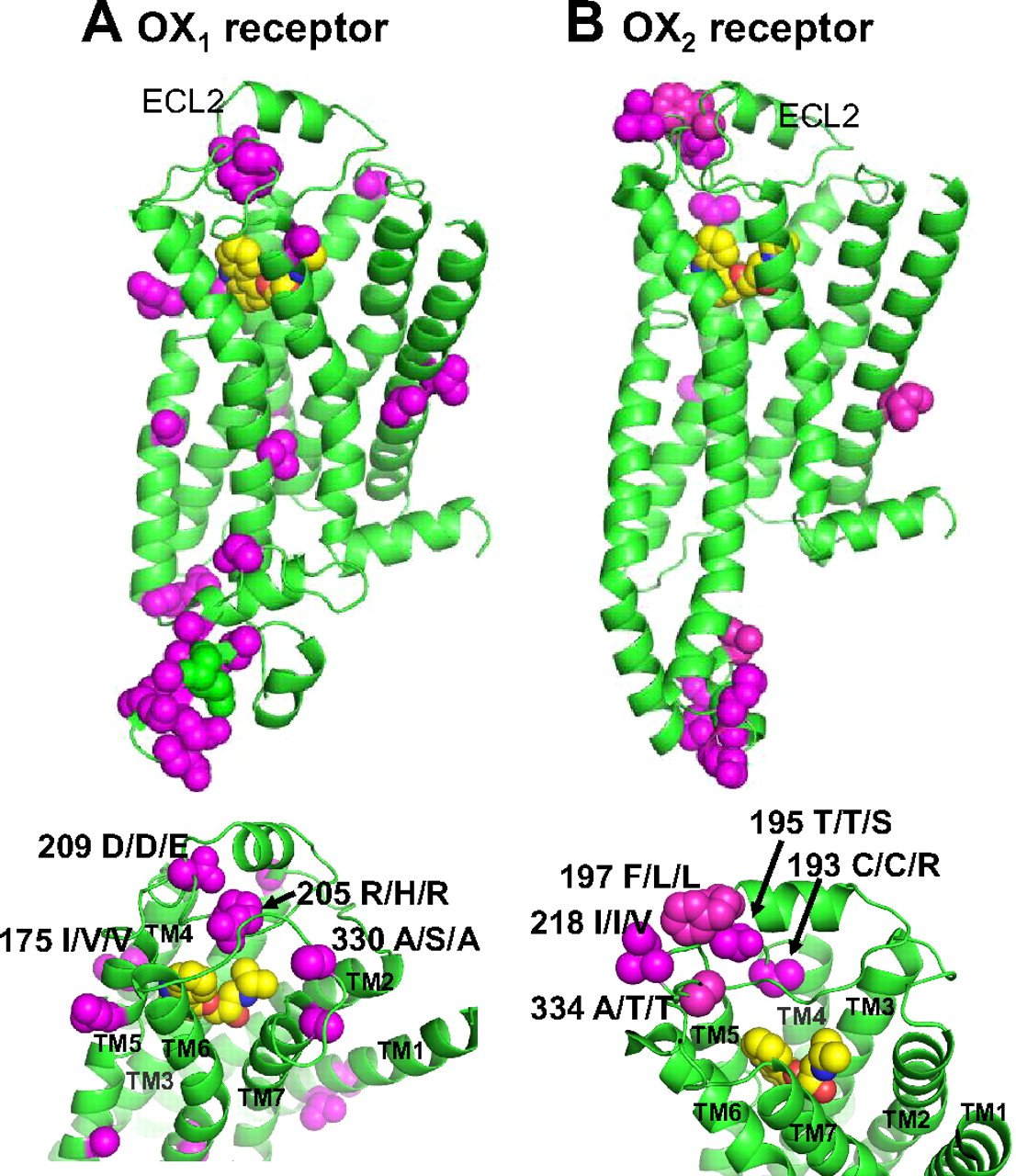
Amino acid divergence in the structures of OX1 and OX2 receptors among human, rat, and canine sequences. Homology models were created using MOE software (Chemical Computing Group, Montreal, QC, Canada) based on the crystal structure of carazolol binding to the β2-adrenergic receptor (Protein Date Bank code 2rh1) (Cherezov et al., 2007; Rosenbaum et al., 2007) used as the template. Sequence alignment of the transmembrane helices and the extracellular loop 2 (ECL2) of OX1 and OX2 receptors with the 2rh1 structure was performed according to Malherbe et al. (2010) and the best model selected from 10 intermediates. Backbone coordinates remained identical to the crystal structure such that no minimization was performed (structural data comparison and figure generated by M. Katharine Holloway, Ph.D., Chemical Modeling and Informatics, Merck Research Laboratories). The predicted structure of OX1 (A) and OX2 (B) receptors along with their ligand binding sites (lower panels) showing the peptide backbone (green) for sequence conserved among human, rat, and dog. Space-filling residues exhibiting sequence divergence are shown in magenta and the amino acid position and sequence substitutions are indicated in the lower panel (human/dog/rat amino acids at these positions). TM, transmembrane helices. The binding site is occupied by carazolol (yellow) used in homology modeling to the β2-adrenergic receptor.
2. Orexin 2 Receptor.
Mammalian OX2 receptor sequences exhibit even greater conservation between species (Table 3), which is probably a consequence of its greater role in mediating orexin's effects on arousal and vigilance state (see section V.E). As with the OX1 receptor, the greatest divergence among rat, dog, and human sequences occurs within the cytoplasmic loop between transmembrane helices five and six (Fig. 3B), suggesting that these positions are not critical for transduction of the ligand activation signal or subsequent G protein signaling. Within transmembrane regions and the predicted ligand-binding region, only conserved amino acid substitutions are observed. The most divergent position seems to be a Phe-for-Leu difference in rat and dog sequences located in the second extracellular loop between transmembrane helices four and five. Although this change represents an aromatic to aliphatic residue substitution, both are hydrophobic and are predicted to be removed from the putative ligand binding pocket.
Mammalian OX2 receptor protein homology
The indicated annotated protein sequences were compared with the human OX2 receptor sequence using a BLASTP algorithm with default comparative parameters (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Identity and similarity values are the percentage of identical or homologous amino acids shared with the core region of the human OX2 receptor.
Among the regions of OX2 receptors that are invariant in human, dog, and rat sequences are those critical for the interaction with OX-A and OX-B peptide ligands as well as small-molecule antagonists to the receptor. The ligand-binding pocket is formed by an interface between transmembrane helices, deep within the extracellular portion of both receptors. As indicated by functional studies, known small-molecule antagonists act in an orthosteric mode in that they compete for binding with residues critical for peptide ligand interaction. Transmembrane domain 3 seems to be the most critical, where exchange of OX2 receptor sequence with that from OX1 receptor switches the ligand binding properties of a chimeric receptor (Putula et al., 2011). The extracellular portion of transmembrane domain 3 also contains Gln134 and Thr135, residues essential for peptide ligand and small molecule interactions, respectively (Malherbe et al., 2010; Tran et al., 2011). It is noteworthy that substitution of Thr135 with an alanine in the OX2 receptor, which matches the Ala135 naturally found in the OX1 receptor, substantially attenuates small-molecule binding but induces a small increase in activity toward OX-B (Tran et al., 2011). Additional residues contained in the extracellular portions of transmembrane domains six and seven also contribute to the small-molecule pocket (Malherbe et al., 2010; Tran et al., 2011).
3. Evolutionary Origins.
Evolutionarily, OX2 receptors seem to be a more ancient addition to class B G-protein-coupled receptors relative to the OX1 receptor, which seems to have arisen from a more recent gene duplication (Wong et al., 2011).Hcrtr1 genes encoding OX1 receptors have not been identified outside of the mammalian class (Fig. 4), suggesting that the function of these proteins represents a refinement of sleep, the control of vigilance or potentially other behavioral functions unique to mammals. Using the human orexin receptor sequences as queries in BLASTP searches, proteins exhibiting similarity to these receptors were identified, including neuropeptide FF receptors 1 and 2 (NPFF receptors 1 and 2), substance K receptors, GPR83, neuropeptide Y receptors, and QRF peptide receptors (QRFP receptor; also known as GPR103). Although NPFF receptors 1 and 2 share a greater number of identical amino acid positions with OX2 receptors (31 and 33%) relative to QRFP receptor (27%), QRFP receptors retain a greater number of conserved or similar amino acid positions. When known QRFP receptors and NPFF receptors are included in phylogenetic analysis, QRFP receptors cluster nearer to orexin receptors (Fig. 4). This sequence similarity and lack of identity suggests that QRFP receptors diverged from orexin receptors earlier than NPFF receptors but experienced selective pressure to retain homologous sequence. Reported responses to central administration of QRF peptides, the ligands for QRFP receptor, include increases in arousal and locomotor activity, increased feeding, energy balance regulation, and the modulation of pain responses (Thuau et al., 2005; Moriya et al., 2006; Takayasu et al., 2006; Yamamoto et al., 2008; Lectez et al., 2009; Yamamoto et al., 2009), functions remarkably similar to that of orexin.

Orexin receptor phylogeny and evolutionary conservation. Human OX1 and OX2 receptor sequences were used in BLASTP algorithm searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to identify orthologous orexin receptor sequences from other species as well as nonorexin receptor sequences sharing sequence identity and homology. Human NPFF receptor 1, NPFF receptor 2, and GPR103 protein sequences were then used to identify orthologous versions of these sequences in other species. Sequences were then aligned, and phylogenetic trees generated by using the Cobalt Phylo Tree tools at NCBI (http://www.ncbi.nlm.nih.gov/tools/cobalt), using human rhodopsin as the out-group. Note: human NPFF receptor 1 and NPFF receptor 2 sequences share greater identity with orexin receptors, whereas GPR103 (QRFP receptor) shares greater sequence similarity (residues of similar property) such that phylogenetic comparison, including GPR103 sequences from multiple species places them nearer orexin receptor sequences.
E. Orexin Receptor G Protein Signaling Modulating Neuronal Excitability
Orexin activation of OX1 and OX2 receptors results in target cell activation, generally reflected in increased intracellular Ca2+ levels and postsynaptic excitation that can last several minutes (Sakurai et al., 1998; van den Pol et al., 1998; Hagan et al., 1999; Bourgin et al., 2000; Van Den Pol et al., 2001; Liu et al., 2002; Arrigoni et al., 2010). Presynaptic modulation of neurotransmitter release and postsynaptic modulation of responses to other neurotransmitters have also been observed, particularly for OX2 receptors, the cellular signaling mechanisms of which seem more diverse. The specific intracellular signaling pathways in which each of these receptors participate probably depend upon the cellular expression and subcellular localization (e.g., presynaptic versus postsynaptic) of other second-messenger signaling components.
Studies performed both in native primary neurons and in immortalized cell systems expressing recombinant orexin receptors indicate that these G-protein-coupled receptors increase intracellular Ca2+ through Gαq/11 activation (Lund et al., 2000; Smart et al., 2000; Kukkonen and Akerman, 2001; Holmqvist et al., 2002; Zhu et al., 2003). Both orexin receptors seem to signal primarily through Gαq/11, but some evidence suggests that both receptors are also capable of modulating cyclic nucleotide levels through Gαs and Gαi/o with varying ligand potencies, even though their predominant intracellular effect is to increase intracellular Ca2+ levels (Karteris et al., 2001; Holmqvist et al., 2002; Zhu et al., 2003). The most direct mechanism through which Gαq/11 induces Ca2+ levels in orexin responsive cells is the activation of phospholipase C (PLC), triggering the liberation of inositol-1,4,5-trisphosphate (IP3) and the release of Ca2+ from intracellular stores through IP3 receptors (Smart et al., 1999). However, the full Ca2+ response mediated by OX2 receptors requires extracellular Ca2+ conducted through either non–voltage-gated cation channels potentiating the PLC response (Lund et al., 2000; Kukkonen and Akerman, 2001; Holmqvist et al., 2002) or through L- and N-type Ca2+ channels, as indicated by a block in OX-A responsiveness of dopaminergic neurons of the ventral tegmental area (VTA) by Ω-conotoxin and nitrendipine (Uramura et al., 2001). Still other work has suggested a mechanism through which diacylglycerol, liberated by PLC, may directly activate transient receptor potential channels responsible for Ca2+ influx (Larsson et al., 2005; Näsman et al., 2006; Louhivuori et al., 2010).
The capacity of OX2 receptors to both increase intracellular Ca2+ and regulate cAMP levels enables orexin signaling to modulate both the presynaptic release of neurotransmitters and the postsynaptic response to other transmitters. Orexin can regulate the presynaptic release of serotonin, GABA or glutamate (van den Pol et al., 1998; Liu et al., 2002). Postsynaptically in the VTA, the substantial Ca2+ responses induced by orexin can induce long-duration changes in N-methyl-d-aspartate receptor expression, thereby potentiating the response of these neurons for several hours (Borgland et al., 2006; Borgland et al., 2008). Orexin neurons also release dynorphin, which attenuates inhibitory postsynaptic potentials, an effect that ultimately potentiates orexin-induced postsynaptic activation of neurons in tuberomammillary nuclei (TMN) (Eriksson et al., 2004; Kantor et al., 2009; Williams and Behn, 2011). This synergistic effect of neuropeptides may provide an explanation for the more pronounced obesity and REM dysregulation phenotype of animal models in which orexin-secreting neurons are genetically ablated relative to Hcrt knockout animals harboring a mutation in the gene encoding the prepro-orexin peptide (Hara et al., 2005; Kantor et al., 2009).
III. Orexin Receptor Reagents and Potential Therapeutics
A. Orexin Receptor Agonists/Potentiators
Advances in the study of orexin receptor agonism have been limited; fewer small-molecule tools are available compared with many antagonist compounds developed to date. Lack of an established positive control complicates any screening strategy to uncover a small-molecule agonist. Studies have thus been limited primarily to synthetic and modified versions of the endogenous neuropeptide agonists OX-A and OX-B.
In contrast to the narcoleptic phenotype in dogs and humans with dysfunctional orexin receptors or deficiencies in endogenous peptide ligands, (Lin et al., 1999; Nishino et al., 2000; Peyron et al., 2000; Wu et al., 2002), studies in rodents have demonstrated that intracerebroventricular administration of the orexin ligands serves to induce arousal and increase wakefulness (Hagan et al., 1999; Akanmu and Honda, 2005). Deadwyler et al. (2007) further reported that systemic and intranasal delivery of OX-A can decrease the effects of sleep deprivation on cognitive performance in nonhuman primates. The observation that systemic administration elicits an effect in these studies supports the finding that orexin-A crosses the blood-brain barrier by diffusion (Kastin and Akerstrom, 1999). No reports of behavioral responses to exogenously applied orexin peptides in humans, however, have appeared in the literature.
In addition to shedding light on the function of the endogenous orexin ligands, work with synthetic, modified peptides has allowed a greater understanding of ligand-receptor interactions. For example, the orexin-B analog [Ala11,d-Leu15]orexin-B was generated by replacing l-leucine residues at positions 11 and 15. These particular substitutions resulted in an enhancement of selectivity toward the OX2 receptor (compared with the OX1 receptor) by approximately 400-fold (Asahi et al., 2003). In the same study, a systematic approach to residue replacement revealed that three leucine residues are important for OX-B's selectivity for the OX2 receptor and determined the minimal peptide sequence required for orexin receptor activation.
In a recent patent disclosure, Yanagisawa (2010) reports a small-molecule agonist for OX2 receptors. This represents the only published account of nonpeptidic orexin ligands to date. The chemical series reportedly induces a robust Ca2+ response in OX2 receptor-expressing Chinese hamster ovary cells. There is one report of a small-molecule allosteric OX2 receptor potentiator, a compound that binds to a site on the receptor other than the ligand-binding site and potentiates the response of the receptor to its cognate ligand (Lee et al., 2010). This compound, N-benzyl-N-(3,4-dimethoxybenzyl)glycyl-N2-(1-phenylethyl)glycinamide (OBPt-9), was identified via a microarray-based, two-color, cell-binding screen and was shown to potentiate the response to orexin-A in both OX1 receptor- and OX2 receptor-expressing cells (Lee et al., 2010). Further characterization of these molecules should prove informative.
B. Dual Orexin Receptor Antagonists
Intensive efforts to identify orexin receptor antagonists began soon after the discovery of these excitatory neuropeptides and their receptors. At least four structurally distinct dual orexin receptor antagonists (DORAs) have entered human trials including almorexant [Actelion Pharmaceuticals, now GlaxoSmithKline (GSK), Brentford, Middlesex, UK], N-[[(2S)-1-[[5-(4-fluorophenyl)-2-methyl-4-thiazolyl]carbonyl]-2-piperidinyl]methyl]-4-benzofurancarboxamide (SB-649868; GSK), suvorexant (Merck, Whitehouse Station, NJ), and MK-6096 (Merck). Others have reported the discovery and characterization of additional DORAs as well as selective OX1 receptor and OX2 receptor antagonists (SORAs) (Coleman and Renger, 2010). Both Actelion and Merck have reported clinical proof of concept for treating primary insomnia with their respective receptor antagonists.
In 2007, Actelion disclosed data for almorexant, a potent DORA, showing this compound to be effective in promoting sleep in preclinical species. When administered to rats, almorexant dose-dependently increased REM and NREM sleep, effects beginning within an hour of dosing and persisting for up to 12 h after treatment (Brisbare-Roch et al., 2007). With repeat dosing at 100 mg · kg−1 · day−1 in rats, no tolerance to the sleep effects were seen, and no rebound was observed upon discontinuation of drug treatment (Brisbare-Roch et al., 2008). It is noteworthy that when dosed during the inactive period, when endogenous levels of orexin are at their lowest, the compound had little effect beyond normal sleep (Brisbare-Roch et al., 2007). In dogs, 100 mg/kg almorexant significantly reduced mobility scores relative to vehicle-treated dogs; the subjects could be easily aroused by the presence of a familiar individual but quickly returned to sleep once the stimulus was withdrawn (Jenck et al., 2007). Cataplexy was not observed in either species at any dose (Brisbare-Roch et al., 2007).
In double-blind, placebo-controlled clinical studies, almorexant was well tolerated, with reports of somnolence, dizziness, disturbed attention, and fatigue at doses above 200 mg. The incidence of somnolence increased with dose over the range of 100 to 1000 mg. As in preclinical studies, no cataplexy-related side effects were reported (Hoever et al., 2010). In double-blind studies with zolpidem as an active control, almorexant increased sleep efficiency, reduced sleep latency, and increased total sleep time at doses greater than 200 mg in healthy volunteers. A phase II study in patients with primary insomnia demonstrated the efficacy of both the 200- and 400-mg doses in improving sleep efficiency. Significant effects on secondary endpoints including latency to persistent sleep (LPS) and wake after sleep onset (WASO) were observed at the 400-mg dose (Dingemanse et al., 2007). Actelion initiated a phase III study in adults with primary insomnia (RESTORA1) in 2007. In this study, almorexant met the primary endpoint of superiority compared with placebo on both objective and subjective measures of WASO. However, an undisclosed human tolerability issue resulted in termination of Phase III clinical development in January 2009 (Almorexant in adult subjects, NCT00608985, http://www.clinicaltrials.gov).
Researchers at GSK also discovered a series of piperidine-derived antagonists, including SB-649868 as potent DORAs. SB-649868 was reported to inhibit both OX1 and OX2 receptor activity and entered clinical trials in 2005. Preclinically, SB-649868 was shown to be sleep-promoting in rodent and primate studies (Di Fabio et al., 2011). In 2007, GSK announced that SB-649868 had advanced to phase II clinical trials. In initial single rising dose studies, SB-649868 was well tolerated and exhibited proportional increases in exposure across the dose range. After administration of SB-649868 to healthy volunteers, there were statistically significant improvements in total sleep time, reduced LPS, and WASO at both doses relative to placebo. Neither dose produced cognitive impairment the morning after evening drug administration (Bettica et al., 2009; Renzulli et al., 2011). Clinical studies, however, revealed that SB-649868 increased exposure of coadministered simvastatin in a drug-drug interaction study, consistent with the potent inhibition of CYP3A4 in vitro (Bettica et al., 2011). Phase II studies of SB-649868 were placed on clinical hold in late 2007 because of the emergence of a reported preclinical toxicity, and GSK entered into a collaborative agreement with Actelion in 2008 to codevelop almorexant and other potential back-up compounds.
Merck has developed a diverse portfolio of orexin receptor antagonists in several distinct structural classes (Coleman et al., 2011a). After completing a screening campaign to identify new leads, researchers at Merck disclosed proline bis-amides including DORA-1 as potent dual orexin receptor antagonists. Intracerebroventricular administration of orexin B to rats placed in a beam-break box produced significant increases in locomotor activity over several hours. When rats were pretreated with DORA-1 by intraperitoneal injection 30 min before neuropeptide administration, DORA-1 produced dose-dependent reductions in this locomotor activity relative to baseline (Bergman et al., 2008).
Merck has also reported N,N-disubstituted-1,4-diazepanes, including suvorexant (MK-4305) and DORA-12, to be potent dual orexin receptor antagonists. Both compounds are selective antagonists with excellent activity in cell-based assays (Cox et al., 2009, 2010). Suvorexant inhibits orexin induced Ca2+ levels in cells expressing human OX1 or OX2 receptors with IC50 values of 50 and 56 nM, respectively, but has >6000-fold selectivity against a panel of 170 receptors and enzymes. Preclinically, suvorexant is orally bioavailable, has good brain penetrance, and demonstrates orexin receptor occupancy in rat brain (Cox et al., 2010; Winrow et al., 2011). An example of the sleep-promoting effects of DORA-12 in mice is seen in Fig. 5A. Active phase treatment is associated with dose-dependent reductions in active wake and augmentation of NREM and REM sleep, effects that diminish abruptly with the onset of the animals' normal inactive phase. These DORA-12-induced changes are mediated through OX1 and OX2 receptors, because these effects are absent in genetically modified mice lacking these receptors (Fig. 5B). In other rodent sleep studies, suvorexant dose dependently reduced active wake and increased REM and NREM sleep when administered orally at 10, 30, and 100 mg/kg. Suvorexant was also highly efficacious in promoting sleep in canines and rhesus monkeys (Winrow et al., 2011). Based on these and other efficacy studies as well as a favorable pharmacokinetic and safety profile, suvorexant was selected to advance into clinical development.

The sleep-promoting effects of DORA-12 are absent in OX1/2 receptor double-knockouts. Electrocorticogram and electromyogram were monitored in wild-type (A) and age-matched mice with targeted ablation of both OX1 and OX2 receptors (B) by radiotelemetry to determine mean time spent in the indicated sleep stages as described previously (Winrow et al., 2012). At times indicated by arrows, vehicle [20% vitamin E TPGS (d-α-tocopheryl polyethylene glycol 1000 succinate), by mouth, closed symbols] or DORA-12 at 60 or 100 mg/kg (open symbols) were administered in a balanced 5-day crossover paradigm. Treatment occurred during the late active phase, approximately 4 h before the onset of the inactive phase (10:00/10:30 AM; zeitgeber time, 08:00/08:30; black arrows). Closed and open bars below each plot represent dark (active) and light (inactive) phases, respectively. Plotted points are the mean time spent in each sleep state during 30-min intervals after treatment over 5 days of consecutive treatment as determined by automated scoring and analysis as described previously (Winrow et al., 2012). Error bars (where visible) depicting the S.E.M. for each point are included, and time points exhibiting significant differences between vehicle and DORA-12 responses are indicated by gray vertical lines and tick marks (short, p < 0.05; medium, p < 0.01; long, p < 0.001; linear mixed effects model for repeated measures applied t test).
Suvorexant was well tolerated in phase I studies, with peak plasma levels achieved at 1.5 to 4 h after dosing and a terminal plasma half-life of 8 to 14 h. In healthy volunteers, dose-dependent observations of somnolence were evident. Next-day residual sedation was not observed when suvorexant was administered at doses of 10 and 50 mg in the evening, whereas these same doses provided significant increases in overall sleep efficiency and reductions in WASO and LPS in a dose-dependent manner. Results from the phase IIb study demonstrated that suvorexant was superior to placebo in improving sleep efficiency on the first night of treatment as well as at the end of 4 weeks in patients with primary insomnia. These improvements in sleep efficiency were noted at all doses (10, 20, 40, and 80 mgs). Suvorexant also showed improvements in the secondary endpoints of reduced WASO at all doses and reduced LPS at 80 mg (Herring et al., 2010). In 2010, Merck announced that suvorexant had entered into phase III trials; it is currently the most advanced orexin antagonist in active clinical development.
Merck has disclosed an additional series of 2,5-disubstituted piperidine carboxamides including DORA-22 and MK-6096 as potent dual orexin receptor antagonists. The structure and preclinical pharmacology of MK-6096, a second dual orexin receptor antagonist in clinical development from Merck, has been disclosed (Coleman et al., 2012; Winrow et al., 2012). MK-6096 is structurally distinct from suvorexant and is highly efficacious in promoting sleep in rats (3–30 mg/kg) and dogs (0.25–0.5 mg/kg) (Winrow et al., 2012). MK-6096 was reported to have entered phase II clinical studies in 2009.
C. Orexin 2 Receptor-Selective Antagonists
Although dual orexin receptor antagonists have been shown to promote sleep in multiple species, SORAs have been used extensively to evaluate the relative role of each receptor subtype in the control of arousal and sleep, as well as other behaviors and physiology. An OX2 SORA [1-(2,4-dibromophenyl)-3-[(4S,5S)-2,2-dimethyl-4-phenyl-1,3-dioxan-5-yl] urea (JNJ-10397049)] has been evaluated for sleep/wake effects, both alone and in conjunction with 1-(6,8-difluoro-2-methyl-quinolin-4-yl)-3-(4-dimethylamino-phenyl)-urea (SB-408124), an OX1 SORA (McAtee et al., 2004; Dugovic et al., 2009). Both compounds are brain-penetrant, with JNJ-10397049 producing ∼80% cortical OX2 receptor occupancy for more than 6 h, an OX2 receptor occupancy matched by almorexant at this dose. Sleep-promoting effects of the OX2 SORA JNJ-10397049 at 30 mg/kg were due to lengthening sleep bouts, whereas treatment with almorexant was associated with sleep-promoting effects marked by an increased number of REM and NREM sleep bouts. Both almorexant and JNJ-10397049 reduced histamine levels in the LH, whereas neither the histamine levels nor sleep parameters were affected by the relatively OX1 receptor-selective SB-408124 (Dugovic et al., 2009).
Researchers at Roche have reported EMPA (N-ethyl-2-[(6-methoxy-pyridin-3-yl)-(toluene-2-sulfonyl)-amino]-N-pyridin-3-ylmethyl-acetamide) as a selective OX2 SORA structurally distinct from JNJ-10397049 that exhibits greater affinity for human OX2 relative to OX1 receptors (approximately 900-fold selective) (Malherbe et al., 2009). Autoradiography using [3H]EMPA in rat brain slices shows high specific binding in hypothalamus, tuberomammillary nuclei, hippocampus, and nucleus accumbens. In vivo, EMPA dose-dependently reversed OX-B–induced hyperlocomotion in mice, achieving full reversal at a dose of 300 mg/kg i.p. It is noteworthy that EMPA induced no psychomotor deficits when evaluated in a rat Rotorod assay at doses as high as 30 mg/kg i.p. (Malherbe et al., 2009). These results are consistent with earlier results reported for structurally distinct DORAs, including almorexant and SB-649868.
D. Orexin 1 Receptor-Selective Antagonists
The need for potent, selective preclinical research tools is highlighted by the wealth of studies using 1-(2-methylbenzoxazol-6-yl)-3-[1,5]naphthyridin-4-yl urea (SB-334867), described as a highly selective OX1 SORA developed by SmithKline Beecham (now part of GlaxoSmithKline). More than 160 papers have reported the use of SB-334867 as an OX1 SORA in behavioral models evaluating addiction, feeding, sleep, and other behaviors. This compound antagonizes OX-A-induced [Ca2+]i signal mediated by OX1 receptors expressed on Chinese hamster ovary cells (KB, 40 nM), and shows ∼50-fold selectivity relative to OX2 receptors evaluated in the same assay (KB, 1995 nM) (Haynes et al., 2000; Smart et al., 2001) (Table 4). These results are consistent with binding affinities observed in our laboratories, which have found it to be ∼45-fold selective for OX1 over OX2 receptors (Ki = 18 and 835 nM, respectively) (A. Gotter, P. Coleman, J. Renger, and C. Winrow, unpublished observations). However, examination of SB-334867 potency against a panel of 170 other enzymes and receptors showed significant interactions with at least seven other targets at concentrations less than 10 μM. These included activities toward the adenosine A2A receptor (Ki, 0.67 μM), 5-HT2C receptor (Ki, 1.2 μM), monoamine transporter (Ki, 1.44 μM), norepinephrine transporter (Ki, 1.58 μM), adenosine transporter (Ki, 2.45 μM), adenosine A3 receptor (Ki, 3 μM), and 5-HT2B receptor (Ki, 3.47 μM) (A. Gotter, P. Coleman, J. Renger, and C. Winrow, unpublished observations). The favorable pharmacokinetic properties and commercial availability of SB-334867 have made this a popular tool compound for studying orexin signaling in vivo, and there is the potential for substantial central nervous system exposure with this compound. For example, in rats administered 20 mg/kg i.p. (25% 2-hydroxypropyl β-cyclodextrin), we observed a sustained concentration of 3.4 μM in plasma at both 30 minutes and 2 h after dosing. Given the selectivity profile of SB-334867, there exists a possibility for antagonism for not only OX1 receptors but also OX2 and several other targets at these doses. It is for these reasons that some caution should be exercised in interpretations of behavioral observations attributed to selective OX1 receptor antagonism in studies using SB-334867 exclusively.
Selectivity and potency of major orexin receptor antagonists
Two other OX1 receptor-selective antagonists appearing in the literature are SB-408124 and 5-bromo-N-[(2S,5S)-1-(3-fluoro-2-methoxybenzoyl)-5-methylpiperidin-2-yl]methyl-pyridin-2-amine (GSK1059865). Disclosed in 2004 by SmithKline Beecham, SB-408124 has a published OX1/OX2 receptor selectivity of 63-fold (Kb, 22 versus 1405 nM in Ca2+ mobilization assays), slightly better than SB-334867 (Langmead et al., 2004). Like SB-334867, however, SB-408124 exhibits significant activity toward other receptors, notably 5-HT2B (0.32 μM), dopamine D1 (1.78 μM), 5-HT2C (1.88 μM), adenosine A2A (2.77 μM), and α2b-adrenergic receptors (3.29 μM). SB-408124 has good pharmacokinetic properties, including bioavailability (∼80%) and brain penetration (1.7%) in rats (A. Gotter, P. Coleman, J. Renger, and C. Winrow, unpublished observations). However, these favorable properties allow SB-408124 to reach levels that may also affect other receptors. GSK1059865, a recently identified OX1-selective antagonist (Gozzi et al., 2011), may offer an improvement over SB compounds, as it has a reported OX1/OX2 receptor selectivity of ∼79-fold (Kb, 1.6 versus 126 nM in IP3 accumulation assays), with no significant activity at concentrations under 1 μM toward a panel of 113 different receptors with the exception of the κ-opioid receptor (Ki, 320 nM) (A. Gotter, P. Coleman, J. Renger, and C. Winrow, unpublished observations). More selective OX1 SORAs will undoubtedly be developed, but currently available reagents used in combination with DORAs, 2-SORAs, and genetic models have nonetheless provided valuable insight into the function of this receptor in the control of vigilance state and other orexin-mediated physiology and behavior.
IV. Orexin Neurons Are a Key Component of Pathways Regulating Sleep
The identification of mutant hcrtr2 genes encoding truncated versions of OX2 receptors responsible for genetically transmitted canine narcolepsy (Lin et al., 1999) and the description of the narcoleptic phenotype of Hcrt knockout mice 1 month later (Chemelli et al., 1999) set off extensive investigations into the mechanisms through which orexins promote arousal and control of vigilance state. These findings also led to focused efforts to develop small-molecule antagonists to probe the function of orexin receptors in sleep and to validate these receptors as targets for the development of pharmacological therapeutics.
A. Neurological Pathways Regulating Sleep
The identification of orexin neuropeptides and their cognate receptors has provided an understanding of the network of neuronal pathways involved in the interplay between sleep and arousal-promoting centers and how these areas interact to control vigilance state. The predominant sleep-promoting influence in the brain is provided by the ventrolateral preoptic (VLPO) and adjacent median preoptic areas. Neurons of the VLPO are most active during NREM sleep, partially active during REM sleep, and silent during wakefulness. They send inhibitory projections to arousal promoting areas, including the tuberomammillary nuclei (TMN), laterodorsal and pedunculopontine tegmental (LDT, PPT) nuclei, the locus ceruleus (LC), dorsal raphe (DR) nuclei, the ventral tegmental area (VTA) (Saper et al., 2005b, 2010; España and Scammell, 2011) and notably, orexinergic neurons of the LH (Sakurai et al., 2005b; Yoshida et al., 2006). This inhibitory influence is primarily mediated by GABAergic influences. In fact, potentiation of GABA signaling from the VLPO is thought to underlie the mechanism of action of currently marketed sleep agents (e.g., zolpidem and eszopiclone) (España and Scammell, 2011).
Orexin-secreting neurons of the LH project to brainstem nuclei involved in promoting arousal (Fig. 6). The activity of these nuclei, which include the TMN, LDT/PPT, DR, and LC, is dependent upon the balance of influence imposed by both inhibitory signals from the VLPO and excitatory ones provided by orexin. Ultimately, the integration of these signals determines arousal and behavioral state. A primary orexinergic projection is sent to the TMN, which preferentially express OX2 over OX1 receptors (Trivedi et al., 1998; Marcus et al., 2001). TMN neurons project broadly to the prefrontal cortex (PFC), thalamus, and other subcortical structures and are normally active during wake and progressively less active during NREM and REM sleep. They represent the primary source of histaminergic (HA) neurons in the brain, and HA receptor agonists and antagonists promote and attenuate arousal, respectively (Monti et al., 1986; Lin et al., 1988; Mochizuki and Scammell, 2003). Up-regulation of this histaminergic activity underlies the mechanism of a new class of wake-promoting drugs that inhibit histamine H3 receptors to counter the reduced HA levels observed in narcoleptics (Lin et al., 2008; España and Scammell, 2011).

Orexin and OxR efferent pathways associated with arousal, vigilance state, and reward pathways. NAc, nucleus accumbens; HA, histaminergic; DA, dopaminergic; ACh, cholinergic; NE, noradrenergic; 5-HT, serotonergic. Green, orexinergic neuron projections; red, preferential OX1 receptor expression; blue, preferential OX2 receptor expression; violet, both OX1 and OX2 receptor expression.
Orexin neurons also send projections to cholinergic tegmental nuclei (the LDT and PPT) as well as noradrenergic LC and serotonergic DR neurons. OX1 receptors are preferentially expressed in the LC, whereas both orexin receptors are detectable in LDT, PPT, and DR (Fig. 6) (Trivedi et al., 1998; Marcus et al., 2001). Along with arousal-promoting influences, these brainstem regions are also responsible for gating between vigilance states, particularly in and out of REM sleep. Orexin influences on neurons of the LDT and PPT also regulate muscular atonia that accompanies REM sleep, through both direct and indirect effects on ventromedial neurons of the medulla, which in turn inhibit spinal motor neurons through GABA projections. More extensive discussions of these mechanisms regulating vigilance state and sleep-dependent motor activity have been reviewed elsewhere (Saper et al., 2010; España and Scammell, 2011; Scammell and Winrow, 2011).
B. Orexin-A and -B Promote Arousal and Modulate Vigilance State
Orexins provide an arousal signal that is both necessary and sufficient for normal sleep/wake regulation. Over the course of the 24-h circadian cycle, changes in arousal match oscillating levels of orexin. In nocturnal animal models, orexin levels rise over the night-time hours, peaking late in the active phase, whereas in primates, OX-A levels in CSF accumulate over daytime hours, peaking just before the dark phase (Taheri et al., 2000; Zeitzer et al., 2003). OX-A applied exogenously (via intracerebroventricular injection) to rats results in increased locomotor activity, grooming and wakefulness, whereas the mean time spent in NREM and REM sleep is diminished. These effects are greatest when OX-A is administered during the inactive phase when endogenous orexin levels are at their lowest and barely detectable when applied during periods of wakefulness, as might be expected of an arousal signal (Hagan et al., 1999; Piper et al., 2000). Exogenously applied OX-A also promotes arousal in mice with selected genetic ablation of orexinergic neurons. In this case, the levels of wakefulness and NREM and REM suppression in mutant animals exceed those seen in wild-type animals treated identically (Mieda et al., 2004), indicating that downstream orexin signaling components, including orexin receptors, are intact and are up-regulated in these mutants and that the neuropeptide alone is sufficient as a wakefulness signal. Similar results are seen in narcoleptic dogs where OX-A rescues the cataplexy and hypersomnolence phenotype of an animal harboring a mutation in the Hcrt gene encoding prepro-orexin but has no effect in dogs with mutations in the gene for the OX2 receptor (Fujiki et al., 2003). Artificial activation of orexin-secreting neurons is sufficient to drive arousal. In an elegant set of experiments, Adamantidis et al. (2007) and later Carter et al. (2009) used mice in which the expression of channelrhodopsin-2 was driven by the Hcrt promoter such that orexin-containing neurons could be photically stimulated by a fiber optic means. Activation of orexigenic neurons in the LH of these mice induced transitions from NREM or REM sleep into wakefulness (Adamantidis et al., 2007), but in the face of increased sleep pressure induced by sleep deprivation, these wake-promoting effects as well as orexin-dependent activation of TMN and LC neurons were diminished, indicating that homeostatic influences converge downstream of orexin neuron activation (Carter et al., 2009).
C. Modulation of Orexin Signaling
In general, the timing and extent of sleep and wakefulness is driven by two different influences; the circadian clock that aligns the timing of an organism's physiology and behavior with daily environmental timing cues, and the homeostatic drive for sleep aligning the restorative properties of rest with physiological and energy needs. The endogenous circadian pacemaker resides in the suprachiasmatic nuclei (SCN) of the hypothalamus, and controls daily cycles of arousal, locomotor activity, gut motility, and the timing of sleep even in the absence of external cues such as ambient lighting changes (Huang et al., 2011). When grown in culture, neurons of the SCN in themselves have periods of electrical activity of ∼24 h (Welsh et al., 1995). The activity of the SCN is primarily entrained to the environment by ambient lighting cues (Klein et al., 1991). The output from the SCN to the sleep network is conveyed through the ventral subparaventricular zone to the dorsomedial nucleus of the hypothalamus (DMH). From here the DMH projects to VLPO neurons via inhibitory GABA projections and sends excitatory connections to orexin containing neurons of the LH (Saper et al., 2005b). The DMH, which is largely active during wakefulness (Saper et al., 2005a), also receives appetite, feeding, and body temperature inputs, such that some level of homeostatic regulation may occur at this level (Saper et al., 2005b). Together, the DMH and the VLPO coordinate the activity of orexin neurons to regulate arousal aligned with circadian environmental cues.
The specific mechanism through which the homeostatic drive for sleep is mediated is less clear, however, but is thought to be associated with energy homeostasis and the need to conserve metabolic energy. The need for sleep accumulates with wakefulness such that the pressure for sleep accumulates with sleep deprivation. Evidence for orexin neurons being an integration site for homeostatic influences are the observations that orexin cell firing is increased by direct acetylcholine, glutamate, and ghrelin application and decreased by leptin, glucose, norepinephrine, 5-HT, and GABA (Burdakov, 2004). One attractive candidate for this regulation is adenosine, because during prolonged wakefulness, ATP is degraded to ADP, AMP, and eventually adenosine, which accumulates in parts of the brain (Porkka-Heiskanen et al., 2000). Orexin neurons express adenosine A1 receptors, and application of the adenosine A1R antagonist 1,3,-dipropyl-8-phenylxanthine into the LH increases wake and suppresses both REM and NREM sleep (Thakkar et al., 2008). Microinfusion of the adenosine A2A agonist 2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine(CGS21680) into the ventral striatum both promotes sleep measures and attenuates c-Fos production in orexin-producing cells, suggesting not only that adenosine's sleep promoting effect may be facilitated by reduced orexinergic cell firing but also that orexin signaling is not necessarily the exclusive mechanism for adenosine-mediated sleep promotion (Satoh et al., 2006).
Corticotropin-releasing factor (CRF) and dopamine also seem to modulate the influence of orexin on arousal. CRF signaling in response to stress represents a possible interaction with orexin signaling and sleep, because elevated levels of the hormone are associated with arousal, and intracerebroventricular administration of CRF induces wakefulness and locomotor activity (Sakurai and Mieda, 2011). Any interaction with CRF signaling, however, appears to be upstream of orexin, because orexin-induced arousal persists in CRF receptor knockouts as well as in the presence of CRF receptor antagonists (Fenzl et al., 2011). Evidence for the modulation of orexin signaling in arousal and vigilance state by dopamine has also recently emerged (Sakurai et al., 2010; Sakurai and Mieda, 2011). In orexin peptide knockout mice, pharmacological D1 receptor activation decreases the prevalence of sleep attacks relative to wild-type animals. Conversely, the hypersomnolence of these animals is exacerbated with D1 antagonism, whereas D2 receptor modulation has little to no effect on arousal (Burgess et al., 2010). Cataplectic attacks, however, are affected by D2 receptor activity; pharmacological D2 activation and inhibition are associated with substantial increases and decreases, respectively, in behavioral arrest (Burgess et al., 2010). These studies further illustrate the existence of distinct pathways for arousal control and the regulation of vigilance state as well as the dopamine receptor subtype involvement in each.
V. Genetic and Pharmacological Dissection of Orexin-Mediated Arousal
Our current understanding of the role of specific orexin signaling components in arousal and vigilance state is based on both pharmacological manipulations using orexin receptor modulators and the evaluation of animals mutant for orexin receptors, the Hcrt gene encoding the prepro-orexin precursor and targeted ablation of orexinergic neurons. Combining the interpretations of both approaches has proven invaluable toward uncovering the role of orexin and its cognate receptors in the control of sleep and arousal.
A. Canine Narcolepsy
In dogs, narcolepsy is manifested as active-phase sleep attacks and, during the inactive phase, as short sleep latency and fragmented sleep with rapid EEG/polysomnographic transitions between sleep stages. In severe cases, cataplectic attacks are characterized by atonia with hind-limb buckling during which the animal retains consciousness, often with open eyes that are able to follow visual stimuli. Consciousness may give way to sleep, typically REM. These episodes are typically triggered by palatable food presentation or play, much as positive emotions do in human patients (Nishino, 2005).
Canine narcolepsy is classified into two forms: familial (genetic), associated with mutations in the OX2 receptor gene, and sporadic, which is typically associated with loss of orexin-secreting neurons. The genetic form is transmitted in an autosomal-recessive fashion with complete penetrance. To date, OX2 receptor mutations include truncations within transmembrane region five or just after transmembrane region six in Doberman pinschers and Labrador retrievers (Lin et al., 1999) and a point mutation in a dachshund family resulting in a glutamate to lysine change at amino acid position 54, rendering a receptor incapable of being bound and activated by OX-A or OX-B (Hungs et al., 2001). CSF and brain levels of OX-A and OX-B are normal in these mutants, and orexin-containing neurons are normal in appearance (Ripley et al., 2001). Although exogenously applied OX-A can reverse narcoleptic symptoms in sporadic narcoleptic dogs deficient in orexin neurons, high doses of intracerebroventricular and intravenous administration of OX-A have no effect on arousal or cataplectic symptoms of OX2 receptor mutant narcoleptic Doberman pinschers (Fujiki et al., 2003). Sporadic canine narcolepsy (poodles, beagles, dachshunds, collies, fox terriers) does not seem to be associated with a single, highly penetrant gene mutation but is typically associated with symptoms more pervasive than the genetic form (Nishino, 2005). Unlike humans in which a leukocyte antigen gene is associated with sporadic narcolepsy, a canine allele specifically associated with the disorder remains elusive (Peyron et al., 2000; Nishino, 2005; De la Herrán-Arita et al., 2011).
B. Genetically Engineered Mouse and Rat Models
Genetic manipulation of rats and mice has been used to mimic the clinical etiology of narcolepsy associated with orexinergic neuron loss and to dissect the role of individual orexin signaling components. Efforts have included transgenic models in which the expression of a cellular toxic transgene is used to specifically ablate orexin-producing neurons to imitate the autoimmune loss of these neurons in patients with narcolepsy. Genetic models also include targeted gene knockouts of the Hcrt gene encoding the prepro-orexin peptide processed into OX-A and OX-B ligands (orexin peptide knockouts), the Hcrtr1 and Hcrtr2 genes encoding OX1 and OX2 receptors (OX1 receptor and OX2 receptor knockouts), and double receptor mutant mice lacking both of these receptors (OX1/2 receptor double knockouts). Together, with pharmacological manipulation using selective receptor agonists and antagonists described in sections III and IV, these mutant animals have provided invaluable information regarding not only the role of orexin signaling in narcolepsy/cataplexy but also the general neuronal pathways regulating sleep and vigilance state (De la Herrán-Arita et al., 2011). Both approaches uncovered the importance of each of these signaling components in arousal, vigilance state, and the regulation of sleep. Overall, the importance of these signaling components to arousal may be summarized as follows: Ox neurons > orexin peptide KOs ≥ OX1/2 receptor double KO > OX2 receptor KOs ≫ OX1 receptor KOs. It should be noted, however, that in many cases, these animal models have been evaluated by different laboratories using different methods (e.g., EEG versus visual observation) often using different criteria to make interpretations regarding the impact of a given mutation on arousal and/or cataplectic behavior. Efforts have been made, however, to standardize the criteria for the cataplectic behavior in mice. Based on observations from orexin peptide knockouts, the following criteria have been established on behalf of the International Working Group on Rodent Models of Narcolepsy (Chemelli et al., 1999; Willie et al., 2003; Scammell et al., 2009): 1) abrupt atonia lasting over 10 s, 2) animal immobility during the episode, 3) quantitative EEG (qEEG) dominated by θ activity during the episode, and 4) behavioral arrest preceded by a period of active wake lasting > 40 s. In addition, cataplectic behavior should be reversible, with administration of clomipramine or other anticataplectic (e.g., monoamine reuptake inhibitors such as desipramine) (Willie et al., 2003; Scammell et al., 2009). Nevertheless, this expression of the importance of these components on arousal can be concluded on the basis of the studies discussing these genetic models below, along with what has been determined from both dogs and humans.
C. Orexin Neurons Are Critical for Both Arousal and Vigilance State Gating
In theory, the best preclinical models for the majority of human cases exhibiting narcolepsy with cataplexy are transgenic mice and rats lacking orexin-producing neurons. These animals (Ox/Atx mice and rats) express an apoptotic Ataxin-3 transgene whose expression is driven by the human Hcrt gene promoter, resulting in programmed cell death of orexin-containing neurons while leaving neighboring neurons, including melanin-concentrating hormone-containing neurons of the LH, intact (Hara et al., 2001; Beuckmann et al., 2004). Despite incomplete loss of orexin neurons in hemizygous animals, OX-A levels within CSF, cortex, and brainstem are as much as 100-fold lower than that of wild-type animals (Zhang et al., 2007). The phenotype of Ox/Atx mice and rats resembles that seen in human patients with narcolepsy/cataplexy active-phase hypersomnolence, behavioral arrest/atonia episodes triggered by excited ambulation or grooming, and frequent wake-to-REM transitions never observed in wild-type animals that often enter REM through an intervening NREM transition (wake to NREM to REM). Inactive phase sleep is fragmented with reduced REM latency and frequent, but short bouts of wake and NREM with transitions occurring rapidly (Hara et al., 2001; Beuckmann et al., 2004). Remarkably, these animals remain sensitive to ectopically expressed or exogenously administered OX-A and OX-B, which reverse behavioral arrests and REM abnormalities, including wake-to-REM transitions of Ox/Atx mice, in some cases to greater effect than in wild-type mice (Mieda et al., 2004). One divergence from the accepted criteria for cataplexy, however, is the presence of quantitative EEG spectral pattern resembling wake during episodes of behavioral arrest in Ox/Atx rats (Beuckmann et al., 2004), as opposed to θ or REM activity characteristic of cataplexy. It remains to be seen whether this is a species-dependent divergence from mice or if more complete loss of orexin neurons in homozygous animals may affect these animals even more profoundly.
D. The Role of the Hcrt Gene and Hcrt Gene Product
Mice with a targeted deletion of the Hcrt gene encoding the prepro-orexin precursor peptide display a narcolepsy phenotype similar to transgenic Ox/Atx animals, with cataplectic episodes satisfying the criteria described above (Chemelli et al., 1999; Fujiki et al., 2009; Scammell et al., 2009). Furthermore, the abrupt behavioral arrests in orexin peptide knockouts are reversed by clomipramine, whereas caffeine only increased wakefulness and actually exacerbated cataplectic symptoms, demonstrating the specificity of this treatment (Willie et al., 2003). Although orexin peptide knockouts clearly exhibit cataplexy, their phenotype is not as pervasive as that observed in Ox/Atx animals. Direct comparisons revealed an even greater number of vigilance state transitions and time spent in REM for Ox/Atx animals (Kantor et al., 2009). These results indicate that orexin-containing neurons provide additional signals beyond orexin itself, perhaps glutamate or dynorphin, which can contribute to narcoleptic symptoms (Sakurai et al., 2005a; Kantor et al., 2009). Modeling of orexin neuron activity suggests that dynorphin is capable of modulating orexin responses, delaying its arousal effects at the sleep/wake transition by affecting the sensitization and firing rate of orexin-sensitive neurons (Williams and Behn, 2011). Nevertheless, orexin peptide knockout animals have provided a useful model in which to study the cataplectic episodes in detail, where both scheduled palatable food and running wheel presentation effectively increase the frequency of cataplectic episodes, presumably mimicking the effect of positive emotions in human patients (España et al., 2007; Clark et al., 2009). Consistent with this observation is the finding that both positive and negative olfactory stimuli (female and coyote urine) are also sufficient to induce narcoleptic episodes in male orexin peptide knockout mice (Morawska et al., 2011). These behavioral patterns indicate that orexin peptide knockouts are a particularly good model for early onset narcolepsy with cataplexy. Mutation of the human HCRT gene affecting peptide trafficking and processing is known to be associated with severe cataplexy (5–20 episodes/day) (Peyron et al., 2000).
E. Role of OX2 Receptors in the Control of Arousal, Vigilance State
Arousal responses to orexin are primarily mediated by OX2 receptors. Intracerebroventricular administration of OX-A, OX-B, or [Ala11]OX-B, a modified signaling peptide having 120-fold selectivity for OX2 receptors, similarly promote wakefulness and decrease the amount of time spent in REM and slow-wave sleep in a dose-dependent manner in rats (Hagan et al., 1999; Piper et al., 2000; Akanmu and Honda, 2005). Conversely, OX2 receptor-selective antagonists have sleep-promoting effects similar to antagonists having equal potencies for both OX1 and OX2 receptors, including attenuated active wake, increased REM and NREM sleep, and decreased latencies to NREM and REM (Dugovic et al., 2008). As might be expected, OX2 receptor knockouts display a narcoleptic phenotype similar to that of orexin peptide knockouts, including hypersomnolence, fragmented wakefulness and NREM sleep, increased active phase NREM sleep, and limited wake-to-REM transitions (Willie et al., 2003). The behavioral arrests exhibited by these mice, however, fall short of the cataplexy criteria established by Scammell et al. (2009). Although clomipramine reverses behavioral arrests exhibited by OX2 receptor knockouts, these episodes are far less frequent than those observed in Hcrt knockouts, are typically more gradual in their onset, and are preceded by quiet wakefulness rather than “emotive” behaviors such as grooming or climbing that precede abrupt behavioral arrests. These gradual arrests are characterized by EEG power spectra similar to that of NREM sleep, as opposed to θ-rich REM sleep typical of abrupt cataplectic episodes (Willie et al., 2003). As such, the behavioral arrests displayed by OX2 receptor knockouts do not seem cataplectic in nature but suggest that other signaling mechanisms may be required for these episodes.
F. Role of Orexin 1 Receptor in the Control of Vigilance State
As the only other known orexin receptor, the incomplete phenotype of OX2 receptor knockouts relative to orexin peptide knockout animals indicates that the OX1 receptor does participate in the mechanism of cataplexy. However, constitutive OX1 receptor mutants reportedly display only a mild sleep phenotype with some increase in fragmentation (Kisanuki et al., 2000). Likewise, OX1 receptor-selective antagonism with compounds of imperfect selectivity elicits little to no effect on sleep architecture (Dugovic et al., 2008) but has been reported to both increase extracellular dopamine in the PFC and to attenuate the sleep promoting effects of OX2 receptor antagonism (Dugovic et al., 2009). Given its expression on locus ceruleus neurons involved in the control of REM and its activating effect on those neurons, a role for OX1 receptor in gating transitions into REM remains possible (Bourgin et al., 2000; Ohno and Sakurai, 2008). In support of this assertion is the observation that small interfering RNA-mediated knockdown of OX1 receptor expression in locus ceruleus is associated with inappropriate increases in REM sleep during the active period of rats for up to 4 days after treatment, a time-course coincident with reduced OX1 receptor mRNA levels. Remarkably, neither wakefulness, NREM sleep, nor the qEEG power spectra of treated animals was affected, suggesting that this effect was specific for vigilance state gating and not a general effect on arousal (Chen et al., 2010).
G. Mechanisms Underlying Narcolepsy/Cataplexy
1. Hypersomnolence.
Disruption in the arousal effects of orexin are clearly mediated through deficiencies in OX2 receptor activity, most likely through histaminergic neurons of the tuberomammillary TMN. In fact, adenovirus-mediated focal expression of OX2 receptors selectively within the TMN of OX2 receptor knockout mice is sufficient to rescue the arousal deficits of these narcoleptic mutants, whereas the sleep fragmentation phenotype was unaffected, indicating that the control of vigilance state gating by orexin resides in brain nuclei exclusive to the TMN (Mochizuki et al., 2011). The hypersomnolence, sleep attacks, and decreased latency to NREM and REM sleep displayed by both OX2 receptor knockout mice (Willie et al., 2001) and OX2 receptor mutant dogs with genetically transmitted narcolepsy (Lin et al., 1999) are clear illustrations of this receptor's role in disrupted arousal pathways. The sleep promoting effects of selective OX2 receptor antagonism is associated with attenuated extracellular histamine levels in the LH (Dugovic et al., 2009), whereas histamine H1 receptor blockade with pyrilamine blocks arousal induced by OX-A (Yamanaka et al., 2002).
Dopamine also seems to modulate orexin-mediated arousal and the prevalence of hypersomnia in narcoleptic models. In orexin peptide knockouts, sleep attack prevalence was decreased with pharmacological D1 receptor activation and increased with inhibition of these receptors, whereas D2 receptor modulation had little to no effect (Burgess et al., 2010). These findings together with the observation that OX1 receptor antagonism has the potential to elevate PFC dopamine levels and attenuate OX2 receptor antagonist-induced sleep (Dugovic et al., 2009) suggests that dopaminergic signaling has the potential to modulate hypersomnolence associated with diminished OX2 receptor activity.
2. Sleep Stage Instability.
A symptom common to all of the model organisms in which components of orexin signaling are disrupted is sleep fragmentation associated with vigilance state instability (Saper et al., 2010). OX1 receptor knockouts exhibit mild sleep fragmentation whereas OX2 receptor knockouts have intermediate instability approaching that of orexin peptide knockouts or Ox/Atx transgenic animals. In the latter cases, the disruptions are more pronounced with rapid transitions between states and inappropriate wake-to-REM transitions. Orexin peptide knockout animals have also been evaluated in constant dark conditions to examine both the circadian control of sleep and sleep architecture in the absence of the masking effects of light. Although the circadian timing of sleep/wake cycles was normal, the absence of light/dark arousal cues revealed unusually rapid and random transitions between sleep states, including wake to REM and short duration bout time, suggesting behavioral state instability with a low threshold for transition (Mochizuki et al., 2004). Using a state space analysis technique, Diniz Behn et al. (2010) analyzed high-resolution quantitative EEG spectra in relation to EEG/EMG polysomographic state of orexin peptide knockout animals to track the rate of movement between sleep states on a second-by-second basis. Although most state transitions were normal relative to wild-type animals, orexin peptide knockouts seemed to experience less stability such that drifting out of wake occurred more readily, ultimately explaining the sleep fragmentation described previously. Overall, Ox mutants spent more time near the wake-to-NREM transition boundary and less time in deep NREM or θ-rich wake than wild-type counterparts. During cataplectic episodes, orexin peptide knockouts also exhibited greater θ activity than that typically observed in REM (Diniz Behn et al., 2010).
3. Cataplexy.
Many of the characteristics of cataplexy indicate that these episodes are related to a REM-like intrusion into wakefulness. Bilateral muscle atonia and a prevalence of θ qEEG power along with the circumstantial propensity for REM sleep and the dysregulation of vigilance state boundary control associated with narcolepsy substantiate this assertion (Beuckmann and Yanagisawa, 2002). However, cataplexy may not be as simple as an intrusion of REM sleep into wakefulness. At the cellular level, histaminergic neurons normally quiescent during both REM and NREM sleep are active during cataplectic episodes (John et al., 2004), which is consistent with the observation that canines and human patients maintain consciousness and are aware of their surroundings (Siegel and Boehmer, 2006). Cholinergic neurons of pedunculopontine nuclei that are normally active during wake and REM have attenuated activity during cataplexy, further indicating that this state may be distinct from REM (Thankachan et al., 2009). Muscarinic acetylcholine-mediated signaling from pedunculopontine nuclei neurons also seems to be involved, because pharmacologically induced acetylcholine activity in this region is associated with increases in behavioral arrest number without affecting mean arrest time in narcoleptic OX1 and OX2 receptor double-knockout animals (Kalogiannis et al., 2010), although the EEG characteristics of behavioral arrest episodes in these animals remain to be determined. Dopamine also seems to influence the prevalence of cataplectic attacks. Unlike hypersomnolence, which is attenuated by D1 receptor subtype activation, cataplectic attacks in orexin peptide knockouts were substantially increased with D2 activation and attenuated with antagonism of this receptor (Burgess et al., 2010).
In Ox/Atx mice, the prevalence of behavioral arrest episodes was attenuated by adenoassociated virus-mediated expression of prepro-orexin selectively in the zona incerta region of the hypothalamus (Liu et al., 2011). Remarkably, this ectopic orexin expression was not associated with a reduction in hypersomnolence, sleep attacks, or any detectable changes in sleep architecture, indicating that activity of the zona incerta has a specific role in stabilizing motor tone that is typically disrupted in cataplectic attacks. Retrograde tracer mapping found these neurons to receive inputs from the amygdala and to innervate the locus ceruleus, suggesting a possible pathway connecting emotional state with muscle paralysis associated with REM sleep (Liu et al., 2011). Together, these studies begin to round out our understanding of the role of orexin signaling in narcolepsy/cataplexy and, in so doing, its function in arousal and vigilance state.
H. Genetic versus Pharmacological Manipulation: Complementary Interpretations
Independently, experiments using mutant animals and pharmacological manipulation of orexin receptor activity are largely complementary but have distinct caveats associated with these approaches. These differences stem from the constitutive nature of gene knockouts relative to acute transient pharmacological manipulation. Constitutive gene knockouts and Ox/ATX transgenics more closely mimic narcolepsy and varying aspects of the disorder than what might be expected from pharmacological manipulation. For example, behavioral arrests have been observed in Hcrt and OX2 receptor knockouts (Chemelli et al., 1999; Willie et al., 2003) and in mice in which both orexin receptors have been mutated (Kalogiannis et al., 2011). Although it is currently unclear whether the behavioral episodes observed in the later model constitute cataplexy (Scammell et al., 2009), cataplectic episodes have not been observed in response to even high doses of dual orexin receptor antagonists across multiple species (Brisbare-Roch et al., 2007).
The fundamental difference between constitutive genetic manipulation and pharmacological perturbation is the likely development of molecular, cellular, neuronal, and/or behavioral mechanisms that compensate for the loss of a targeted gene product or orexin neuron loss. Indeed, the hypersomnolence exhibited by animals with a targeted disruption of the Hcrt gene is still punctuated by periods of wakefulness (Chemelli et al., 1999), and pharmacological orexin receptor antagonism is capable of acutely promoting sleep in wild-type animals to levels exceeding that seen in untreated mutant mice lacking both receptors (Winrow et al., 2012). Because of the likelihood of compensatory mechanisms, interpretations regarding the function of a gene product from genetically manipulated animal models should be made with this caveat in mind. In this regard, the availability of specific reagents targeting orexin receptors provides useful tools to specifically explore the function of orexin signaling within intact animals. Still, the interpretation of pharmacological perturbations depends not only on reagent specificity but also on the time of day at which they are applied, given oscillating endogenous orexin levels. Because OX-A and OX-B levels are highest during waking periods and reach a nadir during the inactive phase (Taheri et al., 2000; Zeitzer et al., 2003), the effectiveness of orexin receptor antagonists is expected to be greatest during periods of behavioral activity, a prediction confirmed experimentally (Brisbare-Roch et al., 2007; Li and Nattie, 2010; Winrow et al., 2011).
Further work combining genetic and pharmacological approaches will be particularly valuable. The most obvious experiments will examine the effectiveness of potential narcolepsy therapeutics in these knockout and Ox/ATX transgenic models of the disorder. These are anticipated to include both orexin agonists and other wake-promoting compounds such as those modulating histamine. Other studies have found orexin receptor knockouts to lack the response to orexin receptor antagonists, demonstrating their selectivity (Winrow et al., 2011, 2012). The use of small-molecule reagents specific for other neuronal targets in these animal models also has the potential to more clearly define the role of orexin in additional neurotransmitter pathways involved in modulating neurophysiology and behavior.
VI. Orexin Function beyond Sleep and Arousal
A. Central Modulation of Behavior and Physiology by Orexin Signaling
1. Feeding.
The name orexin was ascribed by Sakurai et al. (1998) in their initial report on the behavioral effects of administration of the neuropeptide in rodents to reflect the peptide's apparent effects on feeding. In these animals treated with synthetic orexin peptide, increased wakefulness accompanied by feeding behavior was interpreted to reflect an increase in appetite. It is noteworthy that many of the patents covering orexin receptor antagonists indicate that these molecules may have potential therapeutic applications for obesity and metabolic disorders. Early efforts included the characterization of orexin receptor expression and screening for orexin receptor antagonists with the aim of identifying new drugs for treating metabolic disorders (Alvaro et al., 2009, 2011; Liu, 2009). Subsequently, the focus of many investigators shifted to the impact of orexin receptor modulation for regulating sleep and wake using small-molecule antagonists. Although the wake-promoting effects of orexin and the sleep-promoting effects of orexin receptor antagonists have been widely documented, the role of this neurotransmitter in modulating feeding behavior is less established.
In multiple rodent studies, administration of exogenous OX-A has been demonstrated to increase food intake, which is frequently associated with elevated blood glucose levels. On the other hand, pretreatment with the partial OX1 SORA, SB-334867, blocks the effects of OX-A on food intake (Haynes et al., 2000; Rodgers et al., 2000, 2001; Yamada et al., 2000; Thorpe and Kotz, 2005; Thorpe et al., 2005; Yi et al., 2009). The effect of OX-A on food intake, however, is diminished or absent in aged rats, and Western blots suggest that decreased OX1 receptor levels may be responsible for the diminished feeding response (Takano et al., 2004; Kotz et al., 2005). It is noteworthy that acute treatment with SB-334867 or an anti-orexin antibody reduced levels of natural feeding, often leading to a reduction in body weight (Haynes et al., 2000, 2002; Yamada et al., 2000; Rodgers et al., 2001; Ishii et al., 2004, 2005). SB-334867 has further been observed to have anorectic action and to accelerate satiety (Haynes et al., 2000; Rodgers et al., 2001; Ishii et al., 2005). Together, these studies have suggested that orexin receptor antagonists may be useful for the treatment of obesity and eating disorders.
Gene expression studies suggest that orexin levels may predetermine eating preferences as well as respond to eating habits. For example, baseline orexin expression in the perifornical LH is higher in rats prone to overeating a high-fat diet (Morganstern et al., 2010), and expression stimulated by a high-fat diet was closely associated with elevated triglycerides (Wortley et al., 2003). Another study showed that food deprivation induced mRNA expression of transcripts encoding prepro-orexin, OX1, and OX2 receptors in the hypothalamus. In addition, food deprivation led to changes in the G-protein coupling with orexin receptors, altering the relative levels of coupling to Gq, Gs, Go, and Gi (Karteris et al., 2005). Interactions between the orexin pathway and NPY, nitric oxide, serotonin, acetylcholine, and GABA signaling mechanisms have been implicated (Dube et al., 2000; Niimi et al., 2001; Orlando et al., 2001; Farr et al., 2005; Thorpe et al., 2006; Frederick-Duus et al., 2007). Most orexin neurons are glucose-sensitive, and their activity can be modulated by the peptide hormones leptin and ghrelin (Muroya et al., 2001; Yamanaka et al., 2003; González et al., 2008; Louis et al., 2010).
Multiple studies point to the involvement of orexin signaling in reward-based feeding. Administration of orexin has been shown to affect multiple reward-seeking behaviors, including operant responding for high-fat pellets and sucrose, conditioned place preference (CPP) for food, cue-induced reinstatement of extinguished sucrose-seeking, and food-reinforced responding under both variable and progressive ratio schedules of reinforcement (Harris et al., 2005; Cason et al., 2010; Sharf et al., 2010). These studies use the partial OX1 SORA SB-334867 to suggest the involvement of OX1 receptors in reward-based feeding. Studies also suggest orexin's involvement in both dieting and overconsumption of palatable foods, once again suggesting a link to the treatment of obesity. During dieting however, OX1 receptors seem to contribute to operant self-administration but are not involved in relapse to food-seeking behaviors (Nair et al., 2008; Choi et al., 2010).
Rodent and human genetics have helped tease out the role of orexin in energy homeostasis. Orexin/ataxin-3 mice exhibit late-onset obesity despite the fact that food intake is decreased, gaining more weight than wild-type mice on a high-fat diet (Hara et al., 2001, 2005). Transgenic orexin overexpression, on the other hand, renders mice resistant to diet-induced obesity (Funato et al., 2009). Genetic studies and the administration of the OX2 receptor-selective agonist, [Ala11,d-Leu15]OX-B suggest that this receptor is involved primarily in inhibiting the obesity phenotype. In human narcolepsy, weight gain and reduced energy expenditure often accompany the increased risk of type 2 diabetes. Patients exhibiting narcolepsy with cataplexy have a higher incidence of obesity and a higher body mass index than narcoleptics without cataplexy or the general population (Honda et al., 1986; Schuld et al., 2000, 2002; Sonka et al., 2010). The direct role of orexin on feeding in these patients remains in question given the secondary effect of hypersomnolence and the reduced physical activity and energy expenditure associated with the profound phenotype of those with narcolepsy/cataplexy. In support of this idea is the observation that orexin/ataxin-3 mice exhibit reduced wakefulness in response to food deprivation relative to wild-type animals, whose change in vigilance presumably reflects a foraging drive to identify additional food sources (Yamanaka et al., 2003).
In sum, the orexin system is well situated to govern the integration of multiple pathways (metabolic, motivational, sleep/wake) required to maintain energy homeostasis and drive the feeding-sleep cycle (Rodgers et al., 2002; Sakurai, 2002; Burdakov and Alexopoulos, 2005; Rolls et al., 2010). Despite carefully controlled studies, however, many of orexin's short-term effects on feeding seem secondary to its role in promoting arousal. This is supported by the observation that OX-A does not promote feeding when administered during the normal active phase (España et al., 2002). More work will confirm this short-term effect on feeding as well as the possibility that orexin mediates the rewarding properties of food.
2. Reward Pathways and Addiction.
Orexin neurons project from the LH to the VTA and to components of the mesocorticolimbic reward pathway, including the nucleus accumbens, amygdala, and the medial PFC (Fadel and Deutch, 2002). Furthermore, orexin receptors are expressed in brain regions associated with reward pathways, including the VTA and nucleus accumbens (see Fig. 6) (Trivedi et al., 1998; Marcus et al., 2001). Lesion and intracranial self-stimulation experiments have long indicated the LH as important in brain reward function (Olds and Milner, 1954; Velley et al., 1983). Several landmark studies performed between 2003 and 2006 strongly suggest a role for orexin signaling in drug abuse and addictive reward processing. Georgescu et al. (2003) reported activation of orexin neurons in the LH and induction of orexin gene expression in response to long-term morphine administration or morphine withdrawal. Furthermore, the authors found that withdrawal symptoms were attenuated in orexin knockout mice. In 2005, Harris et al. (2005) showed that orexin neurons in the LH were strongly activated by a CPP for morphine, cocaine, or food. Activation of LH orexin cells or intra-VTA administration of OX-A was sufficient to reinstate extinguished drug seeking behavior; this reinstatement was blocked by systemic pretreatment with an OX1 SORA. That same year, Boutrel et al. (2005) reported that intracerebroventricular administration of OX-A reinstated cocaine-seeking and that OX-A elevated intracranial self-stimulation thresholds. They proposed the involvement of orexin in stress pathways linked to addiction, because OX-A reinstatement was blocked by inhibition of either the noradrenergic or the CRF system. Borgland et al. (2006) expanded upon these studies and linked orexin signaling with synaptic plasticity in VTA dopamine neurons and behavioral sensitization to cocaine. On the basis of these studies, a vast body of preclinical research has evolved around orexin signaling in addictive behaviors (Aston-Jones et al., 2010; Lawrence, 2010; Sharf et al., 2010; Zhou et al., 2011). Together, these studies have indicated the potential for blocking drug-seeking behaviors through antagonism of orexin signaling, suggesting orexin receptor antagonists as possible therapeutics for treating addiction to a variety of abusive drugs, including cocaine, amphetamine, alcohol, and morphine.
Harris and Aston-Jones (2006) were the first to formally propose the dichotomy theory of reward versus arousal. That is, they proposed that a functional heterogeneity exists among orexin neurons and their associated circuitries such that the effects of orexins on drug-seeking are mediated via two behavioral systems: 1) reward processing and 2) arousal/stress. Furthermore, these two systems show regional specificity within the hypothalamus; i.e., the LH orexin system affects drug-seeking behavior through activation of reward pathways (mostly through the VTA) and the perifornical-dorsomedial hypothalamus controls drug behavior via activation of arousal and stress pathways (Harris and Aston-Jones, 2006). Finally, the dichotomy in orexin function is likely also attributable to differences in OX1 and OX2 receptor signaling (OX1 in reward seeking and OX2 in arousal). This dichotomy of function was the focus of a more recent publication in which the authors used functional magnetic resonance imaging to show that the two receptors had distinct neuroanatomical patterns of functional inhibition. Gozzi et al. (2011) spatially monitored the modulatory effects of OX1 or OX2 receptor blockade on the regional cerebral blood flow increases produced by d-amphetamine. The study used the highly selective OX1 and OX2 SORAs GSK1059865 and JNJ10397049, respectively. The two compounds showed distinct patterns of inhibition; GSK1059865 modulated the functional response to d-amphetamine in a focal striatal pattern, whereas JNJ10397049 induced widespread attenuation of the response predominantly in the cortex. In an attempt to correlate behavior with these divergent imaging profiles, the authors investigated the effect of the compounds on sleep and cocaine-induced CPP in rats. Robust sleep effects were observed for JNJ10397049 but not GSK1059865, whereas GSK1059865 dose-dependently reduced expression of cocaine-induced CPP (Gozzi et al., 2011). In sum, the authors proposed that OX1 receptor activity in the striatum is specifically involved in drug-related reward behaviors, whereas cortical activity of OX2 receptors is essential for regulating sleep/wake.
The majority of in vivo experiments investigating the role of orexin in addiction have used the partially selective OX1 SORA SB-334867 to suggest that OX1 receptors are involved in drug-seeking behaviors. That said, the high doses of SB-334867 used in some of these studies and the compound's limited specificity suggest that blockade of OX2 receptors may in fact have contributed to the results of these studies (Scammell and Winrow, 2011; Shoblock et al., 2011). Indeed, more recent experiments with OX2 SORAs JNJ10397049 and (2S)-1-(3,4-dihydro- 6,7-dimethoxy- 2(1H)-isoquinolinyl)-3,3-dimethyl-2-[(4-pyridinylmethyl)amino]-1-butanone (TCSOX229) argue for a direct role of OX2 receptor signaling in drug behaviors involving ethanol and morphine (Li et al., 2011; Shoblock et al., 2011). Shoblock et al. (2011) expound upon the potential for an OX2 SORA that might provide treatment for addiction and also serve to treat the comorbid insomnia that often accompanies drug use and contributes to relapse.
Patients with narcolepsy are frequently clinically treated with addictive drugs yet rarely demonstrate addictive behaviors (Nishino and Mignot, 1997). Dimitrova et al. (2011) tested the hypothesis that persons with orexin deficiency exhibit a diminished tendency toward addictive behaviors. They found similar risk-taking behavior in narcoleptics relative to control groups and no significant differences in substance or alcohol abuse. Although these results counter the hypothesis that orexin deficiency affects reward processing, these investigators suggested that orexin-deficient subjects use a different neurological mechanism for these behaviors (Dimitrova et al., 2011). In fact, a recent functional magnetic resonance imaging study suggests altered activity in brain reward circuits in patients exhibiting narcolepsy with cataplexy (Ponz et al., 2010).
The aforementioned studies indicate that orexins and orexin receptors play a role in many models of addiction but that results are variable depending upon the particular addictive substance, animal model, and orexin receptor antagonist used; moreover, results are probably mediated by distinct neuronal populations and circuits. Further studies will more clearly define the role of orexin in these mechanisms, as well as its possible interconnections with arousal pathways.
3. Anxiety.
Much work has been done preclinically in rodent behavioral models to elucidate the role of orexin signaling in anxiety. OX-A was shown to have an anxiogenic effect in the mouse light-dark exploration test and the mouse and rat elevated plus-maze test (Suzuki et al., 2005). The authors of the preclinical study suggest that CRF, 5-HT, and NPY may be involved in mediating the anxiogenic effects of OX-A. Indeed, others have substantiated the involvement of these systems in mediating stress-related orexinergic effects by showing a reduction in orexin-mediated stress behaviors by pretreatment with the CRF antagonist α-helical CRF (Ida et al., 2000), an altered sleep response to restraint stress in serotonin transporter knockout mice (Rachalski et al., 2009), and an inhibition of orexin-induced activation of the hypothalamic-pituitary axis by an NPY antagonist (Jászberényi et al., 2001). Li et al. (2010) investigated the effects of administration of OX-A and OX-B into the paraventricular nucleus of the thalamus, a site characterized by orexin-containing fibers and known to innervate forebrain areas associated with fear and anxiety. Administration of OX-A and OX-B caused an anxiogenic response in the rat elevated plus-maze, whereas κ-opioid or CRF receptor antagonists attenuated the effects of OX-A (Li et al., 2010). Furthermore, endogenous orexins may be involved in producing anxiety in these models, because the OX2 SORA TCSOX229, but not the partial OX1 SORA SB-334867, reduced the anxiogenic response in a foot shock-induced anxiety model. The use of elevated plus-maze and light-dark exploration in the Syrian golden hamster showed an anxiogenic response for orexins in the amygdala and an interaction with GABAergic signals (Avolio et al., 2011). In fact, decreased GABA levels have been reported in those suffering from panic disorder (Goddard et al., 2001). In a rat model of sodium lactate-induced panic attacks, activation of orexin-synthesizing neurons is required for the panic response in panic-prone rats. Accordingly, treatment with Hcrt small interfering RNA or an OX1 antagonist (SB-334867 or SB-408124) blocks the behavioral and cardiovascular effects elicited by sodium lactate challenge associated with the panicked state (Johnson et al., 2010).
In contrast to the reports of anxiogenic responses mediated by orexin, rat studies using a startle paradigm ascribe an anxiolytic effect to the neuropeptides (Singareddy et al., 2006). Exogenous administration of OX-A or OX-B produced an anxiolytic effect in situations reflective of unconditioned, but not conditioned, anxiety. Suzuki et al. suggest that differences in the range of response to orexin (anxiogenic versus anxiolytic) may be attributable to the dose of orexin, interacting signaling pathways, and perhaps differential roles of the orexin receptor subtypes in mediating anxiety-related behaviors (Suzuki et al., 2005).
Clinically, several recent studies have reported on the link between orexinergic signaling and anxiety and stress disorders. In a study of 10 male veterans with post-traumatic stress disorder (PTSD) and 10 healthy men, it was reported that CSF and plasma OX-A levels are significantly lower in the patients suffering from PTSD; furthermore, levels of OX-A in the CSF negatively correlate with the level of severity of the disorder (Strawn et al., 2010). In contrast, Johnson et al. (2010) report higher CSF levels of orexin in subjects exhibiting panic anxiety symptoms compared with subjects without anxiety. These conflicting reports may reflect the differential responses to orexin as described in preclinical anxiety studies, the different forms of anxiety highlighted in the two clinical studies, or may be secondary to disrupted sleep/wake cycles observed in patients with these illnesses. Fortuyn et al. (2010) studied the prevalence of anxiety and mood disorders in narcolepsy. They found that although mood disorders are not increased among narcoleptics, anxiety behaviors, including panic attacks and social phobias, are present in more than half of narcoleptic subjects, 35% of whom are also diagnosed with anxiety disorder. Another more recent study, however, found that rates of both depression and anxiety are higher in narcoleptic subjects (Dimitrova et al., 2011).
4. Depression/Mood.
The activation of orexin-secreting and orexin receptor-expressing neurons in response to stress and orexin activation of stress-related systems, including norepinephrine, dopamine, and CRF, may point to a possible role in PTSD and depression (Berridge et al., 2010). OX-A administration was reported to reduce immobility in the rat forced-swim test and increased hippocampal neurogenesis, with pretreatment of SB-334867 blocking these effects (Ito et al., 2008). Lutter et al. (2008) showed that intact orexin signaling was necessary for the efficacy of caloric restriction in a mouse model of depression. After caloric restriction, wild-type mice show less immobility in the forced swim test compared with orexin peptide knockout mice. Likewise, in a social defeat model, caloric restriction is efficacious in wild-type mice but not in orexin peptide knockout mice. Behavioral profiling studies conducted on mice lacking OX1 and OX2 receptors as well as in wild-type animals treated with the partially selective OX1 receptor antagonist SB-334867 suggest that a balance of orexin receptor activity may differentially affect behavioral despair activities (Scott et al., 2011). In tail suspension and forced swim test models of despair behavior, OX1 receptor-null mice displayed a significant reduction in immobility, suggesting an antidepressant phenotype, and similar results were observed in wild-type animals treated with SB-334867. On the other hand, OX2 receptor-null mice displayed an increase in behavioral despair (Scott et al., 2011). Although this study detected no genotype-dependent changes in anxiety measures, these apparent counter-acting orexin receptor activities are reminiscent of that suggested for orexin signaling in anxiety described above.
Clinical reports on orexin levels in depression are varied across specific affective disorders. In mild-to-moderately depressed subjects, CSF OX-A diurnal variation seems dampened, overall levels seeming higher in patients and modest but significant reductions after treatment with the antidepressant sertraline, a serotonin-reuptake inhibitor (Salomon et al., 2003). In contrast, suicidal patients with major depressive disorder had lower OX-A levels in CSF compared with other suicidal patients (Brundin et al., 2007). A more recent study of patients with mania failed to show an association between severity of disease and levels of orexin. Evaluation of OX-A in CSF from five patients exhibiting manic episodes failed to reveal significant differences compared with age-matched patients with major depressive disorder or healthy control subjects without any psychiatric or neurological disorder (Schmidt et al., 2010). Overall, these studies indicate a potential role for orexin signaling in preclinical depression models; however, additional studies will be needed to better understand the contributions of orexin signaling to mood and anxiety.
B. Orexin Influences on Peripheral Physiology
A growing body of evidence indicates that orexin signaling has the capacity to influence peripheral physiology. Many of these effects seem to be mediated by hypothalamic orexinergic neuron activity, modulating either autonomic nervous system tone or perhaps secondary to the peptides' effects on arousal and vigilance state. Clear demonstrations of the physiological role of orexin receptor activity in peripheral tissues are scarce, but the emergence of increasingly selective orexin receptor antagonists, however, should provide useful reagents to resolve this issue, because their concentrations are typically 10- to 100-fold higher in the periphery relative to the brain after administration. The potential roles for orexin signaling in the periphery, and whether these effects might be autonomic or secondary to its behavioral effects, remain to be determined.
1. Metabolism and Gastrointestinal Motility.
The vast body of research related to orexins and feeding behaviors has prompted a closer look at the role of orexin signaling in feeding-related responses in the periphery. Orexin ligand and receptor expression in the gastrointestinal tract have been investigated in rat, human, dog, and a limited number of other species. Orexin-like immunoreactivity has been observed throughout the gastrointestinal tract and the pancreas in various cell types (e.g., neurons, nerve fibers, smooth muscle, endocrine cells) and is often colocalized with signaling molecules such as vasoactive intestinal peptide, neuronal nitric-oxide synthase, substance P, insulin, or gastrin, although a full understanding of the selectivity of some antibodies has been a challenge (Kirchgessner, 2002; Nakabayashi et al., 2003; Voisin et al., 2003; Heinonen et al., 2008; Dall'Aglio et al., 2008, 2009, 2010). Orexin-containing projections extend from the lateral hypothalamus to the dorsal motor nucleus of the vagus (DMV) and project to target peripheral tissues including stomach, intestine, and pancreas. In a study using whole-cell patch-clamp recordings, it was shown that responsiveness to the orexins is organized in a viscerotopical manner, DMV neurons projecting to gastric fundus and corpus being much more responsive than those projecting to the more distal duodenum and caecum (Grabauskas and Moises, 2003).
Orexin signaling is hypothesized to be a trigger of the gastrointestinal secretion and motility associated with the cephalic phase of feeding in response to the sight, smell, taste, or anticipation of food (Takahashi et al., 1999; Okumura and Takakusaki, 2008). In rats, intracisternal injection of OX-A, but neither intracisternal OX-B nor intraperitoneal OX-A, stimulated gastric acid secretion (Takahashi et al., 1999). This central action was blocked by atropine or vagotomy, suggesting that orexin is affecting the gastrointestinal tract from the brain via the vagal system. Furthermore, the effects of exogenously added or endogenous OX-A on gastric acid secretion were inhibited by the orexin receptor antagonist SB-334867 (Ehrström et al., 2005b; Yamada et al., 2005).
Intestinal bicarbonate secretion is considered another key feature of the cephalic phase of feeding. In contrast to gastric secretion, however, bicarbonate secretion in the duodenum is peripherally rather than centrally mediated and has been shown in rats to be affected by nutritional status (Bengtsson et al., 2007). At the cellular level, fasting was shown to reduce the expression of OX1 receptor mRNA in duodenal enterocytes and to inhibit OX-A induced calcium mobilization (Bengtsson et al., 2009). It has been widely demonstrated in rodent and guinea pig that OX-A and OX-B modulate gastrointestinal motility (Kirchgessner and Liu, 1999; Kobashi et al., 2002; Krowicki et al., 2002; Näslund et al., 2002; Ehrström et al., 2003; Katayama et al., 2003; Bülbül et al., 2010a,b). The effects of orexins vary depending upon location within the gastrointestinal tract and represent a combination of central and peripheral actions (Baccari, 2010). Experiments in rat and one clinical report suggest effects on gastric emptying rate as well (Ehrström et al., 2005a,b; Bülbül et al., 2010a).
Intracerebroventricular injection of OX-A stimulated pancreatic exocrine secretion, demonstrating that in addition to acting on gastric acid secretion, OX-A in the brain modulates rat pancreatic fluid output (Miyasaka et al., 2002). Data suggest that glucose-sensing neurons in the lateral hypothalamus release orexin, which acts on neurons of the DMV to modulate pancreatic vagal pathways (Wu et al., 2004). Studies linking orexin to glucose homeostasis suggest that the orexin system is poised to connect critical physiological systems including metabolism, goal-oriented (reward) behaviors, and arousal (Tsujino and Sakurai, 2009; Tsuneki et al., 2010).
Given its critical function in controlling arousal and sleep, and its presumed role in maintaining energy homeostasis, orexin signaling may be expected to respond to changes in plasma pH associated with fluctuations in energy expenditure. Indeed, changes in evoked and spontaneous firing activity of orexin neurons in organotypic culture are observed in response to changes in extracellular pH (Williams et al., 2007; Gestreau et al., 2008). The role of orexin in modulating pulmonary changes in response to plasma hypoxia and hypercapnia, however, is less clear. Observations of ventilatory responses to these conditions in orexin peptide knockouts and after central administration of orexin peptides has been mixed; some studies have found an involvement of orexin in mediating increased ventilatory responses to hypercapnia (Deng et al., 2007; Nakamura et al., 2007; Dias et al., 2009, 2010; Nattie and Li, 2010), decreased pulmonary activity to hypoxia (Deng et al., 2007; Nakamura et al., 2007), or no detectable response to either condition (Zhang et al., 2005). Conversely, a study of patients with narcolepsy-cataplexy detected attenuated responses to hypoxia but not hypercapnia (Han et al., 2010), which is exactly opposite that found in rodent models. Clearly, more work needs to be done to confirm a role for orexin in mediating ventilatory responses to changing blood gas levels as opposed to controlling sleep/wake cycles to maintain energy homeostasis.
2. Potential Roles in Nociception/Pain.
Orexinergic neurons from the hypothalamus project to numerous supraspinal sites involved in the modulation of pain, including the thalamus, DR, LC, reticular formation, periaqueductal gray, and the trigeminal nucleus caudalis (Peyron et al., 1998). OX-A and OX-B fibers have been identified in the rat spinal cord, and orexinergic projections from the hypothalamus have been shown in rat, mouse, and human to innervate multiple layers of the spinal dorsal horn (van den Pol, 1999; Date et al., 2000). There, they terminate in laminae I and II, the location of peripheral sensory afferent terminals that synapse onto central nociceptive neurons. Despite differential OX1 and OX2 receptor expression, both receptors are present in the aforementioned supraspinal sites involved in descending nociceptive modulation (Marcus et al., 2001). OX-A and OX1 receptor immunoreactivity are localized to the superficial laminae of the spinal dorsal horn and in the dorsal root ganglion (DRG) neurons of peripheral sensory afferents (Bingham et al., 2001; Hervieu et al., 2001).
Work in multiple pain models suggests that supraspinal orexins may be involved in the modulation of pain. Intracerebroventricular administration of OX-A produces an antinociceptive effect in multiple rodent pain models, including acute, chemical, inflammatory, and neuropathic pain models. The antinociceptive effect is blocked by administration of SB-334867, suggesting the effects may be mediated by OX1 receptors. In fact, OX-B is less potent than OX-A in attenuating the pain response in these models (Bingham et al., 2001; Yamamoto et al., 2003a; Mobarakeh et al., 2005b). OX-A may modulate supraspinal histamine release, because the effects of OX-A in acute and chemical pain models are enhanced in H1 or H2 receptor knockout mice or with the administration of H1 or H2 antagonists (Mobarakeh et al., 2005a).
In addition, OX-A and OX-B have a functional role in spinal sensory transmission, according to in vivo and in vitro electrophysiological studies, respectively (Grudt et al., 2002; Peng et al., 2008). OX-B may elicit its effect by facilitating glycine release presynaptically. Behavioral studies support these electrophysiological findings, because intrathecal administration of OX-A and, to a lesser extent, OX-B inhibits withdrawal responses and spontaneous nociceptive behaviors in acute, chemical, inflammatory, neuropathic, and postsurgical pain models (Yamamoto et al., 2002, 2003a,b; Cheng et al., 2003; Kajiyama et al., 2005; Mobarakeh et al., 2005a; Jeong and Holden, 2009). The antinociceptive effects of the orexins are blocked by intrathecally administered SB-334867, once again suggesting that pain modulation may be mediated through OX1 receptors.
In a few instances, administration of SB-334867 alone enhanced sensitivity in the in vivo models, supporting the idea of enhanced or tonic OX1 receptor-mediated inhibitory tone during pain (Bingham et al., 2001; Cheng et al., 2003; Jeong and Holden, 2009). Consistent with this hypothesis of orexin-mediated inhibitory tone, orexin peptide knockout mice show enhanced thermal hypersensitivity in an inflammatory pain model (Watanabe et al., 2005). Furthermore, knockout mice as well as orexin/ataxin transgenic mice, with degeneration and dysfunction of orexinergic neurons, show attenuation of stress-induced analgesia (Xie et al., 2008).
Although the role of the orexins and their receptors in the periphery has not been extensively studied, two studies point to a role for OX1 receptors on peripheral DRG neuron function. Application of OX-A to cultured DRG neurons increases action potentials and levels of intracellular calcium. These effects are blocked by SB-334867 and by the protein kinase C inhibitor chelerythrine, suggesting that peripheral sensory transmission may be modulated by activation of OX1 receptors and subsequent protein kinase C-dependent calcium signaling (Yan et al., 2008; Ozcan et al., 2010).
Orexin signaling in the trigeminal nucleus caudalis seems to modulate nociceptive transmission in the context of facial pain accompanying headache and migraine. OX-A inhibits spontaneous and stimulus-evoked responses in trigeminal nucleus caudalis neurons; antagonist treatment suggests the effect might be mediated via OX1 receptors. On the other hand, OX-B facilitates these same responses suggesting an involvement of OX2 as well (Bartsch et al., 2004; Holland et al., 2005, 2006). In a rat model of dural vasodilation thought to be representative of migraine pathophysiology, intravenous administration of OX-A but not OX-B resulted in the inhibition of vasodilation, and pretreatment with SB-334867 blocked this effect (Holland et al., 2005). Increased levels of OX-A have been reported in the CSF of chronic migraine sufferers, although the significance of this finding is currently unknown (Sarchielli et al., 2008). Clinical studies have identified polymorphisms within the orexin receptor genes that are associated with cluster headache and migraine. A 1246G→A polymorphism in HCRTR2 is significantly associated with cluster headaches but not with treatment response, whereas the same polymorphism is not associated with migraine (Rainero et al., 2004, 2007; Schürks et al., 2006, 2007a,b; Pinessi et al., 2007). More recently, a 1222G→A polymorphism within the HCRTR1 gene has been found to be associated with an increased risk of migraine without aura (Rainero et al., 2011). Further investigations are needed, however, to determine whether these genetic correlations reflect variation around these chromosomal loci or real expression or biochemical changes affecting orexin receptor activity and or signaling function.
3. Influence on Cardiovascular Physiology.
Preclinical studies, mostly in rat, have focused on the cardiovascular effects of exogenously added orexins. In the majority of studies, central administration (intracerebroventricular or intracisternal) of extraphysiological doses of orexin peptides, particularly OX-A, acts to increase blood pressure and heart rate, although differences in experimental paradigms have also found unchanged or decreased cardiovascular parameters with orexin administration (Samson et al., 1999, 2007; Shirasaka et al., 1999; Chen et al., 2000; White et al., 2006). The partial OX1 receptor antagonists SB-408124 and SB-334867 and the OX2 receptor antagonists [Ala11,d-Leu15]OX-B and TCSOX229 either partially or fully blocked the effects of exogenous orexins, suggesting that both receptor subtypes are involved (White et al., 2006; Samson et al., 2007; Huang et al., 2010). Direct administration to specific brain regions evoked either increases or decreases in blood pressure and heart rate, suggesting that stimulation of different structures may indeed differentially modulate cardiovascular function; however, differences in protocol, dose, and state of anesthesia probably also contribute to the disparate results (Chen et al., 2000; Antunes et al., 2001; Machado et al., 2002; Sato-Suzuki et al., 2002; Smith et al., 2002, 2007; Ciriello and de Oliveira, 2003; Ciriello et al., 2003; de Oliveira et al., 2003; Shahid et al., 2011). In fact, the doses used in several of the studies is relatively high compared with the endogenous OX-A levels (0.2–0.4 pmol/ml) in rat CSF (Fujiki et al., 2001), and so the relevance to the role of intrinsic orexins in cardiovascular function is unknown. More relevant to the safety profile of orexin receptor antagonists in the clinic is the finding that central administration of the aforementioned OX1 or OX2 SORAs elicited no changes in heart rate or blood pressure when administered alone in preclinical models (Hirota et al., 2003; White et al., 2006; Samson et al., 2007; Huang et al., 2010; Shahid et al., 2011). Furthermore, preclinical studies in rats and dogs showed no effects on heart rate and blood pressure with oral administration of the dual orexin receptor antagonist, almorexant (ACT-078573) (Brisbare-Roch et al., 2007; Furlong et al., 2009). Oral ascending single-dose and multiple-day dosing of almorexant in the clinic has also caused no changes in heart rate or blood pressure (Brisbare-Roch et al., 2007; Hoever et al., 2010).
The effects of genetic manipulation on cardiovascular function have been studied in both orexin peptide knockout mice and orexin neuron-ablated orexin/ataxin-3 transgenic mice and rats, leading to a conflicting data set regarding the effects on heart rate and blood pressure (Kayaba et al., 2003; Schwimmer et al., 2006; Zhang et al., 2006; Schwimmer et al., 2010; Bastianini et al., 2011). The relevance of either of these genetic models to the pharmacological antagonism of orexin receptors in humans is unknown.
VII. Conclusion
Relative to the current therapies, orexin receptor antagonism represents a novel mechanism for the treatment of insomnia, and provides potential advantages over the current standard of care, which includes “z-drugs” (zolpidem, zaleplon, zopiclone, eszopiclone) that interact with the allosteric site of GABAA receptors. Although they are not benzodiazepines themselves, z-drugs interact with the benzodiazepine binding site on these receptor subunits with improved GABAA subtype specificity. GABAA receptors are inhibitory ligand-gated chloride channels for which conductance is potentiated by z-drugs to provide an inhibitory influence on postsynaptic activity and ultimately CNS depression in areas in which these receptors are expressed (Costa and Guidotti, 1979; Sullivan and Guilleminault, 2009). GABAA receptors exhibit wide expression and function in a number of pathways, including those associated with arousal, anxiety, psychomotor tone, and cognition. As such, z-drugs have the potential to affect behavior and physiology beyond sleep (Ashton, 1994; Hoque and Chesson, 2009). Whereas z-drugs attenuate sleep latency and promote NREM sleep, they also suppress REM and slow-wave components of normal sleep (Lancel, 1999; Bettica et al., 2012). In contrast, sleep architecture induced by DORAs is characterized by increased mean NREM and REM time (Brisbare-Roch et al., 2007; Winrow et al., 2011, 2012; Bettica et al., 2012). The most salient effect of zolpidem in rats is the suppression of both the mean time spent in REM sleep and the number of REM sleep bouts, with comparatively slight reductions in active wake time (Renger et al., 2004). On the other hand, dual orexin receptor antagonists such as suvorexant, MK-6096, DORA-22, SB-649868, and almorexant promote sleep that includes increased NREM and REM sleep in multiple species including humans (Brisbare-Roch et al., 2007; Di Fabio et al., 2011; Winrow et al., 2011, 2012; Bettica et al., 2012). GABAA receptor-induced sleep is also associated with significant impairment of locomotor coordination not seen with DORAs, even when administered at 10-fold above its effective dose (Steiner et al., 2011).
The pharmacology associated with orexin receptors is relatively simple compared with GABAA receptors, which are composed of five subunits arranged in a heteromultimeric complex. As such, the myriad effects potentially elicited by GABAA receptor modulation depend upon subtype specificity, where targeting the α1 subtype has sedative and anticonvulsive effects but is also associated with amnesia and dependence. The α2, α3, and α5 subtypes seem differentially involved in anxiolysis, muscle relaxation, and amnesia (Nutt and Stahl, 2010; Tan et al., 2011). GABAA-mediated amnesia may be an underlying mechanism for anxiolytic and antidepressant effects, because loss of memory for unpleasant events may actually be favorable for these psychiatric outcomes. Amnesia may also underlie the rare side effects of walking, eating, and driving while asleep observed in human patients taking zolpidem (Hoque and Chesson, 2009; Tan et al., 2011). In contrast, the orexin receptor antagonist almorexant has no impact on spatial learning and memory tasks or avoidance retention in rodent models at 300 mg/kg, which is 10-fold above the effective dose required to induce sleep (Dietrich and Jenck, 2010).
Continued clinical investigation will be required to understand the advantages and potential shortcomings of orexin receptor antagonism relative to z-drugs. Parameters to be monitored include sleep-stage dysregulation, next-day sleepiness, and cognitive performance and other physiological responses secondary to the modulation of sleep/wake cycles. Preclinical genetic models mimicking human narcolepsy with chronic constitutive loss of orexin signaling suggest that orexin receptor antagonism may be associated with deficiencies in sleep-stage regulation and active phase cataplexy. As indicated in section V.H, however, effects observed in constitutive knockout models are not necessarily equal to those induced by transient pharmacological manipulation. Nevertheless, cataplexy in response to orexin antagonism is a concern to be closely monitored in both the clinic and in preclinical models. Next-day sleepiness and impaired cognitive impairment are not expected for orexin antagonists but are nonetheless monitored closely in clinical trials. Short-acting orexin receptor antagonists administered before the onset of rest, however, are likely to avoid most all of these potential issues and perhaps even improve some of these measures by virtue of their ability to improve sleep maintenance.
Compared with the current standard of care, the role of orexin signaling in the control of arousal and vigilance state suggests that orexin receptor antagonism represents a selective mechanism capable of effectively promoting sleep associated with increases in both NREM and REM sleep. Orexin signaling is both sufficient to induce arousal and necessary for the normal maintenance of vigilance state (España and Scammell, 2011). Orexinergic neurons are restricted to the lateral hypothalamus and selectively project to arousal-promoting histaminergic neurons of the TMN as well as brainstem nuclei involved in sleep/wake control (Trivedi et al., 1998; Marcus et al., 2001). As an arousal signal, orexin neurons have daily oscillations in activity that give rise to accumulating peptide levels during waking hours and fall silent during the normal inactive period (Taheri et al., 2000; Zeitzer et al., 2003). As such, orexin receptor antagonists have maximal effects late in the active period, precisely at the most therapeutically relevant time, but have little to no effect during the inactive phase (Brisbare-Roch et al., 2007; Li and Nattie, 2010; Winrow et al., 2011). As described in the latter sections of this review, orexin signaling has the potential to affect physiology and behavior beyond the regulation of sleep. How much of this function (e.g., feeding, metabolism, cardiovascular physiology) is a secondary consequence of orexin-mediated arousal relative to direct mechanisms is a matter of ongoing debate and remains to be determined. Taken together, orexin antagonism represents a novel and selective mechanism for the therapeutic treatment of insomnia as well as offering potential opportunities for alternative indications.
Acknowledgments
All authors are employed by Merck Research Laboratories and receive salary and research support from Merck and Co., Inc., and potentially own stock in the company. We are grateful to Cathy Decherney for the collection of patent and literature information as well as Stephanie Born, Joseph Lynch, Douglas MacNeil, Duane Reiss, Anthony Roecker, Shawn Stachel, Mark Urban, and Jason Uslaner, all of Merck Research Laboratories, for helpful discussions of specific areas regarding orexin function.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Gotter, Webber, Coleman, Renger, and Winrow.
Footnotes
A.L.G. and A.L.W. contributed equally to this work.
This article is available online at http://pharmrev.aspetjournals.org.
↵1 Abbreviations:
- 5-HT
- 5-hydroxytryptamine (serotonin)
- CGS21680
- 2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine
- CPP
- conditioned place preference
- CRF
- corticotropin-releasing factor
- CSF
- cerebrospinal fluid
- DMH
- dorsomedial nucleus of the hypothalamus
- DMV
- dorsal motor nucleus of the vagus nerve
- DORA
- dual orexin receptor antagonist
- DR
- dorsal raphe
- DRG
- dorsal root ganglion
- EMPA
- N-ethyl-2-[(6-methoxy-pyridin-3-yl)-(toluene-2-sulfonyl)-amino]-N-pyridin-3-ylmethyl-acetamide;GSK,GlaxoSmithKline
- GSK1059865
- 5-bromo-N-[(2S,5S)-1-(3-fluoro-2-methoxybenzoyl)-5-methylpiperidin-2-yl]methyl-pyridin-2-amine
- HCRT
- hypocretin gene
- HCRTR1
- hypocretin (orexin) receptor 1 gene
- HCRTR2
- hypocretin (orexin) receptor 2 gene
- IP3
- inositol-1,4,5-trisphosphate
- JNJ-10397049
- [1-(2,4-dibromophenyl)-3-[(4S,5S)-2,2-dimethyl-4-phenyl-1,3-dioxan-5-yl] urea
- LC
- locus ceruleus
- LDT
- laterodorsal tegmental nuclei
- LH
- lateral hypothalamus
- LPS
- latency to persistent sleep
- MK-4305
- suvorexant
- MK-6096
- [2(R,5R)-5-{[(5-fluoropyridin-2-yl)oxy]methyl}-2-methylpiperidin-1-yl][5-methyl-2-(pyrimidin-2-yl)phenyl]methanone
- nNOS
- neuronal nitric-oxide synthase
- NPFF
- neuropeptide FF
- NPY
- neuropeptide Y
- NREM
- non–rapid eye movement
- OBPt-9
- N-benzyl-N-(3,4-dimethoxybenzyl)glycyl-N2-(1-phenylethyl)glycinamide
- OX
- orexin
- Ox/Atx
- transgenic orexinergic neuron ablation mutant
- PFC
- prefrontal cortex
- PLC
- phospholipase C
- PPT
- pedunculopontine tegmental nuclei
- PTSD
- post-traumatic stress disorder
- qEEG
- quantitative electroencephalography
- QRFP receptor
- QRF peptide receptor (GPR103)
- REM
- rapid eye movement
- SB-334867
- 1-(2-methylbenzoxazol-6-yl)-3-[1,5]naphthyridin-4-yl urea
- SB-408124
- 1-(6,8-difluoro-2-methyl-quinolin-4-yl)-3-(4-dimethylamino-phenyl)-urea
- SB-649868
- N-[[(2S)-1-[[5-(4-fluorophenyl)-2-methyl-4-thiazolyl]carbonyl]-2-piperidinyl]methyl]-4-benzofurancarboxamide
- SCN
- suprachiasmatic nuclei
- SORA
- selective OX1 receptor and OX2 receptor antagonist
- TCSOX229
- (2S)-1-(3,4-dihydro- 6,7-dimethoxy- 2(1H)-isoquinolinyl)-3,3-dimethyl-2-[(4-pyridinylmethyl)amino]-1-butanone
- TMN
- tuberomammillary nuclei
- VLPO
- ventrolateral preoptic area
- VTA
- ventral tegmental area
- WASO
- wake after sleep onset.
- © 2012 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
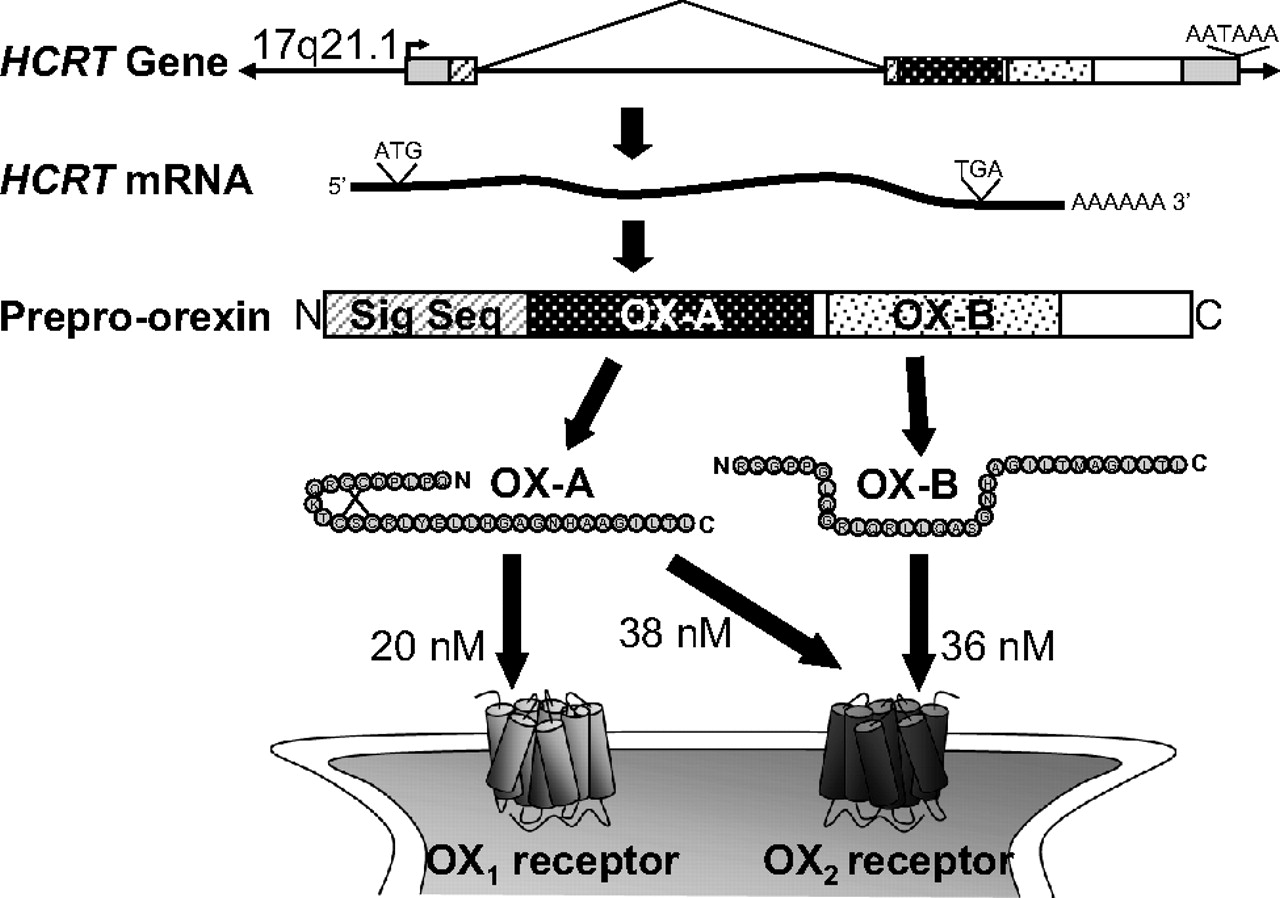{kind=link}
{kind=link}
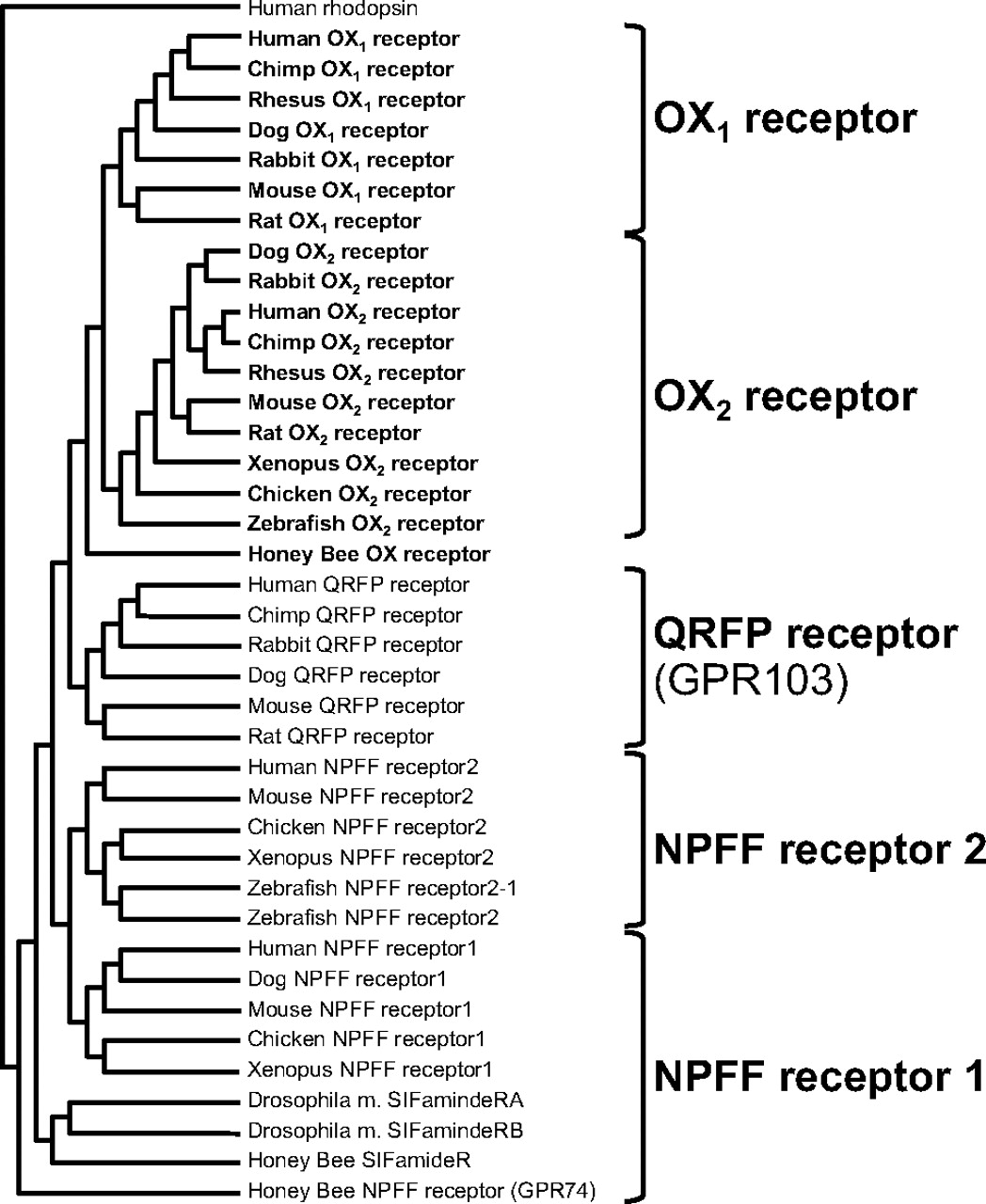{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Discovery and Nomenclature of Orexin Signaling Components
- III. Orexin Receptor Reagents and Potential Therapeutics
- IV. Orexin Neurons Are a Key Component of Pathways Regulating Sleep
- V. Genetic and Pharmacological Dissection of Orexin-Mediated Arousal
- VI. Orexin Function beyond Sleep and Arousal
- VII. Conclusion
- Acknowledgments
- Authorship Contributions
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters